

Abstract
Growth hormone (GH) signaling is essential for postnatal linear bone growth, but the relative importance of GHs actions on the liver and/or growth plate cartilage remains unclear. The importance of liver derived insulin like‐growth factor‐1 (IGF‐1) for endochondral growth has recently been challenged. Here, we investigate linear growth in Suppressor of Cytokine Signaling‐2 (SOCS2) knockout mice, which have enhanced growth despite normal systemic GH/IGF‐1 levels. Wild‐type embryonic ex vivo metatarsals failed to exhibit increased linear growth in response to GH, but displayed increased Socs2 transcript levels (P < 0.01). In the absence of SOCS2, GH treatment enhanced metatarsal linear growth over a 12 day period. Despite this increase, IGF‐1 transcript and protein levels were not increased in response to GH. In accordance with these data, IGF‐1 levels were unchanged in GH‐challenged postnatal Socs2‐/‐ conditioned medium despite metatarsals showing enhanced linear growth. Growth‐plate Igf1 mRNA levels were not elevated in juvenile Socs2‐/‐ mice. GH did however elevate IGF‐binding protein 3 levels in conditioned medium from GH challenged metatarsals and this was more apparent in Socs2‐/‐ metatarsals. GH did not enhance the growth of Socs2‐/‐ metatarsals when the IGF receptor was inhibited, suggesting that IGF receptor mediated mechanisms are required. IGF‐2 may be responsible as IGF‐2 promoted metatarsal growth and Igf2 expression was elevated in Socs2‐/‐ (but not WT) metatarsals in response to GH. These studies emphasise the critical importance of SOCS2 in regulating GHs ability to promote bone growth. Also, GH appears to act directly on the metatarsals of Socs2‐/‐ mice, promoting growth via a mechanism that is independent of IGF‐1. J. Cell. Physiol. 9999: 2796–2806, 2015. © 2015 Wiley Periodicals, Inc.
The anabolic role of growth hormone (GH) in long bones is well accepted. The relative contributions of GH acting on the liver (increasing growth promoting endocrine factors) or directly (on growth plate cartilage) however remain unclear (Lupu et al., 2001). It is likely that both these modes of GH action function in a highly coordinated manner to regulate growth plate function and linear bone growth. GH increases insulin like growth factor‐1 (IGF‐1) production in a number of tissues. Specifically, GH acting on the liver results in an increase in circulating IGF‐1 which functions in an endocrine manner (Sjogren et al., 1999; Yakar et al., 1999). GH induced IGF‐1 production within the growth plate cartilage functions in an autocrine/paracrine manner. Furthermore, GH may also act on the growth plate via IGF‐1 independent mechanisms (Gevers et al., 2002a; Wang et al., 2004).
As expected, studies with Snell (dw/dw) and Ames (df/df) hypopituitary dwarf mice reveal a reduction in body weight and growth retardation. Mice with global inactivated GHR (ghr), GH‐ releasing hormone receptor (ghrhr), IGF‐1 (Igf1), IGF‐1 receptor (IGF‐1R; Igf1r), and insulin receptor substrate‐1 (Irs‐1) have a similar phenotype (Sinha et al., 1975; Smeets and Vanbuuloffers, 1983,1983; Li et al., 1990; Sornson et al., 1996; Wang et al., 2004).
The importance of circulating IGF‐1 on linear growth was challenged by two independent studies. Liver‐specific IGF‐1 deficient (LID) mice have a significant reduction of circulating IGF‐1, but body weight and bone length were essentially normal (Sjogren et al., 1999; Yakar et al., 1999). Furthermore, a phenotypic comparison of LID, acid labile subunit knockout (ALSKO), insulin like growth factor binding protein (IGFBP)‐3 knockout (BP3KO), and triply deficient LID/ALSKO/BP3KO mice indicated that while all had decreased serum IGF‐1 levels this did not predict linear growth potential (Yakar et al., 2009). Compared to WT mice, ALSKO mice (60% reduction in serum IGF‐1) were 8% shorter, whereas BP3KO mice (40% reduction in serum IGF‐1) were 5% longer, and LID mice (80% reduction in serum IGF‐1) were of equal length. Most strikingly, despite virtually undetectable circulating IGF‐1 (2.5% of WT IGF‐1 levels), the triply deficient LID/ALSKO/BP3KO mice exhibited only a modest 6% decrease in body length, comparable to that of the ALSKO mice that had much greater serum IGF‐1 levels (Yakar et al., 2009). However, interpretation of some of these observations is complicated by a marked increase in circulating GH to supraphysiological levels (Yakar et al., 2002) and models with a physiological level of circulating GH maybe more informative.
Some function has been attributed to circulating IGF‐1, and it is possible that a threshold concentration of circulating IGF‐1 is required for normal linear growth (Lupu et al., 2001). The data from the LID/ALSKO/BP3KO mice however are at odds with this concept (Yakar et al., 2002; Yakar et al., 2009). When hepatic IGF‐1 production was achieved in mice lacking Igf1 gene expression in all other tissues, it was found that circulating IGF‐1 contributes to 30% of the adult body size and sustains postnatal development (Stratikopoulos et al., 2008). Similarly, in Igf1 null mice with hepatic over expression of the rat Igf1, serum IGF‐1 production supported normal body growth during and after puberty, despite absence of tissue IGF‐1 (Wu et al., 2009; Elis et al., 2010).
Alternative strategies to delineate between circulating and local IGF‐1 effects on linear bone growth have involved the targeted deletion of Igf1 in epiphyseal chondrocytes. These mice had a 40% reduction in cartilage IGF‐1 expression and normal circulating IGF‐1 levels (Govoni et al., 2007). Linear growth was reduced by 27% between 2 and 4 weeks of age, highlighting that local chondrocyte‐produced IGF‐1 is an important regulator of longitudinal growth. While highly informative, these studies fail to address the possibility of a GH action on the growth plate, which is independent of IGF‐1. Double ghr/igf1 KO mice have a more severe phenotype than ghr or igf1 KO's alone suggesting that there are GH actions on linear bone growth that are independent of IGF‐1 (Lupu et al., 2001).
A role for GH acting directly on growth plate cartilage is also suggested by data from suppressor of cytokine signaling 2 (SOCS2) null mice (Metcalf et al., 2000; MacRae et al., 2009). SOCS2 is expressed by epiphyseal chondrocytes, and is a recognised negative regulator of GH signaling via inhibition of the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway (Hilton, 1999; Greenhalgh et al., 2002; Rico‐Bautista et al., 2006; Flores‐Morales et al., 2006; Pass et al., 2009). Intriguingly, these mice are characterized by increased growth, including increased long bone length and mass, without elevated circulating levels of IGF‐1 or GH (Metcalf et al., 2000; Greenhalgh et al., 2005; MacRae et al., 2009; Dobie et al., 2014). It can therefore be assumed that GH actions directly on growth plate cartilage (IGF‐1 dependent or independent) are driving this increased linear bone growth.
The ex vivo metatarsal culture method has been exploited in many studies as a method for analysing endochondral bone growth. This model provides a more physiological environment than cultured chondrocytes as chondrocyte interactions with each other and the extracellular matrix (ECM) are maintained. (Mushtaq et al., 2004; MacRae et al 2007; Chagin et al 2010). Studies have shown that the growth rate of embryonic bones in culture is similar to that found in vivo (Scheven & Hamilton 1991; Coxam et al., 1996). Furthermore, the ability to grow metatarsals in long‐term cultures without fetal bovine serum allows for the manipulation of medium conditions and investigation of the effects of various treatments. Recently, it has been reported that GH is able to stimulate longitudinal growth of Socs2‐/‐ metatarsals, but not WT bones (Pass et al., 2012). The Socs2‐/‐ metatarsal culture model is therefore a valuable model in investigating the mechanisms by which local GH enhances linear bone growth.
This study aimed to utilize the metatarsal organ culture model to explore the mechanisms by which linear bone growth is enhanced in Socs2‐/‐ mice, and in doing so, establish the mechanisms of GH's control of growth plate function.
Materials and Methods
Mice
Socs2‐/‐ mice were generated as previously described (MacRae et al., 2009). For genotyping, tail or ear biopsied DNA was analyzed by PCR for SOCS2 (WT) or the neocassette (Socs2‐/‐) Primer sequences can be found in Supp. Table S1 (Eurofins MWG Operon, London, UK) (Pass et al., 2012). All animal experiments were approved by Roslin Institute's Animal Users Committee, and the animals were maintained in accordance with Home Office (UK) guidelines for the care and use of laboratory animals.
