

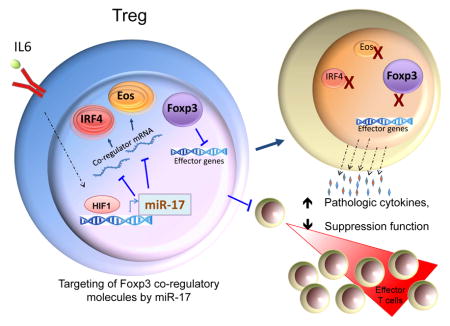
SUMMARY
Regulatory T (Treg) cells are important in maintaining self-tolerance and immune homeostasis. The Treg cell transcription factor Foxp3 works in concert with other co-regulatory molecules, including Eos, to determine the transcriptional signature and characteristic suppressive phenotype of Treg cells. Here, we report that the inflammatory cytokine IL-6 actively repressed Eos expression through the microRNA-17 (miR-17). miR-17 expression increased in Treg cells in the presence of IL-6, and its expression negatively correlated with that of Eos. Treg cell suppressive activity was diminished upon overexpression of miR-17 in vitro and in vivo, and RNAi of miR-17 resulted in enhanced suppressive activity, which was mitigated upon co-expression of an Eos mutant lacking miR-17 target sites. Ectopic expression of miR-17 imparted effector T cell-like characteristics to Treg cells via the de-repression of effector cytokine genes. Thus, miR-17 provides a potent layer of Treg cell control through targeting Eos and additional Foxp3 co-regulators.
eTOC Blurp
T regulatory (Treg) cells play important roles in the resolution of inflammation. Pan and colleagues show that, in turn, inflammatory cues induce the expression of miR-17, which targets transcriptional co-regulators that act in concert with Foxp3 to diminish the suppressive activity of Treg cells.
INTRODUCTION
Regulatory T (Treg) cells maintain self-tolerance and control the magnitude of both protective and pathologic immune responses (Kretschmer et al., 2008; Sakaguchi et al., 2010; Tang and Bluestone, 2008; Vignali et al., 2008). Treg cells can either originate in the thymus or be induced from naïve T cells in the periphery. Both populations typically express the transcription factor Foxp3 that is indispensable for Treg cell development and function (O’Shea and Paul, 2010; Sakaguchi et al., 2013). Inactivating mutations of Foxp3 in humans and mice result in severe autoimmune pathology (Ziegler, 2006), demonstrating the importance of Foxp3+ Treg cells in immune control.
Treg cell plasticity or phenotypic instability is the subject of debate (Sakaguchi et al., 2013). The bulk of Foxp3 expressing Tregs are reported to retain Foxp3 expression during infection and Th1-mediated inflammation (Rubtsov et al., 2010). Additionally, Tregs that do lose Foxp3 expression may regain it and their suppressive function– with phenotypic instability being chiefly seen among induced Tregs (Miyao et al., 2012). Other reports have shown that Foxp3 levels Treg cell functions can be disrupted by inflammatory cues (Floess et al., 2007; Fontenot et al., 2005; Zhou et al., 2009). Indeed cytokines such as IL-4, IL-6, and IL-21 have been shown to bestow Foxp3+ T cells with Teff cell attributes in vitro (Tsuji et al., 2009; Xu et al., 2007; Yang et al., 2008a). Treg cells can also be “reprogrammed” to take on Teff cell-like function while retaining Foxp3 expression (Sharma et al., 2013). Mechanisms of potential plasticity are, however, not well defined.
The importance of the inflammatory cytokine IL-6 in defining the Treg/Th17 cell balance has been established. The presence of this and other Stat3-signaling cytokines during T cell activation can divert developing Tregs cells towards a Th17 fate (Bettelli et al., 2006; Veldhoen et al., 2006; Yang et al., 2008a; Zhou et al., 2008a), it can also inhibit the ability of natural Treg cells to suppress T cell proliferation in vitro (Goodman et al., 2009; Shen and Goldstein, 2009). Although IL-6 mediated inhibition of Foxp3 expression (Gao et al., 2012; Lal et al., 2009; Yang et al., 2008b; Zheng et al., 2008) may account for some of this antagonism, it is possible that IL-6 may impact other molecules important for Treg cell suppressive function.
Foxp3 cooperates with a cadre of co-factors to shape the transcriptional landscape of Treg cells (Fu et al., 2012; Rudra et al., 2012). One such co-regulator, Eos, is essential for Foxp3-mediated control of Treg cell gene expression (i.e. repression of effector T cell genes) and function (Pan et al., 2009). While Treg cells contain high amounts of Eos, only low levels are detected in Th17 cells (Quintana et al., 2012). Furthermore, a subset of ‘reprogrammed’ Treg cells appears prone to loss of Eos expression (Sharma et al., 2013). This suggests that Eos is tightly regulated in developing Treg cells as well as those undergoing conversion to an expanded or Teff cell-like phenotype. Other transcriptional regulators associated with Foxp3 activity include IRF-4 (Zheng et al., 2009), Satb1 (Fu et al., 2012; Rudra et al., 2012), and GATA-1 (Fu et al., 2012). These molecules could share partially redundant co-repressor function that assures silencing of Teff cell genes in Foxp3+ Treg cells (Bettini et al., 2012; Darce et al., 2012; Fu et al., 2012). The mechanisms that regulate the expression of Eos and other co-regulators of Foxp3 activity in Treg cells are not well understood.
