

ABSTRACT
Ageing is a process that inevitably affects most living organisms and involves the accumulation of macromolecular damage, genomic instability and loss of heterochromatin. Together, these alterations lead to a decline in stem cell function and to a reduced capability to regenerate tissue. In recent years, several genetic pathways and biochemical mechanisms that contribute to physiological ageing have been described, but further research is needed to better characterize this complex biological process. Because premature ageing (progeroid) syndromes, including progeria, mimic many of the characteristics of human ageing, research into these conditions has proven to be very useful not only to identify the underlying causal mechanisms and identify treatments for these pathologies, but also for the study of physiological ageing. In this Review, we summarize the main cellular and animal models used in progeria research, with an emphasis on patient-derived induced pluripotent stem cell models, and define a series of molecular and cellular hallmarks that characterize progeroid syndromes and parallel physiological ageing. Finally, we describe the therapeutic strategies being investigated for the treatment of progeroid syndromes, and their main limitations.
KEY WORDS: Ageing, Progeria, Rejuvenation, iPSCs
Summary: This Review defines the molecular and cellular hallmarks of progeroid syndromes according to the main cellular and animal models, and discusses the therapeutic strategies developed to date.
Introduction
The physiological deterioration that accompanies ageing constitutes a major risk factor for the development of human pathologies, such as cancer, cardiovascular disorders and neurodegenerative diseases (Kennedy et al., 2014). Key molecular hallmarks of the ageing phenotype include telomere attrition, genomic instability, loss of proteostasis, epigenetic alterations, mitochondrial dysfunction, deregulated nutrient sensing, stem cell exhaustion, cellular senescence and altered intercellular communication (Lopez-Otin et al., 2013). At the macromolecular level, ageing is characterized by the development of wrinkles, greying and loss of hair, presbyopia, osteoarthritis and osteoporosis, progressive loss of fertility, loss of muscle mass and mobility, decreased cognitive ability, hearing loss, and a higher risk for the development of cancer and heart diseases, among other features (López-Otín et al., 2013).
Progeroid syndromes are a group of very rare genetic disorders that are characterized by clinical features that mimic physiological ageing, such as hair loss, short stature, skin tightness, cardiovascular diseases and osteoporosis. Consequently, they constitute a relevant source of information to understand the molecular mechanisms involved in normal ageing. Progeroid disorders do not show differences in prevalence depending on sex or ethnic origin, and appear at an early age, mainly due to defects in the nuclear envelope and DNA repair mechanisms (Gordon et al., 2014). Affected individuals die at a young age, usually as a consequence of cardiovascular problems and musculoskeletal degeneration.
In this Review, we classify human progeroid syndromes into two main groups according to the mechanisms that underlie the disease. Next, we discuss recent findings in the study of progeroid syndromes, achieved through the use of cellular and animal models. On the basis of these findings, we propose nine candidate hallmarks of premature ageing, and highlight their similarities with those described for physiological ageing. These proposed hallmarks recapitulate the most remarkable characteristics of progeroid syndromes and define the mechanisms underlying their pathogenesis, which could provide ideas for future studies on both physiological and pathological ageing. Finally, we review different therapeutic strategies developed for the treatment of these rare but devastating diseases.
A classification system for human progeroid syndromes
All progeroid syndromes are characterized by similar clinical features (Table 1), but their underlying mechanisms can vary depending on the mutated gene and the pathway that is consequently altered. Below, we have classified progeroid syndromes into two general categories based on the molecular pathway involved. The first group includes those syndromes caused by alterations in components of the nuclear envelope, such as Hutchinson-Gilford progeria syndrome (HGPS), Néstor-Guillermo progeria syndrome (NGPS), atypical progeria syndromes (APSs), restrictive dermopathy (RD) and mandibuloacral dysplasia (MAD). The second group consists of progeroid syndromes induced by mutations in genes involved in DNA-repair pathways, such as Werner syndrome (WS), Bloom syndrome (BS), Rothmund-Thomson syndrome (RTS), Cockayne syndrome (CS), xeroderma pigmentosum (XP), trichothiodystrophy (TTD), Fanconi anaemia (FA), Seckel syndrome (SS), ataxia telangiectasia (AT), ataxia telangiectasia-like disorder (ATLD), cerebroretinal microangiopathy with calcifications and cysts (CRMCC), and Nijmegen breakage syndrome (NBN). A subcategory of this group comprises dyskeratosis congenita (DC) and Hoyeraal-Hreidarsson syndrome (HHS), linked to mutations in components of the telomerase complex (see Box 1 for a glossary of terms) that cause telomere attrition.
Table 1.
Clinical features of progeroid syndromes

Box 1. Glossary.
Emerin: protein encoded by the EMD gene. Present in the inner nuclear membrane in vertebrates, and highly expressed in cardiac and skeletal muscle. Mutations in emerin cause X-linked recessive Emery-Dreifuss muscular dystrophy, cardiac conduction abnormalities and dilated cardiomyopathy.
Cryptic splice site: splice sites that are usually dormant and can be activated by nearby mutations, often causing genetic diseases.
CST complex: a complex involved in termination of telomere elongation, composed of CTC1, STN1 and TEN1.
Double-strand break (DSB) repair: double-strand breaks (DSBs) can lead to genome rearrangements. Three mechanisms exist to repair DSBs: non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ) and homologous recombination (HR).
Interstrand crosslinks (ICLs): highly toxic DNA lesions that prevent transcription and replication by inhibiting DNA strand separation.
Multipotent cell: progenitor cell that has the gene activation potential to differentiate into multiple, but limited, cell types.
Nucleotide excision repair (NER): DNA-repair mechanism that removes DNA damage induced by ultraviolet (UV) light, mostly thymine dimers and 6,4-photoproducts.
Pluripotent cell: stem cell that has the potential to differentiate into any of the three germ layers: endoderm, mesoderm and ectoderm.
RecQ helicase family: helicases are enzymes important in replication and genome maintenance that unwind paired DNA and translocate in the 3′-to-5′ direction.
Shelterin complex: protein complex that protects mammalian telomeres from DNA-repair mechanisms, in addition to regulating telomerase activity. Subunits of shelterin bind to telomeres and induce the formation of a t-loop, a cap structure that prevents DNA-damage-sensing machinery from mistakenly repairing telomeres. The absence of shelterin causes telomere uncapping and thereby activates damage-signalling pathways that can lead to NHEJ, HR, senescence or apoptosis. Shelterin has six subunits: TRF1, TRF2, POT1, RAP1, TIN2 and TPP1.
Somatotropic axis: consists of growth hormone (GH) and insulin-like growth factors (IGF-I and -II) together with their associated carrier proteins and receptors. Regulates metabolism and other physiological processes. Other hormones, such as insulin, leptin, glucocorticoids and thyroid hormones, modulate GH and IGF synthesis and availability.
Telomerase complex: ribonucleoprotein complex encoded by the TERT, TERC, DKC1 and TEP1 genes that enlarges telomeres by adding the polynucleotide ‘TTAGGG’ to their 3′ end thanks to its reverse transcriptase activity, so that they can protect the ends of the chromosomes from deterioration or from fusion with other chromosomes.
Nuclear architecture instability and premature ageing
The nuclear lamina is a highly regulated membrane barrier that separates the nucleus from the cytoplasm in eukaryotic cells, and contains lamins and other proteins involved in chromatin organization and gene regulation (Burke and Stewart, 2013) (Fig. 1). There are two major types of lamin proteins, the A-type, encoded by the gene LMNA, which includes lamins A and C, and the B-type, encoded by the genes LMNB1 and LMNB2, and includes lamins B1, B2 and B3. Lamin A undergoes complex post-translational processing steps, such as farnesylation, cleavage by the zinc metallopeptidase STE24 (ZMPSTE24), carboxyl methylation by the isoprenylcysteine carboxylmethyltransferase (ICMT), and excision of the farnesylated residue (Fig. 1). Lamin A also interacts with many different proteins, such as the barrier to autointegration factor (BAF), to achieve mitotic and post-mitotic nuclear assembly (Jamin and Wiebe, 2015). Mutations in genes that encode nuclear-lamina proteins cause progeroid syndromes, such as HGPS, NGPS, APSs, RD and MAD (Agarwal et al., 2003; Barthelemy et al., 2015; De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003; Navarro et al., 2005; Puente et al., 2011). In this section, we describe the main clinical features of progeroid syndromes caused by mutations in key elements of the nuclear envelope (Table 1).
Fig. 1.

Prelamin A physiological and pathological processing and maturation. Processing of prelamin A in (A) normal cells, leading to the generation of mature lamin A and assembly of the normal nuclear envelope, and (B) HGPS cells, where the HGPS-associated mutation (G608G) in the gene encoding prelamin A, LMNA, activates a cryptic splicing site that results in the deletion of 50 amino acids slightly upstream of the C-terminus of prelamin A, encompassing the final cleavage site for ZMPSTE24 and leading to the accumulation of a toxic form of lamin A named progerin. This leads to disruption of the nuclear envelope, detectable as bulging or ‘nuclear blebbing’, which is shown in the representative images of HGPS and NGPS human fibroblasts illustrated below, in comparison to control cells. Lamin A/C (green) and DAPI (blue) staining is shown. Note that nuclear blebbing in NGPS cells is not due to the accumulation of progerin (see main text), and this image has been included purely to demonstrate the phenotype. FTase, farnesyltransferase; ICMT, isoprenylcysteine carboxyl methyltransferase; HGPS, Hutchinson-Gilford progeria syndrome; NGPS, Nestor-Guillermo progeria syndrome; ZMPSTE24, zinc metalloproteinase STE24.
Hutchinson-Gilford progeria syndrome
HGPS is the most prevalent and widely studied accelerated-ageing syndrome. Most cases of HGPS originate from a de novo heterozygous silent mutation in the LMNA gene (G608G). This mutation activates a cryptic splicing site (Box 1) that results in the deletion of 50 amino acids near the C-terminus of prelamin A, which encompasses the final cleavage site for CAAX prenyl protease 1 homolog (ZMPSTE24) to produce lamin A. This leads to the accumulation of a toxic protein called progerin, which disrupts the integrity of the nuclear envelope (Fig. 1) (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003). Individuals with HGPS display alopecia (hair loss), atherosclerosis, lipodystrophy, heart infarction and death during puberty (Table 1) (Gordon et al., 2014). Cells derived from these individuals present nuclear shape abnormalities known as ‘blebs’ (Fig. 1B) and shortened telomeres, and undergo premature senescence as a consequence of genome instability (Gonzalo and Kreienkamp, 2015). Progerin also accumulates during physiological ageing, reinforcing the parallels between normal and pathological ageing (Scaffidi and Misteli, 2006). Similar to aged individuals, HGPS individuals demonstrate vascular stiffening, atherosclerotic plaques and calcium dysfunction (Gerhard-Herman et al., 2012; Olive et al., 2010). Nonetheless, some basic features of ageing, such as the deterioration of the nervous system, the immune system deficits and the increased susceptibility to cancer, are not recapitulated in HGPS (Gordon et al., 2014). This is due to the low levels of prelamin A expression in the brain (Jung et al., 2012), and to the presence of a tumour protection mechanism mediated by bromodomain containing protein 4 (BRD4) in cells from individuals with HGPS (Fernandez et al., 2014) (discussed in more detail later).
Atypical progeria syndromes
Several lamin A mutations, including A57P, R133L and L140R, which are predicted to alter key protein-protein interaction domains, are associated with APSs. Individuals with APS show many of the clinical features of HGPS (Table 1), but their cells do not accumulate prelamin A or progerin (Barthelemy et al., 2015).
Restrictive dermopathy and mandibuloacral dysplasia
RD is a rare recessive condition caused by ZMPSTE24 mutations that lead to the accumulation of lamin A precursors (Navarro et al., 2005). MAD is characterized by lipodystrophy and skeletal and metabolic abnormalities (Table 1). MAD with type A lipodystrophy (MADA) is induced by the homozygous R527H LMNA mutation, which leads to accumulation of prelamin A and changes in nuclear architecture (Novelli et al., 2002), whereas MAD with type B lipodystrophy (MADB) is caused by compound heterozygous mutations in ZMPSTE24 (Agarwal et al., 2003).
