

Abstract
An emerging number of non-chemokine mediators are found to bind to classical chemokine receptors and to elicit critical biological responses. Macrophage migration inhibitory factor (MIF) is an inflammatory cytokine that exhibits chemokine-like activities through non-cognate interactions with the chemokine receptors CXCR2 and CXCR4, in addition to activating the type II receptor CD74. Activation of the MIF-CXCR2 and -CXCR4 axes promotes leukocyte recruitment, mediating the exacerbating role of MIF in atherosclerosis and contributing to the wealth of other MIF biological activities. Although the structural basis of the MIF-CXCR2 interaction has been well studied and was found to engage a pseudo-ELR and an N-like loop motif, nothing is known about the regions of CXCR4 and MIF that are involved in binding to each other. Using a genetic strain of Saccharomyces cerevisiae that expresses a functional CXCR4 receptor, site-specific mutagenesis, hybrid CXCR3/CXCR4 receptors, pharmacological reagents, peptide array analysis, chemotaxis, fluorescence spectroscopy, and circular dichroism, we provide novel molecular information about the structural elements that govern the interaction between MIF and CXCR4. The data identify similarities with classical chemokine-receptor interactions but also provide evidence for a partial allosteric agonist compared with CXCL12 that is possible due to the two binding sites of CXCR4.
Keywords: chemokine, cytokine, G protein-coupled receptor (GPCR), protein-protein interaction, structure-function, protein mapping
Introduction
G protein-coupled receptors (GPCRs)6 are capable of interacting with a wealth of agonists ranging in diversity from atoms to large proteins (1). Chemokines are small chemotactic cytokines that activate GPCRs to orchestrate phagocyte and lymphocyte recruitment involved in the immune and inflammatory response. In addition, various homeostatic cell migration processes, including hematopoietic progenitor cell release from the bone marrow, are regulated by chemokines and their receptors (2–4). Because of the critical role of chemokines and their receptors in numerous pathophysiological processes, including tumor metastasis, inflammatory disease, infection, and atherosclerosis among other diseases, these proteins are potential therapeutic targets (5–9). Thus far, plerixafor (AMD3100) and maraviroc are Food and Drug Administration-approved drugs targeting CXCR4 and CCR5 for hematopoietic stem cell mobilization and inhibition of HIV cell entry, respectively.
Chemokines and chemokine receptors are classified in the subfamilies C, CC, CXC, and CX3C based on the cysteine motif near the N terminus (10, 11). About 45 human chemokines and 20 chemokine receptors have been identified, indicating a significant degree of redundancy for chemokine agonists. In the CXC subfamily, chemokines are further divided into the subgroups defined by the presence of a Glu-Leu-Arg motif (ELR+) prior to the cysteine motif or its absence (ELR−) (10). CXCR1 and CXCR2 are receptors for the ELR+ subgroup, promote inflammatory and atherogenic recruitment of monocytes and neutrophils, and support angiogenic responses. In contrast, most receptors for ELR− chemokines are angiostatic and serve to regulate homeostatic cell migration (12). The monomeric chemokines from all subfamilies exhibit remarkably conserved tertiary structures but, with some exceptions, can vary dramatically in subfamily-dependent dimeric and oligomeric forms (12). Recent progress in GPCR crystallography has led to the elucidation of the three-dimensional structures of several receptors complexed to agonists, inverse agonists, antagonists, allosteric regulators, and G proteins (13–17). The structure of the chemokine receptor CXCR4 has been solved with two small molecule ligands and the herpesvirus-8 chemokine vMIP-II (18, 19), whereas the structure of CCR5 has been solved with the Food and Drug Administration-approved compound maraviroc (20). The structure of the human chemokine CX3CL1 in complex with the human cytomegalovirus GPCR US28 has also been solved (21).
Most chemokines have a two-site mechanism for binding their receptors (22). Site 1 involves interactions between the chemokine N-loop, which follows the cysteine motif, and the receptor N-terminal region. The interactions for the second site are between the chemokine N-terminal residues prior to the cysteine motif with the receptor extracellular loops (EL) and a transmembrane cavity (19, 22). For CXCR4 and its canonical agonist CXCL12 (SDF-1α), the CXCL12 N-loop is composed of a RFFESH sequence, which interacts with the CXCR4 N-terminal region (19, 23). The disordered N-terminal region of CXCL12 engages mostly with EL2 and penetrates into the transmembrane cavity to activate CXCR4 (site 2) (22, 24). These two binding sites are also observed in the disulfide-bonded co-complex of vMIP-II·CXCR4, although the receptor N-terminal region is not entirely defined presumably due to its flexibility (19).
Macrophage migration inhibitory factor (MIF) is a pleiotropic inflammatory cytokine and a critical upstream mediator of innate immunity. Dysregulated MIF activity exacerbates autoimmune and inflammatory conditions, including septic shock, inflammatory lung diseases, autoimmune diseases, cancer, and atherogenesis (25–31). MIF contains an evolutionarily conserved catalytic cavity similar to microbial tautomerases/isomerases that use Pro-1 as a catalytic base (32). Although a physiological substrate has yet to be defined, MIF uses two pseudo-substrates for similar activities in cavities found between subunits (33, 34). MIF also binds and signals through three receptors: the CD74·CD44 complex, CXCR2, and CXCR4 (35–37). Engagement of MIF with these three different receptors is partly responsible for the varied biological activities associated with MIF. MIF is also unique because it is the only protein that activates both an ELR+ and ELR− chemokine receptor. The determinants that lead to biological function between MIF and CXCR2 or CD74 have been studied (38–40), but far less is known about the interactions with CXCR4.
In this study, mutagenesis, MIF inhibitors, and CXCR4 antagonists were used to determine whether the MIF catalytic site, its N-like loop region, and the CXCR4 transmembrane cavity were important for binding interactions. Peptide sequences from MIF or CXCR4 were studied using a variety of techniques to probe for interactions between peptide regions and the complementary full-length binding partner. MIF activation of CXCR3-CXCR4 hybrid receptors was also used to define regions of CXCR4 that interact with MIF. The results were mapped on the three-dimensional structures of MIF and CXCR4 to provide insight into interactions between the proteins and suggested a partial allosteric site and a unique mechanism of activation. MIF does not activate β-arrestin-2 or inhibit HIV-1 entry by two dual-tropic strains, providing evidence that the allosterism and a unique mechanism does not fully re-capitulate the biological functions of CXCL12-CXCR4.
Results
Functional Interactions of MIF-CXCR4
To eliminate complications from mammalian cells that might express more than one or all MIF cell surface receptors, we used a genetically modified strain of Saccharomyces cerevisiae that replaces the ste2 GPCR with human CXCR4. Additional genetic modifications in S. cerevisiae allow an agonist to activate CXCR4 leading to a signaling cascade that results in β-galactosidase expression from the Fus1-lacZ reporter plasmid. This system has been successfully used to identify a constitutive CXCR4 mutant (41) and two allosteric peptide agonists (42).
The MIF cDNA was cloned in-frame at the 3′-end of the S. cerevisiae α-factor secretion sequence (Fig. 1). The cloning strategy resulted in a double mutant of Pro-1 and Met-2 to valine and serine (P1V/M2S), respectively. (Because of the absence of a secretion sequence in the human MIF cDNA, the N-terminal proline is referred to as Pro-2 in some studies to indicate that it follows the initiating Met (43), although in more recent studies it is Pro-1 to indicate it is the N-terminal residue for the mature protein (39). The P1V/M2S mutations were changed back to wild-type MIF residues, and plasmids containing either wild-type MIF or the P1V/M2S double mutant were transformed into the S. cerevisiae strain. The P1V/M2S mutant allowed us to probe the contribution of Pro-1 and the catalytic cavity in CXCR4 signaling. CXCR4 agonist-induced β-galactosidase activity was measured from lysed cells. These studies verified that wild-type MIF functions as a CXCR4 weak partial agonist (Fig. 1, C and D), as determined previously in mammalian cells (37). The P1V/M2S mutant did not activate CXCR4, suggesting that the MIF catalytic cavity is important for binding or inducing CXCR4 signaling. A dose-response effect using extracellular MIF is observed, but an EC50 value cannot be obtained because a plateau cannot be reached at the higher concentrations. In comparison, exogenous CXCL12 has about a 100-fold increase of the EC50 value in S. cerevisiae relative to mammalian cells (data not shown). Among the possible explanations for the differences in dose response for exogenous protein agonist in S. cerevisiae compared with mammalian cells is access to CXCR4 in the membrane due to the yeast cell wall (44), the absence of a tyrosylprotein sulfotransferase homolog that sulfates CXCR4 tyrosine residues at the N-terminal region and increases affinity for CXCL12 (45), and the absence of a CD74·CXCR4 heterodimeric complex that may have higher affinity for MIF (46, 47). To determine whether there is any competition in the activation of CXCR4 between MIF and CXCL12, we measured the effect of increasing concentrations of MIF in the presence of a constant concentration of CXCL12 and observed a decrease in signaling with increased MIF concentrations (Fig. 1E). The decreased activity is due to displacement of CXCL12 by MIF-induced partial agonism (Fig. 1C) and decreased potency (Fig. 1D) relative to CXCL12.
FIGURE 1.

MIF signaling of CXCR4 in S. cerevisiae. A, pheromone response pathway in S. cerevisiae. Activation of the Ste 2 receptor by pheromone leads to a signaling cascade resulting in transcription of the pheromone response genes. B, CXCR4 replaces the Ste 2 receptor. Gpa1 is modified such that it can couple with CXCR4. Ste 14 and Far 2 are deleted to lead to a more robust signaling response. To measure the robustness of the response, the pheromone response genes are substituted with the lacZ gene, which is produced, and enzymatic activity is measured. C, comparison of the effects of co-expression of CXCR4 with CXCL12/SDF-1α, wild-type MIF, and the double mutant P1V/M2S MIF. D, dose-response effect of exogenous MIF added to CXCR4-expressing S. cerevisiae. The EC50 values cannot be measured because a concentration that reaches the maximum signaling cannot be obtained. E, functional competition between MIF and CXCL12 in activating CXCR4. Dose response of MIF in the presence of a constant concentration of CXCL12 (2 μm) results in a decrease in signaling due to the displacement of CXCL12 by the higher concentrations of the less potent MIF.