MicroRNAs (miRNAs ) impact aspects of immunity, including the function, homeostasis and phenotypic stability of Treg cells (O’Connell et al., 2010). MiRNAs are short (~22 nucleotide), non-coding RNAs produced via sequential processing of primary RNA polymerase II transcripts by the class III RNase enzymes Drosha and Dicer. MiRNAs act on target protein-encoding mRNAs through the RNA-induced silencing complex, marking them for translational repression or degradation (Stefani and Slack, 2008). Different miRNA clusters have been shown to be involved in the immune response (Hou et al., 2009; Li et al., 2007; Xiao et al., 2008; Zhou et al., 2008b). Deletion of Dicer1 and Drosha in Treg cells results in autoimmunity similar to that seen in Scurfy (Foxp3 null) mice although Foxp3 expression levels are not significantly changed (Chong et al., 2008; Liston et al., 2008). Several miRNAs contribute to Treg cell function and phenotypic stability. For instance, miR-146a promotes Treg-mediated control of Th1 responses (Lu et al., 2010); miR-10a prevents acquisition of a Th17-like phenotype by Treg cells (Takahashi et al., 2012); and miR-155 supports Treg cell homeostasis and expansion (Lu et al., 2009) as well as their development (Kohlhaas et al., 2009).
The miR-17-92 miRNA cluster has been implicated in immune regulation and lymphomagenesis. The gene encoding this cluster is located on human chromosome 13q31, in a genomic region that is often amplified in lymphomas, and other cancers that also have high expression of the mature miRNAs of this locus (Ota et al., 2004; Tagawa and Seto, 2005). The inflammatory cytokine IL-6 induces miR-17-92 expression (Brock et al., 2009), and ectopic expression of the miR-17-92 cluster in T cells causes autoimmunity in mice (Xiao et al., 2008).
Studies of miR-17-92 deficient mice have implicated these miRNAs in the regulation of Teff and Treg cell function. One study found that members of this cluster promote IFNγ production by Th1 cells while suppressing the differentiation of iTregs (Jiang et al., 2011). Another found that miR-17-92 deficient T cells were less pathogenic than wild type cells in a model of GVHD – being poor producers of IFNγ more inclined to become Th2 cells and suppressive iTreg cells (Wu et al., 2015). In contrast, another study found that the miR-17-92 cluster supports natural Treg function by promoting expression of the anti-inflammatory cytokine IL-10 (de Kouchkovsky et al., 2013) suggesting that the miRNAs of this cluster may play complex and incompletely visualized roles in the biology of T cell subsets.
Here we report that IL-6 actively suppressed Eos mRNA and protein expression through miR-17. This targeting of Eos transcript and that of other Foxp3 co-regulators including Satb1 and IRF-4 resulted in decreased Treg cell suppressive function and the acquisition of Teff cell characteristics, including the production of effector cytokines.
RESULTS
T cell specific deletion of miR-17-92 enhanced Treg cell function
To assess the role of the miR-17-92 cluster in Treg cell function, we generated T cell-specific miR-17-92 null (mir17-92−/−) mice by breeding mir17-92flox/flox mutants to CD4-Cre+ transgenic mice. In agreement with a previous study using a T cell-restricted mir17-92 deletion approach (Jiang et al., 2011), mir17-92−/− animals showed no obvious abnormalities in T cell subset distribution or indications of altered immune regulation at baseline. However, upon induction of experimental autoimmune encephalomyelitis (EAE), miR-17-92-deficient mice fared better than wild type (WT) littermates. While disease onset was not delayed in mir17-92−/− mice, they recovered extensively relative to WT mice (Figure 1A). In keeping with a more restrained immune response, mir17-92−/− mice harbored lower proportions of IL-17- and IFNγ-producing CD4+ T cells in their CNS during recovery compared to WT controls (Figures 1B and 1C). However, Foxp3+ CD4+ T cell frequencies in the CNS of WT and mir17-92−/− mice (Figures 1D and 1E) were comparable. In line with this, T cell miR-17-92 deficiency did not affect in vitro Treg cell induction or the generation of pathogenic Th17 cells (Figure 2A).
Figure 1. CD4+ T cell specific miR-17-92 deletion mice more effectively control experimental autoimmune encephalomyelitis (EAE).

(A) Wild type (WT) mice and those of a CD4+ specific miR-17-92 deletion strain (mir17-92Flox/Flox/CD4-Cre+, or “mir17-92−/−“) were injected s.c. with 100 μg MOG 35–55 in CFA and 250ng Pertussis Toxin i.p. Disease severity was scored daily. Mean scores for WT mice and mir17-92−/− mice over time are represented (±SEM; p < 0.05, n = 7–10 per group). Shown are combined results of 3 individual experiments. (B and C) On day 45, CNS-infiltrating T cells were recovered and stained for IL-17, IFNγ. The mean percentage of CNS IL-17+CD4+ T cells on day 45 was determined. (D and E) CNS-infiltrating T cells were isolated on day 45 and the percentage of CD4+ T cells that were Foxp3+ was determined. C and E present the mean (± SEM; *p < 0.05) of at least 3 experiments, while B and D are representative analyses.
Figure 2. miR-17-92 deficiency does not impact Foxp3 induction, but enhances Treg cell suppressive function.

(A) In vitro of differentiation of naïve (CD62LhiCD25−CD4+) T cells from WT and mir17-92−/− mice into Th17 and iTreg cell lineages. (B) Analysis of the in vitro function of Treg cells from WT and mir17-92−/− littermates. Treg cells were mixed with CFSE labeled Naïve T lymphocytes from CD45.1+ C57BL/6 mice for three days. CD45.1+ T cells were analyzed for CFSE dilution. (C) The effect of miR-17-92 deficiency on IL-10 expression by Treg cells. mRNA from FACS purified Treg cells was used to generate cDNA for qRT-PCR analysis of IL-10 transcript. Data are representative of 3 independent experiments.
Without a major effect on the generation of Treg Teff cells, we suspected miR-17-92 might interfere with Treg cell function. Indeed, in vitro functional analysis of Treg cell suppression revealed that Treg cells isolated from mice with Foxp3-driven miR-17-92 deficiency (Foxp3yfpCre+/mir17-92flox/flox) were more effective than WT controls at suppressing the proliferation of WT naïve T cells (Figure 2B). MiR-17-92 deficiency in responder cells did not alter this trend (Supplementary Figure S1A).