Growth analysis and growth plate dynamics
Six‐week‐old male WT and Socs2 ‐/‐ mice received a subcutaneous injection of 10 mg/kg calcein (Sigma, Poole, UK) in sodium bicarbonate solution 2 days prior to sacrifice. Tibiae, fixed overnight in 4% paraformaldehyde (PFA) were embedded in methylmethacrylate and sections (5 μm) were cut and processed using standard procedures (Idris et al., 2009). The longitudinal bone growth rate was measured as previously described (Owen et al 2009; Pass et al., 2012). In brief, the distance between the original growth plate mineralization front and the final fluorescing mineralization front within the metaphysis was measured at 10 different points along the width of the section using image analysis software and a Leica DMBR fluorescent microscope. Measurements were divided by the number of days between injection and sacrifice (2 days) to give a bone formation rate per day. Four sections per tibia were measured from 6 mice per group.
Embryonic and postnatal metatarsal organ culture
The middle three metatarsals were isolated from 17‐day‐old WT and Socs2‐/‐ embryos (E17) or 3‐day‐old (PN3) WT and Socs2‐/‐ pups. At both developmental ages the growth plate contains both proliferating and hypertrophic chondrocytes but the primary ossification center, while newly formed in E17 metatarsals, is almost completely developed in PN3 bones (van Loon et al 1995; Reno et al., 2006). Each bone was cultured individually in 1 well of a 24‐well plate (Costar, High Wycombe, UK) in 300 μl αMEM medium (without ribonucleosides) or 300 μl DMEM + F12 medium supplemented with 0.2% BSA (Fraction V), 5 μg/ml L‐ascorbic acid phosphate, 1 mM β‐glycerophosphate, 0.05 mg/ml gentamicin, 1.25 μg/ml fungizone (Invitrogen, Paisley, UK) as previously described (Chagin et al., 2010; Pass et al., 2012). The perichondrium and the primary ossification center was not removed prior to explant culture. Recombinant human (rh)GH and rhIGF‐1 (Bachem, Merseyside, UK; both 100 ng/ml) were added as used previously (Mushtaq et al., 2004a; MacRae et al., 2006a; Pass et al., 2012). Recombinant mouse (rm)IGF‐2 (R&D systems, Minneapolis, MN) was added at the same concentration as rhIGF‐1 (100 ng/ml). One micrometer NVP‐AEW541 (IGF‐1 receptor kinase inhibitor) (Garcia‐Echeverria et al., 2004; Gan et al., 2010) was added 16 h prior to the addition of GH. Bones were incubated in a humidified atmosphere (37°C, 5% CO2), for up to 13 days. Bone lengths were measured from articular surface to articular surface through the middle of the metatarsals using a Nikon eclipse TE300 microscope with a digital camera attached, using Image Tool (Image Tool Version 3.00, San Antonio, TX). For RNA extraction 3–4 bones were pooled in 100 µl Trizol reagent (Invitrogen) at days 7 and 12 of culture and RNA extracted according to the manufacturer's instructions (RNeasy Mini Kit, Qiagen). Conditioned medium was collected at days 5, 7, and 12 and stored at −80°C.
Growth plate micro dissection and RNA extraction
Tibiae were dissected from 7‐week‐old male WT (n = 4) and Socs2‐/‐ mice (n = 3). Bones were briefly immersed in 4% aqueous (wt./vol.) polyvinylalcohol (PVA; Grade GO4/140, Wacker Chemicals, Walton‐on‐Thames, UK), chilled by precipitate immersion in n‐hexane (BDH, Poole, UK; grade low in aromatic hydrocarbons) and stored at −80°C (25;26). Using optimal cutting temperature (OCT) embedding medium (Brights, Huntingdon, UK), 30 μm thick longitudinal sections of the proximal tibia were cut at −30°C (Brights, OT model cryostat), mounted on Superfrost Plus slides (Fischer Scientific, Chicago, IL) before storage at −80°C. Slides were briefly thawed as described previously (Nilsson et al., 2007; Staines et al., 2012), and then dehydrated in graded solutions of ethanol (70%, 95%, and 100%) with sections kept under a xylene droplet throughout the microdissection. The entire growth plate was dissected free from the perichondrium, the secondary ossification zone and the primary spongiosa. Growth plates from both tibae of each mouse were pooled in 2.88 μl β‐mercaptoethanol (Sigma, Poole, Dorset) and 400 μl Solution C (0.322 g guanidine thiocyanate, 377 µl nuclease free water, 23 µl 0.75 M sodium citrate). From each animal approximately 40–60 µg of growth plate tissue was obtained and RNA isolation was performed using proeteinase K digestion followed by phenol:chloroform extraction as previously described (Heinrichs et al., 1994).
Real‐time quantitative PCR (RT‐qPCR)
RNA content was assessed using a nanodrop spectrophotometer (Thermo Scientific, Chicago, IL) by the absorbance at 260 nm and purity by A260/A280 ratio. Reverse‐transcription was completed as described previously (Farquharson et al., 1999; Houston et al., 1999). RT‐qPCR was performed using the SYBR green (Roche) detection method on a Stratagene Mx3000P real‐time qPCR system (Stratagene, CA) using the following programme: 1 cycle at 95°C for 15 min; 40 cycles at 94°C for 15 sec; 55°C for 30 sec; 72°C for 30 sec; 1 cycle at 95°C for 1 min, 55°C for 30 sec and 95°C for 30 sec. Relative gene expression was calculated using the ΔΔCt method (Livak and Schmittgen, 2001). Each cDNA sample was normalized to housekeeping gene gapdh (Primer Design, Southampton, UK) as previously described (Martensson et al 2004). Reactions were performed with gene of interest primers Igf1, Igfbp3, and Igf2 (Invitrogen), as well as Socs1, Socs2, and Socs3 (Supp. Table 1).
Immunohistochemistry
Tibiae were dissected from 6‐week‐old male WT (n = 6) and Socs2‐/‐ mice (n = 6) and fixed in 70% ethanol for 48 h at 4°C before decalcification in 10% EDTA (pH 7.4) for approximately 4 weeks at 4°C with regular changes. Tissues were finally dehydrated and embedded in paraffin wax, using standard procedures, and sectioned at 5 µm. For immunohistochemical analysis, sections were dewaxed in xylene and rehydrated before incubation at 37°C for 30 min in 0.1% trypsin (Sigma) for antigen demasking. Endogenous peroxidases were blocked by 0.03% H2O2 in methanol (Sigma) for 30 min. Antibodies to IGFBP3 and IGF‐2 (both Santa Cruz Biotechnology, Heidelberg, Germany) were diluted to 0.4 μg IgG/ml and 0.5 μg IgG/ml, respectively and incubated for 18 h at 4°C. Control sections were incubated with an equal concentration IgG. A Vectastain Rabbit ABC kit (Vector Laboratories, Peterborough) was then used according to the manufacturer's instructions. The sections were finally dehydrated, counterstained with hematoxylin and mounted in DePeX.
Conditioned medium analysis
Total IGF‐1 and IGFBP3 levels in conditioned medium from metatarsals cultured ± GH were assessed at days 5, 7, and 12 by ELISA (Quantikine, R&D Systems, Minneapolis, MN). IGF‐2 levels in conditioned medium were assessed at day 12 (Ray Biotech, Inc) according to the manufacturer's instructions. IGF‐1 assays included a step to dissociate the potentially interfering binding proteins from the ligand.
Statistical analysis
Direct comparison between two sets of data was done by Student's t‐test or a suitable non‐parametric test if the data was not normally distributed. Time course experiments were analyzed with a repeated measures 2‐way ANOVA for which suitable post‐tests for multiple comparisons were conducted. Analysis was carried out using SigmaPlot v11.0 (Systat Software Inc, London, UK). Data are presented as mean ± standard error of the mean (SEM). P values <0.05 were considered significant.
Results
SOCS2 regulation of GH induced growth in metatarsals
WT metatarsals extracted at E17 did not increase their length in response to GH over the 12 day culture period (Fig. 1A). Transcript analysis at day 7, revealed a 17‐fold increase in Socs2 mRNA expression in the GH‐treated group in comparison to the control group (P < 0.01) (Fig. 1B). Socs1 and Socs3 mRNA expressions were however not enhanced in response to GH (Fig. 1B). Socs2‐/‐ metatarsals growth significantly increased in response to GH from day 7 to day 12 (P < 0.05) (Fig. 1C), similar to previous studies (Pass et al., 2012). IGF‐1 stimulated longitudinal growth in WT and Socs2‐/‐ metatarsals as expected. The level of stimulation in the presence of IGF‐1 was comparable between WT and Socs2‐/‐ metatarsals, indicating that SOCS2 does not regulate IGF‐1 induced growth in this model (Fig. 1A and C). Assessment of WT and Socs2‐/‐ PN3 metatarsals revealed a similar GH response to that noted with the embryonic metatarsal with only the Socs2‐/‐ bones showing enhanced growth in response to GH (P < 0.01) (Supp. Fig. 1A and C). Also, Socs2 but not Socs1 or 3 mRNA expression was increased in response to 12 days of GH treatment (4‐fold; P < 0.01) in WT metatarsals (Supp. Fig. 1B). Taken together, these data indicate that SOCS2 is a major regulator of GH promotion of longitudinal growth in the metatarsal organ culture model.