Pharmacological Studies of MIF-CXCR4 Interactions in S. cerevisiae
To test whether the CXCR4 transmembrane cavity is involved in MIF interactions, we tested the effects of the orthosteric antagonists AMD3100 and IT1t on MIF-induced CXCR4 activation. MIF was used alone or in the presence of either antagonist (Fig. 2A). Interestingly, higher concentrations of AMD3100 and IT1t were necessary to inhibit CXCR4 activation by MIF (Fig. 2A) relative to the more potent CXCL12 (Fig. 2B). The high concentrations of the antagonists suggest MIF binding and/or activation of CXCR4 occurs through a different mechanism than CXCL12. These results are also consistent with a maximum 50% displacement of CXCL12 binding by MIF in mammalian cells (37). The binding mechanism between CXCL12 and CXCR4 consists of a two-step mechanism with initial interactions between chemokine and receptor that subsequently allow the flexible N-terminal region of CXCL12 (23, 24) to enter the CXCR4 transmembrane cavity (22). The N-terminal region of MIF, however, is inflexible as Pro-1 is wedged among residues in the catalytic cavity, and residues 2–6 form a β-strand that is part of a core β-sheet of the protein (43).
FIGURE 2.

Signaling effects (β-galactosidase activity) of CXCR4 antagonists on MIF or CXCL12 agonism and of the MIF inhibitor ISO-1 on CXCR4 signaling. A, 1- or 5-fold excess concentration of IT1t and AMD3100 relative to the MIF concentration shows a dose-response effect that is moderate compared with B, where there is greater response for CXCL12 at equivalent concentrations to IT1t and AMD3100. C, MIF active site inhibitor ISO-1 has a clear dose-response effect at 0.1-, 1-, and 5-fold excess of MIF on CXCR4 signaling, indicating that the active site is involved in binding and/or signaling.
To determine whether inhibitors of the MIF catalytic cavity have any effect on CXCR4-mediated signaling, we used the prototypic MIF active site inhibitor, (SR)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid (ISO-1). We show a clear dose-response effect of ISO-1 with virtually no signaling at a 5-fold excess of ISO-1 relative to the MIF concentration (Fig. 2C). This indicates that the active site of MIF is critically important for activating CXCR4 and agrees with the lack of receptor activation from the MIF double mutant P1V/M2S at the catalytic site.
Characterization of CXCR4 Extracellular Regions Interacting with MIF
Having established that MIF and CXCR4 have a dissimilar component in their interactions relative to classical chemokine-receptor interactions, we investigated whether specific peptides from the three CXCR4 extracellular loops (EL1–3) are involved in MIF interactions. This analysis was similar to the peptide array approach used for probing CXCR2 interactions with MIF (38). With the exception of EL1, the peptide sequences were designed to be positionally shifted by three residues and subsequently immobilized on a glass slide. One peptide was used for the shorter EL1 consisting of residues 97–110. In addition to the extracellular loops, some of these peptides included the extracellular part of transmembrane helices for CXCR4 where interactions occur for vMIP-II based on the structure of CXCR4·vMIP-II complex (19). Five peptides constitute EL2 (residues 176–202), which contain residues for two anti-parallel β-strands and part of helix 5 in CXCR4. Similarly, the three peptides for EL3 (sequence 262–282) represent the loop with sequences from helices 6 and 7.
MIF interacted with peptides immobilized on glass slide-based array from EL1 and EL2(182–196), with an apparent decreasing affinity for peptide EL2(185–199) and minimal interactions for peptide EL2(188–202) (Fig. 3A). In the x-ray structure of CXCR4, residues 182–196 make up part of the second β-strand (from a two-stranded sheet), a loop, and the N-terminal part of transmembrane helix 6 (18, 19). Interestingly, single cysteine mutants on both vMIP-II and CXCR4 were screened to identify a disulfide bridge between the two proteins that would lead to co-crystallization. The disulfide that was crystallized involves a CXCR4 cysteine mutant at position 187 that is within the EL2(182–196) residues, which exhibits binding to MIF in this study (19). Consequently, if vMIP-II and CXCL12 have general similarities in binding CXCR4, these residues may be a source of competitive binding between MIF and CXCL12. There are no apparent interactions for peptides from EL3 (Fig. 3A).
FIGURE 3.

Characterization of CXCR4 extracellular regions interacting with MIF. A, peptide microarray analysis indicates that MIF interacts with extracellular loops (EL) EL1 and EL2 of CXCR4, while EL3 is not involved in the interaction. The interaction of biotinylated full-length recombinant MIF with glass slide-immobilized (“spotted”) peptides corresponding to the indicated sequences of the CXCR4 extracellular regions is shown as relative signal intensity. Gray vertical bar on the right axis indicates to which EL the spotted sequences correspond. B–D, circular dichroism spectropolarimetry confirms the role EL1 (B) and EL2 (C) in the binding interface with MIF, whereas no indication for a role of EL3 (D) was obtained. Recombinant MIF and extracellular loop peptides of CXCR4 were mixed in solution at 1:10 (for EL1) or 1:20 (for EL2 and EL3) molar ratios and spectra compared with the mathematical addition of the individual spectra (“sum of spectra”). Spectra of the individual peptides/proteins and mixtures are presented according to the indicated color code. Conformations and conformational changes in the CD spectra were measured as raw ellipticity versus the wavelength in the far-UV range.
To further confirm these findings under solution conditions and determine whether there are any changes in the secondary structure, we performed circular dichroism (CD) spectroscopy with MIF and CXCR4 extracellular loop peptides. The CD spectrum of MIF is typical of a protein with α-helices and β-sheets. As expected, the CXCR4 extracellular loops do not have secondary structure in the absence of the remaining receptor sequence (Fig. 3, B–D). If the CD spectrum upon mixing MIF and each CXCR4 extracellular loop is different from the additive spectra of MIF and each extracellular loop, it would indicate binding occurred with an induced conformational change. The induced change is likely to be in the CXCR4 loop, as the full-length structure of MIF is stable and unlikely to change its secondary structure upon peptide binding.
The CXCR4 EL1-, EL2-, and EL3-spanning peptide sequences of 100–110, 176–200, and 262–285, respectively, were synthesized by solid-phase peptide synthesis, and each CD spectrum was recorded. CD measurements of each loop peptide mixed with full-length human MIF were compared with the additive CD effects of MIF and each loop. The absence of any difference in the spectra of the MIF·CXCR4 loop complex and the additive CD spectra of MIF and a CXCR4 loop eliminates a change in secondary structure but does not preclude an interaction (48). The experiment and analysis with EL1 indicate this loop interacts with MIF (Fig. 3B). The CD spectrum of the EL2/MIF mixture is also substantially different from the additive CD effects of the single peptide and MIF spectra (Fig. 3C), whereas EL3 does not show any change in CD in the presence of MIF (Fig. 3D). These data suggest that the receptor regions EL1 and EL2 contribute to interactions with MIF. No conclusion can be made about any interaction with EL3 based on CD spectroscopy, although the absence of a change in CD is consistent with the lack of binding using the glass slide-based array approach.
Binding of MIF to the N-terminal Peptide of CXCR4
We examined whether the CXCR4 N-terminal region directly interacts with MIF using binding and functional assays. The 27-mer N-terminal sequence of CXCR4 (CXCR4(1–27) peptide) was probed for in vitro interaction by coating the peptide on a 96-well plate, incubating with 2 μg/ml biotinylated MIF, and competing with increasing concentrations of non-labeled MIF or a lysozyme control. There was a clear decrease of bound biotinylated MIF with increasing concentrations of unlabeled MIF and no decrease with lysozyme as a control (Fig. 4A). In a functional assay, we found the CXCR4(1–27) peptide inhibits peripheral blood mononuclear cell migration by MIF (Fig. 4B).
FIGURE 4.

Binding and functional assays between MIF and CXCR4(1–27). A, CXCR4(1–27) coated on 96-well plates and incubated with 150 μm biotinylated MIF was competed with increasing concentrations of MIF or lysozyme control. These results demonstrate a direct and specific interaction. B, CXCR4(1–27) blocks MIF-induced mononuclear cell migration. 1 × 106 cells/ml PBMCs were placed in the upper chamber of a 24-well cell culture insert. 8 nm hMIF were placed in the lower chamber with or without CXCR4(1–27). After 3 h of incubation, the transmigrated cells were fixed, stained, and counted. C, solution mixture of CXCR4(1–27) peptide with Alexa Fluor-labeled full-length human MIF evokes a conformational change in MIF leading to a change in Alexa fluorescence emission. Peptide was added at 1:1 molar ratio and at an excess of 250-, 500-, 750-, 1000-, and 2000-fold as indicated by color code, and fluorescence spectra were recorded between wavelengths 500 and 600 nm. D, no changes in the secondary structure of MIF and CXCR4(1–27) upon mixing the two molecules. CXCR4(1–27) was mixed with full-length human MIF, and potential changes in secondary structure were assessed by far-UV circular dichroism spectroscopy. The spectrum of the mixed molecules does not differ from that of the mathematical addition of the separate CD spectra.
Fluorescence spectroscopy was used to measure a binding interaction between Alexa-488-bound MIF and CXCR4(1–27). A Kd of >9.7 μm was calculated from the fluorescence changes with different concentrations of CXCR4(1–27) (Fig. 4C). However, CD spectroscopy did not reveal an additive effect between full-length MIF and peptide CXCR4(1–27), suggesting there are no changes in the secondary structure of MIF and CXCR4(1–27) upon mixing the two molecules (Fig. 4D). Residues 1–27 are not visible in any of the CXCR4 crystal structures, suggesting this part of the N-terminal region is flexible and does not have any secondary structure (18, 19). These results indicate that MIF binds to CXCR4(1–27) but does not result in perturbation or formation of any secondary structure.