Néstor-Guillermo progeria syndrome
NGPS is caused by a homozygous mutation (c.34G<A; p.Ala12Thr) in BANF1 (barrier to autointegration Factor 1), which encodes the protein BAF, involved in chromatin organization, nuclear assembly and gene-expression regulation. Fibroblasts from NGPS individuals demonstrate reduced levels of BAF protein, aberrant nuclear morphology, namely blebbing (Fig. 1), and altered subcellular distribution of emerin (see Box 1) (Cabanillas et al., 2011; Puente et al., 2011). The A12T mutation also weakens the binding of BAF to DNA, which is crucial for its many roles in the cell, suggesting that this deficiency contributes to the cellular phenotype observed in NGPS (Paquet et al., 2014). NGPS shares many clinical features with HGPS (Table 1), but, for unknown reasons, lacks cardiovascular defects, which likely contributes to the longer lifespan of NGPS patients (Cabanillas et al., 2011).
Defects in DNA repair, and premature ageing
Damage to nuclear DNA is a direct cause of ageing and cancer, and a decline in DNA repair with age has been described (Vijg and Suh, 2013). Moreover, many progeroid syndromes are caused by defects in DNA repair (Childs et al., 2014; Vermeij et al., 2016). In humans, there are five RecQ helicases – proteins that repair double-strand breaks during DNA replication (Box 1), and maintain genome stability and telomere integrity (Bernstein et al., 2010). Mutations in three of them [WRN (Werner syndrome, RecQ helicase-like), BLM (Bloom syndrome, RecQ helicase-like) and RECQ4 (RecQ helicase-like 4)] are associated with WS, BS and RTS premature-ageing syndromes, respectively. Other DNA-repair proteins altered in progeroid syndromes include the excision-repair cross-complementing (ERCC) family, the FA proteins and the XP proteins, which collaborate to enable DNA nucleotide excision repair (NER) and double-strand-break repair (Fig. 2; Box 1) (Vermeij et al., 2016). Mutations in genes encoding these and other proteins involved in DNA repair are responsible for different premature-ageing disorders, the clinical features of which are summarized below and in Table 1.
Fig. 2.

Mutations in proteins involved in DNA repair lead to premature ageing syndromes. Diagrams of the proteins involved in (A) double-strand break (DSB) repair, (B) interstrand cross-link (ISL) repair, (C) nucleotide excision repair (NER) and (D) telomere elongation and maintenance, including the shelterin complex, the telomerase complex and the CST (CTC1, STN1 and TEN1) complex (Box 1). Proteins encoded by genes mutated in progeroid syndromes are shown in orange, blue, green and red, whereas non-mutated proteins are shown in grey.
Werner syndrome
WS is caused by mutations in the WRN gene that result in the production of WRN truncated proteins (Yu et al., 1996). Individuals with this disorder display a range of premature-ageing symptoms (Table 1) and a high incidence of cancer that might be related to the chromosomal instability, telomere dysfunction and senescent phenotype exhibited by cells lacking WRN (Crabbe et al., 2007).
Bloom syndrome
BS is caused by mutations in the BLM gene, which encodes the RecQ helicase BLM. These mutations lead to the mislocalization of BLM protein or to the impairment of its helicase or ATPase domains (Machwe et al., 2005). This syndrome is characterized by immunodeficiency and predisposition to cancer, among other features (Table 1) (Ellis et al., 2008).
Rothmund-Thomson syndrome
RTS is caused by mutations in RECQL4, which encodes a helicase localized at telomeres and mitochondria (Croteau et al., 2012). This syndrome is mainly characterized by growth deficiency, skin abnormalities and increased susceptibility to cancer (Table 1) (Puzianowska-Kuznicka and Kuznicki, 2005). The loss of RECQL4 in fibroblasts of affected individuals leads to impaired DNA repair after oxidative stress and to growth arrest, which could explain the dwarfism associated with this disorder (Werner et al., 2006).
Cockayne syndrome, xeroderma pigmentosum and trichothiodystrophy
CS, XP and TTD are a group of related disorders associated with defects in NER (Box 1) (Marteijn et al., 2014). CS is a neurodegenerative disorder caused by mutations in the ERCC6 and ERCC8 genes, and is mainly characterized by neural abnormalities and growth failure (Table 1) (Tan et al., 2005). XP arises from mutation of any of many different genes, including XPA, XPB, XPC, XPG, ERCC4, ERCC6, DDB2 and POLH. This syndrome is characterized by an impaired capacity to repair the damage caused by UV light, which leads to increased cancer susceptibility (Cleaver, 2005). TTD is another rare progeroid disorder generated by mutations in XPB, XPD or TFB5, and is characterized by skin photosensitivity, growth retardation and a reduced life expectancy (Itin et al., 2001).
Fanconi anaemia
Mutations in FA genes, which encode proteins that are involved in DNA repair, such as FANCA or BRCA2, can lead to FA disease. Individuals with FA exhibit a higher cancer susceptibility, bone marrow failure, short stature, skin abnormalities and developmental disabilities (Table 1) (Deakyne and Mazin, 2011). Cells from individuals with FA produce high levels of reactive oxygen species (ROS), which can damage telomeric regions (Uziel et al., 2008). This defect, together with an insufficient DNA-repair system, results in single-strand DNA breaks that induce accelerated telomere shortening. Mutant cells show shorter telomeres and an increase in chromosome-end fusions compared to normal controls, resulting in genomic instability and multinucleated cells (Bogliolo and Surralles, 2015).
Seckel syndrome
SS is caused by mutations in the ATR (ataxia telangiectasia and Rad3-related) gene, which codes for the serine/threonine kinase ATR. SS is characterized by intrauterine growth retardation and postnatal dwarfism (Table 1) (Shanske et al., 1997). ATR protein has a crucial role in preventing replicative stress, sensing DNA damage and consequently arresting the cell cycle, and in maintaining telomere integrity (Pennarun et al., 2010).
Ataxia telangiectasia
AT is caused by mutations in the ATM (ataxia telangiectasia mutated) gene, which encodes a serine/threonine kinase that is activated by DNA double-strand breaks. AT individuals show progressive cerebellar degeneration, pigmentary abnormalities, hair greying and increased cancer susceptibility (Table 1). ATM is involved in DNA-damage signalling, cell cycle arrest and telomere maintenance (Sahin and DePinho, 2010).
Dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome
Telomere shortening is observed during normal ageing both in human and mouse cells, acting as a barrier to tumour growth and contributing to cell senescence (Armanios and Blackburn, 2012). Various premature ageing disorders, such as DC and HHS, are linked to mutations in components of the telomerase complex (Box 1). DC is a rare autosomal-dominant disorder caused by mutations in TERC (telomerase RNA component), TERT (telomerase reverse transcriptase), WRAP53 (WD repeat component, antisense to TP53) or CTC1 (CTS telomere maintenance complex component 1), among other genes (Stanley and Armanios, 2015). X-linked DC is caused by mutations in DKC1 (dyskeratosis congenita 1) (Fig. 2) (Yoon et al., 2006). Individuals with DC present bone marrow failure, cancer predisposition, premature hair greying and osteoporosis (Table 1) (Stanley and Armanios, 2015). HHS is a very rare multisystem disorder that phenocopies DC but with an increased severity (Table 1) (Walne et al., 2013). Mutations responsible for HHS have been identified in RTEL1 (regulator of telomere elongation helicase 1), which encodes a helicase that is essential for telomere maintenance, and ACD (adrenocortical dysplasia homolog), which encodes the shelterin TPP1 (Box 1) (Kocak et al., 2014; Walne et al., 2013).
The development of cellular and mouse models for the study of the progeroid syndromes described above has been extremely useful in order to elucidate the mechanisms underlying such diseases. The most relevant models are described in the following sections.
Cellular models of progeroid syndromes
The generation of patient-derived induced pluripotent stem cells (iPSCs) in recent years has enabled researchers to study specific tissues affected by a disease and to discover new tissue-specific drugs (Studer et al., 2015; Tang et al., 2016). Recently, these approaches have also been used to study different progeroid syndromes and to identify barriers to reprogramming in normally aged and prematurely aged cells.
iPSC generation from progeroid cells
iPSCs from several progeroid syndromes have been successfully generated and differentiated along multiple lineages (Table 2). However, progeria-derived iPSCs normally show reduced reprogramming efficiency in comparison to control cells despite being indistinguishable from normal iPSCs in other ways. In line with this, HGPS and NGPS iPSCs lack disease-specific features, such as nuclear blebs, epigenetic changes or, in the case of HPGS iPSCs, progerin expression, demonstrating the erasure of age-associated marks (Lo Cicero and Nissan, 2015; Soria-Valles et al., 2015a; Xiong et al., 2013). Once differentiated, however, the progeroid iPSC-derived cells show age-associated alterations, frequently mimicking the associated pathologies. These iPSC-derived models have been useful for studying the molecular mechanisms of HGPS and NGPS progerias and for screening drug candidates that could be used to treat these disorders (Pitrez et al., 2016; Soria-Valles et al., 2015b). Likewise, when WS fibroblasts are reprogrammed, their telomeres elongate, indicating that telomere function is restored (Shimamoto et al., 2014). The differentiation of WS iPSCs to mesenchymal stem cells (MSCs) results in a recurrence of premature senescence that is associated with telomere attrition. This phenotype can be rescued by the expression of hTERT or by knocking down p53 (Cheung et al., 2014). Another study, using iPSCs generated from CS fibroblasts, has demonstrated that lack of functional ERCC6 increases cell death and ROS production, and upregulates TP53 relative to normal cells (Andrade et al., 2012).
Table 2.
Progeroid-syndrome-derived iPSCs
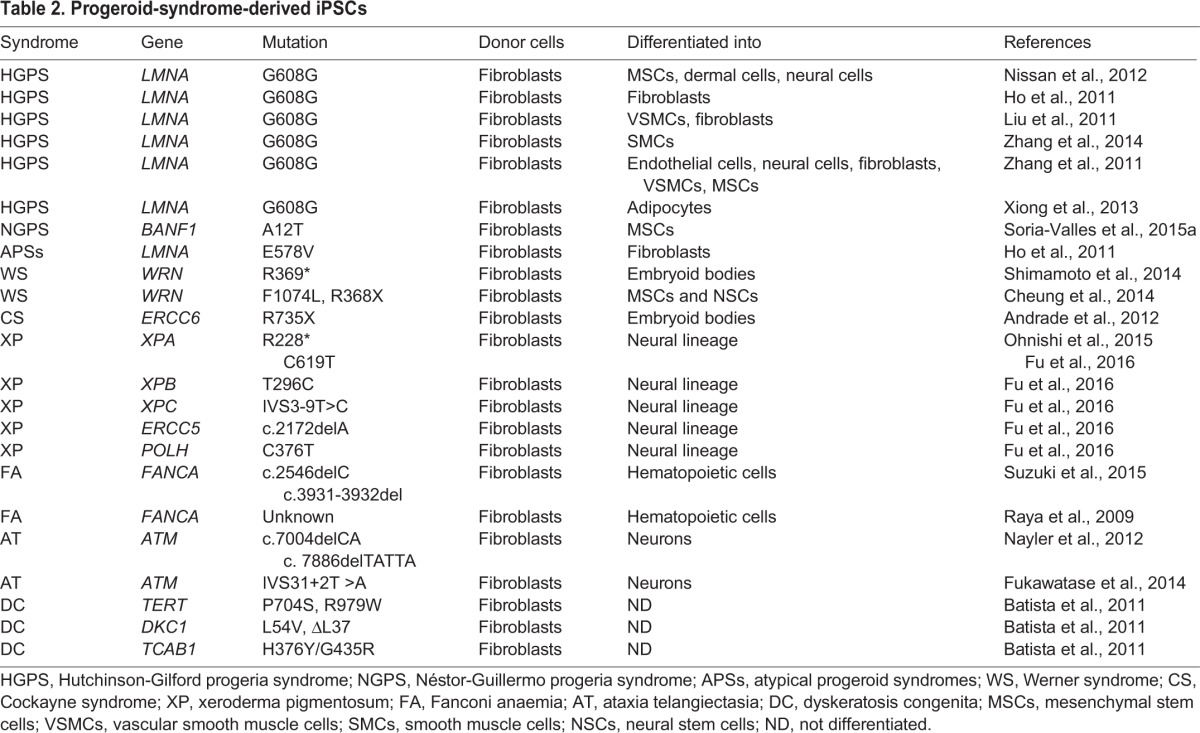
XP iPSCs from patients with mutations in XPA have also been developed to produce in vitro models of neurological disorders (Fu et al., 2016). These iPSCs can be differentiated into neural cells, indicating that the expression of wild-type XPA alleles is not required for iPSC generation and differentiation (Ohnishi et al., 2015). Studies with DC-derived iPSCs have shown that, even in the undifferentiated state, these cells can exhibit substantially reduced telomerase levels or mislocalization of telomerase from Cajal bodies to nucleoli, which disables the telomere elongation that normally accompanies reprogramming (Batista et al., 2011). Notably, iPSCs from fibroblasts of individuals with FANCA mutations have an impaired ability to differentiate towards early hemoangiogenic progenitors, which indicates that the hematopoietic phenotype of individuals with FA originates from an early hematopoietic stage (Suzuki et al., 2015). Correction of the FANCA mutation increases reprogramming and differentiation capacities of the resulting cells (Raya et al., 2009).
