

Abstract
Background & Aims
Acetaminophen (APAP)-induced liver injury is the most frequent cause of acute liver failure in the US and many other countries. Metabolism of APAP results in formation of APAP protein adducts (APAP-AD) in hepatocytes and triggers mitochondrial dysfunction and necrosis. However, the mechanisms for how APAP-AD are removed from hepatocytes remain unknown.
Methods
Mice or primary hepatocytes were treated with APAP. APAP-AD were determined by immunoblot, immunostaining and HPLC-ED analysis.
Results
We found that APAP-AD were detected at 1 hr, peaked at approximately 2 hrs, declined at 6 hrs and almost full removed at 24 hrs post treatment with APAP in mouse livers and in primary mouse hepatocytes. APAP-AD displayed a punctate pattern and were co-localized with GFP-LC3 positive autophagosomes and Lamp1 positive lysosomes in APAP-treated primary hepatocytes. Moreover, isolated autophagosomes and autolysosomes from APAP-treated mouse livers contained APAP-AD, suggesting autophagy may selectively remove APAP-AD. APAP-AD were detected in both detergent soluble and insoluble pools in APAP-treated mouse livers and hepatocytes. More importantly, pharmacological inhibition of autophagy by leupeptin or chloroquine increased whereas induction of autophagy by Torin 1 decreased serum APAP-AD levels in APAP-treated mice, which correlated with alanine aminotransferase levels and liver necrosis. Furthermore, SQSTM1/p62, an autophagy receptor protein, was recruited to APAP-AD. Adenovirus-mediated shRNA knockdown of SQSTM1/p62 led to increased APAP-AD and necrosis in primary hepatocytes.
Conclusions
Our data indicate that APAP-AD are removed through selective autophagy. Pharmacological induction of autophagy may be a novel promising approach for treating APAP-induced liver injury.
Keywords: acetaminophen, autophagy, p62/SQSTM1, acetaminophen protein adducts, liver injury
Graphical Abstract
Introduction
Acetaminophen (APAP) is a safe drug at therapeutic levels, but an overdose can cause severe liver injury and even acute liver failure in animals and man [1]. It is well established that the formation of a reactive metabolite, presumably N-acetyl-p-benzoquinone imine (NAPQI), which causes the depletion of cellular glutathione (GSH) and APAP protein adduct (APAP-AD) formation, is critical for the initiation of toxicity [1–4]. Initially, it was hypothesized that protein binding directly causes cell death [5]. However, this hypothesis was later refined by suggesting that critical proteins within cells were being adducted. Despite substantial efforts and some success in identifying APAP-AD, consequences of protein binding on certain enzyme activities in the cell could not explain the massive cell necrosis observed after APAP overdose [6, 7]. However, it was recognized that APAP overdose causes mitochondrial dysfunction, which is critical to trigger hepatocyte necrosis and subsequent liver injury [8–10]. The effects on mitochondria are thought to be initiated by APAP-AD formation. This hypothesis originated from the comparison between APAP, where mitochondrial APAP-AD and toxicity were observed, and 3′-hydroxyacetanilide (AMAP), a nonhepatotoxic regioisomer of APAP, which shows similar overall protein binding in the mouse liver but no adducts in mitochondria [11]. More importantly, similar to the mouse model, APAP-AD are formed in patients after APAP overdose [12], and we demonstrated that mitochondrial damage is an important feature of APAP-induced liver injury in patients [8, 13]. Thus, APAP-AD formation is the critical initiation event for mitochondrial damage, which is vital for the amplification of cell death signaling events that determine APAP-induced necrosis and liver injury [14]. Therefore, timely removal of APAP-AD and damaged mitochondria to maintain normal mitochondrial function and provide sufficient ATP is a critical process necessary for the recovery of mice and humans from APAP intoxication [15, 16]. Despite the importance of APAP-AD formation, how hepatocytes remove APAP-AD is unknown.
Cells can respond and adapt to various forms of stress, which may limit the extent of cell death. One adaptation and protective mechanism to control cellular stress, protein homeostasis and maintain mitochondrial quality is autophagy. Autophagy is a highly conserved intracellular degradation pathway, which is activated in response to adverse environmental conditions, such as the deprivation of nutrients or growth factors, as a survival mechanism [17, 18]. Autophagy protects cells by selectively removing toxic protein aggregates and damaged mitochondria [19–21]. We previously demonstrated that APAP activates autophagy in primary cultured hepatocytes and in mouse livers to remove damaged mitochondria [21–23]. However, whether APAP-induced autophagy can also help remove APAP-AD is not known. Therefore, our goal in the present study was to assess the role and mechanisms of APAP-induced autophagy in removal of APAP-AD in primary cultured mouse and human hepatocytes as well as in a mouse model.
Materials and Methods
Antibodies and Reagents
Antibodies used in the study were p62 (Abnova), β-Actin (Sigma), GAPDH (Cell Signaling), Lamp1 (Developmental Studies Hybridoma Bank, Iowa City, IA), and the rabbit anti-APAP-adducts antibody was kindly provided by Dr. Lance Pohl (National Heart, Lung and Blood Institute) [24]. The rabbit polyclonal anti-LC3 antibody was described previously [25]. The secondary antibodies and other reagents were described in Supplemental Materials.