Confirming that Treg cells lacking miR-17-92 are functionally enhanced in vivo, we induced EAE in mice specifically lacking this miRNA cluster in Foxp3+ T cells. As with CD4-driven deletion, these mice displayed less severe disease and milder inflammation than WT mice (Supplementary Figure S1B, C). These findings imply that in Treg cells, miR-17-92 expression antagonizes suppressive function. In line with this, mir17-92−/− Tregs did express heightened levels of IL-10 message compared to WT-derived Treg cells (Figure 2C). The mechanism responsible for these effects however remained to be determined.
Identification of Eos as a potential target of miR-17-92
The phenotype observed upon miR-17-92 over-expression in Treg cells resembled that observed upon silencing the Foxp3 co-factor Eos. Previously, we showed that silencing Eos expression perturbed Foxp3-mediated gene regulation and Treg cell suppressive function without impacting the expression of Foxp3 itself (Pan et al., 2009). Other important and possibly redundant molecular collaborators of Foxp3 have also been identified and characterized (Fu et al., 2012; Rudra et al., 2012).
The behavior of mir17-92−/− Treg cells suggests that suppression may be negatively regulated by the miRNAs of this cluster. To test this notion, we screened a panel of miR-17-92 miRNAs for the ability to bind the 3′UTR of the Eos, Irf4, Gata1, Lef1 and Satb1 transcripts bioinformatically. We found miR-17, an individual mature miRNA of the miR-17-92 cluster, to be a likely Eos-targeting candidate (Supplementary Figure S2A). We also found that miR-17 could target expression of other co-regulatory factors including IRF4 and Satb1 (Supplementary Figure S1B,C) suggesting that a miRNA of this cluster could potently undermine Treg cell function by antagonizing multiple factors necessary for Foxp3 activity. These results were supported by the observation that Eos, IRF-4 and Satb1 levels were stunted in Treg cells from miR-17-92 transgenic mice activated in vitro compared to WT Treg cells (Supplementary Figure S2D, E, F).
Regulation of Eos by miR-17 in response to IL-6
Results suggesting that members of the miR-17-92 cluster have a negative role in Treg cell function in vivo and in vitro encouraged further investigation of individual cluster constituents and their regulation in Treg cells. We focused on the role of miRNA in Eos regulation since this Foxp3 co-regulator has been shown to be important for Treg cell gene expression (Pan et al., 2009; Sharma et al., 2013). In vitro activation of Treg cells with anti-CD3/CD28 antibodies decreased endogenous miR17 levels (Supplementary Figure S3A). This was concomitant with elevated Eos mRNA (Figure 3A), supporting that this miRNA cluster regulates Eos.
Figure 3. Identification of miR-17 as an Eos 3′UTR targeting miRNA.

(A) Expression of Eos mRNA in Treg cells after stimulation. CD4+ Foxp3GFP+ Treg cells were isolated by FACS and incubated with anti-CD3/CD28 antibodies for the indicated times and Eos mRNA levels were determined by RT-PCR. (B) The psBZN - iCHECK2 reporter vector (Bozeman, Montana, USA) shown was used to assess the ability of miRNAs to target the Eos 3′UTR. (C) Luciferase reporters containing the WT Eos 3′UTR or mutant Eos 3′ (MU). miR-17-92, miR-17-19a, and miR-17 were screen for inhibition of Eos 3′UTR luciferase activity relative to mock treatment. Rluc =Renilla luciferase; Luc = firefly luciferase; and pA =polyadenylation signal. The pcDNA3 vector was used to transiently express miRNAs. Eos 3′ MU contained mutations at miR-17 and miR-92 sites and served as a negative control for miRNA inhibition.
To verify that miR-17 was indeed the miRNA responsible for targeting Eos expression, a luciferase reporter assay was utilized. Here a reporter containing either the normal 3′UTR of Eos or a seeding region mutant (miR-17 and miR-92 sites) were constructed and co-transfected into 293T cells along with plasmids encoding the entire cluster; miR17-19a; miR20a-92a; miR17; or miR19a. While the mutant Eos 3′UTR was insensitive to inhibition regardless of co-expressed miRNAs, recipients of WT Eos 3′UTR and any miR construct containing miR17 (i.e., miR-17-92, miR-17-19a and miR-17) displayed significantly inhibited Eos 3′UTR luciferase activity. On the other hand, mock treated and miR19a recipients showed no reduction and miR20a-92a only slightly inhibited reporter activity (Figure 3B and 3C). These results identified miR-17 as the miRNA within miR-17-92 responsible for targeting the Eos 3′UTR.
We further characterized the regulation of miR-17 in Treg cells and other T cell subsets. A cytokine reported to down-modulate both Treg cell lineage development and Treg cell suppressive activity is IL-6 (Lal et al., 2009; Yang et al., 2008b). IL-6, produced by antigen-presenting cells (APCs) as a result of Toll-like receptor engagement, has been demonstrated to block the suppressive activity of Treg cells (Pasare and Medzhitov, 2003). Additionally, exposure to IL-6 was reported to induce IL-17 production and Foxp3 loss by Treg cells (Lal et al., 2009; Zhou et al., 2007). However, it is not well understood how IL-6 mitigates Treg cell function. We suspected that inflammatory cues such as IL-6 might undermine Treg cell function through the induction of miR-17.
To test this hypothesis and to obtain insights into the physiological role of miR-17, we stimulated Treg cells in the presence or absence of IL-6. Activation of Treg cells led to a decrease in miR-17 levels. However, in the presence of IL-6, the levels of miR-17 increased (Figure 4A). As previously seen, Eos expression was inversely related to miR-17 levels while increased IL-17 levels were also noted upon Treg cell activation in the presence of IL-6 (Figure 4B, C). IL-10 levels where not significantly affected by IL-6 treatment (Figure 4D).