Binding of MIF Regions to the CXCR4 N-terminal Peptide(1–27)
We next examined MIF peptides that bind to CXCR4(1–27). The glass slide-based array technology was used with immobilized 15-mer MIF peptides, with each peptide positionally shifted by three residues to produce peptides that covered the entire MIF sequence. These peptides were probed for binding to biotinylated CXCR4(1–27). This analysis is unable to probe sites such as the MIF catalytic cavity because the cavity is composed of residues across the entire protein sequence with some residues from an adjacent subunit. Nonetheless, the experiment indicated that a large interface from residues 43 to 98 are involved MIF-CXCR4(1–27) interactions (Fig. 5A). The most pronounced binding was obtained with the peptide containing residues 67–81. A role for residues 67–81 in the MIF-CXCR4 interaction was confirmed in a cellular assay that is based on the ability of chemokine receptors to interfere with the activity of adenylate cyclase through activation of Gαi2. Forskolin triggers cyclic AMP (cAMP) production in CHO cells. Human MIF was able to block forskolin-induced cAMP production in cells stably expressing full-length human CXCR4. Peptide MIF(67–81) was able to partly reverse the inhibitory effect of MIF through competitive binding, but a MIF-derived control peptide (MIF(13–27)) had no effect (Fig. 5B). The residues including the region around 67–81 are mapped to part of the second α-helix and a loop that is adjacent to the catalytic cavity between subunits. The electrostatic potential for these residues are shown in one subunit (for clarity) and indicate there is a positive potential (Fig. 5C).
FIGURE 5.
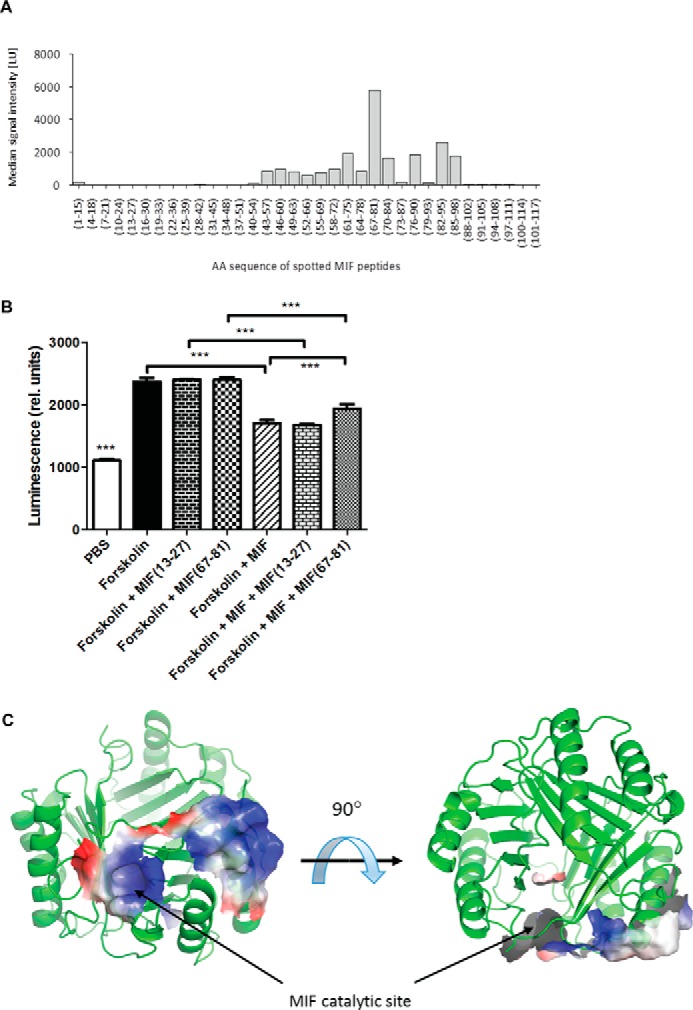
Identification of MIF binding regions to the N-terminal peptide of CXCR4. A, MIF residues 43–98, in particular 67–81, are involved in MIF-CXCR4(1–27) interactions. Peptide array technology was used with immobilized 15-mer MIF peptides, with each peptide positionally shifted by three residues to produce peptides that covered the entire MIF sequence. Peptides were probed for binding to biotinylated CXCR4(1–27). B, cell-based signaling assay confirms a role for residues 67–81 in the MIF-CXCR4 interaction. The ability of peptide 67–81 versus control peptide (residues 13–27) to reverse the inhibitory activity of MIF on forskolin-triggered cAMP production in stable CHO-CXCR4 transfectants was measured by Hit Hunter commercial cAMP test and readout as relative luminescence units. C, residues mapped on the ribbon structure of MIF identified by biochemical experiments in A and B to interact with CXCR4 mapped on a single subunit of MIF (for clarity) with the related electrostatic potential. The identified sites are the catalytic cavity and adjacent region involving part of the second α-helix. ***, p < 0.05.
Confirmation of the Interaction sites in CXCR4 Using Cell-expressed CXCR4/CXCR3 Chimeras
To study the contributions of peptide-protein-binding sites in an in situ three-dimensional context, chimeric receptors consisting of mixed CXCR4 and CXCR3 regions were constructed, and plasmids were transfected into CHO cells. In particular, the roles of CXCR4 extracellular loops and parts of the transmembrane helices that were close to the extracellular regions were evaluated. As seen in Fig. 5B, human MIF was able to markedly block forskolin-induced cAMP production in transfectants expressing full-length wild-type CXCR4. MIF does not bind to CXCR3 (37), thus allowing chimeric constructs to associate the cAMP-inhibitory effect of MIF to specific CXCR4 receptor regions. The chimeras shown in Fig. 6A were tested and compared with the full-length CXCR4 and CXCR3 wild-type receptors. Chimeras feature the following: (i) the N terminus of CXCR4; (ii) the CXCR4 N terminus through part of helix 3, including EL1; and (iii) the CXCR4 N terminus through part of helix 5 (including EL1 and EL2). When full-length CXCR4 was ectopically expressed in these cells, MIF inhibited forskolin-mediated cAMP production by 68% (Fig. 6, B and C). The chimeras featuring the CXCR4 N terminus together with EL1 and EL2 or EL1 alone of CXCR4 supported a 39% inhibitory effect of MIF (Fig. 6, B, D, and E). The chimera covering only the N terminus of CXCR4 still allowed for 28% inhibition by MIF (Fig. 6B and F), whereas MIF was unable to affect forskolin-triggered cAMP generation in an all-CXCR3 transfectant (Fig. 6, B and G) or in empty plasmid-transfected CHO cells (Fig. 6, B and H). Together, this underscores the contribution of the N terminus of CXCR4 to MIF binding and confirms the role of EL1 and/or EL2 in binding.
FIGURE 6.

Confirmation of the interaction sites in CXCR4 using cell-expressed CXCR4/CXCR3 chimeras. A, scheme summarizing the chimeras (N terminus of CXCR4, CXCR4 N terminus through part of helix 3, including EL1, and CXCR4 N terminus through part of helix 5 (including EL1 and EL2)) as well as all-CXCR4 and all-CXCR3 wild-type receptors. B–H, inhibitory effect of MIF on forskolin-induced cAMP production in CHO transfectants expressing various CXCR4/CXCR3 receptor chimeras as indicated and comparison with all-CXCR4 and all-CXCR3 wild-type receptors and empty plasmid-transfected controls. B, summary of experiments represented as relative inhibition of cAMP by MIF. C–H, inhibitory effect of MIF on cAMP production for individual receptor chimera or controls expressed as relative luminescence. C, all-CXCR4 wild-type receptor; D, CXCR4 N terminus through part of helix 5 (including EL1 and EL2); E, CXCR4 N terminus through part of helix 3, including EL1; F, N terminus of CXCR4; G, all-CXCR3 wild-type receptor; H, empty plasmid control. Control refers to CHO-transfectants without forskolin treatment. ***, p < 0.05.
β-Arrestin 2 Activation and Inhibition of HIV-1 Entry by MIF
Because CXCL12 activation of CXCR4 results in recruitment of β-arrestin 2 and endocytosis, MIF was also tested for this activity. The β-arrestin 2 activation experiment was performed in a firefly luciferase complementation assay where the N-terminal fragment of luciferase is fused to the CXCR4 C terminus, and the C-terminal fragment is fused to β-arrestin 2 in the human breast cancer cell line MDA-MB-231. Only when CXCR4 and β-arrestin 2 are in proximity will the N- and C-terminal fragments of luciferase interact and produce bioluminescence (49). MIF did not activate β-arrestin 2, whereas CXCL12 showed significant β-arrestin 2 activation (Fig. 7A) (50). The lack of any interaction is surprising given results from Jurkat T-cells that show 40% CXCR4 internalization by MIF (whereas CXCL12 results in 80% internalization), presumably induced by β-arrestin 2 (37).
FIGURE 7.
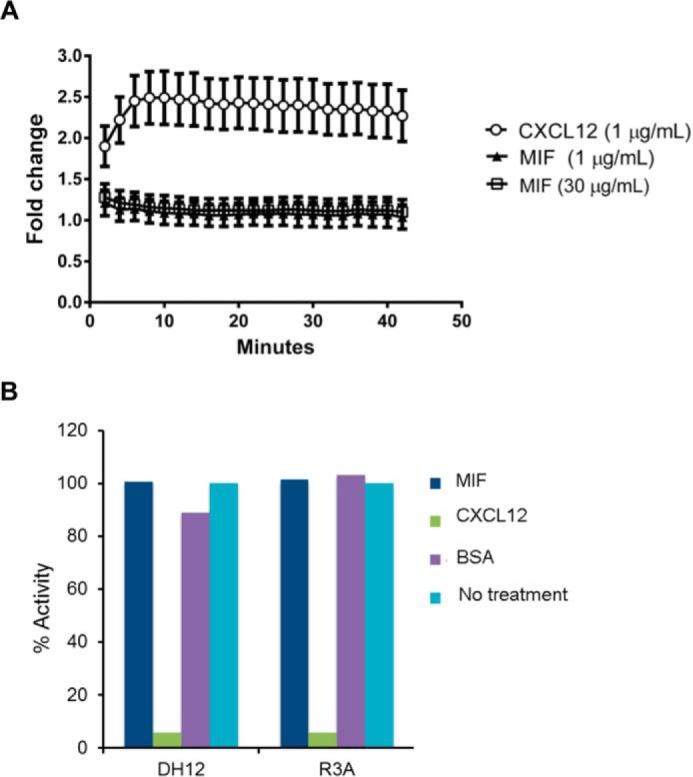
MIF does not re-capitulate CXCR4-mediated β-arrestin activation or CXCL12 inhibition of HIV-1 T-cell entry. A, click beetle luciferase complementation assay for recruitment of β-arrestin 2 to CXCR4 reveals that MIF does not promote interaction of these proteins, whereas CXCL12 drives association of CXCR4 with β-arrestin 2. B, inhibition of U87.CD4.U87 viral entry by MIF, CXCL12 (positive control), and bovine serum albumin (negative control) compared with untreated controls. Cells infected with the viral pseudotypes of the dual-tropic env strains DH12 or R3A with a luciferase reporter gene that is activated upon infection is inhibited by CXCL12 CXCR4 chemokine agonist but not by MIF or the bovine serum control. Results are plotted against the percentage luciferase activity with 100% for the untreated controls for each pseudotype.