Animal experiments
Wild-type mice were maintained in a C57BL/6 background. All animals received humane care and the procedures were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center. Mice were either given saline (i.p.) or APAP (500 mg/kg, i.p.). Mice were sacrificed at 0.5, 1, 2, 6 and 24 hrs. In some experiments, mice were treated with chloroquine (CQ, 60 mg/kg, i.p.) or leupeptin (Leu, 40 mg/kg, i.p.) simultaneously with APAP (500 mg/kg) for 3 and 6 hrs, respectively. For post treatment experiments, mice were either given saline or APAP (500 mg/kg, i.p) for 2 hrs followed by administration of vehicle (4% methyl-β-cyclodextrin in saline, i.p.) or Torin 1 (2 mg/kg, dissolved in 4% methyl-β-cyclodextrin in saline, i.p.) for another 6 hrs. Liver injury was assessed by the determination of serum alanine aminotransferase (ALT) activity and hematoxylin and eosin (H&E) staining of liver sections as we described previously [22]. Total liver lysates were prepared using RIPA buffer (1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl (lauryl) sulfate).
Acquisition of human samples
Blood samples were obtained from patients admitted to the University of Kansas Hospital in Kansas City, Kansas following APAP overdose and from healthy volunteers, as previously described [13].
Hepatic GSH content and APAP-protein adducts measurement
GSH was measured using a modified Tietze assay, as described [26]. APAP-cysteine (APAP-Cys) adducts were measured by high pressure liquid chromatography (HPLC) with electrochemical detection (ED) as we described previously [23]. Namely, sample dialysis was replaced with a rapid gel-filtration method to remove free APAP and APAP-GSH prior to digestion and analysis. Protein levels were determined using a standard bicinchoninic acid assay, and adducts were normalized to total protein in the liver homogenates.
Primary hepatocytes culture
Murine and human hepatocytes were isolated by a retrograde, nonrecirculating perfusion of livers with 0.05% Collagenase Type IV (Sigma) as described previously [27, 28]. All cells were maintained in a 37°C incubator with 5% CO2.
Immunoblot analysis for autophagy markers and APAP-AD from human blood samples
Twenty micrograms of serum proteins from healthy human volunteers and APAP-exposed humans were subjected to immunoblot analysis for APAP-AD and LC3. Human blood samples were obtained from APAP overdose patients and healthy volunteers as described previously [13].
Cellular fractionation for autophagosomes and lysosomes, preparation for detergent soluble and insoluble liver fractions, immunoblot assay, electron microscopy and fluorescence confocal microscopy were described in detail in Supplemental Materials.
Statistical Analysis
Experimental data were subjected to Student t-test or one way analysis of variance analysis with Scheffé’s post hoc test where appropriate.
Results
Time-dependent changes of APAP-AD levels following APAP treatment
Using a specific antibody against APAP-AD and HPLC-ED analysis for APAP-cysteine (APAP-Cys) adducts, we performed a time-course study to determine dynamic changes of APAP-AD formation in the mouse liver following APAP treatment. We found three major APAP-adducted proteins at approximately 32, 45, and 60 kDa in APAP-treated mouse liver, and the 45 kDa protein was the most abundant (Fig. 1A). These bands were specific for APAP because they were barely detected in the control samples, and the pattern was consistent with previous reports using similar antibodies [29, 30]. APAP-AD increased after 1 hr treatment with APAP and reached a maximum around 2 hrs after treatment. APAP-AD began to decline at 6 hrs and further declined at 24 hrs after treatment. These results from western blot analysis were consistent with the results that were obtained by HPLC-ED analysis for APAP-AD (Fig. 1B). Hepatic GSH levels started to decline at 0.5 hr and reached the lowest levels at 2 hrs after APAP administration. The GSH levels started to recover at 6 hrs following APAP administration and were completely recovered at 24 hrs (Fig. 1C). These data indicate that the depletion of GSH after APAP treatment correlates with increased formation of APAP-AD.
Figure 1. The dynamic change of APAP-AD levels in mouse livers.

Male C57BL/6J mice were treated with APAP (500 mg/kg) for different time points and pooled liver lysates from 3–4 mice of each group were subjected to western blot analysis (A) or HPLC-ED analysis (B) for APAP-AD (means ± SE, n=3–4). (C) Mice were treated as in (A) and total hepatic GSH contents were determined and data are presented as means ± SE (n=3–5). * P<0.05. One way anova analysis with Scheffé’s post hoc test.