Figure 1.
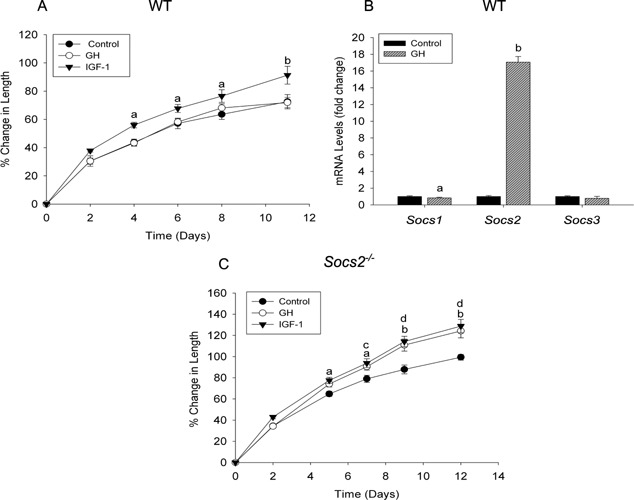
SOCS2 regulation of GH‐induced embryonic metatarsal growth. Graphs showing (A) WT and (C) Socs2‐/‐ E17 metatarsal growth in response to GH or IGF‐1 (100 ng/ml) over a 12 day period. Data are presented as mean ± SEM. Significance denoted by IGF‐1 versus control a P < 0.05, b P < 0.001. GH versus control c P < 0.05, d P < 0.001, (n ≥ 6). (B) Transcript analysis of Socs1, 2, and 3 in WT metatarsals following 7 days GH (100 ng/ml) treatment. Data represented as means ± SEM. Significance from untreated metatarsals denoted by a P < 0.05, b P < 0.01, (n = 3).
Increased growth of Socs2‐/‐ metatarsals in response to GH is not associated with an increase in chondrocyte IGF‐1
We next aimed to determine if the growth promoting effects of GH on Socs2‐/‐ metatarsals were direct or mediated by IGFs. Following 7 days of GH treatment, WT metatarsals showed no increase in longitudinal growth compared to untreated samples, whereas Socs2‐/‐ metatarsal growth increased by 15% (P < 0.01) (Fig. 2A and B). Expression levels of Igf1, Igfbp3, and Igf2 at this time point were unaltered in response to GH in WT and Socs2‐/‐ metatarsals (Fig. 2C). Protein analysis of day 5 conditioned medium revealed increased IGFBP3 protein levels in GH‐treated Socs2‐/‐ (Control 8.2 ± 1.35, GH 23.5 ± 5.76; P < 0.01), but not in corresponding WT conditioned medium (Tables 1 and 2). ELISA analysis of day 7 conditioned medium similarly found no increase in IGF‐1 or IGFBP3 protein levels in GH treated WT metatarsals (Table 1). While IGF‐1 remained at basal level in GH‐stimulated Socs2‐/‐ conditioned medium at day 7, IGFBP3 levels increased significantly (Control 5.2 ± 0.94, GH 22.6 ± 3.34; P < 0.01) (Table 2).
Figure 2.
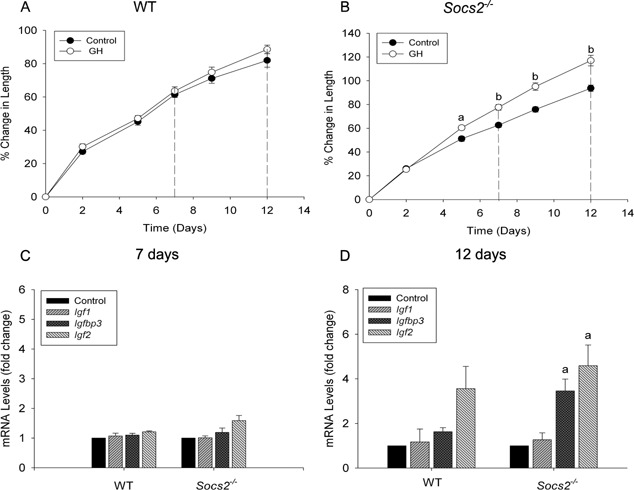
Igf1, Igfbp3, and Igf2 transcript analysis following GH treatment. Graphs showing (A) WT and (B) Socs2‐/‐ E17 metatarsal growth in response to GH (100 ng/ml) over a 12 period. Dotted lines represent points at which Igf1, Igfbp3, and Igf2 expression levels were measured. Data are presented as mean ± SEM. Significance from untreated metatarsals denoted by a P < 0.05, b P < 0.01 (n ≥ 6). Transcript analysis of Igf1, Igfbp3, and Igf2 in WT and Socs2‐/‐ metatarsals following (C)7‐ and (D)12 days GH (100ng/ml) treatment. Data are presented as mean ± SEM. Significance from untreated metatarsals denoted by a P < 0.05, (n = 3)
Table 1.
IGF‐1, IGFBP3, and IGF‐2 protein levels in conditioned medium from E17 WT metatarsals following 5, 7, or 12 days GH (100 ng/ml) treatment
| Day | Treatment | IGF‐1 (ng/ml) | IGFBP3 (ng/ml) | IGF‐2 (ng/ml) |
|---|---|---|---|---|
| 5 | Control | 2.4 ± 0.48 | 9.0 ± 1.43 | nd |
| GH | 2.1 ± 0.23 | 9.7 ± 1.41 | nd | |
| 7 | Control | 6.4 ± 0.86 | 12.2 ± 1.68 | nd |
| GH | 4.0 ± 0.70 | 19.7 ± 4.57 | nd | |
| 12 | Control | 11.6 ± 1.16 | 25.0 ± 5.09 | 7.0 ± 1.39 |
| GH | 9.5 ± 1.06 | 39.8 ± 3.69a | 8.4 ± 0.97 |
Data are presented as mean ± SEM (n ≥ 5). Significance from day matched control samples denoted by a P < 0.05. nd = no data.
Table 2.
IGF‐1, IGFBP3, and IGF‐2 protein levels in conditioned medium from E17 Socs2‐/‐ metatarsals following 5, 7, or 12 days GH (100 ng/ml) treatment
| Day | Treatment | IGF‐1 (ng/ml) | IGFBP3 (ng/ml) | IGF‐2 (ng/ml) |
|---|---|---|---|---|
| 5 | Control | 2.7 ± 0.59 | 8.2 ± 1.35 | nd |
| GH | 2.6 ± 0.26 | 23.5 ± 5.76b | nd | |
| 7 | Control | 3.3 ± 0.39 | 5.2 ± 0.94 | nd |
| GH | 2.8 ± 0.28 | 22.6 ± 3.34b | nd | |
| 12 | Control | 9.9 ± 0.90 | 22.2 ± 3.51 | 1.7 ± 0.34 |
| GH | 7.0 ± 0.39 | 49.6 ± 1.89c | 17.1 ± 1.72c |
Data are presented as mean ± SEM (n ≥ 5). Significance from control day matched control samples denoted by b P < 0.01, c P < 0.001.
At day 12, Socs2‐/‐ metatarsal growth increased by 23% (P < 0.01) in response to GH, while WT metatarsal growth remained at control level (Fig. 2A and B). Similar to the day 7 data; expression levels of Igf1, Igfbp3, and Igf2 were unaltered in response to GH in WT metatarsal (Fig. 2D). Similarly, Igf1 transcript levels remained at basal level in GH‐treated Socs2‐/‐ metatarsals, whereas Igfbp3 and Igf2 transcript levels significantly increased by 3.5‐ (P < 0.05) and 4.6‐ fold (P < 0.05) respectively (Fig. 2D). A similar trend was observed when analyzing protein levels of conditioned medium (Tables 1 and 2). GH failed to have a stimulatory effect on IGF‐1 levels in WT or Socs2‐/‐ conditioned medium. However, GH treatment did result in a 59% (P < 0.01) and 123% (P < 0.001) increase in IGFBP3 levels in WT and Socs2‐/‐ conditioned medium, respectively. IGF‐2 protein levels were also increased in Socs2‐/‐ conditioned medium in response to GH (P < 0.001) (Table 2). No increase in IGF‐2 levels was observed in corresponding WT medium (Table 1).
Experiments carried out on PN3 metatarsals produced comparable results to those observed in E17 metatarsals in terms of IGF‐1 production. Despite there being an increase in growth of Socs2‐/‐ metatarsals in response to GH, IGF‐1 levels in conditioned medium at day 7 and 12 were not preferentially increased in Socs2‐/‐ metatarsals (Supp. Tables 2 and 3). GH stimulated IGFBP3 protein levels in WT‐conditioned medium, an increase that appeared to be enhanced in the absence of SOCS2 at day 12 (Supp. Tables 2 and 3). A comparable increase in IGF‐2 levels in conditioned medium following GH treatment was observed in WT and Socs2‐/‐ metatarsals (Supp. Tables 2 and 3).
