

Abstract
Objective:
To directly estimate the frequency and penetrance of CAG repeat alleles associated with Huntington disease (HD) in the general population.
Methods:
CAG repeat length was evaluated in 7,315 individuals from 3 population-based cohorts from British Columbia, the United States, and Scotland. The frequency of ≥36 CAG alleles was assessed out of a total of 14,630 alleles. The general population frequency of reduced penetrance alleles (36–39 CAG) was compared to the prevalence of patients with HD with genetically confirmed 36–39 CAG from a multisource clinical ascertainment in British Columbia, Canada. The penetrance of 36–38 CAG repeat alleles for HD was estimated for individuals ≥65 years of age and compared against previously reported clinical penetrance estimates.
Results:
A total of 18 of 7,315 individuals had ≥36 CAG, revealing that approximately 1 in 400 individuals from the general population have an expanded CAG repeat associated with HD (0.246%). Individuals with CAG 36–37 genotypes are the most common (36, 0.096%; 37, 0.082%; 38, 0.027%; 39, 0.000%; ≥40, 0.041%). General population CAG 36–38 penetrance rates are lower than penetrance rates extrapolated from clinical cohorts.
Conclusion:
HD alleles with a CAG repeat length of 36–38 occur at high frequency in the general population. The infrequent diagnosis of HD at this CAG length is likely due to low penetrance. Another important contributing factor may be reduced ascertainment of HD in those of older age.
Huntington disease (HD) is a dominant neurodegenerative disorder marked by motor abnormalities, cognitive impairments, and behavioral disturbances.1 A CAG repeat longer than 35 trinucleotides in exon 1 of the Huntingtin gene (HTT) defines an allele capable of causing the HD phenotype, with reduced penetrance at 36–39 CAG repeats.2–4 The frequency of repeat alleles ≥36 CAG in the general population is unknown. Although many control cohorts have been assessed by exome or whole-genome sequencing,5 short-read technologies are unable to accurately assess HTT CAG repeat lengths.6 In this study, we estimate the frequency of individuals in the general population with ≥36 CAG repeats in several large, randomly ascertained samples from 3 Western countries, using current genetic diagnostic standards for HD.4
The smallest CAG repeat length associated with the HD phenotype is 36, but repeat lengths of 36–39 CAG have been found in asymptomatic elderly individuals, indicating that the lower end of the HD CAG repeat range is incompletely penetrant over the average human lifespan.7,8 Using our general population frequency of reduced penetrance CAG repeat alleles, we directly estimate the penetrance of 36–38 CAG repeats among individuals ≥65 years of age. We compare our direct penetrance rates to estimates from clinical cohorts in order to evaluate penetrance bias that may result from clinical ascertainment. To our knowledge, this study offers the first large-scale assessment of ≥36 CAG repeats in a randomly selected control population, and the first attempt to directly assess the penetrance of alleles in the reduced penetrance range.
METHODS
Study populations.
A total of 3,952 saliva-extracted DNA samples were obtained from the Coriell Personalized Medicine Collaborative (CPMC) Community Cohort.9 The CPMC advertised to health care providers at local hospitals as well as to the general public through coverage in local media and by word of mouth. Participants were 18 years of age or older. Participants were not recruited based on the presence or absence of any disease phenotype. Samples used for this study were limited to those who explicitly consented to have their samples used in research beyond the scope of the CPMC. Samples were anonymized before use in this study.
A total of 2,016 anonymous general population samples were obtained from a population-based research DNA sample set held by NHS Grampian, Scotland. All DNA was originally extracted from buccal swabs taken from participants of a historic antenatal cystic fibrosis screening study conducted in Aberdeen, Scotland.
General population controls from British Columbia, Canada, were obtained from a large archived cohort of samples previously collected for the purpose of lipid screening as part of routine clinical bloodwork. Donors were not recruited or collected on the basis of any specific lipid profile or disease and were reported to be representative of British Columbia's general population in that they were largely Caucasian individuals of Northern European ancestry. A subset of 1,600 permanently anonymized samples was randomly selected from the archived collection of approximately 6,000 DNA.
Standard protocol approvals, registrations, and patient consents.