Autophagy compartments are associated with APAP-AD
We next determined dynamic changes of APAP-AD in primary cultured mouse hepatocytes. We found that the levels of APAP-AD peaked at 6 hrs and declined at 24 hrs after APAP treatment in primary cultured mouse hepatocytes (Fig. 2A), which is similar to the changes of APAP-AD that we found in APAP-treated mouse livers (Fig. 1A). APAP treatment also increased autophagic flux in cultured hepatocytes as demonstrated by the time-dependent degradation of the autophagy substrate protein p62. The levels of LC3-II also decreased between 0.5 and 6 hrs, likely due to increased numbers of autolysosomes, which degrade LC3-II along with autophagic cargo after APAP treatment. Interestingly, the level of LC3-II increased again 24 hrs after APAP treatment, which was likely due to increased formation of early autophagosomes (Fig. 2A). Immunostaining for APAP-AD revealed that APAP treatment increased APAP-AD signals compared to non-treated control cells (Fig. 2B). Interestingly, APAP-AD signals displayed a punctate peri-nuclear pattern, where lysosomes are enriched. Indeed, more than 20% of GFP-LC3 puncta were colocalized with APAP-AD signals, which were further increased to approximately 50% in the presence of CQ, indicating that APAP-AD could be enveloped by GFP-LC3 positive AP/autolysosomes (Fig. 2B & C). The increased colocalization of GFP-LC3 puncta with APAP-AD by CQ treatment could be due to blocking of autophagic degradation of APAP-AD. Moreover, we found that APAP-AD signals also colocalized with Lamp1, a lysosomal marker, and this colocalization was enhanced in the presence of CQ (Supplemental Fig 1). These findings indicate that autophagosomal and lysosomal compartments are closely associated with APAP-AD in hepatocytes. To further confirm that APAP-AD can be enveloped in the AP and lysosomes (Ly) in the liver, we purified AP and Ly from saline, APAP, APAP+CQ or CQ-treated mouse livers using a Nycodenz gradient centrifugation approach. These fractions were confirmed by WB analysis for the AP marker (LC3-II) and Ly marker (Lamp1) (Fig. 2D–E). Interestingly, APAP-AD were detected in both AP and Ly fractions in APAP-treated mouse livers but not in control saline-treated mouse livers (Fig. 2D). Moreover, the levels of APAP-AD were higher in Ly fractions isolated from APAP+CQ-treated mice than APAP treatment alone (Fig. 2E). The WB analysis results were also confirmed by HPLC analysis for APAP-Cys using purified Ly fractions, which showed a 2 fold increase of APAP-AD in Ly compartments in the presence of CQ (Fig. 2F). These results indicate that APAP-AD are indeed enclosed in AP and Ly compartments. Blocking lysosomal degradation by CQ further enhanced the colocalization of APAP-AD with autolysosomal compartments
Figure 2. Autophagy compartments are associated with APAP-AD.


(A) Primary cultured mouse hepatocytes were treated with APAP (5 mM) for different time points and total cell lysates were subjected to western blot analysis. (B) Primary mouse hepatocytes were infected with adenovirus GFP-LC3 (10 moi) overnight and then treated with APAP (5 mM) in the presence or absence of CQ (20 μM) for 6 hrs. Cells were fixed and immunostained for APAP-AD followed by confocal microscopy. Representative images are shown. The right panel image was an enlarged photograph from the boxed area. Arrows denote the colocalization of GFP-LC3 positive puncta with APAP-AD. GFP-LC3 puncta that were colocalized with APAP-AD per cell were quantified and data are presented as means ± SE (n=3 independent experiments, more than 15 cells were counted) (C). * P<0.05. One way anova analysis with Scheffé’s post hoc test. (D & E) Male C57BL/6J mice were treated with saline, APAP (300 mg/kg), CQ (60 mg/kg) or APAP (300 mg/kg)+CQ (60 mg/kg) for 4 hrs. Mice were sacrificed and subjected to Nycodenz gradient centrifugation. Total lysates and isolated AP and Ly fractions were subjected to WB analysis. (F) Ly factions (200 μg) were combined from two mice and were further digested with proteinase K followed by HPLC analysis for APAP-Cys adducts.
We next determined the level of APAP-AD when mice were treated with APAP in the presence or absence of leupeptin (Leu) or chloroquine (CQ), which inhibits autophagy degradation by blocking lysosomal proteases or increasing lysosomal pH. We found that serum ALT levels were almost 5-fold higher in APAP and Leu-treated mice compared to APAP alone-treated mice (Fig. 3A), suggesting that inhibition of autophagy by Leu further exacerbated APAP-induced liver injury, which is in agreement with our previous findings using CQ to inhibit autophagy [22]. To our surprise, the total liver APAP-AD levels were very comparable between the APAP and APAP plus Leu treatment groups. Leu alone and APAP plus Leu treatments increased LC3-II and p62 levels, indicating that Leu treatment indeed blocked hepatic autophagy (Fig. 3B). However, we found that APAP treatment increased blood APAP-AD levels, which were markedly enhanced in the presence of Leu likely due to elevated hepatocyte necrosis by Leu (Fig. 3C). Similar results were observed when autophagy was blocked by CQ (Supplemental Fig. 2). These data suggest that blocking autophagy may lead to overall increase of APAP-AD when hepatic and serum APAP-AD are taken into consideration. Interestingly, we found that inhibition of autophagy by Leu enhanced serum ALT levels almost 4-fold higher than APAP treatment alone as early as 3 hours (Supplemental Fig. 3A). Moreover, we also detected increased serum APAP-AD levels in APAP plus Leu treated mice but not in APAP alone-treated mice at such an early time point (Supplemental Fig. 3B). These data suggest that inhibition of autophagy may lower the threshold time to develop APAP-induced liver injury.