AT-derived fibroblasts have also been reprogrammed, albeit at a reduced efficiency (Fukawatase et al., 2014). These iPSCs display hypersensitivity to ionizing radiation, alterations in DNA-damage signalling pathways, metabolic changes and cell cycle checkpoint defects. AT-derived iPSCs have been differentiated into functional neurons, providing a unique model for the study of AT-associated neurodegeneration (Nayler et al., 2012).
Surprisingly, iPSCs have not yet been derived from RD, MAD, RTS, TTD, BS or SS cells, nor from APSs. The reason for this might partly rest with the low reprogramming efficiency of progeroid cells owing to a series of reprogramming barriers that are starting to be elucidated, as discussed below.
Barriers to reprogramming in ageing cells
Several features displayed by aged cells, such as genetic damage, telomere shortening and cell senescence, represent barriers that greatly reduce the efficiency of cell reprogramming. The reprogramming process is slow and inefficient in part because the forced expression of the Yamanaka factors is a stressful mechanism that activates apoptosis and cellular senescence via the upregulation of tumour suppressor proteins, including p53, p16 (INK4a) and p21 (CIP1) (Li et al., 2009). Accordingly, the inhibition of these factors improves reprogramming efficiency (Banito and Gil, 2010; Hong et al., 2009). Metabolic studies also indicate that inhibition of mammalian target of rapamycin (mTOR) notably improves the efficiency of iPSC generation (Chen et al., 2011), identifying mTOR as an important repressor of reprogramming.
Interestingly, NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) activation constitutes another barrier to somatic cell reprogramming in normally and prematurely aged cells (Soria-Valles et al., 2015a). Accordingly, NF-κB inhibition significantly increased the reprogramming efficiency of NGPS and HGPS fibroblasts, and of fibroblasts from advanced-age donors. Progeroid fibroblasts also showed a noticeable overexpression of DOT1L (DOT1-like histone H3-K79 methyltransferase), induced by NF-κB. Consistent with this, DOT1L inhibition increased reprogramming efficiency of progeroid cells and physiologically aged fibroblasts. Remarkably, treatment of progeroid mice with DOT1L inhibitors extends their longevity, implicating DOT1L as a newly identified target for rejuvenation-based approaches (Soria-Valles et al., 2015b).
Cells from individuals with FA failed to be reprogrammed to iPSCs unless the defective FA gene was replaced (Muller et al., 2012; Raya et al., 2009). However, a recent study has revealed that E6 protein from human papillomavirus 16 (HPV16) rescues FA-derived iPSC colony formation via p53 inhibition (Chlon et al., 2014). Consequently, the FA pathway is required for reprogramming through p53-dependent mechanisms, emphasizing the importance of classical tumour suppressors as barriers for cell reprogramming in progeroid syndromes.
Mouse models of progeroid syndromes
Mouse models have been widely used to explore the molecular mechanisms of ageing and progeroid syndromes. Thus, experiments using mice with an extended lifespan or with signs of premature ageing have helped to elucidate the basic processes that affect ageing (Table 3) (Lopez-Otin et al., 2013; Vanhooren and Libert, 2013).
Table 3.
Main mouse models used for the study of progeroid syndromes
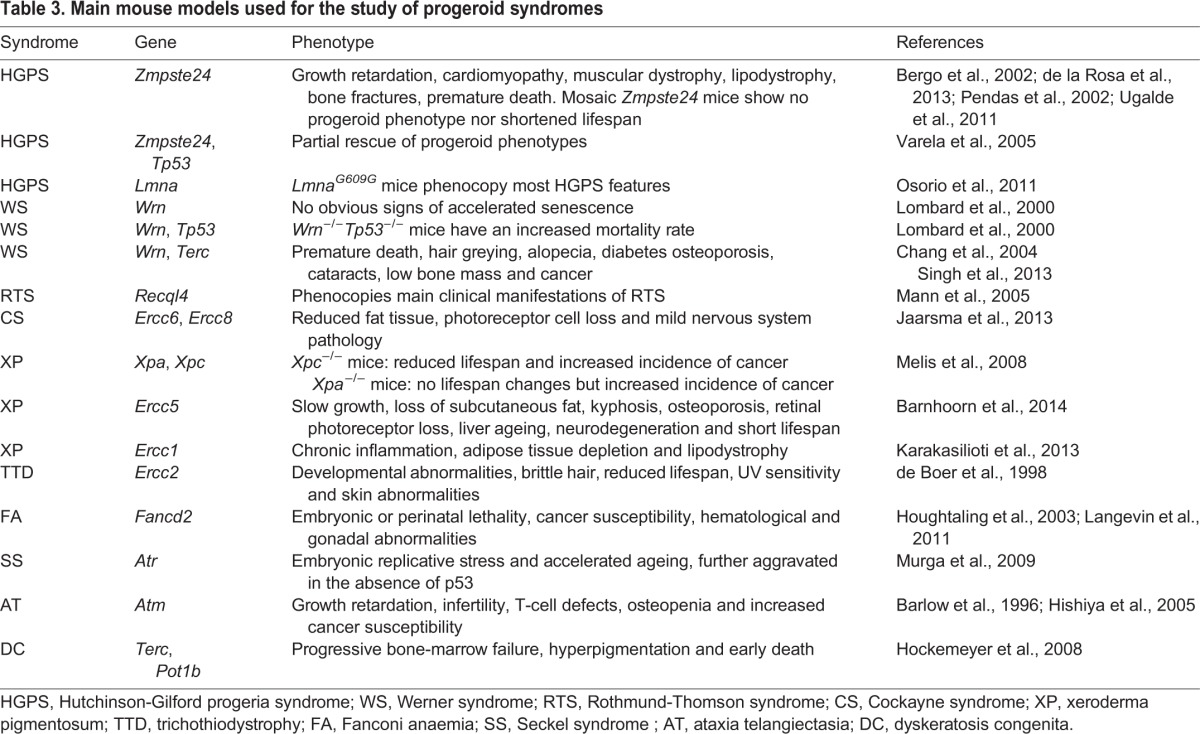
In 2002, Pendás et al. and Bergo et al., developed the first HGPS mouse model – Zmpste24-null (Zmpste24−/−) mice – which showed growth retardation, cardiomyopathy, muscular dystrophy, lipodystrophy and premature death (Bergo et al., 2002; Pendas et al., 2002). Studies of these mice demonstrated that the accumulation of farnesylated prelamin A damages the nuclear envelope, hyperactivates p53 signalling, and causes cellular senescence, stem cell dysfunction and a progeroid phenotype (Osorio et al., 2009; Varela et al., 2005). Zmpste24−/− mice are also defective in DNA repair (Liu et al., 2005), and in the p53-dependent upregulation of miR-29, which represses extracellular matrix genes (Ugalde et al., 2011). Extracellular matrix remodelling is a widely known mechanism that promotes renovation of adult tissues, and, consequently, repression of such genes impairs tissue restoration and promotes ageing (Gutierrez-Fernandez et al., 2015). However, this mouse model cannot be used to study the aberrant splicing of LMNA observed in individuals with HGPS, prompting the generation of a mouse strain that carries the HGPS mutation (LMNAG608G) (Osorio et al., 2011). These mice accumulate progerin and phenocopy the main clinical manifestations of human HGPS, such as shortened lifespan and bone and cardiovascular alterations. This model has been widely used to develop new therapeutic strategies for HGPS.
Other mouse models related to the lamin-A–Zmpste24 system have helped to elucidate why individuals with HGPS are not predisposed to cancer despite their elevated levels of DNA damage (Cau et al., 2014; Gordon et al., 2014). Zmpste24 mosaic mice that contain both Zmpste24-proficient cells and Zmpste24−/− cells have revealed a lower incidence of invasive carcinomas compared to mice that are heterozygous for Zmpste24 (de la Rosa et al., 2013). Parallel studies have described another cancer protective mechanism in HGPS cells involving suppression of cell proliferation and metastatic properties owing to an altered pattern of chromatin binding by the transcription regulator BRD4 (Fernandez et al., 2014). Other mouse models with Lmna mutations have been developed, although many of these do not fully recapitulate the progeroid phenotype observed in HGPS or APSs (Das et al., 2013; Poitelon et al., 2012; Varga et al., 2006). Zmpste24−/− mouse models have been used to study the molecular mechanisms of RD and MADB progeroid syndromes, although this mouse model does not fully recapitulate several features of these diseases (Osorio et al., 2009). In summary, many mouse models for laminopathies have been developed and studied in order to elucidate the mechanisms underlying the pathogenesis of these and related progeroid syndromes.
Several mouse models have also been created to mimic the clinical phenotypes associated with WS. Wrn-knockout mice have no obvious signs of accelerated senescence and fail to recapitulate clinical WS features (Lombard et al., 2000). However, Wrn−/−Terc−/− mice exhibit many of the key phenotypes, in support of a role for WRN in telomere maintenance (Chang et al., 2004). Mouse models of BS have also been developed, but their early lethality has hampered their utility for the in vivo study of this disease (Luo et al., 2000). Likewise, mice bearing the most common mutations found in RTS individuals have been generated. These mice show severe growth retardation, hair loss, dry skin and increased cancer susceptibility (Mann et al., 2005). Mutant mice have been developed for all CS-associated genes, most of which develop mild CS-like symptoms (Jaarsma et al., 2013). Mice mutant for Xpa−/− and Xpc−/− have also been reported. Xpc−/− mice demonstrate a reduced lifespan and an increased rate of lung cancer, whereas Xpa−/− mice show no lifespan alterations but exhibit a higher rate of liver carcinomas (Hosseini et al., 2015; Melis et al., 2008). Xpg-deficient mice also display many progeroid features, including loss of subcutaneous fat, osteoporosis, neurodegeneration and a short lifespan (Barnhoorn et al., 2014). Furthermore, DNA damage in mice carrying an Ercc1-Xpf DNA-repair defect triggers a chronic inflammatory response, leading to lipodystrophy (Karakasilioti et al., 2013). Finally, a mouse model of TTD recapitulates the human disorder, demonstrating developmental defects, brittle hair, UV sensitivity, skin abnormalities and reduced lifespan (de Boer et al., 1998).
There are several FA mouse models, all of which are characterized by embryonic or perinatal lethality, gonadal abnormality and associated infertility (Bakker et al., 2013). Nevertheless, most FA mouse models do not show any apparent hematological abnormalities, in contrast with humans with FA, who are affected by life-threatening anaemia. An exception to this is the Fancp mutant mouse, which has lowered numbers of white blood cells and platelets (Langevin et al., 2011). Hematopoietic stem cells (HSCs) from other FA mouse mutants (Fancc, Fancd2 and Fancg) have a reduced repopulating ability and fail to maintain hematopoietic homeostasis under stress conditions (Bakker et al., 2013). Fancd2-, Fancf- or Fancm-deficient mice also exhibit an increased incidence of tumorigenesis (Bakker et al., 2013). The SS mouse model, characterized by ATR deficiency, shows high levels of replicative stress during embryogenesis and exhibits accelerated ageing, which is further aggravated in a Tp53-null background (Murga et al., 2009). AT mouse models recapitulate many of the characteristics observed in humans with AT, such as growth retardation, infertility, immune defects and increased susceptibility to lymphomas, although neurodegeneration is not observed (Barlow et al., 1996). Additionally, Atm-deficient mice show increased cancer susceptibility and a severe osteopenic phenotype, which is partly caused by a stem cell defect due to decreased expression of IGF, an important regulator of proliferation (Hishiya et al., 2005).