These data reveal that increased longitudinal growth in response to GH observed in the Socs2‐/‐ metatarsal model is not mediated by increased IGF‐1 expression. They also highlight the possible importance of IGFBP3 and IGF‐2 in this model as secondary messengers to GH in promoting longitudinal bone growth in Socs2‐/‐ metatarsals.
Altered IGF‐2 and IGFBP3 levels in the growth plates of Socs2‐/‐ mice
Examination of growth plates from WT mice indicated that both IGF‐2 and IGFBP3 protein were preferentially localized to the proliferating zone of the growth plate with little or no staining in the hypertrophic chondrocytes. The staining in the proliferating chondrocytes of the Socs2‐/‐ growth plates of both IGF‐2 and IGFBP3 appeared similar to that observed in similarly stained WT growth plates. In contrast, growth plates from Socs2‐/‐ mice contained a greater number of hypertrophic chondrocytes that stained positively for both IGF‐2 and IGFBP3 (Fig. 3). All control sections showed no staining (data not shown).
Figure 3.
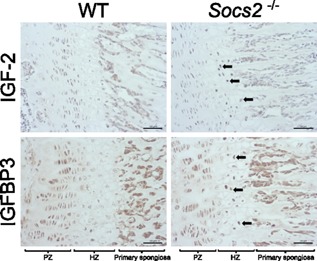
Immunohistochemical localization of IGF‐2 and IGFBP3 to the proliferating and hypertrophic chondrocytes of WT and Socs2‐/‐ growth plates. In contrast to WT growth plates, hypertrophic chondrocytes within the Socs2‐/‐ growth plate expressed high levels of both IGF‐2 and IGFBP3 (arrows). Scale bars = 50μm
No additive effects of GH and IGF‐1 action in the promotion of metatarsal linear growth
Having shown that both GH and IGF‐1 stimulated longitudinal growth in Socs2‐/‐ metatarsals, and that GH stimulation was independent of increased IGF‐1 levels, we next aimed to determine if the actions of GH and IGF‐1 on longitudinal growth were additive. Dual treatment with GH and IGF‐1 in WT metatarsals significantly increased growth in comparison to control samples (P < 0.001) (Fig. 4A). This level of increase did not differ from that observed with IGF‐1 treatment alone. In Socs2‐/‐ metatarsals, growth was significantly increased in response to both IGF‐1 and GH treatment in comparison to control samples (P < 0.01, P < 0.05 respectively; Fig. 4B). There was no additional increase in metatarsal growth upon dual treatment (Fig. 4B).
Figure 4.
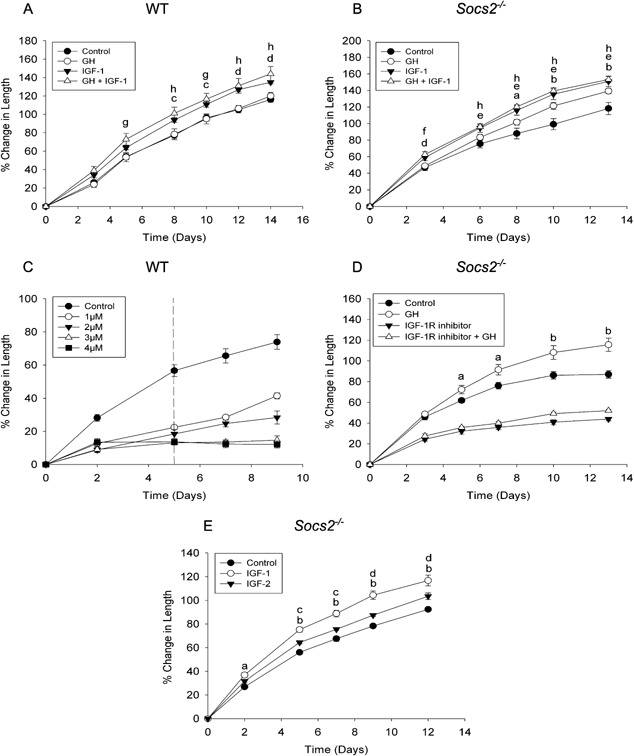
Effect of IGF‐1R inhibitor, IGF‐2, and combined IGF‐I + GH on metatarsal growth. Graphs showing (A) WT and (B) Socs2‐/‐ E17 metatarsal growth in response to GH or IGF‐1 (100ng/ml) or GH + IGF‐1 over a 13–14 day period. Data are presented as mean ± SEM. Significance denoted by GH versus control a P < 0.05, b P < 0.001. IGF‐1 versus control c P < 0.05, d P < 0.01, e P < 0.001, GH+IGF‐1 versus control f P < 0.05, g P < 0.01, h P < 0.001, (n ≥ 6). (C) Graph showing WT E17 metatarsal growth in response to NVP‐AEW481 (1–4 μM) for 5 days with 4 days recovery. Data are presented as mean ± SEM. (n ≥ 6). (D) Graph showing Socs2‐/‐ E17 metatarsal growth in response to GH (100 ng/ml), NVP‐AEW481 (1 μM) or GH + NVP‐AEW481. Data are presented as mean ± SEM. Significance of GH versus control denoted by a P < 0.05, b P < 0.01, (n ≥ 6). (E) Graph showing Socs2‐/‐ E17 metatarsal growth in response to IGF‐1 or IGF‐2 (100 ng/ml). Data are presented as mean ± SEM. Significance denoted by IGF‐1 versus control a P < 0.01, b P < 0.001. IGF‐2 versus control c P < 0.05 d P < 0.01, (n ≥ 6).
The IGF‐1R is critical for GH‐induced growth
The cellular responses of IGF‐1 and IGF‐2 are both mediated through the IGF‐1R. To investigate the importance of this receptor in mediating the effects of GH on longitudinal bone growth, we assessed the growth of Socs2‐/‐ metatarsals in the presence of GH and an IGF‐1R inhibitor (NVP‐AEW541). Initially, WT metatarsals were treated with varying concentrations of NVP‐AEW541 for 5 days before the inhibitor was removed from the medium (Fig. 4C). All concentrations of NVP‐AEW541 (1–4 μM) inhibited metatarsal growth confirming that endogenous IGF signaling is important for metatarsal growth. One micrometer of NVP‐AEW541 was selected for future experiments as on the removal of the inhibitor, metatarsals showed increased growth suggesting that the inhibitor at this concentration (cf 3 and 4 μM) had no lasting toxic effects on growth potential. Socs2‐/‐ metatarsals treated with the NVP‐AEW541 showed significantly decreased growth compared to control samples (P < 0.05, Fig. 4D). GH treatment was unable to stimulate growth in the presence of the IGF‐1R inhibitor, suggesting that the effects of GH on longitudinal bone growth are mediated through the IGF‐1R.
IGF‐2 stimulation of embryonic Socs2‐/‐ metatarsals
As GH‐induced growth in Socs2‐/‐ metatarsals was associated with an increase in IGF‐2 levels (Table 2), we next determined if IGF‐2 could stimulate longitudinal growth. IGF‐2 treatment significantly increased longitudinal growth of Socs2‐/‐ metatarsals from day 5 to 12 (P < 0.05; Fig. 4E). This enhanced growth by IGF‐2 was however, significantly less than that stimulated by equimolar IGF‐1, indicating that IGF‐1 is the more potent of the two IGF ligands in promoting linear bone growth (Fig. 4E). Similar results were observed in WT metatarsals (data not shown).
Igf1 mRNA levels in the Socs2‐/‐ growth plate
Calcein labeling of 6‐week‐old Socs2 ‐/‐ mice revealed that tibial growth over a 2 day period of Socs2‐/‐ mice (44.5 ± 1.8μm) was increased in comparison to WT mice (36.8 ± 1.57 μm) (21%; p<0.05) (Fig. 5A). Igf1 levels were also assessed in the growth plates of WT and Socs2‐/‐ mice and found not to be significantly altered from expression levels of WT mice. Therefore, despite increased bone growth rates in Socs2‐/‐ mice there was no elevation in growth plate Igf1 transcript levels (Fig. 5B).
Figure 5.
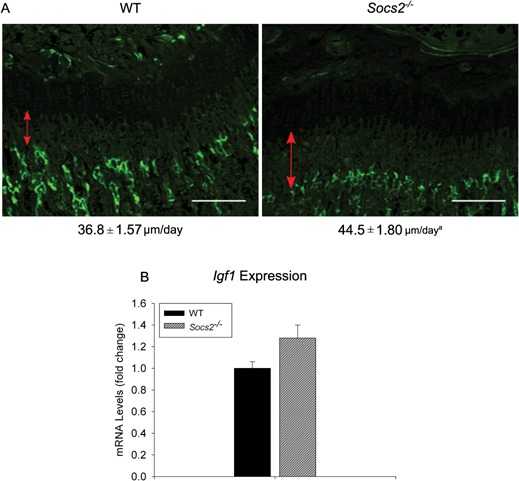
Normal IGF‐1 mRNA expression in Socs2‐/‐ growth plate. (A) Analysis of longitudinal bone growth rate by dynamic histomorphometry of 6‐week‐old male WT and Socs2‐/‐ mice. The red lines indicate the distance between the original growth plate mineralization front at the proximal end of the tibia and the fluorescing mineralization front. The numbers under the images indicate longitudinal bone formation rate (μm/day). Scale bars = 100 μm. Data presented as mean ± SEM. Significance denoted by a P < 0.05, (n ≥ 6). (B) Transcript analysis of Igf1 expression in WT and Socs2‐/‐ growth plates, micro‐dissected from 7‐week‐old male mice. Data are presented as fold change relative to WT expression as mean ± SEM, (n ≥ 3).