Figure 3. Inhibition of autophagy by Leu increases serum APAP-AD levels and liver injury in mice.


Male C57BL/6J mice were treated with APAP (500 mg/kg), or APAP+Leu (40 mg/kg) or Leu (40 mg/kg) for 6 hrs. (A) Serum ALT levels were analyzed and data are presented as means ± SE (n=3–5). * P<0.05. One way anova analysis with Scheffé’s post hoc test. (B) Total liver lysates and (C) serum were subjected to western blot analysis for APAP-AD. Data (means ± SE) were from the densitometry analysis of LC3-II levels compared to the control groups. The membrane for the serum samples was further stained with Poncea red to serve as the loading control. Detergent soluble (D) and insoluble fractions (E) were prepared from the liver tissues and subjected to western blot analysis for APAP-AD.
We only detected 34 kDa APAP-AD on isolated mitochondria but not the 45 and 60 kDa APAP-AD, suggesting the 34 kDa APAP-AD may be specific for mitochondria. Furthermore, we found 45 and 60 kDa APAP-AD were the predominant forms of APAP-AD, whereas only a minimal amount of 34 kDa APAP-AD and no mitochondrial proteins (VDAC and Tom20) were detected in the serum (Supplemental Fig. 4A & B). These data suggest that autophagic removal of APAP-AD could be a specific separate event rather than just serving as a surrogate marker for removal of damaged mitochondria that are adducted by APAP. Since autophagy mainly degrades intracellular protein aggregates that are often detergent insoluble [20], we wondered whether APAP-AD could also form detergent insoluble aggregates in hepatocytes. Indeed, we found that APAP treatment induced both detergent soluble and insoluble APAP-AD, and the detergent insoluble APAP-AD levels were more predominant than the detergent soluble levels (Supplemental Fig. 5). Leu plus APAP treatment slightly decreased the detergent soluble APAP-AD and p62 levels but markedly increased the detergent insoluble APAP-AD and p62 levels (Fig. 3D & E), suggesting that blocking autophagy degradation results in selective accumulation of detergent insoluble APAP-AD and p62. Collectively, these data suggest that APAP-AD are associated with the autophagy-lysosomal pathway, and blocking autophagy degradation increases insoluble APAP-AD levels and necrosis in the liver as well as blood APAP-AD levels in mice.
Post- and co-treatment with Torin 1 protects against APAP-induced liver injury
To determine whether pharmacological stimulation of autophagy after the peak of adduct formation can attenuate APAP-induced liver injury by accelerated removal of APAP-AD, mice were treated with Torin 1 (an mTOR inhibitor and autophagy inducer) 2 hrs after APAP administration. Therapeutic Torin 1 treatment markedly decreased blood ALT and APAP-AD levels at 8 hrs (Fig. 4A & D) and improved necrosis in the liver (Fig. 4B) although did not affect the total hepatic APAP-AD levels (Fig. 4C). The levels of phosphorylated 4E-BP1 decreased by APAP or Torin 1 treatment (Fig. 4C), suggesting that Torin 1 inhibits mTOR in the mouse livers. Co-treatment of Torin 1 with APAP reduced blood APAP-AD to almost undetectable levels, but it did not affect hepatic APAP-AD levels. As a result, Torin 1 also significantly reduced APAP-induced increased serum ALT levels (Supplemental Fig. 6). These results suggest that pharmacological induction of autophagy may be beneficial for treating APAP-induced liver injury even after APAP is metabolized and APAP-AD is generated.
Figure 4. Activation of autophagy by Torin 1 decreases serum APAP-AD levels and liver injury in a post-APAP mouse model.

Male C57BL/6J mice were first treated with APAP (500 mg/kg) for 2 hrs, then some mice were further treated with Torin 1 (2 mg/kg) or vehicle (4% methyl-β-cylcodextran in saline, i.p.) for another 6 hrs. (A) Serum ALT levels were analyzed and data are presented as means ± SE (n=4–5). * P<0.05. One way anova analysis with Scheffé’s post hoc test. (B) Representative H & E staining images were shown. (C) Total liver lysates and (D) serum were subjected to western blot analysis for APAP-AD. The membrane for the serum samples was further stained with Poncea red to serve as the loading control.
p62 promotes autophagic removal of APAP-AD to reduce APAP-induced hepatotoxicity
We found that a small portion of APAP-AD stained positive for p62 in APAP-treated hepatocytes, which was markedly enhanced in the presence of CQ (arrows, Fig. 5A). These results together with the colocalization of APAP-AD with GFP-LC3 puncta (Fig. 2B–C) suggest that p62 could serve as a receptor for selective removal of APAP-AD by autophagy. Knockdown of p62 using an Adenovirus shRNA against p62 markedly reduced endogenous p62 levels in primary cultured hepatocytes but markedly enhanced APAP-AD levels in the soluble fractions but not in the insoluble fractions following APAP treatment (Fig. 5B). Knockdown of p62 also significantly reduced the colocalization of APAP-AD with GFP-LC3 puncta (Supplemental Fig. 7A–C). Moreover, knockdown of p62 significantly enhanced APAP-induced necrosis in primary mouse hepatocytes compared with the shRNA control vector infected cells (Fig. 5C & D). These results suggest that p62 is associated with selective autophagic degradation of APAP-AD, which is an important cellular protective mechanism against APAP-induced cell death.