CXCL12 prevents HIV-1 T-cell infection by the co-receptor CXCR4. We also studied whether MIF would prevent HIV-1 T-cell entry. We tested the dual-tropic strains DH12 and R3A, and found that MIF does not interfere with HIV-1 entry using the U87.CD4.CXCR4 cell line (Fig. 7B). The most likely explanation for the inability to block HIV-1 infection is due to the lower affinity of MIF (19 nm) to CXCR4 compared with CXCL12 (3.6 nm) (23, 37). A correlation between the Kd value for binding to CXCR4 by CXCL12 mutants and the IC50 value for inhibition of T-cell entry is well known (23). Even an increase in Kd from 3.6 nm for wild-type CXCL12 to 20 nm for residues CXCL12(2–67) (deletion of the N-terminal residue) leads to undetectable inhibition of HIV-1 infection (23). The Kd value for wild-type MIF is similar to that of CXCL12(2–67).
Discussion
MIF biology involves disparate functions and is associated with unrelated diseases such as inflammation (26), cancer (51), diabetes (52), atherosclerosis (29, 53), autoimmune disease (54), parasitic (55), and microbial infections (56) among other disorders. These distinct activities are likely due to activities of the constitutively expressed MIF in the cytosol (57) and the exported MIF by activated cells (58). Among the intracellular activities for MIF are inhibition of p53 (59), redox quenching (60), and a potential enzymatic activity (33, 34, 61). The extracellular activities involve binding and activation of CD74 and two chemokine receptors CXCR2 and CXCR4, which has led to the designation of MIF as both a cytokine and a non-cognate ligand of chemokine receptors. Other proteins that do not have any sequence or structural similarity to chemokines but activate chemokine receptors have been termed chemokine-like function chemokines or atypical chemokines, and they vary in their chemokine receptor specificity (25, 26). Human β-defensin-1 binds to CCR6 and is chemotactic for immature dendritic cells; tyrosyl-tRNA synthetase is processed into a secreted agonist for CXCR1, histidyl-tRNA synthetase activates CCR5, and seryl-tRNA synthetase induces the migration of CCR3-transfected cells (62–64). β-Defensin-3, ubiquitin, peptucin analogs, as well as a complex between HMGB1 and CXCL12 have been reported as CXCR4 agonists or antagonists (65–68). The structural basis underlying the engagement of chemokine receptors by these atypical chemokines is poorly understood and is likely to differ for each chemokine-like function because the structure of each protein does not share similarity with others (25).
Mechanistic insights into MIF-receptor interactions for the chemokine receptor CXCR2 have been reported (38, 40). MIF binding to CXCR2 has similarities to CXCL8-CXCR2 interactions. For example, there is a two-binding site model with the peptide MIF(47–56) that resembles the chemokine N-loop and interacts with the CXCR2 N-terminal peptide(19–33) as well as peptides EL1(108–120) and EL2(184–198), which most likely include binding site 1. There also is a pseudo-ELR motif consisting of Asp-44–Xaa–Arg-11 that binds at the second site composed of peptide EL3(286–300) that includes binding site 2. Given the focus on the pseudo-ELR motif of MIF (38, 40) and the use of peptides to probe for interactions on the CXCR2 N-terminal loop and extracellular domains (38, 40), the presumed transmembrane cavity was not probed for interactions. One of the interesting structural aspects is that the pseudo-ELR motifs and the N-loops are adjacent to each other and surround the solvent channel of MIF along the 3-fold axis of the trimer (38, 40). Using homology modeling and molecular dynamics simulations combined with binding free energy calculation methods, a model of a MIF·CXCR2 complex was produced (69). The model agrees with many of the results from the experimental studies and provides more insight based on the use of full-length MIF and CXCR2 instead of peptide arrays of each protein. For example, CXCR2 N-terminal residues 29–34 and 36–37 interact with the MIF N-like loop residues 46–49 and 55, and the β4 strand (Pro-91, Val-94, and, Ile-96). Hydrophobic residues from CXCR2 EL2 interact with MIF residues Met-101, Ala-103, Ala-104, Val-106, Gly-107, and Trp-108 at the C-terminal end. The pseudo-ELR motif residues Arg-11 form interactions with EL2 residues Asn-202, Asn-203, and Asn-206, whereas Asp-44 interacts with EL3 residue Gln-280. Interestingly, none of these MIF residues are part of the catalytic site, and the model does not identify occupancy by any MIF residues in the CXCR2 transmembrane cavity.
The results of this study indicate there is a partial overlap in the interactions between the MIF·CXCR2 and MIF·CXCR4 complexes, but most of the interactions are different. Both receptors use the N-terminal region to interact with MIF. Fluorescent studies indicate the N-terminal CXCR4(1–27) peptide binds to MIF with a Kd of >10 μm. A tendency of the peptide to aggregate at higher concentrations prevents a more precise determination of the binding constant. Differences occur in locations of the MIF N-loop that interact with the N-terminal region of each receptor. The N-loop of MIF for CXCR2 consists of residues 47–56. The N-loop for CXCR4 interactions, however, consists of a large region of MIF (residues 43–98) with a particularly strong interaction between CXCR4(1–27) and the peptide containing residues 67–81. The other major difference involves the topology of binding for the first and second sites of each receptor. For CXCR2, the binding of the MIF pseudo-ELR motifs and the N-loops surrounds the MIF solvent channel. In contrast, a catalytic inhibitor, as well as a mutation of the catalytic site, blocks MIF-induced signaling of CXCR4 indicating the MIF-CXCR4 contacts are at a different location than that of MIF-CXCR2. Insight into a potential interaction between the MIF catalytic cavity and CXCR4 is based on MIF-ligand co-crystal structures revealing chemical moieties in the MIF active site. The inhibitor ISO-1 (70), the pseudo-substrate 4-hydroxylphenylpyruvate (71), and most other small molecules that interact with aromatic/hydrophobic MIF cavity (72–75) contain an aromatic group, suggesting an aromatic side chain from one of the CXCR4-binding sites occupies this site.
The mapped sites for both proteins are supported by complementarity of the electrostatic potential. Residues at the surface of the catalytic cavity and MIF residues 67–81 have a positive electrostatic potential (Fig. 5B). The CXCR4 EL1 (the entire loop) and residues 182–196 from EL2 have an electronegative potential (Fig. 8). The CXCR4 N-terminal sequence is not visible in the x-ray structures, but the sequence has a net charge of −6 (without any sulfo-tyrosines) (46). These regions for CXCR4 interactions with MIF are also supported by experiments using CXCR4/CXCR3 hybrid receptors (Fig. 6).
FIGURE 8.
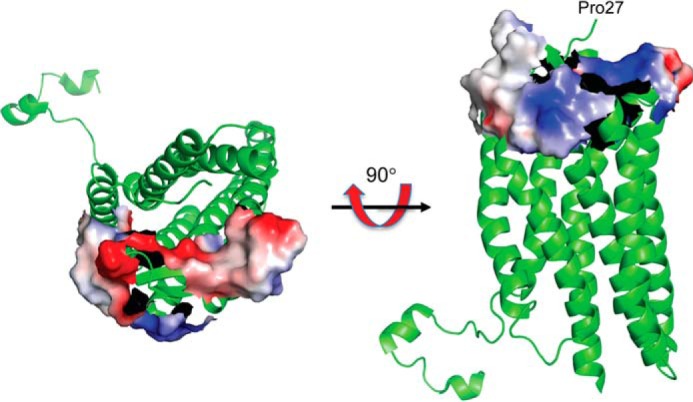
Regions of CXCR4 that interact with MIF mapped on the ribbon diagram of CXCR4. Only the electrostatic potential of extracellular loop 1 and residues 182–196 from extracellular loop 2 are shown because the structure of CXCR4(1–27) is not visible in any of the CXCR4 structures. The two orientations show both positive and negative potentials. The negative potential is complementary to the positive potential shown in Fig. 5C. Note that the CXCR4(1–27) has a net charge of −6 without accounting for the three tyrosine residues that are sulfated (45).
It is of interest to compare binding interactions between MIF-CXCR4 with the known interactions of CXCR4 with chemokines. Recently, the three-dimensional structure of the complex between the herpesvirus-8 vMIP-II and CXCR4 covalently stabilized by a disulfide between the two proteins was determined (19). This structure indicates that the CXCR4 N-terminal region and EL2 residues are important for interactions with vMIP-II, similar to what is found with MIF in this study. However, there are differences in some of the residues from CXCR4 N-terminal region that interact with vMIP-II or MIF. A more significant difference involves the interactions with the CXCR4 transmembrane cavity. The vMIP-II N-terminal residues occupy the transmembrane cavity. Interactions by agonists with a transmembrane cavity of chemokine receptors and class A GPCRs, in general, are known to be very important for receptor activation and signaling. However, the N-terminal residue (Pro-1) of MIF is at an immobile location in the catalytic cavity followed by a β-strand that is part of an internal β-sheet. For a transmembrane interaction to occur with the N-terminal region of MIF, the trimer would need to dissociate into monomers and partially unfold. To date, a stable MIF monomer has not been observed. The structure of the chemokine agonist CXCL12 with CXCR4 has yet to be determined, but interactions have been extensively studied (22). The results of these studies are similar to the conclusions of the vMIPII-CXCR4 structure with the exception that CXCL12 has interactions in the transmembrane cavity that activates the receptor.