Figure 4. IL-6 mediates miR-17 up-regulation through HIF-1α.

(A–D) Expression of miR-17, Eos mRNA, IL-17a, and IL-10 mRNA in Treg cells. CD4+ Foxp3GFP+ cells were stimulated with plate-bound anti-CD3/CD28 in the presence or absence of 20ng/ml IL-6 for 24h. mRNA levels were assessed. Expression of miR17, Eos and IL-10 without stimulation was set to 1. (E) qRT-PCR analysis of miR-17 in T cells. Naïve T cells were activated in the presence of TGFβ and IL-6, or TGFβ alone or media for 48h. Expression without TFGβ and IL-6 was set to 1. (F) Western blot analysis of Eos levels in T cells. Naive T cells were activated as above in the presence of TGFβ or TGFβ and IL-6 for the indicated times. Collected cells were subjected to SDS-PAGE, and Western blot with the indicated antibodies. Shown is a representative blot from at least 3 independent experiments. (G, H) miR-17 and Eos mRNA expression were measured during the course of EAE. Foxp3GFP+ Treg cells were isolated from WT and mir17-92−/− mice crossed to a Foxp3GFP reporter background at the pre-initiation, peak, and recovery phase of disease, and RT-PCR was carried out to measure miR17 and Eos levels. A cohort of mice from either group received anti-IL-6 blocking antibodies. Treg cells were recovered by FACS during the recovery phase of EAE and Eos and miR-17 mRNA was measured. (I) miR-17 expression in Treg cells from WT or CD4-specific HIF-1α null (HIF-1Flox/Flox/CD4-Cre+; HIF-1KO) mice. Treg cells isolated from WT or HIF-1 KO mice were activated with anti-CD3/CD28 for 24h. (J) Expression of miR-17 in T cells stimulated with IL-6 under normoxia and hypoxia. Treg cells were isolated and activated at atmospheric oxygen levels (normoxia) or at 2% oxygen (hypoxia) for 24h. For A–E, G–J miR17, Eos and Il-10 expression in each group was determined by RT-PCR.
This phenotype was reminiscent of that seen upon Eos silencing (Pan et al., 2009), which de-represses Teff cell cytokine genes in Treg cells. In line with this, activation of isolated nTreg cells with IL-6 led to the up-regulation of IL-17. However, in Treg cells lacking miR-17-92 expression, this uncharacteristic production of the proinflammatory cytokine was prevented (Supplementary Figure S3B). Additionally, IL-6 also regulated miR-17 in activated naïve T cells. Here we treated naïve T cells with TGFβ alone or in combination with IL-6. In the latter, miR-17 expression was higher (Figure 4E). Furthermore, Eos protein expression was consistently lower in naïve CD4+ T cells upon IL-6 and TGFβ treatment compared to TGFβ treatment alone (Figure 4F).
In keeping with these observations, during EAE WT Treg cells progressively increased miR-17 levels while losing Eos mRNA. In contrast, Treg cells recovered from mir17-92−/− mice displayed high and sustained levels of Eos during EAE (Figure 4G). These findings suggest that the intense inflammation of EAE dampens Eos expression by inducing miR-17. Also, they suggest that preventing this could enhance the in vivo performance of Treg cells by stabilizing Eos expression. Since IL-6 is an inflammatory cytokine capable of inducing miR-17, and the beneficial effects of neutralizing this cytokine during EAE have been demonstrated (Gijbels et al., 1995), we tested whether this could also alter miR-17 and Eos expression in vivo. Indeed, antibody-mediated blockade of IL-6 during EAE effectively negated expression of miR-17 in WT Treg cells and preserved Eos mRNA levels to a degree seen in mir17-92−/− Treg cells (Figure 4G, H).
Exploring how IL-6 induces miR-17, we examined the upstream promoter region of the miR-17-92 for binding sites for downstream mediators of IL-6 signaling. STAT3 has been reported to interact with the mir17-92 promoter (Brock et al., 2009). However, while we did not find a canonical STAT3 binding site, a conserved, putative site for another IL-6/STAT3–activated factor, HIF-1α was (Supplementary Figure S4A; Supplementary Figure S4B). Chromatin immunoprecipitation (ChIP) analysis confirmed that HIF-1α indeed binds the mir17-92 promoter (Supplementary Figure S4C). Supporting the notion that HIF-1 induces expression of miR-17, levels of the miRNA were lower in IL-6/TCR activated Treg cells from T cell-specific HIF-1α deficient mice than they were in Treg cells from control mice (Figure 4I). In addition, stimulation of WT Treg cells in a hypoxic chamber to stabilize HIF-1 levels increased miR-17 expression by these cells (Figure 4J). These data demonstrate that IL-6 augments miR-17 expression through HIF-1.
Forced expression of miR-17 antagonized Treg cells suppressive activity in vitro
We found that miR-17-92 negatively affected Treg cell function, and particularly miR-17 was chiefly responsible for targeting the Eos transcript. In order to characterize the impact of modulating miR-17 on Treg cell function, an in vitro suppression assay was performed. Treg cells transduced with either a miR-17 expression construct or a control vector were co-cultured with CFSE-labeled naïve CD45.1+ CD4+ cells and irradiated APC and were activated. The suppressive capacity of Treg cells ectopically expressing miR-17 (confirmed in Supplementary Figure S5A) was significantly reduced compared to empty vector controls (Figure 5A). Conversely, antisense silencing of miR-17 (“miRZip17”; Supplementary Figure S5B) improved the ability of Tregs to stymie responder cell proliferation beyond that of controls (Figure 5B). As expected, miR-17 over-expression and silencing were confirmed to reduce and enhance Eos protein levels, respectively, in Treg cells relative to empty vector controls (Supplementary Figure S5C). These complimentary findings suggest that miR-17 antagonizes the suppressive function of Treg cells.