Figure 5. p62 is associated with APAP-AD and knockdown of p62 impairs the clearance of APAP-AD and enhanced APAP-induced hepatotoxicity.


(A) Primary cultured mouse hepatocytes were treated with APAP (5 mM) in the presence or absence of CQ (20 μM) for 6 hrs. Cells were fixed and immunostained for APAP-AD and p62 followed by confocal microscopy. Representative images are shown. The right panel images were enlarged photographs from the boxed areas. Arrows denote the co-localization of p62 proteins with APAP-AD. (B) Primary cultured mouse hepatocytes were infected with either an adenovirus negative shRNA or adenovirus p62 shRNA for 24 hrs and then treated with APAP (5 mM) for 6 hrs. Total cell lysates, detergent soluble and insoluble fractions were subjected to western blot analysis. The levels of APAP-AD were quantified by densitometry analysis and showed below the blot (n=3 independent experiments). (C) Experiments were performed as in (B) and the cells were further treated with APAP (5 mM) for 24 hrs. The cells were stained with PI (1 μg/mL) followed by fluorescence microscopy. Representative images for the overlayed phase-contrast and PI stained images are shown. The number of PI positive cells were quantified and data are presented as means ± SE (n=3 independent experiments, more than 100 cells were counted in each experiment) (D). * P<0.05. One way anova analysis with Scheffé’s post hoc test.
Autophagy regulates APAP-AD in human hepatocytes
We found that APAP treatment induced typical necrosis (APAP 33% ± 3 vs control 11% ± 2) as demonstrated by propidium iodide (PI) staining in primary human hepatocytes (Fig. 6A & B). Similar to murine hepatocytes, APAP treatment increased the level of APAP-AD and inhibition of autophagy by CQ further enhanced the levels of APAP-AD in primary cultured human hepatocytes (Fig. 6C). These results suggest that the role of autophagy in removing APAP-AD is conserved from mouse to human.
Figure 6. Autophagy regulates APAP-AD in human hepatocytes.

Primary cultured human hepatocytes were treated with APAP (10 mM) for 24 hrs. The cells were further stained with PI (1 μg/mL) followed by fluorescence microscopy. Representative images for the overlayed phase-contrast and PI stained images are shown (A). The number of PI positive necrotic cells were quantified and data are presented as means ± SE (n=3 independent experiments, more than 100 cells were counted in each experiment) (B). * P<0.05. One way anova analysis with Scheffé’s post hoc test. (C) Primary cultured human hepatocytes were treated with APAP (10 mM) in the presence or absence of CQ for 24 hrs. Total cell lysates were subjected to western blot analysis.
Serum levels of APAP-AD and autophagy markers in APAP overdosed humans
Since we found that modulating autophagy could affect the serum levels of APAP-AD in mice, we next determined the serum levels of APAP-AD in APAP overdose human patients that had low and high ALT values. We found that the serum levels of APAP-AD increased in APAP overdose humans with higher serum ALT levels (range: 3,131 – 5,365 U/L). APAP-AD were almost undetectable in APAP overdose patients with lower ALT levels (6 – 47 U/L) who did not develop liver injury. While we did not detect any LC3-II proteins in the blood samples from healthy human volunteers, we found LC3-II levels increased in all APAP overdose patients regardless of the ALT values (Fig. 7A). These results suggest that the serum levels of APAP-AD and LC3-II may be used as biomarkers to monitor APAP-induced liver injury. The possible molecular events for how autophagy regulates APAP-AD levels and APAP-induced liver injury are summarized in Fig. 7B.
Figure 7. Serum levels of APAP-AD and autophagy proteins in humans.


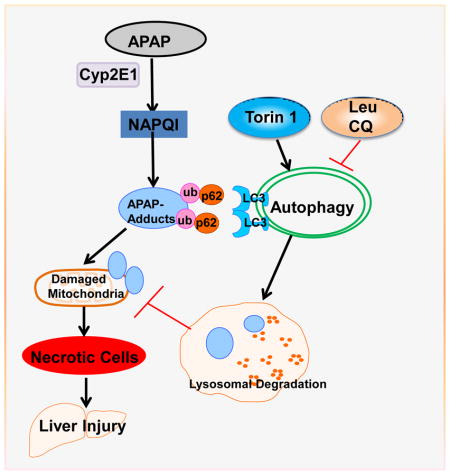
(A) Forty micrograms of serum from healthy volunteers or APAP overdose patients with either low or high ALT values were subjected to western blot analysis. The membrane was further stained with Poncea red to serve as the loading control. Lysates from 293 cells were used as a positive control for LC3-II. (B) A proposed model for the regulation of APAP-AD and APAP-induced hepatotoxicity by autophagy. In hepatocytes, APAP is first metabolized through the cytochrome P450 enzymes (mainly via CYP2E1) and generates reactive metabolites NAPQI, which bind to cellular and mitochondrial proteins to form APAP-AD. Mitochondrial APAP-AD can trigger mitochondrial damage. Damaged mitochondria can lead to necrotic cell death and subsequent liver injury. The autophagy receptor protein p62 is recruited to APAP-AD, which may facilitate APAP-AD transition to the detergent insoluble form and allow their recognition and enwrappment by autophagosomes. Pharmacological induction of autophagy (Torin 1) or inhibition of autophagy (Leu or CQ) improves or impairs autophagic removal of APAP-AD and results in protection or exacerbation of APAP-induced necrosis and liver injury, respectively.