Mouse models of telomere dysfunction in progeroid syndromes have also been generated. The first mutant mouse with telomerase deficiency failed to recapitulate the clinical features of DC. However, when POT1b−a component of the shelterin complex that protects mammalian telomeres−is removed in mice (Box 1), these animals show clinical features of DC (Hockemeyer et al., 2008). Mouse models of APS, HHS, NBN, ATLD or CRMCC are yet to be developed.
Hallmarks of progeroid syndromes
Based on the evidence obtained through experimentation with cellular and animal models, we propose a set of hallmarks for defining the main features of progeroid syndromes, and discuss their relatedness and differences with those of normal ageing (Fig. 3). Defining these molecular hallmarks could help in the design and interpretation of future studies of both physiological and premature ageing, and could also provide clinical benefit by guiding the diagnosis of or therapy for progeroid diseases.
Fig. 3.
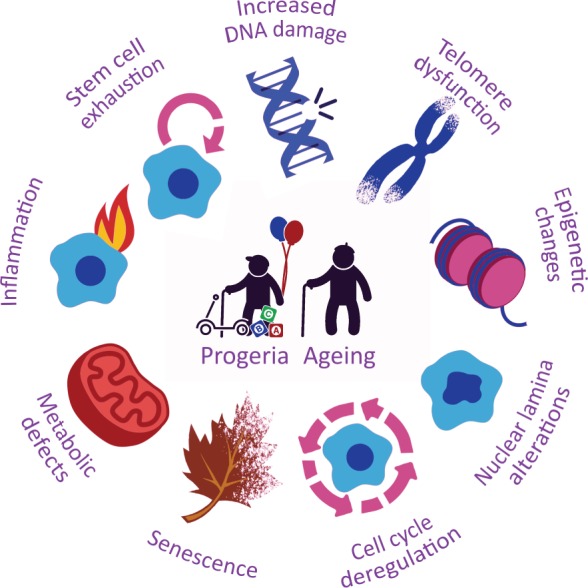
The molecular and cellular hallmarks of progeroid syndromes. These nine proposed hallmarks recapitulate the most remarkable features common to different progeroid syndromes and define the mechanisms underlying the pathogenesis of these diseases.
Increased DNA damage and defective DNA repair
Both physiologically aged and progeroid cells accumulate genomic damage throughout life (Vijg and Suh, 2013). Somatic mutations and other forms of DNA damage progressively compile within cells from both aged humans and model organisms, affecting essential genes and resulting in dysfunctional cells and impaired organismal homeostasis (Lopez-Otin et al., 2013). Many other age-related changes can also affect DNA repair mechanisms, leading to the accumulation of more genomic damage (Gorbunova et al., 2007). HGPS and RD progeroid cells are particularly susceptible to DNA damage induced by ROS or by ionizing radiation, and show a severely impaired capacity to repair DNA damage (Camozzi et al., 2014). Many progeroid syndromes also present repair defects owing to mutations in RecQ helicases (Box 1), or in proteins belonging to the ERCC and XP families (Bernstein et al., 2010; Marteijn et al., 2014).
Telomere dysfunction
Telomere shortening is observed during normal ageing in both human and mouse cells, and has been linked to a decreased lifespan (Blasco et al., 1997). As discussed earlier, progeroid syndromes such as DC and HHS are caused by mutations in different components of the telomerase complex. Other ageing-related diseases, such as WS, FA, SS, AT, ATLD, NBN and CRMCC, are characterized by telomere dysfunction caused by mutations in genes involved in telomere maintenance and DNA repair (Armanios and Blackburn, 2012). HGPS cells also exhibit accelerated telomere shortening during proliferation in culture (Decker et al., 2009). Notably, the ectopic expression of telomerase in these cells increases their proliferation and lifespan by decreasing progerin-induced activation of the p53 and retinoblastoma (Rb) pathways (Kudlow et al., 2008). Additionally, the association of the shelterin TRF2 (telomere repeat-binding factor 2) with telomeric sequences is stabilized by its colocalization with A-type lamins. LMNA mutations lead to the impaired association of these lamins with TRF2, resulting in telomere loss (Wood et al., 2014). Furthermore, the ectopic expression of progerin in normal fibroblasts results in the accumulation of DNA damage at telomeres and in cell senescence (Cao et al., 2011). These studies suggest that telomere dysfunction and associated defective DNA repair contribute to genomic instability and premature senescence of cells in progeroid syndromes.
Changes in epigenetics and chromatin structure
Chromatin remodelling plays a central role in the regulation of gene expression. As such, altered chromatin structure can lead to aberrant gene expression patterns, altering normal cellular functions and contributing to both normal and premature ageing (Chandra et al., 2015). Aged cells also accumulate epigenetic alterations that alter normal gene expression profiles (Beerman and Rossi, 2015). Accordingly, aged cells exhibit increased levels of histone modifications that promote transcription and global histone loss, inducing chromatin relaxation and the misregulation of gene expression. Such epigenetic changes are present also in cells from prematurely ageing individuals and likely contribute to the progression of the associated diseases (Lopez-Otin et al., 2013). In addition, the downregulation of chromatin remodelling factors, such as HP1α (heterochromatin protein 1 alpha), polycomb proteins and the NuRD complex, occurs during normal ageing and leads to global heterochromatin loss (Pollina and Brunet, 2011). In the context of premature-ageing syndromes, the absence of WRN (in WS) or the accumulation of progerin (in HGPS) also causes loss of heterochromatin and telomere attrition (Shumaker et al., 2006; Zhang et al., 2015). Likewise, cells from NGPS individuals exhibit profound changes in chromatin organization (Loi et al., 2016). In cells from individuals with HGPS, the epigenetic mark H3K27me3 is lost on the inactive X chromosome of affected females, as a consequence of downregulation of EZH2 (enhancer of Zeste homolog 2), the methyltransferase responsible for this mark. HGPS cells also exhibit the downregulation of H3K9me3 but increased levels of H4K20me3, which are epigenetic modifications that are associated with global repression of transcription (Shumaker et al., 2006). However, in a different study, increased levels of H3K9me3 were linked to accelerated senescence and compromised genome maintenance (Liu et al., 2013).
Sirtuin 1 (SIRT1) is thought to contribute to telomere maintenance through deacetylation of epigenetic marks such as H4K16ac (Palacios et al., 2010). By contrast, low levels of acetylation at H4K16 have been shown to correlate with a defective DNA-damage response (DDR) and double-strand-break repair to ionizing radiation (Sharma et al., 2010). In line with this, Zmpste24−/− mice show hypoacetylation of histones H2B and H4 (Osorio et al., 2010), likely due to the diminished association of the histone acetyltransferase Mof (male absent on the first) with the nuclear matrix. Rescue experiments performed either by Mof overexpression or by histone deacetylase inhibition promoted repair protein recruitment to DNA damage sites and substantially ameliorated aging-associated phenotypes in these mice (Krishnan et al., 2011).
Heterochromatin alterations have also been observed in other syndromes, such as an active heterochromatinization process mediated by SIRT1 in XP or CS cells after UV irradiation (Velez-Cruz et al., 2013), and the presence of unstable heterochromatin in FA cells (Edelman and Lin, 2001). Although epigenetic changes have not yet been identified in all progeroid syndromes, the frequent occurrence of both epigenetic and chromatin alterations tempt us to speculate that such defects are general features of these diseases.
Aberrant nuclear architecture
Several progeroid syndromes−including HGPS, NGPS, APSs, RD and MAD−are associated with significant perturbations in nuclear organization and loss of nuclear envelope stability. In the case of HGPS, the presence of the farnesyl group in progerin, owing to the aberrant processing of lamin A, leads to progerin association with the inner nuclear membrane, generating defects in nuclear shape (Fig. 1) (Goldman et al., 2004). Such features are also observed in healthy ageing individuals owing to the sporadic activation of the same cryptic splice site in LMNA as in HGPS, whereas inhibition of this splice site reverses the nuclear defects associated with ageing (Righolt et al., 2011; Scaffidi and Misteli, 2006). Several studies have suggested that alterations in the nuclear lamina might lead to premature ageing by affecting the transcription profile of adult epithelial and mesenchymal stem cells and thereby interfering with their ability to retain an undifferentiated state (Espada et al., 2008; Scaffidi and Misteli, 2008). Progeroid fibroblasts obtained from NGPS individuals also show profound abnormalities in the nuclear lamina (Fig. 1) that can be rescued by the ectopic expression of wild-type BANF1 (Puente et al., 2011; Soria-Valles et al., 2015a). Furthermore, cells from individuals with RD show an abnormal nuclear shape and heterogeneous deposits of unprocessed prelamin A (Columbaro et al., 2010). Severe nuclear morphological abnormalities are also observed in MAD and APS cells (Barthelemy et al., 2015). Collectively, these findings highlight the relevance of nuclear lamina aberrations as important features of different progeroid syndromes.
Defects in cell cycle and mitosis
Cell cycle regulation is critically important for repairing genetic damage and preventing uncontrolled cell division. Changes in cell cycle dynamics, such as upregulation of p16 and other cell cycle inhibitors, are observed during physiological ageing and cellular senescence, and consequently represent valuable markers for the ageing process (Coppe et al., 2011). Nuclear envelope changes during cell division play a major role in the control of cell cycle progression (Camozzi et al., 2014). Accordingly, the targeting of nuclear lamina components into daughter cell nuclei in early G1 and cytokinesis progression is impaired in HGPS cells owing to the abnormal processing of lamin A (Dechat et al., 2007). FA proteins are essential for arresting the cell cycle until DNA damage is restored; thus, cells from FA patients have defects in DNA repair (Bogliolo and Surralles, 2015). Individuals with SS display an impaired G2/M checkpoint arrest and an increased number of centrosomes in mitotic cells (Griffith et al., 2008). Moreover, insufficiency of BubR1, a key mitotic checkpoint protein, causes aneuploidy, short lifespan, cachectic dwarfism, cataracts and other progeroid features (Baker et al., 2004).
Cellular senescence
Cellular senescence is a state of stable cell cycle arrest and loss of replicative capacity that mainly results from DNA damage, oxidative stress and telomere shortening (van Deursen, 2014). In HGPS cells, progerin-induced senescence has been partially linked to p53 activation, a feature that is recapitulated in mouse models of this progeroid syndrome (Varela et al., 2005). Cells from NGPS patients also show premature cellular senescence characterized by markers such as senescence-associated β-galactosidase (SA-β-gal) staining, senescence-associated heterochromatin foci, increased levels of p16 and significant loss of lamin B1 (Soria-Valles et al., 2015a). Individuals affected by APS also present senescence phenotypes, driven by a dramatic reduction in lamin B1 expression (Bercht Pfleghaar et al., 2015). Likewise, fibroblasts from WS and SS patients show premature cellular senescence induced by replicative stress and faulty DNA repair, primarily due to the activation of stress kinase p38 (Davis et al., 2007; Tivey et al., 2013). Increased SA-β-gal staining and higher expression of p16 and p21 are also observed in BLM-, WRN- and RECQL4-depleted human fibroblasts (Lu et al., 2014). Furthermore, cells from Xpc−/− mice show increased β-galactosidase activity and high levels of progerin, p16 and ROS (Hosseini et al., 2015). Moreover, a mouse model of SS, characterized by a severe deficiency in ATR, shows accelerated ageing that is further aggravated by the loss of p53 (Murga et al., 2009). Hematopoietic stem cell senescence has also been reported in DC and FA (Stockklausner et al., 2015; Zhang et al., 2007).
Metabolic defects
The ageing process is accompanied by many metabolic alterations, such as insulin resistance and physiological decline in growth hormone (GH), insulin-like growth factor-1 (IGF-1) and sex steroids. Accordingly, the insulin and IGF-1 signalling (IIS) pathway is extensively involved in ageing (Lopez-Otin et al., 2013). Moreover, dietary restriction (DR) increases healthspan in many organisms, further supporting a role for metabolism in ageing (Barzilai et al., 2012). Progeroid syndromes are also accompanied by metabolic changes. Zmpste24−/− mice exhibit profound transcriptional alterations in circulating levels of GH and IGF-1 (Marino et al., 2010), as well as changes in metabolic pathways that are associated with autophagy induction (Marino et al., 2008). Metabolic alterations are also present in progeroid syndromes that feature defects in DNA repair. Wrn-deficient mice exhibit hypertriglyceridemia and insulin resistance (Lebel and Leder, 1998). ERCC1-null mice show a shift toward anabolism and decreased GH-IGF-1 signalling (Niedernhofer et al., 2006), whereas CS mouse models show systemic suppression of the GH-IGF-I somatotropic axis (Box 1), increased antioxidant responses and hypoglycaemia (van der Pluijm et al., 2007). These studies suggest that unrepaired DNA damage induces a highly conserved metabolic response that is mediated by the IIS pathway, which redistributes resources from growth and proliferation for the preservation and protection of somatic integrity (Liao and Kennedy, 2014; Schumacher et al., 2009). Furthermore, mice with telomere dysfunction exhibit a marked deficiency in IGF-I-mTOR signalling, impaired energy homeostasis and suppressed mitochondrial biogenesis (Missios et al., 2014). Notably, mitochondrial dysfunction has been linked to the pathogenesis of progeroid syndromes such as HGPS and CS (Rivera-Torres et al., 2013; Scheibye-Knudsen et al., 2013). Cells derived from HGPS patients show a marked downregulation of mitochondrial oxidative phosphorylation proteins, accompanied by mitochondrial dysfunction (Rivera-Torres et al., 2013). These metabolic and mitochondrial alterations have inspired the development of different strategies for the treatment of progeroid syndromes (discussed below).