Discussion
It is widely understood that GH action on growth plate chondrocytes is an important regulator of linear growth (Ahmed and Farquharson, 2010). The intimate relationship between GH and IGF‐1 does however make it difficult to attribute specific actions to GH, or those resulting through IGF‐1. Tibial growth is reduced in both ghr and Igf1 null mice. This reduction in growth becomes more severe in the double ghr/igf1 mutants, suggesting that GH and IGF‐1 have independent functions (Lupu et al., 2001). In agreement with this, mice deficient of GH show a much higher reduction in longitudinal bone growth than Igf1 null mice (Sims et al., 2000; Mohan et al., 2003; Wang et al., 2004). The present study demonstrates that despite no elevation in IGF‐1 gene and protein expression levels in Socs2‐/‐ metatarsals following GH treatment, GH enhances linear bone growth. This indicates that the growth promoting effects of GH are not accompanied by a rise in IGF‐1 expression by chondrocytes or cells of the perichondrium, which previous studies have indicated is a major source of Igf1 in the growing bone (Parker et al., 2007). This was supported by the observation of normal Igf1 expression levels in the growth plates of Socs2‐/‐ tibia which are characterized by wider growth plates and longer bones (MacRae et al., 2009; Pass et al., 2012).
In addition to their normal GH and IGF‐1 serum concentrations, Socs2 ‐/‐ mice also have normal carbohydrate metabolism compared with ALSKO, BP3KO, LID, and IGF‐1 null mice (Rico‐Bautista et al., 2005; Yakar et al., 2009). Thus, the Socs2 ‐/‐ mouse may represent an important model to study local autocrine/paracrine pathways that promote linear bone growth without leading to insulin resistance and altered carbohydrate metabolism.
Several studies have used the fetal mouse metatarsal culture methods to investigate the anabolic effects of IGF‐1 on linear bone growth. Increased growth of metatarsals in response to IGF‐1 is coupled with an increase in proliferation and an increase in hypertrophic zone size (Mushtaq et al., 2004; MacRae et al., 2006, 2007; Chagin et al., 2010). The effects of GH on metatarsal growth are however less clear, with GH being shown to either stimulate or have no effect on growth (Scheven and Hamilton, 1991; Mushtaq et al., 2004; Pass et al., 2012). In chondrocytes, GH is able to stimulate SOCS2 expression (Pass et al., 2012) and we have now further shown that SOCS2 is a negative regulator of GH's capability to promote longitudinal bone growth in the metatarsal organ culture model. This observation is in agreement with previous reports where mice missing the Socs2 gene have increased linear bone growth due to increased signaling through the GHR (Metcalf et al., 2000; Greenhalgh et al., 2005; MacRae et al., 2009). Although, it is well recognized that GH signaling is also modulated by CIS and SOCS1 and 3 we have reported previously that GH does not stimulate chondrocyte SOCS 1 or 3 protein levels (Pass et al., 2009, 2012). Furthermore, Socs1 ‐/‐ and Socs3 ‐/‐ mice are perinatal and embryonic lethal, respectively and little data exists on their ability to regulate bone growth (Greenhalgh and Alexander, 2004). While further studies are required to investigate GH ability to stimulate chondrocyte CIS expression it is of interest to note that Cis ‐/‐ mice exhibit no obvious growth phenotype (Greenhalgh and Alexander, 2004).
It is widely established that the prenatal growth promoting effects of IGF‐1 are GH independent, and that GH has little or no role in promoting growth in embryonic development (Waters and Kaye, 2002). Therefore, these key studies were also completed using postnatal metatarsals, which may be more sensitive to the growth promoting actions of GH. No evidence for this was however observed in our experiments and older postnatal bones may have to be studied to see a GH response. However, this was not possible as our pilot studies (data not shown) indicated that 3‐day‐old postnatal metatarsals was the oldest age we could study to get measurable growth. Even at postnatal day 3, bone growth was much less than embryonic metatarsals and this is in accord with previous observations (Macrae et al. and 2006; Chagin et al., 2010). It is therefore tantalizing to speculate that variable SOCS2 expression in normal animals may “fine tune” the chondrocytes ability to respond to GH. Such a regulatory mechanism may permit postnatal bones at a specific developmental age to respond to GH and further studies are required to investigate this.
Socs2‐/‐ mice have increased linear bone growth consistent with increased signaling through the GHR (Metcalf et al., 2000; Greenhalgh et al., 2005; Lorentzon et al., 2005; MacRae et al., 2009; Pass et al., 2012). As Socs2‐/‐ mice are devoid of higher circulating IGF‐1 levels, it is likely that the increased linear bone growth and structural alterations within Socs2‐/‐ growth plate are a direct consequence of GH acting directly on growth plate cartilage (Lorentzon et al., 2005; MacRae et al., 2009). The broad distribution of both the GHR and IGF‐1R within the growth plate suggests GH/IGF‐1 may play a role in chondrocyte proliferation, differentiation, and hypertrophy (Lupu et al., 2001; Gevers et al., 2002b; Wang et al., 2011). The germinal zone of the growth plates are significantly enlarged in Igf1 null mice, supporting the idea that GH stimulates longitudinal bone growth independent of IGF‐1 (by stimulating the differentiation of growth plate precursor cells), and via an IGF‐1 dependent mechanism (by inducing Igf1 expression in differentiating chondrocytes which then in‐turn stimulate clonal expansion) (Isaksson et al., 1987; Wang et al., 2004). Although IGF‐1 is expressed in all maturational growth plate zones, it is understood to act mainly on the hypertrophic zone. Igf1‐/‐ mice have attenuation of chondrocyte hypertrophy with no changes in proliferation (Wang et al., 1999). Addition of IGF‐1 to metatarsals leads to an increase in hypertrophic zone lengths as well as increased size of individual chondrocytes (Mushtaq et al., 2004). These mechanistic observations are important as a large part of growth of cultured metatarsals is due to hypertrophic differentiation but it is recognized these observation may not translate directly to the in vivo situation where matrix production, proliferation and hypertrophy all contribute to linear bone growth (Wilsman et al., 1996).
The GH signaling mechanisms responsible for increasing linear bone growth have been recently clarified in studies where GH was found to stimulate the phosphorylation of STATs 1, 3, and 5 in chondrocyte cultures (Pass et al., 2012). The activation of chondrocyte STAT signaling was both increased and prolonged in chondrocytes from Socs2‐/‐ mice in comparison to cells from WT mice (Pass et al., 2012). This explains the observed GH stimulation of linear growth of Socs2‐/‐ embryonic metatarsals and the proliferation of chondrocytes within (Pass et al., 2012).
Both IGF‐1 and IGF‐2 are important regulators of skeletal growth. While IGF‐1 is important for both fetal and postnatal growth, IGF‐2 is thought to function only in the former. IGF‐2 is highly expressed in a variety of tissues in the embryo, and is dramatically down‐regulated shortly after birth (Lui et al., 2008). Disruption of the Igf2 gene results in progeny that are 60% smaller than WT littermates, however postnatal growth is comparable to WT (Dechiara et al., 1990). Although it is widely understood that IGF‐2 actions are independent of GH regulation, GH has been shown to regulate IGF2 transcription in the human liver (von Horn et al., 2002). In this present study, both during fetal and early postnatal linear growth, IGF‐2 levels are regulated by GH. It is conceivable that the increase in linear growth observed in Socs2‐/‐ metatarsals are a result of elevated IGF‐2 levels. IGF‐2 can promote the expansion of fetal hypertrophic chondrocytes in cultured limb explants, possibly through activation of the PI3K and TGF‐ß pathways and downstream elevation of transcription factors (Hamamura et al., 2008; Chen et al., 2010). Both IGF‐1 and IGF‐2 bind to the IGF‐1R, and the cellular responses of both ligands are mediated through this receptor. As inhibition of this receptor abrogates the growth promoting effects of GH, it stands to reason that IGF's are responsible for mediating the increase in linear growth. As there is no increase in the expression of IGF‐1 in response to GH, it is possible that IGF‐2 and not IGF‐1 is responsible for the GH promoted linear growth of the Socs2‐/‐ metatarsals. In agreement with this, IGF‐2 was able to stimulate longitudinal bone growth in metatarsals from WT and Socs2‐/‐ mice.