Discussion
Autophagy plays a critical role in liver physiology and various liver diseases including non-alcoholic and alcoholic liver diseases, viral hepatitis, protein aggregates, drug-induced liver injury and liver tumorigenesis [19]. We have previously demonstrated that pharmacological induction of autophagy protects against APAP-induced liver injury, likely via removal of APAP-induced damaged mitochondria [21, 22]. The purpose of the present study was to investigate whether autophagy is involved in removing APAP-AD.
It is generally agreed that the metabolism of APAP mainly occurs in the liver, and the majority of APAP (approximately 90%) is linked to sulfide or glucuronide and secreted via the kidneys. The rest, approximately 5–10% is further metabolized via cytochrome P450 enzymes (mainly Cyp2E1) to generate NAPQI. NAPQI further conjugates with cellular GSH without obvious harmful effects on hepatocytes under therapeutic dosing. Upon exposure to APAP overdose, cellular GSH is depleted and NAPQI then forms covalent links with the sulfhydryl group on cysteine components of many proteins to form APAP-AD. NAPQI adducted proteins may alter the functions of these proteins and contribute to APAP-induced liver injury. Indeed, the serum levels of APAP-AD are associated with APAP-induced liver injury in both mice and humans [15, 31]. Therefore, many early works have attempted to identify the key APAP-adducted proteins using either immunoblot analysis for subcellular liver fractions or mass spectrometry-based proteomic analysis of liver lysates from APAP overdose mice [30, 32]. Many of the proteins adducted by APAP identified from these earlier studies are cytoplasmic enzymes. However, the activities of these enzymes were only marginally affected by APAP adduction and seem not to be essential for APAP-induced hepatotoxicity [33]. Moreover, 3′-hydroxyacetanilide or N-acetyl-meta-aminophenol (AMAP), a regioisomer of APAP, can form protein adducts in mouse and human hepatocytes but only causes hepatotoxicity in humans [8]. Compared with APAP, fewer mitochondrial protein adducts are induced by AMAP in mice, but the amount of mitochondrial protein adducts are significantly increased by AMAP in primary cultured human hepatocytes [8]. Rats are resistant to APAP-induced liver injury because rats have fewer mitochondrial protein adducts compared with mice after APAP overdose despite almost the same amount of hepatic protein adducts are induced [34]. Thus, it appears that it is not the total amount of protein adducts formed but the sub-cellular location of the protein adducts that is important for hepatotoxicity triggered either by AMAP or APAP. Specifically, the formation of mitochondrial protein adducts is critical for mitochondrial damage and subsequent necrosis. Therefore, understanding the mechanisms by which APAP-AD are regulated after formation in hepatocytes may help to attenuate APAP-induced liver injury.
In the present study, we reported for the first time that APAP-AD were detected in isolated autophagosomes and autolysosmes from APAP-treated mouse livers. APAP-AD were closely associated with GFP-LC3 positive autophagosomes and Lamp1 positive lysosomes in primary cultured mouse hepatocytes. Moreover, pharmacological inhibition of lysosomal functions increased the retention of APAP-AD in autophagosomes and autolysosomes. Furthermore, APAP-AD had both detergent soluble and insoluble portions in the liver and cultured hepatocytes. Results from other cellular protein aggregate studies have revealed that cellular soluble protein aggregates can be removed by the ubiquitin proteasome system, but the cellular insoluble protein aggregates can only be removed via autophagy [20]. Interestingly, we found that inhibition of lysosomal functions by Leu markedly increased the portion of detergent insoluble APAP-AD in APAP-treated mouse livers. Taken together, our data suggest that autophagy is an important mechanism for the regulation of APAP-AD levels in hepatocytes.
One of the intriguing findings in our present study was that modulation of hepatic autophagy (either by Torin 1-mediated induction of autophagy or by Leu- or CQ-mediated inhibition of autophagy) did not alter the total hepatic APAP-AD levels. However, induction of autophagy (Torin 1 treatment) significantly decreased whereas inhibition of autophagy (Leu or CQ treatment) increased blood APAP-AD levels. Increased blood APAP-AD levels have been detected in APAP overdose mice and humans as well as in humans with a therapeutic dose of APAP, which is likely due to the leakage of APAP-AD into the blood as a result of APAP-induced hepatocyte necrosis [15, 31, 34]. Indeed, mice that were treated with APAP and Torin 1 had decreased blood APAP-AD and ALT levels and less liver necrotic areas. Conversely, mice that were treated with either Leu or CQ had markedly increased blood APAP-AD and ALT levels and more liver necrotic areas. Our results thus suggest that the levels of blood APAP-AD but not the hepatic APAP-AD levels seem to correlate with blood ALT levels and liver injury. In addition to the protective mitophagy that we previously demonstrated [21], results from the present study indicate that autophagy may also protect against APAP-induced liver injury by removing APAP-AD.