Figure 5. miR-17 modulates Treg cell suppressive activity.

(A) The effect of miR-17 overexpression on the in vitro function of Treg cells was explored by transducing them with either a bicistronic retrovirus expressing miR-17 or the GFP-containing empty vector (EV) prior to co-culture with CFSE labeled CD4+ naïve T cells from CD45.1+ mice at the indicated ratios in the presence of anti-CD3/CD28 beads for 4 days. CD45.1+ T cells were analyzed by flow cytometry for CFSE dilution (blue line = Tnaive only; red line= Treg cells and Tnaive). (B) The in vitro suppressive capacities of Treg cells transduced with either the antisense construct, miRZip17 or a control vector were determined as in (A). (C–F) The impact of miR-17 on Foxp3 induction and pro-inflammatory cytokine expression by iTreg cells. Naïve T cells from WT mice were transduced as in (A), followed by in vitro Treg skewing for 72h. Intracellular levels of Foxp3 (C) or Foxp3 and IFNγ were determined by flow cytometry after restimulation with PMA and ionomycin (D). Data are representative of 4 independent experiments. (E, F) qRT-PCR analysis of IFNγ and Eos mRNA in Treg cells from (C). Shown are mean +/− SEM from three independent experiments.
Since both IL-6 and HIF-1 have been shown to negatively impact the generation of Foxp3+ Treg cells from naïve CD4+ precursors (Dang et al., 2011; Zhou et al., 2008a), we suspected that miR-17 induction – an event closely tied to this pathway – can suppress de novo supplementation of the iTreg cell pool as well as the function of established Treg cells. Therefore, we studied the effects of miR-17 during the in vitro induction of Treg cells by TGFβ and IL-2 (iTreg cell differentiation). Here, over-expression of miR-17 in naïve CD4+ cells, as before, was achieved by lentiviral transduction prior to iTreg cells skewing. As with miR-17-92 modulation in established Treg cells, miR-17 over-expression did not alter Foxp3 expression, which was similarly induced in both groups. However, miR-17 over-expression did increase the frequency of IFNγ producing cells in both the Foxp3+ and Foxp3- populations in these cultures compared to empty vector treatment (Figure 5D). Similar trends were seen at the transcript level for IFNγ (Figure 5E) and, as expected, ectopic expression of miR-17 significantly decreased Eos mRNA in iTreg cells (Figure 5F). Together, these findings demonstrate that miR-17 can destabilize the suppressive functions and gene expression of established and differentiating Treg cells.
miR-17 modulated the suppressive activity of Treg cells in vivo
Our in vitro findings that miR-17 negatively impacted Treg cell function prompted us to examine the in vivo role of miR-17 in a Treg cell-dependent colitis model. In this model, transfer of naïve CD4+CD25–CD62LHigh T cells induces progressive colitis into Rag2−/− mice. The co-transfer of CD4+CD25+ Treg cells, however, is sufficient to prevent disease. These protective Treg cells were found to progressively repress miR-17 levels throughout the course of the experiment – a trend coinciding with effective suppression of colon inflammation (Supplementary Figure S6A). This and our in vitro findings suggest that robust expression of miR-17 should impair the ability of Treg cells to prevent colitis.
As expected, control Treg cells effectively prevented disease development and weight loss. However, ectopic expression of miR-17 in Treg cells abolished protection. The ability of these defective Treg cells to control colitis was mostly restored by co-transduction with a miR-17-resistant Eos construct (Figure 6A). This trend was also seen at the level of colon pathology. MiR-17 replete Treg cells, unlike their control counterparts, failed to prevent robust colon infiltration and pathology. Treg cells carrying both Eos and miR-17 suppressed the accumulation of leukocytes and immunopathology similar to controls (Figure 6B, C). miR-17 over-expressing Treg cells also saw increased CD4+ Teff cell infiltration of the colon resembling mice receiving no Treg cells. Meanwhile empty vector and Eos/miR-17 carrying Treg cells equally blocked Teff cell accumulation (Figure 6D, blue bars). Treg cells numbers in the lamina propria (LP) were not affected by miR-17 or Eos over-expression (Figure 6D, red bars). However, cytokine staining revealed that excess miR-17 allowed greater IL-17- and IFNγ-producing Teff cell frequencies in recipient mice. Treg cells receiving either empty vector or co-delivery of miR-17 and Eos constructs effectively controlled the pathologic T cells response (Figures 6E–H). A similar trend was observed upon staining for IL-17 in Teff cells recovered from the LP (Figure 6I, J).
Figure 6. Effects of miR-17 overexpression on Treg cell function in vivo.

(A) Percentage weight loss induced in Rag2−/− mice injected i.v. with naïve CD4+ T cells alone (no Treg cells) or these cells and CD45.1+Treg cells co-transduced with a bicistronic retroviral miR-17 expression construct, Treg cells carrying empty vector or Treg cells co-transduced with Eos and miR-17 expression constructs. 8–10 mice were used in each group, and the means ± SD of 3 independent experiments are shown. (B) Representative photomicrography of the distal colon of Rag2−/− mice after adoptive transfers described above. 8 weeks post-transfer, colons were harvested and processed for standard H/E staining and histological analysis. (C) H/E slides were scored in a blinded fashion and colon pathology was scored. Shown are the mean scores for each treatment group +/− SD from at least 3 independent experiments. (D) Colon infiltration by CD4+ T cells during colitis. Lamina propria-infiltrating leukocytes were recovered and the absolute number of original naïve T cells and congenically distinct Treg cells were determined and are represented as the mean +/− SEM. (E to J) Pro-inflammatory cytokine production by naïve T cells co-transferred with the indicated Treg cells. Intracellular IL-17 or IFNγ levels in naive CD4+ (CD45.2+) cells from the (E,F) spleen, (G,H) mesenteric LNs, and (I,J) lamina propria of recipient mice were observed after re-stimulation of the recovered cells. Panels F, H, and J present the mean (± SEM) of three trials, while E, G, and I are representative dot plots.