Confirmation of increased expression of IGF‐2 and IGFBP3 by the hypertrophic chondrocytes of growth plates in response to unrestricted GH signaling was revealed by immunohistochemical analysis of Socs2‐/‐ growth plates. The presence of IGF‐2 and IGFBP3 in hypertrophic chondrocytes is consistent with GHR expression in these cells of the growth plate and our previous observation of increased numbers of phosphorylated STAT‐5–positive hypertrophic chondrocytes of Socs2‐/‐ growth plates (Gevers et al., 2002b; Pass et al., 2012). The staining in the proliferating chondrocytes of the Socs2‐/‐ growth plates appeared similar to that observed in the WT growth plates however it was not possible, because of the nonquantitative nature of immunohistochemistry, to compare levels of IGF‐2 and IGFBP3 in the proliferating chondrocytes. These data suggest that SOCS2 can influence hypertrophic chondrocyte IGF‐2 and IGFBP3 availability and thereby modulate longitudinal bone growth.
The growth rate of tibia from ghr/Igf1 null mice are almost identical to the sum of growth deficit observed in the single ghr and igf1r mutants suggesting that GH and IGF‐1 have independent roles in promoting linear growth (Lupu et al., 2001). Local administration of GH+IGF‐1 however failed to have an additive effect in hypophysectomised animals (Isgaard et al., 1986). In agreement with this, we show that in Socs2‐/‐ metatarsals, dual treatment with GH and IGF‐1 does not promote longitudinal growth beyond individual stimulation. This suggests that GH is promoting linear growth through a common pathway with IGF‐1. It is however plausible that the increase in IGF‐2 levels observed in Socs2‐/‐ metatarsals observed in this study are competing with the exogenous IGF‐1. As IGF‐1 exhibits a higher binding affinity to the IGF‐1R than IGF‐2 it is likely that the growth promoting effects of IGF‐2 would be masked (Danielsen et al., 1990; Germainlee et al., 1992; Oh et al., 1993).
In the present study GH stimulation also causes significant increase in IGFBP3 levels although, like IGF‐1, the source of this IGFBP3 maybe both chondrocytes and/or the surrounding perichondrium (Parker et al., 2007). The increase is more apparent in Socs2‐/‐ metatarsals, where growth is promoted. IGFBP3 is produced in a number of tissues and is regulated mainly by GH, but also to some degree by IGF‐1 (Kiepe et al., 2005). It is the principal carrier of IGFs in serum, where it functions to control tissue IGF concentrations and reduce bioavailability (Clemmons, 1998). Depending on incubation time and dose, it can inhibit or potentiate the actions of IGFs (Demellow and Baxter, 1988; Stewart et al., 1993; Bagi et al., 1994). IGFBP3 may have pro‐apoptotic functions on chondrocytes, independent of IGFs, which is likely mediated through its own cell surface bound receptor (Oh et al., 1993; Spagnoli et al., 2002). The complex relationship between IGF's and IGFBP3 make it difficult to deduce what its role may be in this system. As there appears to be a correlation between increased IGFBP3 and increased growth in response to GH, it is unlikely that the effects of IGFBP3 are inhibitory in this model.
In conclusion, using the murine metatarsal model, this study underscores the critical role of SOCS2 in controlling GH anabolic effects on linear bone growth. This study also provides compelling evidence to support the notion that in the absence of SOCS2, GH can regulate linear growth via local mechanisms that are not accompanied by a rise in local or circulating IGF‐1.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information Figure S1: SOCS2 regulation of GH induced postnatal metatarsal growth. Graphs showing (A) WT and (C) Socs2‐/‐ postnatal (PN) 3 metatarsal growth in response to GH (100 ng/ml) over a 12 day period. Dotted lines indicate points at which conditioned medium analysed. Data are presented as mean ± SEM. Significance GH versus control denoted by a P < 0.05, b P < 0.01, (n ≥ 6). (B) Transcript analysis of Socs1, 2, and 3 in WT metatarsals following 12 days GH (100 ng/ml) treatment. Data represented as means ± SEM. Significance from untreated metatarsals denoted by b P < 0.01, (n = 3).
Supporting Information Table S1: Primers used for genotyping and PCR analysis.
Supporting Information Table S2: IGF‐1, IGFBP3, and IGF‐2 protein levels in conditioned medium from PN3 WT metatarsals following 7, or 12 days GH (100 ng/ml) treatment.
Supporting Information Table S3.
Acknowledgments
The authors would like to thank Elaine Seawright for her contributions to the experiments. The authors would also like to thank Darren Smith and Alex Robertson of the Small Animal Unit. This project was funded by the Biotechnology and Biological Sciences Research Council (BBSRC) UK and Ipsen (UK) through a CASE studentship award (RD), and Institute Strategic Programme Grant Funding from the BBSRC (CF, VEM), BBSRC Institute Career Path Fellowship funding from the BBSRC (VEM).
Literature Cited
- Ahmed SF, Farquharson C. 2010. The effect of GH and IGF1 on linear growth and skeletal development and their modulation by SOCS proteins. J Endocrinol 206:249–259. [DOI] [PubMed] [Google Scholar]
- Bagi CM, Brommage R, Deleon L, Adams S, Rosen D, Sommer A. 1994. Benefit of systemically administered RhIGF‐I and RhIGF‐I/IGFBP‐3 on cancellous bone in ovariectomized rats. J Bone Miner Res 9:1301–1312. [DOI] [PubMed] [Google Scholar]
- Chagin AS, Karimian E, Sundstrom K, Eriksson E, Savendahl L. 2010. Catch‐up growth after dexamethasone withdrawal occurs in cultured postnatal rat metatarsal bones. J Endocrinol 204:21–29. [DOI] [PubMed] [Google Scholar]
- Chen LA, Jiang W, Huang JY, He BC, Zuo GW, Zhang WL, Luo Q, Shi QO, Zhang BQ, Wagner ER, Luo JY, Tang M, Wietholt C, Luo XJ, Bi Y, Su YX, Liu B, Kim SH, He CJ, Hu YW, Shen JK, Rastegar F, Huang EY, Gao YH, Gao JL, Zhou JZ, Reid RR, Luu HH, Haydon RC, He TC, Deng ZL. 2010. Insulin‐like growth factor 2 (IGF‐2) potentiates BMP‐9‐induced osteogenic differentiation and bone formation. J Bone Miner Res 25:2447–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemmons DR 1998. Role of insulin‐like growth factor binding proteins in controlling IGF actions. Mol Cell Endocrinol 140:19–24. [DOI] [PubMed] [Google Scholar]
- Coxam V, Miller MA, Bowman MB, Miller, SC. 1996. Ontogenesis of IGF regulation of longitudinal bone growth in rat metatarsal rudiments cultured in serum‐free medium. Arch Physiol Biochem 104:173–179. [DOI] [PubMed] [Google Scholar]
- Danielsen A, Larsen E, Gammeltoft S. 1990. Chromaffin cells express 2 types of insulin‐like growth‐factor receptors. Brain Res 518:95–100. [DOI] [PubMed] [Google Scholar]
- Dechiara TM, Efstratiadis A, Robertson EJ. 1990. A growth‐deficiency phenotype in heterozygous mice carrying an insulin‐like growth factor II gene disrupted by targeting. Nature 345:78–80. [DOI] [PubMed] [Google Scholar]
- Demellow JSM, Baxter RC. 1988. Growth hormone‐dependent insulin‐like growth‐factor (Igf) binding‐protein both inhibits and potentiates Igf‐I‐stimulated DNA synthesis in human‐skin fibroblasts. Biochem Bioph Res Comm 156:199–204. [DOI] [PubMed] [Google Scholar]
- Dobie R, MacRae VE, Huesa C, Van't Hof, Farquharson SF. 2014. Direct stimulation of bone mass by increased GH signalling in osteoblasts of Socs2‐/‐ mice.J. Endocrinol 223:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elis S, Courtland HW, Wu YJ, Rosen CJ, Sun H, Jepsen KJ, Majeska RJ, Yakar S. 2010. Elevated serum levels of IGF‐1 are sufficient to establish normal body size and skeletal properties even in the absence of tissue IGF‐1. J Bone Miner Res 25:1257–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquharson C, Lester D, Seawright E, Jefferies D, Houston B. 1999. Microtubules are potential regulators of growth‐plate chondrocyte differentiation and hypertrophy. Bone 25:405–412. [DOI] [PubMed] [Google Scholar]