Accumulating evidence now supports that selective autophagic removal of protein aggregates and organelles is mediated by an autophagy-cargo receptor complex, which includes ubiquitin and p62 [35, 36]. Mechanistically, p62 directly binds to poly- or mono-ubiquitin through its C-terminal ubiquitin-associated domain (UBA). p62 also binds directly with LC3 via its LC3-interaction region (LIR), and thus acts as a cargo adapter for ubiquitinated proteins and links them to autophagy degradation [37]. We found that p62 was co-localized with APAP-AD in primary hepatocytes. Moreover, detergent insoluble APAP-AD levels decreased whereas detergent soluble APAP-AD levels increased in APAP-treated p62 knockdown hepatocytes. These results suggest that p62 may bind to APAP-AD and facilitate APAP-AD transition to the detergent insoluble “protein aggregate” like structure, which can only be removed by autophagy. Thus, p62 may serve as a selective receptor for APAP-AD for their selective autophagic removal. Increased necrosis in APAP-treated p62 knockdown cells suggests that p62-mediated selective autophagy for APAP-AD plays a role in protection against APAP-induced necrosis. However, in addition to p62, several other autophagy receptor proteins such as NBR1 (Neighbor of BRCA gene 1) and ALFY (autophagy linked FYVE protein) have been reported to play a role in autophagic removal of protein aggregates [38, 39]. Whether NBR1 or ALFY also plays a role in autophagic removal of APAP-AD in hepatocytes remains to be studied.
In conclusion, we demonstrated that APAP-AD co-localized with GFP-LC3 positive autophagosomes, and isolated autophagosomes and autolysosomes contained APAP-AD in mouse livers. p62 was associated with APAP-AD, and knockdown of p62 increased hepatic APAP-AD levels and necrosis. Pharmacological modulation of autophagy can regulate the turnover of APAP-AD.
Supplementary Material
Lay Summary.
Acetaminophen overdose can form acetaminophen protein adducts and mitochondria damage in hepatocytes resulting in liver injury. Activation of autophagy-lysosomal degradation pathway can help to remove acetaminophen protein adducts. Pharmacological induction of autophagy may be a novel promising approach for treating APAP-induced liver injury.
Acknowledgments
Funding
This study was supported in part by the NIH funds R01 AA020518 (W.X.D), R01 DK102142 (W.X.D. & H.J), National Center for Research Resources (5P20RR021940), the National Institute of General Medical Sciences (8P20 GM103549), T32 ES007079, and an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20 GM103418).
List of Abbreviations
- AP
Autophagosome
- APAP
Acetaminophen
- APAP-AD
Acetaminophen protein adducts
- AMAP
3′-hydroxyacetanilide
- ALT
Alanine aminotransferase
- CQ
Chloroquine
- CYP2E1
Cytochrome P450 2E1
- GSH
Glutathione
- HPLC-ED
High pressure liquid chromatography with electrochemical detection
- LC3
Microtubule-associated protein light chain 3
- Leu
Leupeptin
- Ly
Lysosome
- NAPQI
N-acetyl-p-benzoquinone imine
- mTOR
Mechanistic target of rapamycin
- NAC
N-acetylcysteine
- PI
propidium iodide
Footnotes
Contributions of the authors: H.M.N, M.M.R., C.X., D.K., W.J.A., X.Y. and W.X.D performed experiments. W.X.D and H.J. conceived the idea and supervised and wrote the manuscript. Everyone read this manuscript and approved for submission. There is no conflict of interests to claim for all the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis. 2007;11:525–548. vi. doi: 10.1016/j.cld.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 2.McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 2013;30:2174–2187. doi: 10.1007/s11095-013-1007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol. 2010:369–405. doi: 10.1007/978-3-642-00663-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson SD. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin Liver Dis. 1990;10:267–278. doi: 10.1055/s-2008-1040482. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther. 1973;187:185–194. [PubMed] [Google Scholar]
- 6.Cohen SD, Khairallah EA. Selective protein arylation and acetaminophen-induced hepatotoxicity. Drug Metab Rev. 1997;29:59–77. doi: 10.3109/03602539709037573. [DOI] [PubMed] [Google Scholar]
- 7.Pumford NR, Halmes NC, Hinson JA. Covalent binding of xenobiotics to specific proteins in the liver. Drug Metab Rev. 1997;29:39–57. doi: 10.3109/03602539709037572. [DOI] [PubMed] [Google Scholar]
- 8.Xie Y, McGill MR, Du K, Dorko K, Kumer SC, Schmitt TM, et al. Mitochondrial protein adducts formation and mitochondrial dysfunction during N-acetyl-m-aminophenol (AMAP)-induced hepatotoxicity in primary human hepatocytes. Toxicol Appl Pharmacol. 2015 doi: 10.1016/j.taap.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol Sci. 2001;62:212–220. doi: 10.1093/toxsci/62.2.212. [DOI] [PubMed] [Google Scholar]