For the CPMC, saliva samples were collected in person following a group informed consent session. Only individuals who consented to share their DNA were included in this analysis. The CPMC protocol was reviewed and approved by the institutional review board (IRB) of the Coriell Institute for Medical Research. In addition, the use of samples in this analysis was also reviewed and approved by the IRB of the Coriell Institute for Medical Research. Ethical approval to share fully anonymized material from the archived University of Aberdeen cohort was obtained from North of Scotland Research Ethics Committee (13/NS/0025). Ethical approval for the secondary use of archived anonymous samples from British Columbia was obtained from the University of British Columbia Children's and Women's Health Centre Clinical Research Ethics Board (UBC C&W CREB) (H06-70356). Ethical approval to examine CAG size distributions in all 3 populations was obtained from the UBC C&W CREB (H05-70532).
CAG and CCG repeat sizing.
All samples in this study were sized by an identical assay at the Centre for Molecular Medicine and Therapeutics at UBC in Vancouver, Canada. CAG and CCG repeat sizes were determined as previously described10 using fluorescently labeled primers flanking the CAG repeat (HD344F, 5'-HEX-CCTTCGAGTCCCTCAAGTCCTTC-3' and HD450R, 5'-GGCGGCGGTGGCGGCTGTTG-3') and CCG repeat (HD419F, 5'-AGCAGCAGCAGCAACAGCC-3' and HD482R, 5'-6FAM-GGCTGAGGAAGCTGAGGAG-3'). A third PCR encompassing both CAG and CCG sequences (HD344F, 5'-HEX-CCTTCGAGTCCCTCAAGTCCTTC-3' and HD482R, 5'-GGCTGAGGAAGCTGAGGAG-3') was used to exclude allelic dropout and to phase CAG and CCG sizes from the first 2 sizing assays.3,4,11 Sizing was performed relative to a control panel of sequenced CAG and CCG repeat lengths. CAG repeat alleles were classified as normal (<27 CAG), intermediate (27–35 CAG), or HD-associated (≥36 CAG). HD-associated CAG repeat alleles were classified as reduced penetrance (36–39 CAG) or full penetrance (≥40 CAG). From the original DNA sample sets, 46 CPMC samples, 201 University of Aberdeen samples, and 6 UBC samples failed CAG repeat sizing.
Haplotype analysis.
General population individuals with at least one CAG repeat ≥36 were genotyped at a minimum of 63 single nucleotide polymorphisms across the Huntington gene as previously described.12 Reconstructed risk haplotypes (A1, A2, or A3a) were phased to CAG repeat length by haplotype-specific CCG and CAG repeat length associations previously described10,12 or by haplotype homozygosity.
Frequency and age-dependent penetrance estimates.
Bayesian binomial 95% confidence intervals (CI) for all CAG allelic and genotypic frequencies were calculated using binom and ggplot2 in R. For allelic frequency differences between general population samples, χ2 tests were performed using contingency tables. The number of individuals ≥65 years old in British Columbia was derived from Statistics Canada for the 2012 prevalence year and multiplied by the genotypic frequency of CAG 36, CAG 37, and CAG 38 in our general population sample to estimate the number of individuals ≥65 years old in British Columbia with each genotype. Patients ascertained in our 2012 British Columbia prevalence cohort with diagnostic CAG repeat lengths confirmed by the Molecular Genetics Laboratory at the British Columbia Children and Women's Hospital were included for estimates of age-dependent penetrance of CAG 36–39 alleles.13 To derive age-dependent penetrance, the number of clinically ascertained, genetically confirmed patients ≥65 years old with each reduced penetrance CAG repeat length was divided by the number of individuals ≥65 years old expected to have that repeat length in British Columbia in 2012. Extrapolated clinical penetrance rates for CAG 36, 37, and 38 by age 65 years were taken from multicenter data published by Langbehn et al.14
RESULTS
We assessed HTT CAG repeat length in 3,906 individuals from the adult general population of the United States, 1,815 individuals from the North of Scotland, and 1,594 previously reported individuals from British Columbia, Canada.10 Out of a total of 7,315 individuals (14,630 alleles), 18 individuals had HD-associated CAG repeat lengths ≥36 (0.246%, 95% CI 0.151%–0.380%) (table 1). Among individuals with HD-associated alleles, CAG 36–37 genotypes were the most common (36, 0.096%; 37, 0.082%; 38, 0.027%; 39, 0.000%; ≥40, 0.041%) (figure 1A). Approximately 1 in 16 individuals (6.20%) have an IA genotype (453/7,315; 95% CI 5.66%–6.76%), a small fraction of whom carry 2 IAs (5/7,315; 0.068%). Across all 3 populations, IAs occurred at an allelic frequency of 3.13% (458/14,630; 95% CI 2.86%–3.42%). HD-associated allele frequency (p = 0.485, χ2) and intermediate allele frequency (p = 0.080, χ2) did not significantly differ between samples from the United States, Scotland, and Canada.