Inflammation
Chronic inflammation is associated with normal and pathological ageing (Lopez-Otin et al., 2013). In line with this, a pro-inflammatory phenotype, named ‘inflammageing’, has been observed in mammals during ageing (Salminen et al., 2012). Moreover, senescent cells exhibit a senescence-associated secretory phenotype (SASP) characterized by the secretion of increasing levels of factors that alter their microenvironment in a paracrine manner, reinforcing senescence and activating immune surveillance. This phenomenon is mainly regulated by NF-κB (Acosta et al., 2013; Chien et al., 2011). We have recently found that the aberrant activation of NF-κB by ATM in mouse models of progeroid laminopathies induces the overexpression of pro-inflammatory cytokines and contributes to the pathogenesis of these syndromes (Osorio et al., 2012). Likewise, knocking down Nfkb1 in mice causes premature ageing. Accordingly, the accumulation of senescent cells in Nfkb1−/− tissues is blocked by the administration of anti-inflammatory drugs to these mice (Jurk et al., 2014). A recent study has also demonstrated that age-associated NF-κB hyperactivation impairs the generation of iPSCs by evoking the reprogramming repressor DOT1L, which promotes senescence signals and downregulates pluripotency genes (Soria-Valles et al., 2015a). An inflammatory phenotype associated with premature ageing has also been linked to WS (Goto et al., 2012). Similarly, mouse models of FA show inflammation-mediated upregulation of Notch signalling, which correlates with a decreased self-renewal capacity of FA HSCs (Du et al., 2013).
Stem cell exhaustion
The ageing process is accompanied by a decrease in tissue regeneration and homeostasis, due to a decline in stem cell functions as a consequence of DNA damage, changes in tissue environment and alterations in tumour suppressor gene expression (Behrens et al., 2014). Because stem cells regenerate many adult tissues, changes in these cells likely contribute to the development of age-related diseases and to accelerated ageing syndromes. Consistent with this, progerin accumulation reduces the proliferative and differentiation capacity of pluripotent and multipotent (Box 1) mouse and human cells (Pacheco et al., 2014; Rosengardten et al., 2011). Furthermore, muscle-derived stem/progenitor cells (MDSPCs) from old and progeroid mice have proliferation and differentiation defects, whereas intraperitoneal administration of MDSPCs from young wild-type mice to progeroid mice confers a significant extension of lifespan and health via the secretion of certain factors that act systemically (Lavasani et al., 2012). Hematopoietic stem cell senescence has been described in the context of many progeroid syndromes. For example, TERT mutations that cause DC lead to cellular senescence and to the loss of CD34+ hematopoietic stem cells (Stockklausner et al., 2015). Moreover, TNFα induces premature senescence in bone marrow HSCs and in other tissues of FA mouse models. This induction correlates with ROS accumulation and oxidative DNA damage (Zhang et al., 2007). Together, these studies demonstrate the close relationship between progeroid syndromes and stem cell exhaustion, highlighting the importance of using cellular models to further understand the underlying pathobiology of these diseases and to guide the development of appropriate therapies.
Therapeutic and rejuvenation strategies
Our improved understanding of the molecular basis of the progeroid syndromes has guided the development of therapeutic strategies for these disorders, particularly for HGPS (Gordon et al., 2014). Because progerin is permanently farnesylated, the first therapeutic strategy for treating HGPS involved the use of farnesyltransferase inhibitors (FTIs), such as lonafarnib, which has been used as a potential anticancer drug and had tolerable side effects in children. However, lonafarnib led to only limited improvements of symptoms in HGPS patients (Gordon et al., 2012), in part because, following FTI treatment, progerin is geranylgeranylated and gives rise to another toxic form of prelamin A (Varela et al., 2008). Consequently, an efficient blockade of progerin farnesylation must prevent both farnesylation and geranylgeranylation modifications, which provides the rationale behind a combined therapeutic approach that uses statins and aminobisphosphonates (Varela et al., 2008). This strategy has been successfully tested in Zmpste24−/− mice and is currently being evaluated for the treatment of individuals with HGPS (clinical trials: NCT00425607, NCT00879034 and NCT00916747).
Since these first strategies for HGPS treatment were described, additional therapies have been tested, with promising results, in mouse and cellular models of premature ageing. Many of these interventions derive from recent anti-ageing approaches designed to address alterations in cellular processes, such as nutrient-sensing, mitochondrial efficiency and autophagy, which are, as highlighted above, also deficient in progeria (de Cabo et al., 2014; Lopez-Otin et al., 2013). One such approach is treatment with the mTOR inhibitor rapamycin, which can suppress nuclear structure defects and postpone senescence in HGPS fibroblasts in vitro by inducing progerin clearance through autophagy (Cao et al., 2011). Additionally, the SIRT1 activator resveratrol rescues adult stem cell decline, slows down body weight loss and extends lifespan in Zmpste24−/− progeroid mice (Liu et al., 2012). Likewise, the administration of IGF-I to these mice has proven to positively affect both their health and lifespan (Marino et al., 2010).
Beyond these metabolic interventions, and because HGPS is caused by the activation of an alternative splice site, RNA therapy is another option for the treatment of progeria. Morpholino antisense oligonucleotides targeted to the altered splice site were first applied in HGPS fibroblasts to reduce progerin synthesis (Scaffidi and Misteli, 2005). This approach was also successfully used in the LmnaG609G HGPS mouse model, in which it led to a significant lifespan extension (Osorio et al., 2011). More recently, DOT1L inhibitors have proven to be effective as a rejuvenation strategy for both physiologically aged and HGPS and NGPS progeroid human cells (Soria-Valles et al., 2015a), and the treatment of Zmpste24−/− mice with DOT1L inhibitors increases their longevity and ameliorates their progeroid phenotypes (Soria-Valles et al., 2015a). Moreover, treatment of HGPS cells with a small molecule called ‘remodelin’, which targets the acetyltransferase NAT10, improves nuclear architecture and overall fitness of these progeroid cells (Larrieu et al., 2014). Other potentially beneficial HGPS treatments studied in cell-based and animal models include the administration of sodium salicylate, which inhibits the IκB kinase (IKK) complex (Osorio et al., 2012), pyrophosphate (Villa-Bellosta et al., 2013), methylene blue (Xiong et al., 2016) and Icmt inhibitors (Ibrahim et al., 2013).
Treatment with rapamycin has also been proposed as a therapeutic strategy for MAD because it efficiently triggers lysosomal degradation of farnesylated prelamin A (Camozzi et al., 2014). Resveratrol has proven to be efficient in reversing some of the clinically relevant phenotypes in Wrn-deficient mice, such as insulin resistance and liver steatosis, although it did not improve hypertriglyceridemia or inflammatory stress, nor extend the lifespan of these mice. Resveratrol-treated mutant mice also exhibited an increase in the frequency of different tumours (Labbe et al., 2011), and failed to show improvements in HGPS-associated bone defects (Strandgren et al., 2015). A high-fat diet rescues the metabolic, transcriptomic and behavioural phenotypes of a CS mouse model, whereas β-hydroxybutyrate, PARP (poly ADP ribose polymerase) inhibition and NAD+ supplementation can also rescue CS-associated phenotypes through activation of SIRT1, which is involved in cell cycle regulation and response to stressors (Scheibye-Knudsen et al., 2014). The reversal of mitochondrial defects in CS-derived cells using serine protease inhibitors has also been described (Chatre et al., 2015). Similarly, a glucose-enriched diet ameliorates the impaired energy homeostasis phenotype observed in mice with telomere dysfunctions, through the activation of glycolysis, mitochondrial biogenesis and oxidative glucose metabolism (Missios et al., 2014).
Recently, the clearance of senescent cells has been proposed as a promising strategy to promote healthy ageing, mainly through the administration of so-called ‘senolytic’ pharmacological agents that induce the death of senescent cells (Roos et al., 2016). Clearance of such cells in BubR1 hypomorphic progeroid mice delayed the onset of their ageing phenotype and attenuated its progression (Childs et al., 2015). Selective clearance of senescent cells was also shown to be an effective strategy for rejuvenation of tissue stem cells in normally ageing mice (Chang et al., 2016).
Read-through of premature termination codons in cells from WS and XP patients has been achieved using aminoglycosides: this restores WRN functionality in WS cells (Agrelo et al., 2015) and increases XPC protein production in XP cells (Kuschal et al., 2015). Genetic approaches have also been used to correct XP human cells (Dupuy and Sarasin, 2015), whereas genetic depletion of one allele of the p65 subunit of NF-κB or treatment with IKK inhibitors delays the age-related symptoms and pathologies of XFEPS (XPF-ERCC1 progeroid syndrome) mice, which harbour mutations in ERCC1 (involved in DNA excision repair) (Tilstra et al., 2012).
Another therapeutic option is the synthetic steroid danazol, which has anti-gonadotropic and anti-estrogenic activities and is capable of slowing down the progression of pulmonary fibrosis in individuals with DC (Zlateska et al., 2015). In the case of FA, treatment with p38 MAP-kinase inhibitors has improved the repopulating ability of Fancc-deficient HSCs (Saadatzadeh et al., 2009). Resveratrol also partially rescues the reduced repopulating ability of Fancd2-deficient HSCs (Zhang et al., 2010). Treating SS fibroblasts with p38 inhibitors can also restore their reduced replicative capacity in vitro and ameliorate their aged morphology (Tivey et al., 2013).
Importantly, recent studies suggest that the rate of ageing cannot only be modified by environmental and genetic factors, but also reversed (Freije and Lopez-Otin, 2012; Rando and Chang, 2012). Many rejuvenation strategies are based on epigenetic reprogramming, such as depletion of the methyltransferase Suv39h1, which reduces H3K9me3 levels, improves DNA repair capacity and delays senescence in progeroid cells, and also extends lifespan in progeroid mice (Ibrahim et al., 2013; Liu et al., 2013). In the future, strategies developed to treat progeroid syndromes could also be considered as treatments for pathologies associated with physiological ageing, as long as they share the same mechanisms. This would certainly expand the benefits of these discoveries to a wider medical community. Although many treatment options for individuals with ageing pathologies have been successfully tested in cellular and animal models, initiating therapeutic trials for rare diseases such as progeroid syndromes is an exceptionally difficult task because of the absence of longitudinal studies on different cohorts of patients. Also, the occurrence of only a low number of cases in any given country requires the setting up of clinical trials that follow identical protocols in different parts of the world (Osorio et al., 2011).
Conclusions
Over recent years, advancements in our understanding of the genetic and molecular bases of premature aging disorders through the use of cellular and mouse models have led to a better understanding of the onset and progression of their clinical manifestations. On the basis of these studies, we have herein classified progeroid syndromes in two main categories, depending on the key molecular mechanisms involved. This classification scheme could provide a framework for the better understanding of the aetiology, biology and pathogenesis of progeroid syndromes. We have also appraised the latest findings made through the use of in vitro and in vivo models, which have helped to elucidate the processes that contribute to pathological and physiological ageing, and to characterize the cellular and organismal phenotypes of progeroid syndromes. Consequently, we have proposed nine hallmarks that characterize most progeroid syndromes, which extensively correlate with the nine hallmarks of ageing recently proposed (Lopez-Otin et al., 2013). Such hallmarks compile the most notable characteristics of progeroid syndromes and define the mechanisms underlying their pathogenesis, which might contribute to laying the groundwork for future studies on physiological and pathological ageing.