- 10.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283:13565–13577. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tirmenstein MA, Nelson SD. Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3′-hydroxyacetanilide, in mouse liver. J Biol Chem. 1989;264:9814–9819. [PubMed] [Google Scholar]
- 12.James LP, Alonso EM, Hynan LS, Hinson JA, Davern TJ, Lee WM, et al. Detection of acetaminophen protein adducts in children with acute liver failure of indeterminate cause. Pediatrics. 2006;118:e676–681. doi: 10.1542/peds.2006-0069. [DOI] [PubMed] [Google Scholar]
- 13.McGill MR, Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122:1574–1583. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev. 2012;44:88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGill MR, Lebofsky M, Norris HR, Slawson MH, Bajt ML, Xie Y, et al. Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications. Toxicol Appl Pharmacol. 2013;269:240–249. doi: 10.1016/j.taap.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heard KJ, Green JL, James LP, Judge BS, Zolot L, Rhyee S, et al. Acetaminophen-cysteine adducts during therapeutic dosing and following overdose. BMC Gastroenterol. 2011;11:20. doi: 10.1186/1471-230X-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Czaja MJ, Ding WX, Donohue TM, Jr, Friedman SL, Kim JS, Komatsu M, et al. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–150. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- 21.Williams JA, Ni HM, Haynes A, Manley S, Li Y, Jaeschke H, et al. Chronic Deletion and Acute Knockdown of Parkin Have Differential Responses to Acetaminophen-induced Mitophagy and Liver Injury in Mice. J Biol Chem. 2015;290:10934–10946. doi: 10.1074/jbc.M114.602284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–232. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ni HM, Boggess N, McGill MR, Lebofsky M, Borude P, Apte U, et al. Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol Sci. 2012;127:438–450. doi: 10.1093/toxsci/kfs133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryan PM, Bourdi M, Korrapati MC, Proctor WR, Vasquez RA, Yee SB, et al. Endogenous interleukin-4 regulates glutathione synthesis following acetaminophen-induced liver injury in mice. Chem Res Toxicol. 2012;25:83–93. doi: 10.1021/tx2003992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding WX, Ni HM, Gao W, Chen X, Kang JH, Stolz DB, et al. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Mol Cancer Ther. 2009;8:2036–2045. doi: 10.1158/1535-7163.MCT-08-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther. 1990;255:935–941. [PubMed] [Google Scholar]
- 27.Ding WX, Ni HM, DiFrancesca D, Stolz DB, Yin XM. Bid-dependent generation of oxygen radicals promotes death receptor activation-induced apoptosis in murine hepatocytes. Hepatology. 2004;40:403–413. doi: 10.1002/hep.20310. [DOI] [PubMed] [Google Scholar]
- 28.Ni HM, Du K, You M, Ding WX. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am J Pathol. 2013;183:1815–1825. doi: 10.1016/j.ajpath.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welch KD, Wen B, Goodlett DR, Yi EC, Lee H, Reilly TP, et al. Proteomic identification of potential susceptibility factors in drug-induced liver disease. Chem Res Toxicol. 2005;18:924–933. doi: 10.1021/tx050011b. [DOI] [PubMed] [Google Scholar]
- 30.Pumford NR, Hinson JA, Benson RW, Roberts DW. Immunoblot analysis of protein containing 3-(cystein-S-yl)acetaminophen adducts in serum and subcellular liver fractions from acetaminophen-treated mice. Toxicol Appl Pharmacol. 1990;104:521–532. doi: 10.1016/0041-008x(90)90174-s. [DOI] [PubMed] [Google Scholar]
- 31.James LP, Farrar HC, Sullivan JE, Givens TG, Kearns GL, Wasserman GS, et al. Measurement of acetaminophen-protein adducts in children and adolescents with acetaminophen overdoses. J Clin Pharmacol. 2001;41:846–851. doi: 10.1177/00912700122010744. [DOI] [PubMed] [Google Scholar]
- 32.Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem. 1998;273:17940–17953. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- 33.Pumford NR, Halmes NC, Martin BM, Cook RJ, Wagner C, Hinson JA. Covalent binding of acetaminophen to N-10-formyltetrahydrofolate dehydrogenase in mice. J Pharmacol Exp Ther. 1997;280:501–505. [PubMed] [Google Scholar]
- 34.McGill MR, Williams CD, Xie Y, Ramachandran A, Jaeschke H. Acetaminophen-induced liver injury in rats and mice: comparison of protein adducts, mitochondrial dysfunction, and oxidative stress in the mechanism of toxicity. Toxicol Appl Pharmacol. 2012;264:387–394. doi: 10.1016/j.taap.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manley S, Williams JA, Ding WX. Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp Biol Med (Maywood) 2013;238:525–538. doi: 10.1177/1535370213489446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015 doi: 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- 37.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Isakson P, Holland P, Simonsen A. The role of ALFY in selective autophagy. Cell Death Differ. 2013;20:12–20. doi: 10.1038/cdd.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.