Table 1.
Huntington disease (HD)–associated and intermediate allele frequency in the general population
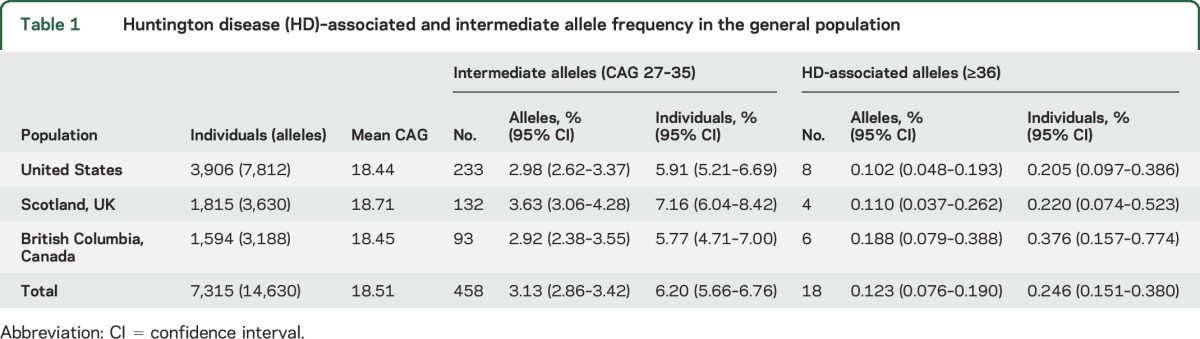
Figure 1. Distribution and haplotype of general population CAG repeat alleles in the Huntington disease (HD) range (≥36 CAG).

(A) HD CAG repeat distribution and frequency out of 14,630 total general population alleles. (B) Haplotypes of HD alleles in the general population conform to risk haplotypes (A1, A2, or A3a) of clinically ascertained, disease-causing HD mutations in patients of European ancestry.
We additionally sought to determine whether the randomly ascertained ≥36 CAG repeat alleles found in our general population sample occur on the same genetic background as those found in patients with clinically manifest HD. We performed HTT haplotype analysis in all 18 general population individuals with alleles ≥36 CAG repeats. All but one general population ≥36 CAG chromosome conformed to our previously established HTT haplotypes of disease-causing mutations in patients with manifest HD of European ancestry, A1, A2, or A3a (figure 1B),12,15 suggesting that HD-associated CAG repeat alleles in the general population occur on a similar cis-genetic background to those in patients from the clinic.
Fifteen of 18 (83.3%) incidentally ascertained ≥36 CAG repeats are considered HD alleles of reduced penetrance (CAG 36–39) by current American College of Medical Genetics diagnostic criteria (table 2).4 However, in our recent multisource prevalence study of HD in British Columbia, Canada, a much smaller proportion (15/353; 4.25%) of genetically diagnosed patients with validated CAG repeat lengths have CAG repeats in this range (figure 2A). Onset of HD typically occurs in adulthood, with length of the expanded CAG repeat inversely related to mean age at disease onset.14,16 Average age at onset is latest in patients with CAG repeats between 36 and 41, suggesting that individuals with reduced penetrance alleles are more likely to manifest HD in old age. Indeed, among symptomatic patients with a confirmed reduced penetrance genotype in British Columbia, 80% (12/15) were ≥65 years of age as of the April 1, 2012, prevalence date. We therefore sought to estimate the minimum penetrance of this allele class among individuals ≥65 years of age by dividing the number of clinically ascertained, symptomatic patients with each reduced penetrance CAG genotype by the expected number of individuals with each genotype in British Columbia (figure 2B).
Table 2.