Not only were miR-17 over-expressing Treg cells unable to restrain the activation of colitogenic Teff cells, they also expressed the Teff cell cytokine IFNγ. In contrast, little-to-no IFNγ mRNA was detected in control Treg cells (Supplementary Figure S6B). Collectively, these findings suggest that modulation of miR-17 expression in Treg cell can drastically alter their in vivo function.
DISCUSSION
Post-transcriptional regulation of gene expression by microRNAs is important for the control of numerous cellular events. Processes critical for the activation and control of the immune system are no exception. Particularly, this mode of regulation is crucial for controlling Treg cell differentiation and function (Liston et al., 2008). In the present study we identified a miRNA, miR-17 that negatively regulated the suppressive function of Treg cells by targeting expression of Foxp3’s transcriptional co-regulators such as Eos, IRF-4 and Satb1 that shape the transcriptional landscape responsible for the Treg cell phenotype (Fu et al., 2012).
The miR-17-92 cluster has been previously reported as important for Th1 differentiation (Jiang et al., 2011). This study found that miR-17-92 deficient naïve CD4+ T cells are less efficient at Th1 differentiation in vitro displaying lower levels of IFNγ and T-bet. Over-expression of individual miRNA cluster constituents revealed that miR-19b alone could enhance IFNγ in differentiating Th1 cells suggesting that Th1 differentiation is promoted by cluster members other than miR-17. In vivo, these authors also found that antigen specific CD4+ T cells transduced with miR-19b significantly increased IFNγ-production while miR-17 over-expression resulted in only modest increases (Jiang et al., 2011). While the expedient recovery of mir17-92−/− in our EAE experiments could reflect deficient Th1 responses, the modest impact of miR-17 on Th1 differentiation and the similar behavior of mice specifically lacking miR-17-92 in Treg cells suggests that a large part of this phenotype can be attributed to altered Treg cell function.
Jiang et al. also found that deletion of miR-17-92 enhances in vitro Foxp3 up-regulation during iTreg cell skewing of naïve CD4+ T cells particularly when TGFβ levels are low (Jiang et al., 2011). In our study, however, we failed to see a significant contribution of miR-17-92 or miR-17 alone to the generation of iTreg cell; this could reflect subtle differences in our in vitro differentiation approaches (i.e. stimuli and TGFβ levels). Compatible with our observations, as well as those of Jiang et al., is the finding that miR-17 (specifically miR-17-5p) is expressed in the T cells of multiple sclerosis patients, implicating this miRNA in human autoimmunity (Lindberg et al., 2010).
Recently, de Kouchkovsky et al. reported that the miR-17-92 cluster positively contributes to Treg cell function. They found that in the absence of the microRNA cluster, Treg cells are capable of proliferation, trafficking, and enforcing immune homeostasis at baseline. However, in this study, miR-17-92 deficient Treg cells are deficient in IL-10 production (de Kouchkovsky et al., 2013). While we also did not see baseline defects in the absence of miR-17-92, we found that the cluster, and particularly miR-17, adversely affected Treg cell function by targeting Eos. The apparent incongruity may stem from unique traits of the conditional deletion mice used to uncover the effects of miR-17-92 deficiency. Further study of the factors governing expression of individual miR-17-92 cluster members and their paralog counterparts may yield additional insights into their potentially varied contributions to Treg cell biology.
IL-6 is crucial for Th17 differentiation but antagonizes Treg cells. In vitro, IL-6 is a key factor in the balance between Foxp3+ Treg and Th17 cells, inhibiting TGF-β-induced generation of Treg cells and promoting Th17 cell differentiation (Bettelli et al., 2006; Zhou et al., 2007). Our data showed that Treg cell activation reduces miR-17 levels while IL-6 enhances them. The resultant decrease in Eos level is likely responsible for some of the effects of IL-6 on Treg cell function while additional mechanisms may impact Foxp3 expression.
In general, TCR activation can enhance the ability of Treg cells to curtail immune activation. Therefore, the increased level of Eos mRNA seen after activation (Figure 3A) is compatible with our previous findings that Eos is important for the execution of Foxp3-mediated gene regulation (Pan et al., 2009). Eos expression has also been associated with a stable suppressive Treg cell phenotype (Sharma et al., 2013). Additionally, the importance of Eos in establishing the Treg cell gene expression profile and phenotype was also highlighted in a study identifying it as a member of a crucial quintet of Foxp3 transcriptional collaborator. In this study, however, modulating individual co-regulators did not substantially disrupt Foxp3-mediated gene regulation, suggesting redundancies in their function. However, when multiple members of the quintet were modified, patterns of Treg cell gene expression were disrupted (Fu et al., 2012).
While Foxp3 co-factors may be redundant and therefore compensate for the deficiencies of each other, our present findings suggest that, under certain conditions, this may not be possible. For instance, since multiple co-regulators are sensitive to regulation by miR-17, conditions leading to robust expression of this miRNA are expected to dampen expression of multiple Foxp3 co-regulators. Moreover, in our studies we found that Eos expression levels were higher than other members of Fu et al.’s quintet. This could suggest that a hierarchy of importance may exist (at least under certain conditions) within the Foxp3 co-regulator pool.
Certain miRNAs such as miR-155 and miR-146a contribute Treg cells and immune control (Lu et al., 2010; Lu et al., 2009), and microRNAs in general are absolutely needed for stable Treg cell function (Liston et al., 2008). Our study adds another wrinkle to the relationship between miRNAs and Treg cells. Specifically, we demonstrated a level of control over Treg cells that hinges upon the important co-regulators of Foxp3. With a growing number of documented Foxp3-interacting and collaborating molecules being brought to light, characterizing the factors governing their expression, including miRNAs, will expand the list of immunoregulatory junctions that may be exploited by novel immunotherapeutic interventions.