This study presents the first analysis of the MIF-CXCR4 binding interactions and their effects on receptor activation and signaling. There are a number of important conclusions based on the results. 1) There are no direct interactions between MIF and the transmembrane cavity. The absence of such an interaction is consistent with the inability of MIF to block HIV-1 infection as 13-mer CXCR4 peptides are sufficient to block HIV-1 entry (76). This strongly suggests there is a cryptic CXCR4-binding site for MIF necessary for signaling. 2) There is a different N-loop for MIF and CXCL12 that binds to the CXCR4 N-terminal region at different residues that partially overlap or are in close proximity. 3) High concentrations of CXCR4 antagonists AMD3100 and IT1t are required to inhibit MIF-mediated signaling compared with lower doses of each antagonist that completely abolishes binding and signaling by the more potent CXCL12. This result supports the absence of direct interactions between MIF and the CXCR4 transmembrane cavity and suggests that MIF binding induces a conformational change at the transmembrane cavity can be reversed by high concentrations of the CXCR4 antagonists. 4) Points 1–3 argue that MIF is a partial allosteric agonist. This is only possible because there are at least two distinct binding sites in CXCR4. Short N-terminal CXCR4 or vMIP-II peptides that do not include the N-loop that interacts with the CXCR4 N-terminal region have been shown to be weak agonists or antagonist, respectively (42, 77–80). Consequently, MIF and CXCL12 compete for binding with the CXCR4 N-terminal region, which is consistent with the 50% maximum displacement of 125I-CXCL12 by 1 μm MIF relative to 100 nm CXCL12 (37). The other property that contributes to the displacement is the koff for the CXCL12 N-terminal residues from the transmembrane cavity, which is MIF-independent. 5) The results that MIF activates G protein signaling in mammalian cells expressing CXCR3/CXCR4 hybrid receptors or in S. cerevisiae cells expressing CXCR4 indicate that the cryptic site also induces a conformational change in transmembrane helix 6 to accommodate G protein interactions necessary for signaling (81). Our experiments do not detect any CXCR4-β-arrestin 2 interactions induced by MIF indicating a possible biased G protein signaling, but this needs further investigation (82, 83). 6) Finally, the MIF catalytic cavity is involved in MIF-CXCR4 interactions that can be targeted for therapeutic intervention without affecting the homeostatic function of CXCL12 and CXCR4.
Experimental Procedures
Cell Culture, Recombinant Proteins, and Reagents
Chinese hamster ovary (CHO) cells were cultured in DMEM/F-12 (1:1) medium supplemented with GlutaMAX, 10% fetal calf serum (FCS), and 1% penicillin/streptavidin. Miscellaneous cell culture reagents were bought from Invitrogen (Karlsruhe, Germany) and PAA (Pasching, Austria). Biologically active recombinant human MIF (rMIF) was expressed, purified, and catalytically assayed essentially as described (39, 84). Labeling of recombinant human MIF with Alexa Fluor-488 was performed according to the manufacturer's recommendation (Molecular Probes/Thermo Fisher Scientific, Eugene, OR) and as described previously (85). Procedures for expression, purification, and re-folding of SDF-1α/CXCL12 were as described (86). All other reagents were obtained from Sigma, Merck, Roth, or Calbiochem and were of the highest purity degree available. IT1t was a kind gift from Dr. Raymond Stevens (The Scripps Research Institute).
Peptide Synthesis
The N-terminal peptide CXCR4(1–27) was custom-synthesized by SciLight Biotechnology, LLC (China). The peptides representing the extracellular loops of CXCR4 (EL1, EL2, and EL3 peptides) and the peptide MIF(67–81) were custom-synthesized by Peptide Specialty Laboratories GmbH (Heidelberg, Germany). All peptides were produced by Fmoc (N-(9-fluorenyl)methoxycarbonyl) solid-phase synthesis and purified by HPLC. The quality of the peptides was checked by mass spectrometry analysis.
Experiments with S. cerevisiae Strain CY12946
The S. cerevisiae strain (CY12946) expressing a functional CXCR4 has been previously described (42). Upon CXCR4 activation, MAPK signaling transcribes and translates β-galactosidase (lacZ), which is quantified by an enzymatic assay. The strain CY12946 was transformed with plasmid Cp6160 that expresses either wild-type human MIF (hMIF), the P1V/M2S double mutant MIF, or wild-type human CXCL12 and contains a Ura-selectable marker. In addition, the plasmids Cp4181 containing the gene for CXCR4 (constitutively expressed) with a Leu-selectable marker and Cp1584 with the β-galactosidase gene under the Fus1 promoter and a Trp-selectable marker were also transformed in CY12946. Strain CY12946 expressing CXCL12 or hMIF but not CXCR4 was used as a negative control. Experimental colonies were transferred from plates to appropriate selective minimal liquid media and grown overnight at 30 °C to express and localize each protein to the periplasmic space for activation of CXCR4 along with colonies representing positive and negative controls. Cells were diluted to 0.01 OD in fresh medium and grown for 1.5 h. β-Galactosidase activity from lysed cells was measured using the Beta Glo kit (Promega, Madison, WI).
To study CXCR4 signaling by extracellular MIF along with pharmacological studies disrupting MIF-CXCR4 interactions, the CY12946 strain was transformed with CXCR4 in Cp4181 and β-gal in Cp1584. Transformed cells were grown overnight in selective medium. The cells were diluted to 0.01 OD and incubated with MIF (117 μm) alone or in the presence of CXCR4 antagonist AMD3100 or IT1t at the indicated fold excess of MIF. In the control study, the transformed cells were incubated with CXCL12 at 2 μm alone or in the presence of AMD3100 at its IC50 (37 nm) or It1T at its IC50 (0.2 nm), and the activation of CXCR4 was quantitated. The data shown are the mean representative of three different experiments done in quadruplicate.
Glass Slide-based Peptide Array
Peptide microarray analysis using glass slide technology was custom-made by JPT Peptide Technologies GmbH (Berlin, Germany). One microarray is composed of three identical subarrays each carrying 753 different peptide sequences spotted in triplicates. Peptides were stepwise synthesized (SPOT-synthesis) on a cellulose membrane, and a reactivity tag was coupled to the N terminus of the peptides. Peptides were cleaved from the cellulose support and transferred into a microtiter plate. The peptides were dispensed on an activated glass surface using a droplet-depositing system. Target peptides were immobilized chemo-selectively and purified by reaction of the peptides with the modified glass surface resulting in the formation of a covalent bond, which allowed the removal of all truncated and acetylated sequences by subsequent washing steps. After all peptides were arrayed on the glass surface, active residues were passivated. Analysis of peptide-protein and peptide-peptide interactions was performed using a TECAN HS4800 microarray processing station. The microarrays were incubated either with biotinylated recombinant human MIF or biotinylated CXCR4(1–27) peptide. For determination of false-positives, one microarray was incubated with fluorescently labeled streptavidin only. After incubation with 200 μl of sample in blocking buffer (10 μg/ml MIF, 10 μg/ml receptor peptides) for 30 min and several washing steps with Tris-buffered saline (TBS) buffer containing 0.1% Tween 20, the array was developed with Cy5-streptavidin, washed again, and dried with a nitrogen stream. Scanning at an appropriate wavelength with the scanning system Axon GenePix 4200AL generated images, showing the signal intensity as a single measurement for each peptide. These grayscale images were analyzed with the GenePixPro 6.0 software, quantifying the intensity of each fluorescent spot on the scanned microarray slide. Each spot feature was analyzed for total intensity and background intensity and corrected for background by the software. Data shown represent the mean values of the corrected median of signal intensities from three identical subarrays on each microarray image.
In Vitro Competitive Assay
MIF-CXCR4 protein interactions were investigated by in vitro coating of 1 μm CXCR4(1–27) in a 96-well plate overnight followed by addition of 150 nm biotinylated MIF. Increasing concentrations of non-labeled MIF or control were added to the CXCR4(1–27) wells to displace biotinylated MIF and washed three times. The remaining biotinylated MIF bound to CXCR4(1–27) at each MIF dose was detected by streptavidin-conjugated alkaline phosphatase with p-nitrophenyl phosphate. The data are the mean representative of three different experiments analyzed using Graphpad Prism.
Peripheral Blood Monocytic Cell Migration
Migration assays of blood peripheral mononuclear cells (PBMCs) were performed in a transmigration well as described previously (87). Briefly, 8 nm MIF was preincubated in the presence or absence of varying excess amounts of CXCR4(1–27) in the lower chamber of a 24-well cell culture insert with 8-μm pore size (Falcon). Experiments were performed in triplicates. Isolated human PBMCs from whole blood were washed and resuspended in RPMI 1640 medium to 1 × 106 cells/ml and were placed in the upper chamber. After incubation for 3 h at 37 °C, the bottom wells containing the migrated cells were methanol-fixed, stained with Giemsa, and counted under light microscopy.
Circular Dichroism Spectroscopy
Far-UV circular dichroism (CD) spectra were recorded in a Jasco 700 CD spectropolarimeter (Jasco Labor-und Datentechnik GmbH, Gross-Umstadt, Germany). Scans were recorded at 25 °C between 195 and 250 nm as an average of three scans and smoothed to obtain the final data. Spectra were collected at 1.0-nm intervals with a bandwidth of 1 nm in a buffer containing 50 mm sodium phosphate, 50 mm sodium chloride, pH 7.2. CD spectra in the presence of ligand were performed as described previously (38, 40). Briefly, rMIF was prepared in 50 mm sodium phosphate, 50 mm sodium chloride buffer. Spectra of rMIF (5 μm) and CXCR4 receptor peptide (100 μm) alone in solution as well as the MIF/receptor peptide mixtures (1:20 ratio for MIF/EL2, MIF/EL3, and MIF/N terminus, as well as 1:10 for MIF/EL1) were recorded in a 1-cm quartz cuvette. CD spectra are presented as a plot of the measured raw ellipticity versus the wavelength because molar ellipticities cannot be accurately determined for protein complexes and mixtures. Predicted spectra were obtained by mathematical addition of the individual spectra of rMIF and the CXCR4 peptides. Dynode voltage values generally were below 800 and did not interfere with CD measurements. The selected CXCR4-EL1 peptide had unfavorable solubility properties. Therefore, spectra were recorded in the presence of 1% hexafluoroisopropanol, a co-solvent previously used in CD spectroscopy of MIF (60).