- Flores‐Morales A, Greenhalgh CJ, Norstedt G. Rico‐Bautista E. 2006. Negative regulation of growth hormone receptor signaling. Mol Endocrinol 20:241–253. [DOI] [PubMed] [Google Scholar]
- Gan YJ, Zhang Y, DiGirolamo DJ, Jiang J, Wang XD, Cao XM, Zinn KR, Carbone DP, Clemens TL, Frank SJ. 2010. Deletion of IGF‐I receptor (IGF‐IR) in primary osteoblasts reduces GH‐induced STAT5 signaling. Mol Endocrinol 24:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Echeverria C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, Gao JP, Brueggen J, Capraro HG, Cozens R, Evans DB, Fabbro D, Furet P, Porta DG, Liebetanz J, Martiny‐Baron G, Ruetz S, Hofmann F. 2004. In vivo antitumor activity of NVP‐AEW541‐A novel, potent, and selective inhibitor of the IGF‐IR kinase. Cancer Cell 5:231–239. [DOI] [PubMed] [Google Scholar]
- Germainlee EL, Janicot M, Lammers R, Ullrich A, Casella SJ. 1992. Expression of A Type‐I insulin‐like growth‐factor receptor with low affinity for insulin‐like growth factor‐II. Biochem J 281:413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers EF, Loveridge N, Robinson IC. 2002a. Bone marrow adipocytes: A neglected target tissue for growth hormone. Endocrinology 143:4065–4073. [DOI] [PubMed] [Google Scholar]
- Gevers EF, Van Der Eerden BCJ, Karperien M, Raap AK, Robinson ICAF, Wit JM. 2002b. Localization and regulation of the growth hormone receptor and growth hormone‐binding protein in the rat growth plate. J Bone Miner Res 17:1408–1419. [DOI] [PubMed] [Google Scholar]
- Govoni KE, Lee SK, Chung YS, Behringer RR, Wergedal JE, Baylink DJ, Mohan S. 2007. Disruption of insulin‐like growth factor‐I expression in type II alpha I collagen‐expressing cells reduces bone length and width in mice. Physiol Geno 30:354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhalgh CJ, Alexander WS. 2004. Suppressors of cytokine signalling and regulation of growth hormone action. Growth Horm IGF Res 14:200–206. [DOI] [PubMed] [Google Scholar]
- Greenhalgh CJ, Bertolino P, Asa SL, Metcalf D, Corbin JE, Adams TE, Davey HW, Nicola NA, Hilton DJ, Alexander WS. 2002. Growth enhancement in suppressor of cytokine signaling 2 (SOCS‐2)‐deficient mice is dependent on signal transducer and activator of transcription 5b (STAT5b). Mol Endocrinol 16:1394–1406. [DOI] [PubMed] [Google Scholar]
- Greenhalgh CJ, Rico‐Bautista E, Lorentzon M, Thaus AL, Morgan PO, Willson TA, Zervoudakis P, Metcalf D, Street I, Nicola NA, Nash AD, Fabri LJ, Norstedt G, Ohlsson C, Flores‐Morales A, Alexander WS, Hilton DJ. 2005. SOCS2 negatively regulates growth hormone action in vitro and in vivo. J Clin Invest 115:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamura K, Zhang P, Yokota H. 2008. IGF2‐driven PI3 kinase and TGF beta signaling pathways in chondrogenesis. Cell Bio Int 32:1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrichs C, Yanovski JA, Roth AH, Yu YM, Domene HM, Yano K, Cutler GB, Baron J. 1994. Dexamethasone increases growth‐hormone receptor messenger‐ribonucleic‐acid levels in liver and growth‐plate. Endocrinology 135:1113–1118. [DOI] [PubMed] [Google Scholar]
- Hilton DJ. 1999. Negative regulators of cytokine signal transduction 8. Cell Mol Life Sci 55:1568–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston B, Seawright E, Jefferies D, Hoogland E, Lester D, Whitehead C, Farquharson C. 1999. Identification and cloning of a novel phosphatase expressed at high levels in differentiating growth plate chondrocytes. BBA‐Mol Cell Res 1448:500–506. [DOI] [PubMed] [Google Scholar]
- Idris AI, Sophocleous A, Landao‐Bassonga E, Canals M, Milligan G, Baker D, van't Hof. 2009. Cannabinoid receptor Type 1 protects against age‐related osteoporosis by regulating osteoblast and adipocyte differentiation in marrow stromal cells. Cell Metab 10:139–147. [DOI] [PubMed] [Google Scholar]
- Isaksson OGP, Lindahl A, Nilsson A, Isgaard J. 1987. Mechanism of the stimulatory effect of growth‐hormone on longitudinal bone‐growth. Endocr Rev 8:426–438. [DOI] [PubMed] [Google Scholar]
- Isgaard J, Nilsson A, Lindahl A, Jansson JO, Isaksson OG. 1986. Effects of local administration of GH and IGF‐1 on longitudinal bone growth in rats. Am J Physiol Endocrinol Metab 250:E367–E372. [DOI] [PubMed] [Google Scholar]
- Kiepe D, Ciarmatori S, Hoeflich A, Wolf E, Tonshoff B. 2005. Insulin‐like growth factor (IGF)‐I stimulates cell proliferation and induces IGF binding protein (IGFBP)‐3 and IGFBP‐5 gene expression in cultured growth plate chondrocytes via distinct signaling pathways. Endocrinology 146:3096–3104. [DOI] [PubMed] [Google Scholar]
- Li S, Crenshaw EB, Rawson EJ, Simmons DM, Swanson LW, Rosenfeld MG. 1990. Dwarf locus mutants lacking 3 pituitary cell‐types result from mutations in the POU‐domain gene Pit‐1. Nature 347:528–533. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(T)(‐Delta Delta C) method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Lorentzon M, Greenhalgh CJ, Mohan S, Alexander WS, Ohlsson C. 2005. Reduced bone mineral density in SOCS‐2‐deficient mice. Pediatr Res 57:223–226. [DOI] [PubMed] [Google Scholar]
- Lui JC, Finkielstain GP, Barnes KM, Baron J. 2008. An imprinted gene network that controls mammalian somatic growth is down‐regulated during postnatal growth deceleration in multiple organs. AM J Physiol‐Reg I 295:R189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A. 2001. Roles of growth hormone and insulin‐like growth factor 1 in mouse postnatal growth. Dev Biol 229:141–162. [DOI] [PubMed] [Google Scholar]
- MacRae VE, Burdon T, Ahmed SF, Farquharson C. 2006. Ceramide inhibition of chondrocyte proliferation and bone growth is IGF‐I independent. J Endocrinol 191:369–377. [DOI] [PubMed] [Google Scholar]
- MacRae VE, Ahmed SF, Mushtaq T, Farquharson C. 2007. IGF‐I signalling in bone growth: Inhibitory actions of dexamethasone and IL‐1 beta. Growth Horm IGF Res 17:435–439. [DOI] [PubMed] [Google Scholar]
- MacRae VE, Horvat S, Pells SC, Dale H, Collinson RS, Pitsillides AA, Ahmed SF, Farquharson C. 2009. Increased bone mass, altered trabecular architecture and modified growth plate organization in the growing skeleton of SOCS2 deficient mice 1. J Cell Physiol 218:276–284. [DOI] [PubMed] [Google Scholar]
- Martensson K, Chrysis D, Sävendahl L. 2004. Interleukin‐1beta and TNF‐alpha act in synergy to inhibit longitudinal growth in fetal rat metatarsal bones. J Bone Miner Res 19:1805–1812. [DOI] [PubMed] [Google Scholar]
- Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA, Hilton DJ, Alexander WS. 2000. Gigantism in mice lacking suppressor of cytokine signalling‐2. Nature 405:1069–1073. [DOI] [PubMed] [Google Scholar]
- Mohan S, Richman C, Guo RQ, Amaar Y, Donahue LR, Wergedal J, Baylink DJ. 2003. Insulin‐like growth factor regulates peak bone mineral density in mice by both growth hormone‐dependent and ‐independent mechanisms. Endocrinology 144:929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushtaq T, Bijman P, Ahmed SF, Farquharson C. 2004. Insulin‐like growth factor‐I augments chondrocyte hypertrophy and reverses glucocorticoid‐mediated growth retardation in fetal mice metatarsal cultures. Endocrinology 145:2478–2486. [DOI] [PubMed] [Google Scholar]
- Nilsson O, Parker EA, Hegde A, Chau M, Barnes KM, Baron J. 2007. Gradients in bone morphogenetic protein‐related gene expression across the growth plate. J Endocrinol 193:75–84. [DOI] [PubMed] [Google Scholar]
- Oh Y, Muller HL, Lee DY, Fielder PJ, Rosenfeld RG. 1993. Characterization of the Affinities of insulin‐like growth‐factor (Igf)‐binding proteins 1‐4 for Igf‐I, Igf‐II, Igf‐I/insulin hybrid, and Igf‐I analogs. Endocrinology 132:1337–1344. [DOI] [PubMed] [Google Scholar]
- Oh Y, Muller HL, Pham H, Rosenfeld RG. 1993. Demonstration of receptors for insulin‐like growth‐factor binding protein‐3 on Hs578T human breast‐cancer cells. J Bio Chem 268:26045–26048. [PubMed] [Google Scholar]