Finally, we have highlighted emerging therapies that could help to ameliorate the accelerated ageing phenotypes that underpin progeroid syndromes. It is well known that setting up of therapeutic trials for rare diseases is a difficult task owing to the lack of clinical correlational studies with similar evaluation guidelines and sufficient cohorts of patients (Osorio et al., 2011). This can present an obstacle to the testing of therapeutic approaches developed based on findings in cell- and animal-based studies. Nonetheless, a deeper characterization of the genetics and mechanisms underlying progeroid syndromes might enable earlier diagnosis and a better understanding of how the disease will progress in specific individuals, providing opportunities for earlier intervention and personalized treatment. Moreover, research focused on delaying or ameliorating physiological ageing could also contribute to developing a suitable therapeutic approach for progeroid syndromes.
Acknowledgements
We acknowledge M. Carrero and all the members of López-Otín's lab for their support and constructive discussions during the preparation of this manuscript. We apologize for omission of relevant works due to space constraints.
Footnotes
Competing interests
C.L.-O. is an Investigator of the Botin Foundation supported by Banco Santander through its Santander Universities Global Division.
Author contributions
D.C., C.S.-V. and C.L.-O. contributed to the design and writing of the review, and D.C. created the figures.
Funding
The work was supported by grants from Ministerio de Economía y Competitividad - Spain [grant number SAF2014-52413-R]; Gobierno del Principado de Asturias; Instituto de Salud Carlos III (RTICC) [grant number RD12/0036/0067] Spain; and Energias de Portugal (EDP) Foundation. We also thank the generous support by J. I. Cabrera. The Instituto Universitario de Oncología is supported by Fundación Bancaria Caja de Ahorros de Asturias.
References
- Acosta J. C., Banito A., Wuestefeld T., Georgilis A., Janich P., Morton J. P., Athineos D., Kang T.-W., Lasitschka F., Andrulis M. et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978-990. 10.1038/ncb2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A. K., Fryns J.-P., Auchus R. J. and Garg A. (2003). Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 12, 1995-2001. 10.1093/hmg/ddg213 [DOI] [PubMed] [Google Scholar]
- Agrelo R., Sutz M. A., Setien F., Aldunate F., Esteller M., Da Costa V. and Achenbach R. (2015). A novel Werner Syndrome mutation: pharmacological treatment by read-through of nonsense mutations and epigenetic therapies. Epigenetics 10, 329-341. 10.1080/15592294.2015.1027853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade L. N. d. S., Nathanson J. L., Yeo G. W., Menck C. F. M. and Muotri A. R. (2012). Evidence for premature aging due to oxidative stress in iPSCs from Cockayne syndrome. Hum. Mol. Genet. 21, 3825-3834. 10.1093/hmg/dds211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armanios M. and Blackburn E. H. (2012). The telomere syndromes. Nat. Rev. Genet. 13, 693-704. 10.1038/nrg3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D. J., Jeganathan K. B., Cameron J. D., Thompson M., Juneja S., Kopecka A., Kumar R., Jenkins R. B., de Groen P. C., Roche P. et al. (2004). BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 36, 744-749. 10.1038/ng1382 [DOI] [PubMed] [Google Scholar]
- Bakker S. T., de Winter J. P. and te Riele H. (2013). Learning from a paradox: recent insights into Fanconi anaemia through studying mouse models. Dis. Model. Mech. 6, 40-47. 10.1242/dmm.009795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banito A. and Gil J. (2010). Induced pluripotent stem cells and senescence: learning the biology to improve the technology. EMBO Rep. 11, 353-359. 10.1038/embor.2010.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C., Hirotsune S., Paylor R., Liyanage M., Eckhaus M., Collins F., Shiloh Y., Crawley J. N., Ried T., Tagle D. et al. (1996). Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86, 159-171. 10.1016/S0092-8674(00)80086-0 [DOI] [PubMed] [Google Scholar]
- Barnhoorn S., Uittenboogaard L. M., Jaarsma D., Vermeij W. P., Tresini M., Weymaere M., Menoni H., Brandt R. M. C., de Waard M. C., Botter S. M. et al. (2014). Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genet. 10, e1004686 10.1371/journal.pgen.1004686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelemy F., Navarro C., Fayek R., Da Silva N., Roll P., Sigaudy S., Oshima J., Bonne G., Papadopoulou-Legbelou K., Evangeliou A. E. et al. (2015). Truncated prelamin A expression in HGPS-like patients: a transcriptional study. Eur. J. Hum. Genet. 23, 1051-1061. 10.1038/ejhg.2014.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzilai N., Huffman D. M., Muzumdar R. H. and Bartke A. (2012). The critical role of metabolic pathways in aging. Diabetes 61, 1315-1322. 10.2337/db11-1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista L. F. Z., Pech M. F., Zhong F. L., Nguyen H. N., Xie K. T., Zaug A. J., Crary S. M., Choi J., Sebastiano V., Cherry A. et al. (2011). Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 474, 399-402. 10.1038/nature10084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerman I. and Rossi D. J. (2015). Epigenetic control of stem cell potential during homeostasis, aging, and disease. Cell Stem Cell 16, 613-625. 10.1016/j.stem.2015.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A., van Deursen J. M., Rudolph K. L. and Schumacher B. (2014). Impact of genomic damage and ageing on stem cell function. Nat. Cell Biol. 16, 201-207. 10.1038/ncb2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bercht Pfleghaar K., Taimen P., Butin-Israeli V., Shimi T., Langer-Freitag S., Markaki Y., Goldman A. E., Wehnert M. and Goldman R. D. (2015). Gene-rich chromosomal regions are preferentially localized in the lamin B deficient nuclear blebs of atypical progeria cells. Nucleus 6, 66-76. 10.1080/19491034.2015.1004256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergo M. O., Gavino B., Ross J., Schmidt W. K., Hong C., Kendall L. V., Mohr A., Meta M., Genant H., Jiang Y. et al. (2002). Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. USA 99, 13049-13054. 10.1073/pnas.192460799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein K. A., Gangloff S. and Rothstein R. (2010). The RecQ DNA helicases in DNA repair. Annu. Rev. Genet. 44, 393-417. 10.1146/annurev-genet-102209-163602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisserbe A., Tertian G., Buffet C., Turhan A., Lambotte O., Nasser G., Alvin P., Tardieu M., Riant F., Bergametti F. et al. (2015). Cerebro-retinal microangiopathy with calcifications and cysts due to recessive mutations in the CTC1 gene. Rev. Neurol. 171, 445-449. 10.1016/j.neurol.2015.01.566 [DOI] [PubMed] [Google Scholar]
- Blasco M. A., Lee H.-W., Hande M. P., Samper E., Lansdorp P. M., DePinho R. A. and Greider C. W. (1997). Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91, 25-34. 10.1016/S0092-8674(01)80006-4 [DOI] [PubMed] [Google Scholar]
- Bogliolo M. and Surralles J. (2015). Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 33, 32-40. 10.1016/j.gde.2015.07.002 [DOI] [PubMed] [Google Scholar]
- Burke B. and Stewart C. L. (2013). The nuclear lamins: flexibility in function. Nat. Rev. Mol. Cell Biol. 14, 13-24. 10.1038/nrm3488 [DOI] [PubMed] [Google Scholar]
- Cabanillas R., Cadiñanos J., Villameytide J. A. F., Pérez M., Longo J., Richard J. M., Álvarez R., Durán N. S., Illán R., González D. J. et al. (2011). Néstor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am. J. Med. Genet. A 155, 2617-2625. 10.1002/ajmg.a.34249 [DOI] [PubMed] [Google Scholar]
- Camozzi D., Capanni C., Cenni V., Mattioli E., Columbaro M., Squarzoni S. and Lattanzi G. (2014). Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies. Nucleus 5, 427-440. 10.4161/nucl.36289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K., Blair C. D., Faddah D. A., Kieckhaefer J. E., Olive M., Erdos M. R., Nabel E. G. and Collins F. S. (2011). Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J. Clin. Invest. 121, 2833-2844. 10.1172/JCI43578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cau P., Navarro C., Harhouri K., Roll P., Sigaudy S., Kaspi E., Perrin S., De Sandre-Giovannoli A. and Lévy N. (2014). Nuclear matrix, nuclear envelope and premature aging syndromes in a translational research perspective. Semin. Cell Dev. Biol. 29, 125-147. 10.1016/j.semcdb.2014.03.021 [DOI] [PubMed] [Google Scholar]
- Chandra T., Ewels P. A., Schoenfelder S., Furlan-Magaril M., Wingett S. W., Kirschner K., Thuret J.-Y., Andrews S., Fraser P. and Reik W. (2015). Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 10, 471-483. 10.1016/j.celrep.2014.12.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S., Multani A. S., Cabrera N. G., Naylor M. L., Laud P., Lombard D., Pathak S., Guarente L. and DePinho R. A. (2004). Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat. Genet. 36, 877-882. 10.1038/ng1389 [DOI] [PubMed] [Google Scholar]
- Chang J., Wang Y., Shao L., Laberge R.-M., Demaria M., Campisi J., Janakiraman K., Sharpless N. E., Ding S., Feng W. et al. (2016). Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78-83. 10.1038/nm.4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatre L., Biard D. S. F., Sarasin A. and Ricchetti M. (2015). Reversal of mitochondrial defects with CSB-dependent serine protease inhibitors in patient cells of the progeroid Cockayne syndrome. Proc. Natl. Acad. Sci. USA 112, E2910-E2919. 10.1073/pnas.1422264112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T., Shen L., Yu J., Wan H., Guo A., Chen J., Long Y., Zhao J. and Pei G. (2011). Rapamycin and other longevity-promoting compounds enhance the generation of mouse induced pluripotent stem cells. Aging Cell 10, 908-911. 10.1111/j.1474-9726.2011.00722.x [DOI] [PubMed] [Google Scholar]
- Cheung H.-H., Liu X., Canterel-Thouennon L., Li L., Edmonson C. and Rennert O. M. (2014). Telomerase protects werner syndrome lineage-specific stem cells from premature aging. Stem Cell Rep. 2, 534-546. 10.1016/j.stemcr.2014.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y., Scuoppo C., Wang X., Fang X., Balgley B., Bolden J. E., Premsrirut P., Luo W., Chicas A., Lee C. S. et al. (2011). Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 25, 2125-2136. 10.1101/gad.17276711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs B. G., Baker D. J., Kirkland J. L., Campisi J. and van Deursen J. M. (2014). Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 15, 1139-1153. 10.15252/embr.201439245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs B. G., Durik M., Baker D. J. and van Deursen J. M. (2015). Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat. Med. 21, 1424-1435. 10.1038/nm.4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlon T. M., Hoskins E. E., Mayhew C. N., Wikenheiser-Brokamp K. A., Davies S. M., Mehta P., Myers K. C., Wells J. M. and Wells S. I. (2014). High-risk human papillomavirus E6 protein promotes reprogramming of Fanconi anemia patient cells through repression of p53 but does not allow for sustained growth of induced pluripotent stem cells. J. Virol. 88, 11315-11326. 10.1128/JVI.01533-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver J. E. (2005). Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat. Rev. Cancer 5, 564-573. 10.1038/nrc1652 [DOI] [PubMed] [Google Scholar]