Reduced penetrance and full penetrance Huntington disease (HD) allele frequency in the general population
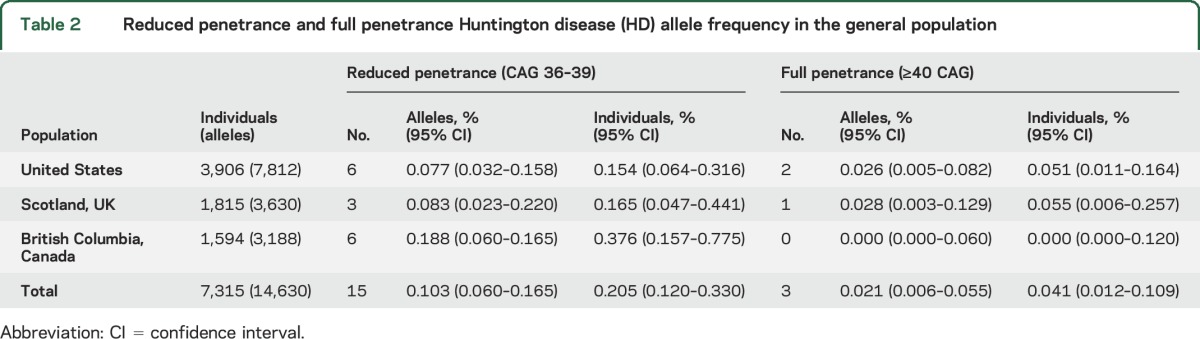
Figure 2. Estimated penetrance of CAG repeats in the CAG 36–39 range at ≥65 years of age.

(A) Frequency of alleles with reduced penetrance for Huntington disease in a multisource ascertainment of patients from British Columbia (BC), Canada. Proportions of patients ≥65 years of age are shown in black. (B) Estimated age-dependent penetrance rates of CAG 36–38 alleles in the general population as compared to clinical penetrance rates. Extrapolated clinical penetrance rates by 65 years of age derive from previously reported survival analysis of clinical onset data.14 Direct penetrance rates are calculated as the number of symptomatic patients ≥65 years of age in BC in 2012 with each CAG repeat genotype divided by the number of individuals ≥65 years of age in BC expected to have each expanded CAG repeat genotype.
CAG 37 is penetrant for HD in a minimum of 0.2% (95% CI 0.1%–0.4%) of individuals ≥65 years of age. CAG 38 is penetrant for HD in a minimum of 2.0% (95% CI 0.6%–9.8%) of individuals ≥65 years of age. No patient with a validated CAG 36 genotype was identified in our clinical prevalence cohort, precluding a penetrance estimate at this CAG length. However, one deceased patient with confirmed CAG 36 has been observed in the province since genetic testing began in 1995, and 7 CAG 36 alleles were observed in our general population sample, suggesting that the penetrance of this allele for the HD phenotype is less than CAG 37 but greater than zero. No CAG 39 alleles were observed in our general population control sample, and thus no expected frequency of CAG 39 genotypes among individuals ≥65 years of age could be calculated for a penetrance estimate. However, two-thirds (10/15) of genetically diagnosed British Columbia patients with validated 36–39 CAG repeat lengths were CAG 39, suggesting that the penetrance of this allele for HD is greater than for CAG 38. Extrapolated CAG 36–38 penetrance rates for HD by 65 years of age, derived from survival analysis of a large, multicenter clinical cohort, exceed our direct penetrance estimates across the entire reduced penetrance allele class, at 6%, 10%, and 19% by age 65 years for CAG 36, 37, and 38, respectively (figure 2B).14
DISCUSSION
We report an unexpectedly high frequency of HD-associated alleles in the reduced penetrance range of 36–39 CAG repeats. These data suggest that approximately 1 in 400 individuals have an expanded CAG repeat associated with HD in Western populations and that approximately 1 in 2,500 have an HD allele in the full penetrance range.