- Owen HC, Ahmed SF, Farquharson C. 2009. Chondrocyte p21WAF1/CIP1 expression is increased by Dexamethasone but does not contribute to Dexamethasone‐induced growth retardation in vivo. Calcified Tissue International 85:326–334. [DOI] [PubMed] [Google Scholar]
- Parker EA, Hegde A, Buckley M, Barnes KM, Baron J, Nilsson O. 2007. Spatial and temporal regulation of GH‐IGF‐related gene expression in growth plate cartilage. J Endocrinol 194:31–40. [DOI] [PubMed] [Google Scholar]
- Pass C, MacRae VE, Ahmed SF, Farquharson C. 2009. Inflammatory cytokines and the GH/IGF‐I axis: Novel actions on bone growth. Cell Biochem Func 27:119–127. [DOI] [PubMed] [Google Scholar]
- Pass C, MacRae VE, Huesa C, Ahmed SF, Farquharson C. 2012. SOCS2 is the critical regulator of GH action in murine growth plate chondrogenesis. J Bone Miner Res 27:1055–1066. [DOI] [PubMed] [Google Scholar]
- Reno PL, McBurney DL, Lovejoy CO, Horton WE, Jr . 2006. Ossification of the mouse metatarsal: Differentiation and proliferation in the presence/absence of a defined growth plate. Anat Record 288:104–118. [DOI] [PubMed] [Google Scholar]
- Rico‐Bautista E, Flores‐Morales A, Fernandez‐Perez L. 2006. Suppressor of cytokine signaling (SOCS) 2, a protein with multiple functions. Cytokine Growth Factor Rev 17:431–439. [DOI] [PubMed] [Google Scholar]
- Rico‐Bautista E, Greenhalgh CJ, Tollet‐Egnell P, Hilton DJ, Alexander WS, Norstedt G, Flores‐Morales A. 2005. Suppressor of cytokine signaling‐2 deficiency induces molecular and metabolic changes that partially overlap with growth hormone‐dependent effects. Mol Endocrinol 19:781–793. [DOI] [PubMed] [Google Scholar]
- Scheven BAA, Hamilton NJ. 1991. Longitudinal bone‐growth in vitro—effects of insulin‐like growth factor‐i and growth‐hormone. Acta Endocrinol 124:602–607. [DOI] [PubMed] [Google Scholar]
- Sims NA, Clement‐Lacroix P, Da PF Bouali, Moriggl N, Goffin R, Coschigano V, Gaillard‐Kelly K, Kopchick M, Baron J, Kelly R. 2000. Bone homeostasis in growth hormone receptor‐null mice is restored by IGF‐I but independent of Stat5. J Clin Invest 106:1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha YN, Salocks CB, Vanderlaan WP. 1975. Pituitary and serum concentrations of prolactin and Gh in snell dwarf mice. P Soc Exp Biol Mede 150:207–210. [DOI] [PubMed] [Google Scholar]
- Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Tornell J, Isaksson OGP, Jansson JO, Ohlsson C. 1999. Liver‐derived insulin‐like growth factor I (IGF‐I) is the principal source of IGF‐I in blood but is not required for postnatal body growth in mice. P Natl Acad Sci USA 96:7088–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets T, Vanbuuloffers S. 1983. A morphological‐study of the development of the tibial proximal epiphysis and growth plate of normal and dwarfed snell mice. Growth 47:145–159. [PubMed] [Google Scholar]
- Smeets T, Vanbuuloffers S. 1983. The influence of growth‐hormone, somatomedins, prolactin and thyroxine on the morphology of the proximal tibial epiphysis and growth plate of snell dwarf mice. Growth 47:160–173. [PubMed] [Google Scholar]
- Sornson MW, Wu W, Dasen JS, Flynn SE, Norman DJ, Carriere I, Ryan C, Miller AK, Zuo AP, Gleiberman L, Andersen AS, Beamer B, Rosenfeld WG. 1996. Pituitary lineage determination by the Prophet of Pit‐1 homeodomain factor defective in Ames dwarfism. Nature 384:327–333. [DOI] [PubMed] [Google Scholar]
- Spagnoli A, Torello M, Nagalla SR, Horton WA, Pattee P, Hwa V, Chiarelli F, Roberts CT, Rosenfeld RG. 2002. Identification of STAT‐1 as a molecular target of IGFBP‐3 in the process of chondrogenesis. J Bio Chem 277:18860–18867. [DOI] [PubMed] [Google Scholar]
- Staines KA, Mackenzie NCW, Clarkin CE, Zelenchuk L, Rowe PS, MacRae VE, Farquharson C. 2012. MEPE is a novel regulator of growth plate cartilage mineralization. Bone 51:418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CEH, Bates PC, Calder TA, Woodall SM, Pell JM. 1993. Potentiation of insulin‐like growth factor‐I (Igf‐I) activity by an antibody‐supportive evidence for enhancement of Igf‐I bioavailability in vivo by Igf binding proteins. Endocrinology 133:1462–1465. [DOI] [PubMed] [Google Scholar]
- Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A, Efstratiadis A. 2008. The hormonal action of IGF1 in postnatal mouse growth. P Matl Acad Sci USA 105:19378–19383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loon JJ, Bervoets DJ, Burger EH, Dieudonné SC, Hagen JW, Semeins CM, Doulabi BZ, Veldhuijzen JP. 1995. Decreased mineralization and increased calcium release in isolated fetal mouse long bones under near weightlessness. J Bone Miner Res 10:550–557. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhou J, Bondy CA. 1999. Igf1 promotes longitudinal bone growth by insulin‐like actions augmenting chondrocyte hypertrophy. Faseb Journal 13:1985–1990. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. 2004. Evidence supporting dual, IGF‐I‐independent and IGF‐I‐dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol 180:247–255. [DOI] [PubMed] [Google Scholar]
- Wang YM, Cheng ZQ, Elalieh HZ, Nakamura E, Nguyen MT, Mackem S, Clemens TL, Bikle DD, Chang WH. 2011. IGF‐1R Signaling in Chondrocytes Modulates Growth Plate Development by Interacting With the PTHrP/Ihh Pathway. J Bone Miner Res 26:1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters MJ, Kaye PL. 2002. The role of growth hormone in fetal development. Growth Horm IGF Res 12:137–146. [DOI] [PubMed] [Google Scholar]
- Wilsman NJ, Farnum CE, Leiferman EM, Fry M, Barreto C. 1996. Differential growth by growth plates as a function of multiple parameters of chondrocytic kinetics. J Orthopaed Res 14:927–936. [DOI] [PubMed] [Google Scholar]
- Wu YJ, Sun H, Yakar S, LeRoith D. 2009. Elevated levels of insulin‐like growth factor (IGF)‐I in serum rescue the severe growth retardation of IGF‐I null mice. Endocrinology 150:4395–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. 1999. Normal growth and development in the absence of hepatic insulin‐like growth factor I. P Natl Acad Sci USA 96:7324–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakar S, Rosen CJ, Beamer WG, Ackert‐Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR, LeRoith D. 2002. Circulating levels of IGF‐1 directly regulate bone growth and density. J Clin Invest 110:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakar S, Rosen CJ, Bouxsein ML, Sun H, Mejia W, Kawashima Y, Wu Y, Emerton K, Williams V, Jepsen K, Schaffler MB, Majeska RJ, Gavrilova O, Gutierrez M, Hwang D, Pennisi P, Frystyk J, Boisclair Y, Pintar J, Jasper H, Domene H, Cohen P, Clemmons D, LeRoith D. 2009. Serum complexes of insulin‐like growth factor‐1 modulate skeletal integrity and carbohydrate metabolism. FASEB J 23:709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Horn H, Ekstrom C, Ellis E, Olivecrona H, Einarsson C, Tally M, Ekstrom TJ. 2002. GH is a regulator of IGF2 promoter‐specific transcription in human liver. J Endocrinol 172:457–465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supporting Information Figure S1: SOCS2 regulation of GH induced postnatal metatarsal growth. Graphs showing (A) WT and (C) Socs2‐/‐ postnatal (PN) 3 metatarsal growth in response to GH (100 ng/ml) over a 12 day period. Dotted lines indicate points at which conditioned medium analysed. Data are presented as mean ± SEM. Significance GH versus control denoted by a P < 0.05, b P < 0.01, (n ≥ 6). (B) Transcript analysis of Socs1, 2, and 3 in WT metatarsals following 12 days GH (100 ng/ml) treatment. Data represented as means ± SEM. Significance from untreated metatarsals denoted by b P < 0.01, (n = 3).
Supporting Information Table S1: Primers used for genotyping and PCR analysis.
Supporting Information Table S2: IGF‐1, IGFBP3, and IGF‐2 protein levels in conditioned medium from PN3 WT metatarsals following 7, or 12 days GH (100 ng/ml) treatment.
Supporting Information Table S3.