- Columbaro M., Mattioli E., Schena E., Capanni C., Cenni V., Levy N., Navarro C. L., Del Coco R., Squarzoni S., Camozzi D. et al. (2010). Prelamin A processing and functional effects in restrictive dermopathy. Cell Cycle 9, 4766-4768. 10.4161/cc.9.23.14210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe J.-P., Rodier F., Patil C. K., Freund A., Desprez P.-Y. and Campisi J. (2011). Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 286, 36396-36403. 10.1074/jbc.M111.257071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe L., Jauch A., Naeger C. M., Holtgreve-Grez H. and Karlseder J. (2007). Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc. Natl. Acad. Sci. USA 104, 2205-2210. 10.1073/pnas.0609410104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau D. L., Rossi M. L., Canugovi C., Tian J., Sykora P., Ramamoorthy M., Wang Z. M., Singh D. K., Akbari M., Kasiviswanathan R. et al. (2012). RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell 11, 456-466. 10.1111/j.1474-9726.2012.00803.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A., Grotsky D. A., Neumann M. A., Kreienkamp R., Gonzalez-Suarez I., Redwood A. B., Kennedy B. K., Stewart C. L. and Gonzalo S. (2013). Lamin A Deltaexon9 mutation leads to telomere and chromatin defects but not genomic instability. Nucleus 4, 410-419. 10.4161/nucl.26873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis T., Wyllie F. S., Rokicki M. J., Bagley M. C. and Kipling D. (2007). The role of cellular senescence in Werner syndrome: toward therapeutic intervention in human premature aging. Ann. N. Y. Acad. Sci. 1100, 455-469. 10.1196/annals.1395.051 [DOI] [PubMed] [Google Scholar]
- de Boer J., de Wit J., van Steeg H., Berg R. J. W., Morreau H., Visser P., Lehmann A. R., Duran M., Hoeijmakers J. H. J. and Weeda G. (1998). A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol. Cell 1, 981-990. 10.1016/S1097-2765(00)80098-2 [DOI] [PubMed] [Google Scholar]
- de Cabo R., Carmona-Gutierrez D., Bernier M., Hall M. N. and Madeo F. (2014). The search for antiaging interventions: from elixirs to fasting regimens. Cell 157, 1515-1526. 10.1016/j.cell.2014.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa J., Freije J. M. P., Cabanillas R., Osorio F. G., Fraga M. F., Fernández-García M. S., Rad R., Fanjul V., Ugalde A. P., Liang Q. et al. (2013). Prelamin A causes progeria through cell-extrinsic mechanisms and prevents cancer invasion. Nat. Commun. 4, 2268 10.1038/ncomms3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., Lyonnet S., Stewart C. L., Munnich A., Le Merrer M. et al. (2003). Lamin a truncation in Hutchinson-Gilford progeria. Science 300, 2055 10.1126/science.1084125 [DOI] [PubMed] [Google Scholar]
- Deakyne J. S. and Mazin A. V. (2011). Fanconi anemia: at the crossroads of DNA repair. Biochemistry 76, 36-48. 10.1134/S0006297911010068 [DOI] [PubMed] [Google Scholar]
- Dechat T., Shimi T., Adam S. A., Rusinol A. E., Andres D. A., Spielmann H. P., Sinensky M. S. and Goldman R. D. (2007). Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc. Natl. Acad. Sci. USA 104, 4955-4960. 10.1073/pnas.0700854104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker M. L., Chavez E., Vulto I. and Lansdorp P. M. (2009). Telomere length in Hutchinson-Gilford progeria syndrome. Mech. Ageing Dev. 130, 377-383. 10.1016/j.mad.2009.03.001 [DOI] [PubMed] [Google Scholar]
- Du W., Amarachintha S., Sipple J., Schick J., Steinbrecher K. and Pang Q. (2013). Inflammation-mediated notch signaling skews fanconi anemia hematopoietic stem cell differentiation. J. Immunol. 191, 2806-2817. 10.4049/jimmunol.1203474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy A. and Sarasin A. (2015). DNA damage and gene therapy of xeroderma pigmentosum, a human DNA repair-deficient disease. Mutat. Res. 776, 2-8. 10.1016/j.mrfmmm.2014.08.007 [DOI] [PubMed] [Google Scholar]
- Edelman J. R. and Lin Y. J. (2001). Translocation of unstable heterochromatin as the mechanism of sister chromatid exchange formation: a proposed hypothesis. Cytobios 106, 171-191. [PubMed] [Google Scholar]
- Ellis N. A., Sander M., Harris C. C. and Bohr V. A. (2008). Bloom's syndrome workshop focuses on the functional specificities of RecQ helicases. Mech. Ageing Dev. 129, 681-691. 10.1016/j.mad.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Eriksson M., Brown W. T., Gordon L. B., Glynn M. W., Singer J., Scott L., Erdos M. R., Robbins C. M., Moses T. Y., Berglund P. et al. (2003). Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293-298. 10.1038/nature01629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espada J., Varela I., Flores I., Ugalde A. P., Cadiñanos J., Pendás A. M., Stewart C. L., Tryggvason K., Blasco M. A., Freije J. M. P. et al. (2008). Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J. Cell Biol. 181, 27-35. 10.1083/jcb.200801096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez P., Scaffidi P., Markert E., Lee J.-H., Rane S. and Misteli T. (2014). Transformation resistance in a premature aging disorder identifies a tumor-protective function of BRD4. Cell Rep. 9, 248-260. 10.1016/j.celrep.2014.08.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernet M., Gribaa M., Salih M. A. M., Seidahmed M. Z., Hall J. and Koenig M. (2005). Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum. Mol. Genet. 14, 307-318. 10.1093/hmg/ddi027 [DOI] [PubMed] [Google Scholar]
- Freije J. M. P. and Lopez-Otin C. (2012). Reprogramming aging and progeria. Curr. Opin. Cell Biol. 24, 757-764. 10.1016/j.ceb.2012.08.009 [DOI] [PubMed] [Google Scholar]
- Fu L., Xu X., Ren R., Wu J., Zhang W., Yang J., Ren X., Wang S., Zhao Y., Sun L. et al. (2016). Modeling xeroderma pigmentosum associated neurological pathologies with patients-derived iPSCs. Protein Cell 7, 210-221. 10.1007/s13238-016-0244-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukawatase Y., Toyoda M., Okamura K., Nakamura K.-I., Nakabayashi K., Takada S., Yamazaki-Inoue M., Masuda A., Nasu M., Hata K. et al. (2014). Ataxia telangiectasia derived iPS cells show preserved x-ray sensitivity and decreased chromosomal instability. Sci. Rep. 4, 5421 10.1038/srep05421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard-Herman M., Smoot L. B., Wake N., Kieran M. W., Kleinman M. E., Miller D. T., Schwartzman A., Giobbie-Hurder A., Neuberg D. and Gordon L. B. (2012). Mechanisms of premature vascular aging in children with Hutchinson-Gilford progeria syndrome. Hypertension 59, 92-97. 10.1161/HYPERTENSIONAHA.111.180919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman R. D., Shumaker D. K., Erdos M. R., Eriksson M., Goldman A. E., Gordon L. B., Gruenbaum Y., Khuon S., Mendez M., Varga R. et al. (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 101, 8963-8968. 10.1073/pnas.0402943101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S. and Kreienkamp R. (2015). DNA repair defects and genome instability in Hutchinson-Gilford Progeria Syndrome. Curr. Opin. Cell Biol. 34, 75-83. 10.1016/j.ceb.2015.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbunova V., Seluanov A., Mao Z. and Hine C. (2007). Changes in DNA repair during aging. Nucleic Acids Res. 35, 7466-7474. 10.1093/nar/gkm756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon L. B., Kleinman M. E., Miller D. T., Neuberg D. S., Giobbie-Hurder A., Gerhard-Herman M., Smoot L. B., Gordon C. M., Cleveland R., Snyder B. D. et al. (2012). Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 109, 16666-16671. 10.1073/pnas.1202529109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon L. B., Rothman F. G., López-Otín C. and Misteli T. (2014). Progeria: a paradigm for translational medicine. Cell 156, 400-407. 10.1016/j.cell.2013.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M., Sugimoto K., Hayashi S., Ogino T., Sugimoto M., Furuichi Y., Matsuura M., Ishikawa Y., Iwaki-Egawa S. and Watanabe Y. (2012). Aging-associated inflammation in healthy Japanese individuals and patients with Werner syndrome. Exp. Gerontol. 47, 936-939. 10.1016/j.exger.2012.08.010 [DOI] [PubMed] [Google Scholar]
- Griffith E., Walker S., Martin C.-A., Vagnarelli P., Stiff T., Vernay B., Al Sanna N., Saggar A., Hamel B., Earnshaw W. C. et al. (2008). Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat. Genet. 40, 232-236. 10.1038/ng.2007.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Fernandez A., Soria-Valles C., Osorio F. G., Gutierrez-Abril J., Garabaya C., Aguirre A., Fueyo A., Fernandez-Garcia M. S., Puente X. S. and Lopez-Otin C. (2015). Loss of MT1-MMP causes cell senescence and nuclear defects which can be reversed by retinoic acid. EMBO J. 34, 1875-1888. 10.15252/embj.201490594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennekam R. C. M. (2006). Hutchinson-Gilford progeria syndrome: review of the phenotype. Am. J. Med. Genet. A 140A, 2603-2624. 10.1002/ajmg.a.31346 [DOI] [PubMed] [Google Scholar]
- Hishiya A., Ito M., Aburatani H., Motoyama N., Ikeda K. and Watanabe K. (2005). Ataxia telangiectasia mutated (Atm) knockout mice as a model of osteopenia due to impaired bone formation. Bone 37, 497-503. 10.1016/j.bone.2005.05.012 [DOI] [PubMed] [Google Scholar]
- Ho J. C. Y., Zhou T., Lai W.-H., Huang Y., Chan Y.-C., Li X., Wong N. L. Y., Li Y., Au K.-W., Guo D. et al. (2011). Generation of induced pluripotent stem cell lines from 3 distinct laminopathies bearing heterogeneous mutations in lamin A/C. Aging 3, 380-390. 10.18632/aging.100277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Palm W., Wang R. C., Couto S. S. and de Lange T. (2008). Engineered telomere degradation models dyskeratosis congenita. Genes Dev. 22, 1773-1785. 10.1101/gad.1679208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H., Takahashi K., Ichisaka T., Aoi T., Kanagawa O., Nakagawa M., Okita K. and Yamanaka S. (2009). Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 460, 1132-1135. 10.1038/nature08235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini M., Mahfouf W., Serrano-Sanchez M., Raad H., Harfouche G., Bonneu M., Claverol S., Mazurier F., Rossignol R., Taieb A. et al. (2015). Premature skin aging features rescued by inhibition of NADPH oxidase activity in XPC-deficient mice. J. Invest. Dermatol. 135, 1108-1118. 10.1038/jid.2014.511 [DOI] [PubMed] [Google Scholar]
- Houghtaling S., Timmers C., Noll M., Finegold M. J., Jones S. N., Meyn M. S. and Grompe M. (2003). Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 17, 2021-2035. 10.1101/gad.1103403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Lee L., Hanson N. B., Lenaerts C., Hoehn H., Poot M., Rubin C. D., Chen D.-F., Yang C.-C., Juch H. et al. (2006). The spectrum of WRN mutations in Werner syndrome patients. Hum. Mutat. 27, 558-567. 10.1002/humu.20337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M. X., Sayin V. I., Akula M. K., Liu M., Fong L. G., Young S. G. and Bergo M. O. (2013). Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science 340, 1330-1333. 10.1126/science.1238880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itin P. H., Sarasin A. and Pittelkow M. R. (2001). Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes. J. Am. Acad. Dermatol. 44, 891-924; quiz 921-924 10.1067/mjd.2001.114294 [DOI] [PubMed] [Google Scholar]
- Jaarsma D., van der Pluijm I., van der Horst G. T. J. and Hoeijmakers J. H. J. (2013). Cockayne syndrome pathogenesis: lessons from mouse models. Mech. Ageing Dev. 134, 180-195. 10.1016/j.mad.2013.04.003 [DOI] [PubMed] [Google Scholar]