Previous attempts to calculate the frequency of the HD mutation have relied upon indirect methods of extrapolating heterozygote frequency from the prevalence of diagnosed HD.17 Our direct estimates show that the heterozygote frequency of ≥36 CAG in the general population is up to 10-fold higher than indirect estimates, but that the majority of these are HD alleles of reduced penetrance (CAG 36–39). The frequency of reduced penetrance alleles in the general population thus appears to be underestimated by clinical ascertainment, and individuals at risk of developing HD may be more numerous than previously believed. Low clinical ascertainment of CAG 36–38 alleles is supported by an inferred ascertainment rate of ≤5% in mutational flow models.18 It is notable that the frequency of fully penetrant HD alleles in our sample (0.041%; 95% CI 0.012%–0.109%) is compatible with prior indirect estimates.17 For example, we estimate that approximately 1 in 2,500 individuals have a full penetrance HD allele in the general population, or approximately 3.0 times the multisource prevalence rate for British Columbia.13
Our data also suggest the penetrance of CAG 36–39 for the typical HD phenotype may be lower than estimated from clinical datasets. CAG-specific penetrance rates of HD alleles have been attempted using clinical cohorts of mutation-positive individuals both with and without clinical signs of HD,14,19 but these estimates are thought to be biased for symptomatic individuals, resulting in a corresponding bias toward higher penetrance rates.20 Analysis of clinical records of manifest and premanifest individuals with 36–39 CAG approximated a 60% chance of clinical diagnosis by 65 years of age.19 We show a much higher frequency of 36–39 CAG repeat alleles in our general population sample than are clinically ascertained at this rate. For example, 15.8% of the population of British Columbia was ≥65 years of age in 2012. Given a 0.205% prevalence rate of 36–39 CAG genotypes in the general population, approximately 1,400 British Columbians ≥65 years of age would have had a 36–39 CAG allele in 2012, and at least 840 would have shown signs of HD given a 60% penetrance rate by age 65 years. In contrast, our comprehensive 2012 prevalence study of HD in British Columbia revealed only 15 affected patients with CAG 36–39, comprising only 4.25% of all verified genetic diagnoses of HD in the province, 13 of which were in patients ≥65 years of age.13 This modest proportion of 36–39 CAG alleles among genetically confirmed patients is comparable to clinical cohorts in Portugal, the Netherlands, and COHORT sites across the United States, Australia, and Canada.21–23
Expanded CAG repeats between 36 and 41 result in the oldest average age at onset in clinical cohorts, with many patients manifesting motor symptoms when older than 60 years.14,16 Late-onset HD may also be characterized by milder signs and slower progression, which elude diagnosis as HD and may be confounded by comorbid diseases of aging.24,25 The absence of family history or overt motor features may further prevent clinicians from considering a diagnosis of HD in late-onset cases. It is therefore possible that some individuals with reduced penetrance alleles escape diagnosis or do not seek medical services for early signs of HD. Given the high frequency of reduced penetrance alleles in our study, genetic testing for HD should be considered in those who present with suggestive but mild clinical features, particularly among the elderly. In premanifest individuals with ≥36 CAG repeats, clinical diagnosis of HD is known to be preceded by a prodromal stage of subtle motor and behavioral signs associated with neuronal loss in the caudate nucleus.26 At present, only individuals with clinically ascertained ≥36 CAG repeats have been evaluated for progressive prodromal phenotypes.27,28 It remains unclear how many elderly asymptomatic individuals with CAG 36–39 genotypes show prodromal signs of HD, and whether such individuals would inevitably progress to HD with increased longevity.
As sequencing technology improves, premanifest individuals without a family history of HD could incidentally discover the presence of an HD-associated allele in their genome. In the absence of population-based penetrance estimates for incidentally ascertained HD alleles, particularly in the 36–39 CAG repeat range, it will be challenging to counsel these individuals about their chance of developing HD.21 In this study, we estimate that 0.2% of individuals with CAG 37 and 2.0% of individuals with CAG 38 are symptomatic for HD among individuals ≥65 years of age. It is crucial to note that the number of individuals who manifest HD with a reduced penetrance allele will change with age structure, both within the ≥65 years of age category and in the population as a whole. Our penetrance estimate should therefore be considered in the context of the demographic structure of a specific population in a specific year. Additionally, our penetrance estimates only consider patients with a CAG 36–38 repeat allele confirmed within British Columbia. Some patients in our prevalence cohort with strictly clinical diagnoses or out-of-province genetic tests may have CAG 36–38 but are not included in our penetrance estimates. Our data nonetheless show a bias toward higher penetrance estimates derived from clinical onset data, and argue that the age-specific penetrance of ≥36 CAG repeat alleles for HD, particularly alleles CAG 36–38, may be lower than previously reported.
The frequency of intermediate alleles reported in this study is comparable to smaller samples.10,22 It is established that rates of paternal germline CAG repeat instability increase across the intermediate allele range, leading to a higher risk of expansion to ≥36 CAG repeat alleles in offspring.29,30 The unexpectedly high frequency of individuals with CAG 36–37 in this study suggests that fathers with reduced penetrance alleles, while at relatively low risk of developing HD themselves, may play a larger role in transmitting full penetrance HD alleles to the next generation than previously appreciated.