EXPERIMENTAL PROCEDURES
Animals
All mice were housed under pathogen-free conditions in the animal facility of Johns Hopkins Animal Resource Center. All animal experiments were performed in specific-pathogen-free, Helicobacter-free facilities following national, state and institutional guidelines.
T cell culture
Naive T cells were purified using a FACS Aria sorter prior to stimulation with anti-CD3/CD28 antibodies in a 24-well plate (1 and 4 mg/well, respectively; Biolegend) for 3–7 days. Th17-skewing conditions consisted of IMDM media supplemented with 5% FBS, 20 ng/ml IL-6, 2.5 ng/ml TGFβ (Peprotech), and 10 μg/ml neutralizing antibodies against IFN-γ, IL-4, and IL-12 (Biolegend). For hypoxia experiments, cells were cultured in a GasPak Plus anaerobic chamber (1% O2) for 24 hr cycles interrupted by normoxic rest.
EAE induction
Six- to eight-week-old, sex-matched WT and mir17-92−/− littermates were injected s.c in the rear flank with 100 μg MOG35–55 peptide (2HNMEVGWYRSPFSRVVHLYRNGK-COOH) in complete Freund’s adjuvant (Sigma) on day 0, and 250 ng pertussis toxin (List Biological) was injected i.p on day 0 and day 1 post-induction. Mice were monitored and disease severity was scored daily.
Isolation of lamina propria leukocytes (LPL) and FACS analysis
Mice (pooled from 4–6 mice per group) were euthanized and the colons excised and placed in ice-cold PBS. After extensive washing of the colonic lumen with PBS, colons were minced into 0.3–0.5 cm pieces, and incubated in HBSS containing 10% FCS and 5 mM EDTA. Remaining tissue was digested (Liberase, DNaseI) at 37ºC for 1 h, and the LPL were recovered by gradient centrifugation, washed, and stained for surface and intracellular markers. Flow cytometric analysis was performed on FACSCalibur (BD Bioscience) and data was analyzed using Flow Jo software (Tree Star Inc.).
In vitro proliferation/suppression assays of Treg cell function
Sorted CD45.1+CD4+CD25− CD62Lhi Naïve T cells were incubated with 2.5 μM CFSE for 7 minutes at 37°C, and excess dye was washed away. So rted CD45.2+ CD4+CD25+Treg cells were cultured alone or in titrated numbers with 2x 104 CFSE-labeled Naïve T cells stimulated with anti-CD3/CD28 Dynabeads at 2:1 cells to beads ratio. After 4 days, CD45.1+ CD4+ responder cells were analyzed for CFSE dilution.
Reporter Assay
The luciferase reporter gene assay reagents were obtained from Promega, and the assay was performed per manufacturer’s instructions.
Real-time RT-PCR
RNA was isolated by a miniRNA extraction kit (QIAGEN). The cDNA archival kit (Applied Biosystem) was used per the manufacturer’s instruction. Triplicate reactions were run using an ABI Prism 7500. mRNA levels were determined by comparative CT method and normalized to b-actin or 18s rRNA expression
microRNA Real time PCR
To measure mature miRNA, cDNA was synthesized using the Taqman MicroRNA RT Kit, and qRT-PCR was performed using the MicroRNA Taqman assay. U6 snRNA was used as internal control to normalize miRNA.
Statistical Analysis
An unpaired Student’s t test was used to determine significance (*p < 0.05).
Supplementary Material
Highlights.
miR-17 targets Eos and other Foxp3 co-regulators via 3′UTR recognition
IL-6 drives miR-17 transcription in Treg cells via HIF-1α. Treg cells lacking miR-17 exhibit increased suppressive function in vitro and in vivo
miR-17 overexpression in Treg cells exacerbates pathology in a murine colitis model
Acknowledgments
Our research is supported by grants from the Melanoma Research Alliance, the National Institutes of Health (RO1AI099300, RO1AI089830 and P30CA006873), National Natural Science Foundations of China (No. 81271054, 81470673), Shanghai Committee of Science and Technology (No. 14DZ2260300), Chang Gung Memorial Hospital (CMRPG3D0551), Ministry of Science and Technology in Taiwan (No. 104-2314-B-182A-110), “Kelly’s Dream” Foundation, the Janey Fund, and the Seraph Foundation, and gifts from Bill and Betty Topecer and Dorothy Needle. FP is a Stewart Trust Scholar; JB was supported by a CCFA Research Fellowship. We are grateful to Dr. Yu, Alice Lin-Tsing, Drs. Ya-Hui Wang and Dr. Chun-Yen Lin (Chang Gung Memorial Hospital) for reagent and mice contribution.
Footnotes
Supplementary Information includes six figures and Supplementary Experimental Procedures can be found with this article online.