Fluorescence Spectroscopy
Fluorescence spectroscopy titrations were recorded in quartz cuvettes in a FluoroLog 3 spectrofluorimeter (Horiba Scientific, Jobin Yvon GmbH, Unterhaching, Germany). MIF-CXCR4 interactions were probed by titrating CXCR4 peptide(1–27) against Alexa Fluor-488-hMIF. MIF was applied at a concentration of 10 nm in 20 mm sodium phosphate buffer, pH 7.4, and the peptide was added at ratios of 1:1, 1:250, 1:500, 1:1000, and 1:2000 in the same buffer. Changes in Alexa Fluor-488 emission were recorded between 500 and 600 nm wavelength.
Generation of Chimeric CXCR4/CXCR3 Receptors Expressed in CHO Cells
CHO cells were cultivated in DMEM/F-12 (1:1) (1×) medium containing GlutaMAX at 37 °C and 5% CO2 in a humidified incubator. Plasmids encoding CXCR4, CXCR3, and chimeric receptor constructs were transfected into CHO cells by PolyFect transfection reagent (Qiagen, Valencia, CA). Two days later, the medium was replaced with fresh medium containing 800 μg/ml G418/neomycin. Only cells with successful incorporation of the gene of interest had integrated the neomycin resistance gene, which allowed for antibiotic selection of cells stably expressing the receptor constructs. Stable transfectants were selected, and expression of the receptor proteins was verified by flow cytometric analysis.
Cyclic AMP Assay and Inhibition by MIF
The cAMP assay was used to analyze the responsiveness of the cloned receptor chimeras to MIF. cAMP levels were measured with the HitHunterTM cAMP XS+ kit (GE Healthcare Europe GmbH, Freiburg, Germany). The assay is based on the separation of the enzyme β-galactosidase into the two fragments EA (enzyme acceptor) and ED (enzyme donor). Re-association of these subunits results in activation of the enzyme and hydrolysis of a given substrate. In this assay, a cAMP antibody inhibits complement fragmentation through binding to the ED-cAMP conjugate. Cell-derived cAMP competes with ED-cAMP for the antibody. Increased cell-derived cAMP generates additional free ED-cAMP and induces a stronger signal. The readout signal is chemiluminescence. The signal intensity is proportional to the intracellular cAMP concentration. Stimulation of cells with forskolin activates adenylate cyclase. Simultaneous application of 3-isobutyl-1-methylxanthine, a phosphodiesterase inhibitor, blocks cAMP degradation. The assay was conducted with the CHO-chemokine receptor transfectants (see above) according to the manufacturer's protocol. Forskolin-triggered cAMP production is inhibited by activation of Gi-coupled chemokine receptors. cAMP levels of non-treated cells were compared with cells treated with forskolin and with cells simultaneously treated with forskolin and rMIF. 20,000 cells were resuspended in phosphate-buffered saline (PBS), pH 7.4, with 3-isobutyl-1-methylxanthine and seeded in a 96-well plate. 100 μm forskolin was applied to various CHO transfectants (wild-type and chimeras) in the presence or absence of MIF. This experiment also used forskolin and MIF with the MIF(13–27) or MIF(67–81) peptides to determine whether there is any competitive inhibition with MIF. After incubation for 30 min at 37 °C and 5% CO2, chemiluminescence was measured in a standard microtiter plate reader.
β-Arrestin Complementation Assay
To measure ligand-dependent association of CXCR4 with the cytosolic adapter protein β-arrestin 2, we used MDA-MB-231 human breast cancer cells stably expressing luciferase complementation reporters CXCR4-CBGN and β-arrestin-2-CBC (50). We seeded cells in 96-well black-walled plates at a density of 15,000 cells per well in DMEM supplemented with 10% fetal bovine serum, 1% glutamine, and 0.1% penicillin/streptomycin/gentamicin. After incubation overnight at 37 °C and 5% CO2, we gently aspirated medium from each well and exchanged it with 50 μl of phenol red-free DMEM (Life Technologies, Inc.) containing 0.2% probumin (EMD Millipore, Billerica, MA). After an additional 5 min, we added 14 μl of phenol red-free DMEM containing 0.2% probumin and 1 μg/ml CXCL12-α (R&D Systems, Minneapolis, MN) or rMIF. We then imaged bioluminescence on an IVIS Lumina LT (PerkinElmer Life Sciences) to acquire 20 consecutive images with open filter, large binning, and a 2-min exposure. Data were graphed as mean values ± S.E. for fold change in bioluminescence relative to vehicle control at each time point to account for dynamics of the luciferin-luciferase reaction (n = 4 per condition) (50). We present data representative of two independent experiments.
Inhibition of T-tropic HIV-1-CXCR4 Interactions
The viral pseudotypes DH12 and R3A were produced from transfected 293T cells as described previously and contain different env genes isolated from patients (88). These two different pseudotypes were used for experiments with MIF. After 2–3 days of incubating transfected cells, supernatants were harvested and filtered with a Corning 0.45-μm syringe filter. Measurement of the p24 antigen was used to determine the viral titer of each pseudotype. Ten nanograms of p24 of each pseudotype was used to infect U87.CD4.CXCR4 cells in the presence of MIF, CXCL12 (positive control), or BSA (negative control). The cells were incubated at 37 °C for 3 days and lysed with 0.1% Triton X-100 in PBS, and viral infection was measured by luciferase activity (88).
Molecular Modeling and Electrostatic Potential
High resolution structures of CXCR4 (Protein Data Bank code 3ODU) and MIF (Protein Data Bank code 3DJH) were used to create models of the protein-binding sites and calculate the electrostatic potential of the sites. The coordinates were deleted for water molecules, the CXCR4 antagonist IT1t, and all ions and molecules derived from the crystallization buffer or cryo-cooling prior to the calculation of the electrostatic potential using PyMOL (89).
Statistical Analysis
Data not specifically mentioned in the experiments above are expressed as means ± S.E. Student's t tests (two-sided, unpaired) were performed to compare experimental groups. Differences with a value of p < 0.05 were considered statistically significant.
Author Contributions
A. K., E. L., and J. B. conceived and designed the project and with G. L. wrote the manuscript. D. R., S. G., C. S., S. Z., N. D., M. B., K. K., A. M., H. L., and A. X. performed the experiments and analyzed the data. C. W. and G. L. were involved in experimental guidance and assisted in interpretation of the some of the results. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank M. Dewor for assistance with peptide preparations and the Robert Dom laboratory (University of Pennsylvania) for the HIV-1 assay.
This work was supported by Deutsche Forschungsgemeinschaft Grants SFB1123/A03 (to J. B. and A. K.), SFB1123/A01 (to C. W.), and DFG-IRTG1508 (to M. B. and J. B.) and National Institutes of Health Grants R01CA170198 and R01CA196018 (to G. L.) and AI065029 and a Pilot Grant from the Yale Cancer Center (to E. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GPCR
- G protein-coupled receptor
- MIF
- migration inhibitory factor
- EL
- extracellular loop
- rMIF
- recombinant human MIF
- hMIF
- human MIF
- ISO-1
- (SR)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid
- PBMC
- peripheral mononuclear cell.