It is unclear why some individuals bearing HD alleles with reduced penetrance (36–39 CAG) manifest the classic HD phenotype as early as midlife, while others remain asymptomatic through advanced ages.19 Age at onset can vary widely between patients with the same expanded CAG repeat allele.16,31 The comparatively high frequency of reduced penetrance alleles in the general population suggests that additional genetic and environmental factors may modify the likelihood of manifesting HD from these alleles within a normal human lifespan.32 Given the small proportion of individuals with CAG 36–38 repeat alleles who manifest with clinical HD, genetic background may play a major role in the likelihood of developing the disease in patients with reduced penetrance genotypes.
We have shown an unexpectedly high frequency of HD alleles with reduced penetrance (CAG 36–39) in the general population. The manifestation of HD in individuals with CAG repeat alleles in the reduced penetrance range is likely low, but prodromal disease phenotypes may be underascertained among the elderly. Individuals with reduced penetrance alleles may also have an increased role in the expansion of CAG repeats into full penetrance alleles in their offspring.
Supplementary Material
GLOSSARY
- CI
confidence interval
- CPMC
Coriell Personalized Medicine Collaborative
- HD
Huntington disease
- IRB
institutional review board
- UBC C&W CREB
University of British Columbia Children's and Women's Health Centre Clinical Research Ethics Board
Footnotes
Editorial, page 247
AUTHOR CONTRIBUTIONS
Study design: C. Kay, Dr. Miedzybrodzka, Dr. Madore, and Dr. Hayden. Acquisition and consenting of samples: J.A. Collins, Dr. Miedzybrodzka, Dr. Madore, E.S. Gordon, Dr. Gerry, and M. Davidson. Genotyping and analysis: C. Kay, J.A. Collins, R.A. Slama. Manuscript and figures: C. Kay, J.A. Collins, Dr. Miedzybrodzka, E.S. Gordon, Dr. Hayden.
STUDY FUNDING
Funding for this study was provided by the Canadian Institutes of Health Research (CIHR: MOP-84438).
DISCLOSURE
C. Kay is funded by a Canadian Institutes of Health Research Doctoral Research Award. J. Collins, Z. Miedzybrodzka, and S. Madore report no disclosures relevant to the manuscript. E. Gordon is an employee of 23andMe. N. Gerry, M. Davidson, and R. Slama report no disclosures relevant to the manuscript. M. Hayden is President of Global R&D and Chief Scientific Officer of TEVA with financial interests in Teva Pharmaceuticals, and is funded by an operating grant from the Canadian Institutes of Health Research. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Bates GP, Dorsey R, Gusella JF, et al. . Huntington disease. Nat Rev Dis Primers 2015;1:15005. [DOI] [PubMed] [Google Scholar]
- 2.Kremer B, Goldberg P, Andrew SE, et al. . A worldwide study of the Huntington's disease mutation: the sensitivity and specificity of measuring CAG repeats. New Engl J Med 1994;330:1401–1406. [DOI] [PubMed] [Google Scholar]
- 3.ACMG/ASHG statement. Laboratory guidelines for Huntington disease genetic testing: The American College of Medical Genetics/American Society of Human Genetics Huntington Disease Genetic Testing Working Group. Am J Hum Genet 1998;62:1243–1247. [PMC free article] [PubMed] [Google Scholar]
- 4.Bean L, Bayrak-Toydemir P. American College of Medical Genetics and Genomics standards and guidelines for clinical genetics laboratories, 2014 edition: technical standards and guidelines for Huntington disease. Genet Med 2014;16:e2. [DOI] [PubMed] [Google Scholar]
- 5.Auton A, Brooks LD, Durbin RM, et al. . A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McIver LJ, Fondon JW III, Skinner MA, Garner HR. Evaluation of microsatellite variation in the 1000 Genomes Project pilot studies is indicative of the quality and utility of the raw data and alignments. Genomics 2011;97:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubinsztein DC, Leggo J, Coles R, et al. . Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am J Hum Genet 1996;59:16–22. [PMC free article] [PubMed] [Google Scholar]
- 8.McNeil SM, Novelletto A, Srinidhi J, et al. . Reduced penetrance of the Huntington's disease mutation. Hum Mol Genet 1997;6:775–779. [DOI] [PubMed] [Google Scholar]
- 9.Keller MA, Gordon ES, Stack CB, et al. . Coriell Personalized Medicine Collaborative: a prospective study of the utility of personalized medicine. Personalized Med 2010;7:301–317. [DOI] [PubMed] [Google Scholar]