AUTHOR CONTRIBUTIONS
HY and CW performed most experiments, analyzed data and assisted in the preparation of the manuscript; JB, YZ, PDAV, XW, JT, BVP, SB, LN, XN, XY, KC, RW, JZ and CY performed experiments and assisted with the revision of manuscript. DP, HL and FP provided lab space and support, designed experiments, reviewed the manuscript. HY, JB, HL and FP designed experiments, wrote and submitted the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bettini ML, Pan F, Bettini M, Finkelstein D, Rehg JE, Floess S, Bell BD, Ziegler SF, Huehn J, Pardoll DM, Vignali DA. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity. 2012;36:717–730. doi: 10.1016/j.immuni.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res. 2009;104:1184–1191. doi: 10.1161/CIRCRESAHA.109.197491. [DOI] [PubMed] [Google Scholar]
- Chong MM, Rasmussen JP, Rudensky AY, Littman DR. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J Exp Med. 2008;205:2005–2017. doi: 10.1084/jem.20081219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darce J, Rudra D, Li L, Nishio J, Cipolletta D, Rudensky AY, Mathis D, Benoist C. An N-terminal mutation of the Foxp3 transcription factor alleviates arthritis but exacerbates diabetes. Immunity. 2012;36:731–741. doi: 10.1016/j.immuni.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kouchkovsky D, Esensten JH, Rosenthal WL, Morar MM, Bluestone JA, Jeker LT. microRNA-17-92 regulates IL-10 production by regulatory T cells and control of experimental autoimmune encephalomyelitis. J Immunol. 2013;191:1594–1605. doi: 10.4049/jimmunol.1203567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Fu W, Ergun A, Lu T, Hill JA, Haxhinasto S, Fassett MS, Gazit R, Adoro S, Glimcher L, Chan S, et al. A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nat Immunol. 2012;13:972–980. doi: 10.1038/ni.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Gao Y, Li Z, Chen Z, Lu D, Tsun A, Li B. Synergy between IL-6 and TGF-beta signaling promotes FOXP3 degradation. Int J Clin Exp Pathol. 2012;5:626–633. [PMC free article] [PubMed] [Google Scholar]
- Gijbels K, Brocke S, Abrams JS, Steinman L. Administration of neutralizing antibodies to interleukin-6 (IL-6) reduces experimental autoimmune encephalomyelitis and is associated with elevated levels of IL-6 bioactivity in central nervous system and circulation. Mol Med. 1995;1:795–805. [PMC free article] [PubMed] [Google Scholar]
- Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD. IL-6 signaling in psoriasis prevents immune suppression by regulatory T cells. J Immunol. 2009;183:3170–3176. doi: 10.4049/jimmunol.0803721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. 2009;183:2150–2158. doi: 10.4049/jimmunol.0900707. [DOI] [PubMed] [Google Scholar]
- Jiang S, Li C, Olive V, Lykken E, Feng F, Sevilla J, Wan Y, He L, Li QJ. Molecular dissection of the miR-17-92 cluster’s critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood. 2011;118:5487–5497. doi: 10.1182/blood-2011-05-355644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol. 2009;182:2578–2582. doi: 10.4049/jimmunol.0803162. [DOI] [PubMed] [Google Scholar]
- Kretschmer K, Apostolou I, Verginis P, von Boehmer H. Regulatory T cells and antigen-specific tolerance. Chem Immunol Allergy. 2008;94:8–15. doi: 10.1159/000154846. [DOI] [PubMed] [Google Scholar]
- Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, Reid SP, Levy DE, Bromberg JS. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–273. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Lindberg RL, Hoffmann F, Mehling M, Kuhle J, Kappos L. Altered expression of miR-17-5p in CD4+ lymphocytes of relapsing-remitting multiple sclerosis patients. Eur J Immunol. 2010;40:888–898. doi: 10.1002/eji.200940032. [DOI] [PubMed] [Google Scholar]
- Liston A, Lu LF, O’Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med. 2008;205:1993–2004. doi: 10.1084/jem.20081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142:914–929. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, Loeb GB, Lee H, Yoshimura A, Rajewsky K, Rudensky AY. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, Huehn J, Hori S. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–275. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]
- O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–1102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, Seto M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004;64:3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- Pan F, Yu H, Dang EV, Barbi J, Pan X, Grosso JF, Jinasena D, Sharma SM, McCadden EM, Getnet D, et al. Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science. 2009;325:1142–1146. doi: 10.1126/science.1176077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, Rangachari M, Zhu C, Xiao S, Seavitt J, et al. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat Immunol. 2012;13:770–777. doi: 10.1038/ni.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, Rudensky AY. Stability of the regulatory T cell lineage in vivo. Science. 2010;329:1667–1671. doi: 10.1126/science.1191996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, Leslie C, Shaffer SA, Goodlett DR, Rudensky AY. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol. 2012;13:1010–1019. doi: 10.1038/ni.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol. 2013;13:461–467. doi: 10.1038/nri3464. [DOI] [PubMed] [Google Scholar]
- Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H, Pan F, Blazar BR, Pardoll DM, Mellor AL, et al. An inherently bifunctional subset of Foxp3+ T helper cells is controlled by the transcription factor eos. Immunity. 2013;38:998–1012. doi: 10.1016/j.immuni.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Goldstein DR. IL-6 and TNF-alpha synergistically inhibit allograft acceptance. J Am Soc Nephrol. 2009;20:1032–1040. doi: 10.1681/ASN.2008070778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- Tagawa H, Seto M. A microRNA cluster as a target of genomic amplification in malignant lymphoma. Leukemia. 2005;19:2013–2016. doi: 10.1038/sj.leu.2403942. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Kanno T, Nakayamada S, Hirahara K, Sciume G, Muljo SA, Kuchen S, Casellas R, Wei L, Kanno Y, O’Shea JJ. TGF-beta and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat Immunol. 2012;13:587–595. doi: 10.1038/ni.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, Fagarasan S. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science. 2009;323:1488–1492. doi: 10.1126/science.1169152. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Heinrichs J, Bastian D, Fu J, Nguyen H, Schutt S, Liu Y, Jin J, Liu C, Li QJ, et al. MicroRNA-17-92 controls T-cell responses in graft-versus-host disease and leukemia relapse in mice. Blood. 2015;126:1314–1323. doi: 10.1182/blood-2015-02-627356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008a;454:350–352. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008b;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGF-beta are resistant to Th17 conversion by IL-6. J Immunol. 2008;180:7112–7116. doi: 10.4049/jimmunol.180.11.7112. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, Corcoran L, Treuting P, Klein U, Rudensky AY. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008a;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol. 2009;21:281–285. doi: 10.1016/j.coi.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, Bluestone JA. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med. 2008b;205:1983–1991. doi: 10.1084/jem.20080707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006;24:209–226. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.