References
- 1. Katritch V., Cherezov V., and Stevens R. C. (2013) Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thelen M. (2001) Dancing to the tune of chemokines. Nat. Immunol. 2, 129–134 [DOI] [PubMed] [Google Scholar]
- 3. Charo I. F., and Ransohoff R. M. (2006) The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621 [DOI] [PubMed] [Google Scholar]
- 4. Moser B., and Loetscher P. (2001) Lymphocyte traffic control by chemokines. Nat. Immunol. 2, 123–128 [DOI] [PubMed] [Google Scholar]
- 5. Bleul C. C., Farzan M., Choe H., Parolin C., Clark-Lewis I., Sodroski J., and Springer T. A. (1996) The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382, 829–833 [DOI] [PubMed] [Google Scholar]
- 6. Endres M. J., Clapham P. R., Marsh M., Ahuja M., Turner J. D., McKnight A., Thomas J. F., Stoebenau-Haggarty B., Choe S., Vance P. J., Wells T. N., Power C. A., Sutterwala S. S., Doms R. W., Landau N. R., and Hoxie J. A. (1996) CD4-independent infection by HIV-2 is mediated by fusin/CXCR4. Cell 87, 745–756 [DOI] [PubMed] [Google Scholar]
- 7. Lapham C. K., Ouyang J., Chandrasekhar B., Nguyen N. Y., Dimitrov D. S., and Golding H. (1996) Evidence for cell-surface association between fusin and the CD4-gp120 complex in human cell lines. Science 274, 602–605 [DOI] [PubMed] [Google Scholar]
- 8. Libby P. (2002) Inflammation in atherosclerosis. Nature 420, 868–874 [DOI] [PubMed] [Google Scholar]
- 9. Oberlin E., Amara A., Bachelerie F., Bessia C., Virelizier J. L., Arenzana-Seisdedos F., Schwartz O., Heard J. M., Clark-Lewis I., Legler D. F., Loetscher M., Baggiolini M., and Moser B. (1996) The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 382, 833–835 [DOI] [PubMed] [Google Scholar]
- 10. Murphy P. M., Baggiolini M., Charo I. F., Hébert C. A., Horuk R., Matsushima K., Miller L. H., Oppenheim J. J., and Power C. A. (2000) International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 52, 145–176 [PubMed] [Google Scholar]
- 11. Bachelerie F., Ben-Baruch A., Burkhardt A. M., Combadiere C., Farber J. M., Graham G. J., Horuk R., Sparre-Ulrich A. H., Locati M., Luster A. D., Mantovani A., Matsushima K., Murphy P. M., Nibbs R., Nomiyama H., et al. (2014) International Union of Basic and Clinical Pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 66, 1–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fernandez E. J., and Lolis E. (2002) Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 42, 469–499 [DOI] [PubMed] [Google Scholar]
- 13. Wang C., Jiang Y., Ma J., Wu H., Wacker D., Katritch V., Han G. W., Liu W., Huang X. P., Vardy E., McCorvy J. D., Gao X., Zhou X. E., Melcher K., Zhang C., et al. (2013) Structural basis for molecular recognition at serotonin receptors. Science 340, 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H.-J., Kuhn P., Weis W. I., Kobilka B. K., and Stevens R. C. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaakola V.-P., Griffith M. T., Hanson M. A., Cherezov V., Chien E. Y., Lane J. R., Ijzerman A. P., and Stevens R. C. (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322, 1211–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu W., Chun E., Thompson A. A., Chubukov P., Xu F., Katritch V., Han G. W., Roth C. B., Heitman L. H., IJzerman A. P., Cherezov V., and Stevens R. C. (2012) Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., and Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qin L., Kufareva I., Holden L. G., Wang C., Zheng Y., Zhao C., Fenalti G., Wu H., Han G. W., Cherezov V., Abagyan R., Stevens R. C., and Handel T. M. (2015) Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan Q., Zhu Y., Li J., Chen Z., Han G. W., Kufareva I., Li T., Ma L., Fenalti G., Li J., Zhang W., Xie X., Yang H., Jiang H., Cherezov V., Liu H., Stevens R. C., Zhao Q., and Wu B. (2013) Structure of the CCR5 chemokine receptor–HIV entry inhibitor maraviroc complex. Science 341, 1387–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burg J. S., Ingram J. R., Venkatakrishnan A. J., Jude K. M., Dukkipati A., Feinberg E. N., Angelini A., Waghray D., Dror R. O., Ploegh H. L., and Garcia K. C. (2015) Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kofuku Y., Yoshiura C., Ueda T., Terasawa H., Hirai T., Tominaga S., Hirose M., Maeda Y., Takahashi H., Terashima Y., Matsushima K., and Shimada I. (2009) Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G protein-coupled receptor CXCR4. J. Biol. Chem. 284, 35240–35250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crump M. P., Gong J. H., Loetscher P., Rajarathnam K., Amara A., Arenzana-Seisdedos F., Virelizier J. L., Baggiolini M., Sykes B. D., and Clark-Lewis I. (1997) Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 16, 6996–7007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dealwis C., Fernandez E. J., Thompson D. A., Simon R. J., Siani M. A., and Lolis E. (1998) Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1α, a potent ligand for the HIV-1 “fusin” coreceptor. Proc. Natl. Acad. Sci. U.S.A. 95, 6941–6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tillmann S., Bernhagen J., and Noels H. (2013) Arrest functions of the MIF ligand/receptor axes in atherogenesis. Front. Immunol. 4, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bernhagen J., Calandra T., Mitchell R. A., Martin S. B., Tracey K. J., Voelter W., Manogue K. R., Cerami A., and Bucala R. (1993) MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 365, 756–759 [DOI] [PubMed] [Google Scholar]
- 27. Calandra T., and Roger T. (2003) Macrophage migration inhibitory factor: a regulator of innate immunity. Nat. Rev. Immunol. 3, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morand E. F., Leech M., and Bernhagen J. (2006) MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat. Rev. Drug Discov. 5, 399–410 [DOI] [PubMed] [Google Scholar]
- 29. Burger-Kentischer A., Goebel H., Seiler R., Fraedrich G., Schaefer H. E., Dimmeler S., Kleemann R., Bernhagen J., and Ihling C. (2002) Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation 105, 1561–1566 [DOI] [PubMed] [Google Scholar]
- 30. Schober A., Bernhagen J., Thiele M., Zeiffer U., Knarren S., Roller M., Bucala R., and Weber C. (2004) Stabilization of atherosclerotic plaques by blockade of macrophage migration inhibitory factor after vascular injury in apolipoprotein E-deficient mice. Circulation 109, 380–385 [DOI] [PubMed] [Google Scholar]
- 31. Zernecke A., Bernhagen J., and Weber C. (2008) Macrophage migration inhibitory factor in cardiovascular disease. Circulation 117, 1594–1602 [DOI] [PubMed] [Google Scholar]
- 32. Andreeva A., Howorth D., Chothia C., Kulesha E., and Murzin A. G. (2014) SCOP2 prototype: a new approach to protein structure mining. Nucleic Acids Res. 42, D310–D314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rosengren E., Aman P., Thelin S., Hansson C., Ahlfors S., Björk P., Jacobsson L., and Rorsman H. (1997) The macrophage migration inhibitory factor MIF is a phenylpyruvate tautomerase. FEBS Lett. 417, 85–88 [DOI] [PubMed] [Google Scholar]
- 34. Rosengren E., Bucala R., Aman P., Jacobsson L., Odh G., Metz C. N., and Rorsman H. (1996) The immunoregulatory mediator macrophage migration inhibitory factor (MIF) catalyzes a tautomerization reaction. Mol. Med. 2, 143–149 [PMC free article] [PubMed] [Google Scholar]
- 35. Leng L., Metz C. N., Fang Y., Xu J., Donnelly S., Baugh J., Delohery T., Chen Y., Mitchell R. A., and Bucala R. (2003) MIF signal transduction initiated by binding to CD74. J. Exp. Med. 197, 1467–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi X., Leng L., Wang T., Wang W., Du X., Li J., McDonald C., Chen Z., Murphy J. W., Lolis E., Noble P., Knudson W., and Bucala R. (2006) CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 25, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bernhagen J., Krohn R., Lue H., Gregory J. L., Zernecke A., Koenen R. R., Dewor M., Georgiev I., Schober A., Leng L., Kooistra T., Fingerle-Rowson G., Ghezzi P., Kleemann R., McColl S. R., et al. (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 13, 587–596 [DOI] [PubMed] [Google Scholar]
- 38. Kraemer S., Lue H., Zernecke A., Kapurniotu A., Andreetto E., Frank R., Lennartz B., Weber C., and Bernhagen J. (2011) MIF-chemokine receptor interactions in atherogenesis are dependent on an N-loop-based 2-site binding mechanism. FASEB J. 25, 894–906 [DOI] [PubMed] [Google Scholar]
- 39. Pantouris G., Syed M. A., Fan C., Rajasekaran D., Cho T. Y., Rosenberg E. M. Jr., Bucala R., Bhandari V., and Lolis E. J. (2015) An analysis of MIF structural features that control functional activation of CD74. Chem. Biol. 22, 1197–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weber C., Kraemer S., Drechsler M., Lue H., Koenen R. R., Kapurniotu A., Zernecke A., and Bernhagen J. (2008) Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc. Natl. Acad. Sci. U.S.A. 105, 16278–16283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang W. B., Navenot J. M., Haribabu B., Tamamura H., Hiramatu K., Omagari A., Pei G., Manfredi J. P., Fujii N., Broach J. R., and Peiper S. C. (2002) A point mutation that confers constitutive activity to CXCR4 reveals that T140 is an inverse agonist and that AMD3100 and ALX40–4C are weak partial agonists. J. Biol. Chem. 277, 24515–24521 [DOI] [PubMed] [Google Scholar]
- 42. Sachpatzidis A., Benton B. K., Manfredi J. P., Wang H., Hamilton A., Dohlman H. G., and Lolis E. (2003) Identification of allosteric peptide agonists of CXCR4. J. Biol. Chem. 278, 896–907 [DOI] [PubMed] [Google Scholar]
- 43. Sun H. W., Bernhagen J., Bucala R., and Lolis E. (1996) Crystal structure at 2.6-A resolution of human macrophage migration inhibitory factor. Proc. Natl. Acad. Sci. U.S.A. 93, 5191–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Nobel J. G., and Barnett J. A. (1991) Passage of molecules through yeast cell walls: a brief essay–review. Yeast 7, 313–323 [DOI] [PubMed] [Google Scholar]
- 45. Seibert C., Veldkamp C. T., Peterson F. C., Chait B. T., Volkman B. F., and Sakmar T. P. (2008) Sequential tyrosine sulfation of CXCR4 by tyrosylprotein sulfotransferases. Biochemistry 47, 11251–11262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Farzan M., Babcock G. J., Vasilieva N., Wright P. L., Kiprilov E., Mirzabekov T., and Choe H. (2002) The role of post-translational modifications of the CXCR4 amino terminus in stromal-derived factor 1α association and HIV-1 entry. J. Biol. Chem. 277, 29484–29489 [DOI] [PubMed] [Google Scholar]