- 10.Semaka A, Kay C, Doty CN, Collins JA, Tam N, Hayden MR. High frequency of intermediate alleles on Huntington disease-associated haplotypes in British Columbia's general population. Am J Med Genet B Neuropsychiatr Genet 2013;162:864–871. [DOI] [PubMed] [Google Scholar]
- 11.Potter NT, Spector EB, Prior TW. Technical standards and guidelines for Huntington disease testing. Genet Med 2004;6:61–65. [DOI] [PubMed] [Google Scholar]
- 12.Kay C, Collins JA, Skotte NH, et al. . Huntingtin haplotypes provide prioritized target panels for allele specific silencing in Huntington disease patients of European ancestry. Mol Ther 2015;23:1759–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fisher ER, Hayden MR. Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord 2014;29:105–114. [DOI] [PubMed] [Google Scholar]
- 14.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet 2004;65:267–277. [DOI] [PubMed] [Google Scholar]
- 15.Warby SC, Montpetit A, Hayden AR, et al. . CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am J Hum Genet 2009;84:351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JM, Ramos EM, Lee JH, et al. . CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 2012;78:690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walker DA, Harper PS, Wells CE, Tyler A, Davies K, Newcombe RG. Huntington's chorea in South Wales: a genetic and epidemiological study. Clin Genet 1981;19:213–221. [DOI] [PubMed] [Google Scholar]
- 18.Falush D, Almqvist EW, Brinkmann RR, Iwasa Y, Hayden MR. Measurement of mutational flow implies both a high new-mutation rate for Huntington disease and substantial underascertainment of late-onset cases. Am J Hum Genet 2001;68:373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quarrell OW, Rigby AS, Barron L, et al. . Reduced penetrance alleles for Huntington's disease: a multi-centre direct observational study. J Med Genet 2007;44:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langbehn DR, Hayden MR, Paulsen JS. CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet 2010;153B:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maat-Kievit A, Losekoot M, Van Den Boer-Van Den Berg H, et al. . New problems in testing for Huntington's disease: the issue of intermediate and reduced penetrance alleles. J Med Genet 2001;38:E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sequeiros J, Ramos EM, Cerqueira J, et al. . Large normal and reduced penetrance alleles in Huntington disease: instability in families and frequency at the laboratory, at the clinic and in the population. Clin Genet 2010;78:381–387. [DOI] [PubMed] [Google Scholar]
- 23.Dorsey E. Characterization of a large group of individuals with Huntington disease and their relatives enrolled in the COHORT study. PLoS One 2012;7:e29522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.James CM, Houlihan GD, Snell RG, Cheadle JP, Harper PS. Late-onset Huntington's disease: a clinical and molecular study. Age Ageing 1994;23:445–448. [DOI] [PubMed] [Google Scholar]
- 25.Lipe H, Bird T. Late onset Huntington disease: clinical and genetic characteristics of 34 cases. J Neurol Sci 2009;276:159–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross CA, Aylward EH, Wild EJ, et al. . Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014;10:204–216. [DOI] [PubMed] [Google Scholar]
- 27.Paulsen JS, Long JD, Ross CA, et al. . Prediction of manifest Huntington's disease with clinical and imaging measures: a prospective observational study. Lancet Neurol 2014;13:1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tabrizi SJ, Scahill RI, Owen G, et al. . Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol 2013;12:637–649. [DOI] [PubMed] [Google Scholar]
- 29.Semaka A, Kay C, Doty C, et al. . CAG size-specific risk estimates for intermediate allele repeat instability in Huntington disease. J Med Genet 2013;50:696–703. [DOI] [PubMed] [Google Scholar]
- 30.Semaka A, Creighton S, Warby S, Hayden MR. Predictive testing for Huntington disease: interpretation and significance of intermediate alleles. Clin Genet 2006;70:283–294. [DOI] [PubMed] [Google Scholar]
- 31.Wexler NS, Lorimer J, Porter J, et al. . Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci USA 2004;101:3498–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J-M, Wheeler Vanessa C, Chao Michael J, et al. . Identification of genetic factors that modify clinical onset of Huntington's disease. Cell 2015;162:516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.