- 47. Schwartz V., Lue H., Kraemer S., Korbiel J., Krohn R., Ohl K., Bucala R., Weber C., and Bernhagen J. (2009) A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 583, 2749–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kleemann R., Kapurniotu A., Mischke R., Held J., and Bernhagen J. (1999) Characterization of catalytic centre mutants of macrophage migration inhibitory factor (MIF) and comparison to Cys81Ser MIF. Eur. J. Biochem. 261, 753–766 [DOI] [PubMed] [Google Scholar]
- 49. Luker K. E., Gupta M., and Luker G. D. (2008) Imaging CXCR4 signaling with firefly luciferase complementation. Anal. Chem. 80, 5565–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Coggins N. L., Trakimas D., Chang S. L., Ehrlich A., Ray P., Luker K. E., Linderman J. J., and Luker G. D. (2014) CXCR7 controls competition for recruitment of β-arrestin 2 in cells expressing both CXCR4 and CXCR7. PLoS ONE 9, e98328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bifulco C., McDaniel K., Leng L., and Bucala R. (2008) Tumor growth-promoting properties of macrophage migration inhibitory factor. Curr. Pharm. Des. 14, 3790–3801 [DOI] [PubMed] [Google Scholar]
- 52. Toso C., Emamaullee J. A., Merani S., and Shapiro A. M. (2008) The role of macrophage migration inhibitory factor on glucose metabolism and diabetes. Diabetologia 51, 1937–1946 [DOI] [PubMed] [Google Scholar]
- 53. Pan J. H., Sukhova G. K., Yang J. T., Wang B., Xie T., Fu H., Zhang Y., Satoskar A. R., David J. R., Metz C. N., Bucala R., Fang K., Simon D. I., Chapman H. A., Libby P., and Shi G. P. (2004) Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 109, 3149–3153 [DOI] [PubMed] [Google Scholar]
- 54. Leech M., Metz C., Santos L., Peng T., Holdsworth S. R., Bucala R., and Morand E. F. (1998) Involvement of macrophage migration inhibitory factor in the evolution of rat adjuvant arthritis. Arthritis Rheum. 41, 910–917 [DOI] [PubMed] [Google Scholar]
- 55. McDevitt M. A., Xie J., Ganapathy-Kanniappan S., Shanmugasundaram G., Griffith J., Liu A., McDonald C., Thuma P., Gordeuk V. R., Metz C. N., Mitchell R., Keefer J., David J., Leng L., and Bucala R. (2006) A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J. Exp. Med. 203, 1185–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weiser J. N., Roche A. M., Hergott C. B., LaRose M. I., Connolly T., Jorgensen W. L., Leng L., Bucala R., and Das R. (2015) Macrophage migration inhibitory factor (MIF) is detrimental in pneumococcal pneumonia and a target for therapeutic immunomodulation. J. Infect. Dis. 212, 1677–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Panstruga R., Baumgarten K., and Bernhagen J. (2015) Phylogeny and evolution of plant macrophage migration inhibitory factor/d-dopachrome tautomerase-like proteins. BMC Evol. Biol. 15, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Swope M. D., and Lolis E. (1999) Macrophage migration inhibitory factor: cytokine, hormone, or enzyme? Rev. Physiol. Biochem. Pharmacol. 139, 1–32 [DOI] [PubMed] [Google Scholar]
- 59. Hudson J. D., Shoaibi M. A., Maestro R., Carnero A., Hannon G. J., and Beach D. H. (1999) A proinflammatory cytokine inhibits p53 tumor suppressor activity. J. Exp. Med. 190, 1375–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kleemann R., Kapurniotu A., Frank R. W., Gessner A., Mischke R., Flieger O., Jüttner S., Brunner H., and Bernhagen J. (1998) Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol-protein oxidoreductase. J. Mol. Biol. 280, 85–102 [DOI] [PubMed] [Google Scholar]
- 61. Lolis E. (2001) Glucocorticoid counter regulation: macrophage migration inhibitory factor as a target for drug discovery. Curr. Opin. Pharmacol. 1, 662–668 [DOI] [PubMed] [Google Scholar]
- 62. Howard O. M., Dong H. F., Yang D., Raben N., Nagaraju K., Rosen A., Casciola-Rosen L., Härtlein M., Kron M., Yang D., Yiadom K., Dwivedi S., Plotz P. H., and Oppenheim J. J. (2002) Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J. Exp. Med. 196, 781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wakasugi K., and Schimmel P. (1999) Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science 284, 147–151 [DOI] [PubMed] [Google Scholar]
- 64. Yang D., Chertov O., Bykovskaia S. N., Chen Q., Buffo M. J., Shogan J., Anderson M., Schröder J. M., Wang J. M., Howard O. M., and Oppenheim J. J. (1999) β-Defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science 286, 525–528 [DOI] [PubMed] [Google Scholar]
- 65. Feng Z., Dubyak G. R., Lederman M. M., and Weinberg A. (2006) Cutting edge: human β defensin 3–a novel antagonist of the HIV-1 coreceptor CXCR4. J. Immunol. 177, 782–786 [DOI] [PubMed] [Google Scholar]
- 66. Saini V., Marchese A., and Majetschak M. (2010) CXC chemokine receptor 4 is a cell surface receptor for extracellular ubiquitin. J. Biol. Chem. 285, 15566–15576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tchernychev B., Ren Y., Sachdev P., Janz J. M., Haggis L., O'Shea A., McBride E., Looby R., Deng Q., McMurry T., Kazmi M. A., Sakmar T. P., Hunt S. 3rd, and Carlson K. E. (2010) Discovery of a CXCR4 agonist pepducin that mobilizes bone marrow hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 107, 22255–22259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schiraldi M., Raucci A., Muñoz L. M., Livoti E., Celona B., Venereau E., Apuzzo T., De Marchis F., Pedotti M., Bachi A., Thelen M., Varani L., Mellado M., Proudfoot A., Bianchi M. E., and Uguccioni M. (2012) HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 209, 551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu L., Li Y., Li D., Xu P., Tian S., Sun H., Liu H., and Hou T. (2015) Exploring the binding mechanisms of MIF to CXCR2 using theoretical approaches. Phys. Chem. Chem. Phys. 17, 3370–3382 [DOI] [PubMed] [Google Scholar]
- 70. Lubetsky J. B., Dios A., Han J., Aljabari B., Ruzsicska B., Mitchell R., Lolis E., and Al-Abed Y. (2002) The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J. Biol. Chem. 277, 24976–24982 [DOI] [PubMed] [Google Scholar]
- 71. Lubetsky J. B., Swope M., Dealwis C., Blake P., and Lolis E. (1999) Pro-1 of macrophage migration inhibitory factor functions as a catalytic base in the phenylpyruvate tautomerase activity. Biochemistry 38, 7346–7354 [DOI] [PubMed] [Google Scholar]
- 72. Cournia Z., Leng L., Gandavadi S., Du X., Bucala R., and Jorgensen W. L. (2009) Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J. Med. Chem. 52, 416–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ouertatani-Sakouhi H., El-Turk F., Fauvet B., Cho M.-K., Pinar Karpinar D., Le Roy D., Dewor M., Roger T., Bernhagen J., Calandra T., Zweckstetter M., and Lashuel H. A. (2010) Identification and characterization of novel classes of macrophage migration inhibitory factor (MIF) inhibitors with distinct mechanisms of action. J. Biol. Chem. 285, 26581–26598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cho Y., Vermeire J. J., Merkel J. S., Leng L., Du X., Bucala R., Cappello M., and Lolis E. (2011) Drug repositioning and pharmacophore identification in the discovery of hookworm MIF inhibitors. Chem. Biol. 18, 1089–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cho Y., and Lolis E. (2012) in The MIF Handbook (Bucala R., ed) pp. 101–118, World Scientific Publishing Co., Singapore [Google Scholar]
- 76. Heveker N., Montes M., Germeroth L., Amara A., Trautmann A., Alizon M., and Schneider-Mergener J. (1998) Dissociation of the signalling and antiviral properties of SDF-1-derived small peptides. Curr. Biol. 8, 369–376 [DOI] [PubMed] [Google Scholar]
- 77. Crump M. P., Elisseeva E., Gong J., Clark-Lewis I., and Sykes B. D. (2001) Structure/function of human herpesvirus-8 MIP-II (1–71) and the antagonist N-terminal segment (1–10). FEBS Lett. 489, 171–175 [DOI] [PubMed] [Google Scholar]
- 78. Elisseeva E. L., Slupsky C. M., Crump M. P., Clark-Lewis I., and Sykes B. D. (2000) NMR studies of active N-terminal peptides of stromal cell-derived factor-1. Structural basis for receptor binding. J. Biol. Chem. 275, 26799–26805 [DOI] [PubMed] [Google Scholar]
- 79. Heveker N., Tissot M., Thuret A., Schneider-Mergener J., Alizon M., Roch M., and Marullo S. (2001) Pharmacological properties of peptides derived from stromal cell-derived factor 1: study on human polymorphonuclear cells. Mol. Pharmacol. 59, 1418–1425 [DOI] [PubMed] [Google Scholar]
- 80. Loetscher P., Gong J. H., Dewald B., Baggiolini M., and Clark-Lewis I. (1998) N-terminal peptides of stromal cell-derived factor-1 with CXC chemokine receptor 4 agonist and antagonist activities. J. Biol. Chem. 273, 22279–22283 [DOI] [PubMed] [Google Scholar]
- 81. Rasmussen S. G., Choi H. J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., et al. (2011) Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rankovic Z., Brust T. F., and Bohn L. M. (2016) Biased agonism: an emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett. 26, 241–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wootten D., Christopoulos A., and Sexton P. M. (2013) Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug Discov. 12, 630–644 [DOI] [PubMed] [Google Scholar]
- 84. Bernhagen J., Mitchell R. A., Calandra T., Voelter W., Cerami A., and Bucala R. (1994) Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF). Biochemistry 33, 14144–14155 [DOI] [PubMed] [Google Scholar]
- 85. Schwartz V., Krüttgen A., Weis J., Weber C., Ostendorf T., Lue H., and Bernhagen J. (2012) Role for CD74 and CXCR4 in clathrin-dependent endocytosis of the cytokine MIF. Eur. J. Cell Biol. 91, 435–449 [DOI] [PubMed] [Google Scholar]
- 86. Murphy J. W., Yuan H., Kong Y., Xiong Y., and Lolis E. J. (2010) Heterologous quaternary structure of CXCL12 and its relationship to the CC chemokine family. Proteins 78, 1331–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kamir D., Zierow S., Leng L., Cho Y., Diaz Y., Griffith J., McDonald C., Merk M., Mitchell R. A., Trent J., Chen Y., Kwong Y. K., Xiong H., Vermeire J., Cappello M., et al. (2008) A Leishmania ortholog of macrophage migration inhibitory factor modulates host macrophage responses. J. Immunol. 180, 8250–8261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Harrison J. E., Lynch J. B., Sierra L. J., Blackburn L. A., Ray N., Collman R. G., and Doms R. W. (2008) Baseline resistance of primary human immunodeficiency virus type 1 strains to the CXCR4 inhibitor AMD3100. J. Virol. 82, 11695–11704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.8, DeLano Scientific, Palo Alto, CA [Google Scholar]