- Jamin A. and Wiebe M. S. (2015). Barrier to Autointegration Factor (BANF1): interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr. Opin. Cell Biol. 34, 61-68. 10.1016/j.ceb.2015.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H.-J., Coffinier C., Choe Y., Beigneux A. P., Davies B. S. J., Yang S. H., Barnes R. H. II, Hong J., Sun T., Pleasure S. J. et al. (2012). Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. USA 109, E423-E431. 10.1073/pnas.1111780109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D., Wilson C., Passos J. F., Oakley F., Correia-Melo C., Greaves L., Saretzki G., Fox C., Lawless C., Anderson R. et al. (2014). Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2, 4172 10.1038/ncomms5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakasilioti I., Kamileri I., Chatzinikolaou G., Kosteas T., Vergadi E., Robinson A. R., Tsamardinos I., Rozgaja T. A., Siakouli S., Tsatsanis C. et al. (2013). DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in NER progeria. Cell Metab. 18, 403-415. 10.1016/j.cmet.2013.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy B. K., Berger S. L., Brunet A., Campisi J., Cuervo A. M., Epel E. S., Franceschi C., Lithgow G. J., Morimoto R. I., Pessin J. E. et al. (2014). Geroscience: linking aging to chronic disease. Cell 159, 709-713. 10.1016/j.cell.2014.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocak H., Ballew B. J., Bisht K., Eggebeen R., Hicks B. D., Suman S., O'Neil A., Giri N., Maillard I., Alter B. P. et al. (2014). Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev. 28, 2090-2102. 10.1101/gad.248567.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V., Chow M. Z. Y., Wang Z., Zhang L., Liu B., Liu X. and Zhou Z. (2011). Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 108, 12325-12330. 10.1073/pnas.1102789108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudlow B. A., Stanfel M. N., Burtner C. R., Johnston E. D. and Kennedy B. K. (2008). Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol. Biol. Cell 19, 5238-5248. 10.1091/mbc.E08-05-0492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuschal C., Khan S. G., Enk B., DiGiovanna J. J. and Kraemer K. H. (2015). Readthrough of stop codons by use of aminoglycosides in cells from xeroderma pigmentosum group C patients. Exp. Dermatol. 24, 296-297. 10.1111/exd.12655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe A., Garand C., Cogger V. C., Paquet E. R., Desbiens M., Le Couteur D. G. and Lebel M. (2011). Resveratrol improves insulin resistance hyperglycemia and hepatosteatosis but not hypertriglyceridemia, inflammation, and life span in a mouse model for Werner syndrome. J. Gerontol. A Biol. Sci. Med. Sci. 66A, 264-278. 10.1093/gerona/glq184 [DOI] [PubMed] [Google Scholar]
- Langevin F., Crossan G. P., Rosado I. V., Arends M. J. and Patel K. J. (2011). Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53-58. 10.1038/nature10192 [DOI] [PubMed] [Google Scholar]
- Larrieu D., Britton S., Demir M., Rodriguez R. and Jackson S. P. (2014). Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 344, 527-532. 10.1126/science.1252651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavasani M., Robinson A. R., Lu A., Song M., Feduska J. M., Ahani B., Tilstra J. S., Feldman C. H., Robbins P. D., Niedernhofer L. J. et al. (2012). Muscle-derived stem/progenitor cell dysfunction limits healthspan and lifespan in a murine progeria model. Nat. Commun. 3, 608 10.1038/ncomms1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel M. and Leder P. (1998). A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc. Natl. Acad. Sci. USA 95, 13097-13102. 10.1073/pnas.95.22.13097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Collado M., Villasante A., Strati K., Ortega S., Cañamero M., Blasco M. A. and Serrano M. (2009). The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460, 1136-1139. 10.1038/nature08290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C.-Y. and Kennedy B. K. (2014). Mouse models and aging: longevity and progeria. Curr. Top. Dev. Biol. 109, 249-285. 10.1016/B978-0-12-397920-9.00003-2 [DOI] [PubMed] [Google Scholar]
- Liu B., Wang J., Chan K. M., Tjia W. M., Deng W., Guan X., Huang J.-D., Li K. M., Chau P. Y., Chen D. J. et al. (2005). Genomic instability in laminopathy-based premature aging. Nat. Med. 11, 780-785. 10.1038/nm1266 [DOI] [PubMed] [Google Scholar]
- Liu G.-H., Barkho B. Z., Ruiz S., Diep D., Qu J., Yang S.-L., Panopoulos A. D., Suzuki K., Kurian L., Walsh C. et al. (2011). Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 472, 221-225. 10.1038/nature09879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Ghosh S., Yang X., Zheng H., Liu X., Wang Z., Jin G., Zheng B., Kennedy B. K., Suh Y. et al. (2012). Resveratrol rescues SIRT1-dependent adult stem cell decline and alleviates progeroid features in laminopathy-based progeria. Cell Metab. 16, 738-750. 10.1016/j.cmet.2012.11.007 [DOI] [PubMed] [Google Scholar]
- Liu B., Wang Z., Zhang L., Ghosh S., Zheng H. and Zhou Z. (2013). Depleting the methyltransferase Suv39h1 improves DNA repair and extends lifespan in a progeria mouse model. Nat. Commun. 4, 1868 10.1038/ncomms2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Cicero A. and Nissan X. (2015). Pluripotent stem cells to model Hutchinson-Gilford progeria syndrome (HGPS): current trends and future perspectives for drug discovery. Ageing Res. Rev. 24, 343-348. 10.1016/j.arr.2015.10.002 [DOI] [PubMed] [Google Scholar]
- Loi M., Cenni V., Duchi S., Squarzoni S., Lopez-Otin C., Foisner R., Lattanzi G. and Capanni C. (2016). Barrier-to-Autointegration Factor (BAF) involvement in prelamin A-related chromatin organization changes. Oncotarget. 7, 15662-15677. 10.18632/oncotarget.6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard D. B., Beard C., Johnson B., Marciniak R. A., Dausman J., Bronson R., Buhlmann J. E., Lipman R., Curry R., Sharpe A. et al. (2000). Mutations in the WRN gene in mice accelerate mortality in a p53-null background. Mol. Cell. Biol. 20, 3286-3291. 10.1128/MCB.20.9.3286-3291.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C., Blasco M. A., Partridge L., Serrano M. and Kroemer G. (2013). The hallmarks of aging. Cell 153, 1194-1217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Fang E. F., Sykora P., Kulikowicz T., Zhang Y., Becker K. G., Croteau D. L. and Bohr V. A. (2014). Senescence induced by RECQL4 dysfunction contributes to Rothmund-Thomson syndrome features in mice. Cell Death Dis. 5, e1226 10.1038/cddis.2014.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G., Santoro I. M., McDaniel L. D., Nishijima I., Mills M., Youssoufian H., Vogel H., Schultz R. A. and Bradley A. (2000). Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat. Genet. 26, 424-429. 10.1038/82548 [DOI] [PubMed] [Google Scholar]
- Machwe A., Xiao L., Groden J., Matson S. W. and Orren D. K. (2005). RecQ family members combine strand pairing and unwinding activities to catalyze strand exchange. J. Biol. Chem. 280, 23397-23407. 10.1074/jbc.M414130200 [DOI] [PubMed] [Google Scholar]
- Mann M. B., Hodges C. A., Barnes E., Vogel H., Hassold T. J. and Luo G. (2005). Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum. Mol. Genet. 14, 813-825. 10.1093/hmg/ddi075 [DOI] [PubMed] [Google Scholar]
- Marino G., Ugalde A. P., Salvador-Montoliu N., Varela I., Quiros P. M., Cadinanos J., van der Pluijm I., Freije J. M. P. and Lopez-Otin C. (2008). Premature aging in mice activates a systemic metabolic response involving autophagy induction. Hum. Mol. Genet. 17, 2196-2211. 10.1093/hmg/ddn120 [DOI] [PubMed] [Google Scholar]
- Marino G., Ugalde A. P., Fernandez A. F., Osorio F. G., Fueyo A., Freije J. M. P. and Lopez-Otin C. (2010). Insulin-like growth factor 1 treatment extends longevity in a mouse model of human premature aging by restoring somatotroph axis function. Proc. Natl. Acad. Sci. USA 107, 16268-16273. 10.1073/pnas.1002696107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn J. A., Lans H., Vermeulen W. and Hoeijmakers J. H. J. (2014). Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 15, 465-481. 10.1038/nrm3822 [DOI] [PubMed] [Google Scholar]
- Melis J. P. M., Wijnhoven S. W. P., Beems R. B., Roodbergen M., van den Berg J., Moon H., Friedberg E., van der Horst G. T. J., Hoeijmakers J. H. J., Vijg J. et al. (2008). Mouse models for xeroderma pigmentosum group A and group C show divergent cancer phenotypes. Cancer Res. 68, 1347-1353. 10.1158/0008-5472.CAN-07-6067 [DOI] [PubMed] [Google Scholar]
- Missios P., Zhou Y., Guachalla L. M., von Figura G., Wegner A., Chakkarappan S. R., Binz T., Gompf A., Hartleben G., Burkhalter M. D. et al. (2014). Glucose substitution prolongs maintenance of energy homeostasis and lifespan of telomere dysfunctional mice. Nat. Commun. 5, 4924 10.1038/ncomms5924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller L. U. W., Milsom M. D., Harris C. E., Vyas R., Brumme K. M., Parmar K., Moreau L. A., Schambach A., Park I.-H., London W. B. et al. (2012). Overcoming reprogramming resistance of Fanconi anemia cells. Blood 119, 5449-5457. 10.1182/blood-2012-02-408674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M., Bunting S., Montaña M. F., Soria R., Mulero F., Cañamero M., Lee Y., McKinnon P. J., Nussenzweig A. and Fernandez-Capetillo O. (2009). A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 41, 891-898. 10.1038/ng.420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro C. L., Cadinanos J., De Sandre-Giovannoli A., Bernard R., Courrier S., Boccaccio I., Boyer A., Kleijer W. J., Wagner A., Giuliano F. et al. (2005). Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum. Mol. Genet. 14, 1503-1513. 10.1093/hmg/ddi159 [DOI] [PubMed] [Google Scholar]
- Nayler S., Gatei M., Kozlov S., Gatti R., Mar J. C., Wells C. A., Lavin M. and Wolvetang E. (2012). Induced pluripotent stem cells from ataxia-telangiectasia recapitulate the cellular phenotype. Stem Cells Transl. Med. 1, 523-535. 10.5966/sctm.2012-0024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer L. J., Garinis G. A., Raams A., Lalai A. S., Robinson A. R., Appeldoorn E., Odijk H., Oostendorp R., Ahmad A., van Leeuwen W. et al. (2006). A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444, 1038-1043. 10.1038/nature05456 [DOI] [PubMed] [Google Scholar]
- Nissan X., Blondel S., Navarro C., Maury Y., Denis C., Girard M., Martinat C., De Sandre-Giovannoli A., Levy N. and Peschanski M. (2012). Unique preservation of neural cells in Hutchinson- Gilford progeria syndrome is due to the expression of the neural-specific miR-9 microRNA. Cell Rep. 2, 1-9. 10.1016/j.celrep.2012.05.015 [DOI] [PubMed] [Google Scholar]
- Novelli G., Muchir A., Sangiuolo F., Helbling-Leclerc A., D'Apice M. R., Massart C., Capon F., Sbraccia P., Federici M., Lauro R. et al. (2002). Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 71, 426-431. 10.1086/341908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi H., Kawasaki T., Deguchi T. and Yuba S. (2015). Generation of xeroderma pigmentosum-A patient-derived induced pluripotent stem cell line for use as future disease model. Cell Reprog. 17, 268-274. 10.1089/cell.2014.0104 [DOI] [PubMed] [Google Scholar]
- Olive M., Harten I., Mitchell R., Beers J. K., Djabali K., Cao K., Erdos M. R., Blair C., Funke B., Smoot L. et al. (2010). Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler. Thromb. Vasc. Biol. 30, 2301-2309. 10.1161/ATVBAHA.110.209460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio F. G., Obaya A. J., López-Otín C. and Freije J. M. P. (2009). Accelerated ageing: from mechanism to therapy through animal models. Transgenic Res. 18, 7-15. 10.1007/s11248-008-9226-z [DOI] [PubMed] [Google Scholar]
- Osorio F. G., Varela I., Lara E., Puente X. S., Espada J., Santoro R., Freije J. M. P., Fraga M. F. and López-Otín C. (2010). Nuclear envelope alterations generate an aging-like epigenetic pattern in mice deficient in Zmpste24 metalloprotease. Aging Cell 9, 947-957. 10.1111/j.1474-9726.2010.00621.x [DOI] [PubMed] [Google Scholar]
- Osorio F. G., Navarro C. L., Cadinanos J., Lopez-Mejia I. C., Quiros P. M., Bartoli C., Rivera J., Tazi J., Guzman G., Varela I. et al. (2011). Splicing-directed therapy in a new mouse model of human accelerated aging. Sci. Transl. Med. 3, 106ra107 10.1126/scitranslmed.3002847 [DOI] [PubMed] [Google Scholar]
- Osorio F. G., Barcena C., Soria-Valles C., Ramsay A. J., de Carlos F., Cobo J., Fueyo A., Freije J. M. P. and Lopez-Otin C. (2012). Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 26, 2311-2324. 10.1101/gad.197954.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco L. M., Gomez L. A., Dias J., Ziebarth N. M., Howard G. A. and Schiller P. C. (2014). Progerin expression disrupts critical adult stem cell functions involved in tissue repair. Aging 6, 1049-1063. 10.18632/aging.100709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios J. A., Herranz D., De Bonis M. L., Velasco S., Serrano M. and Blasco M. A. (2010). SIRT1 contributes to telomere maintenance and augments global homologous recombination. J. Cell Biol. 191, 1299-1313. 10.1083/jcb.201005160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet N., Box J. K., Ashton N. W., Suraweera A., Croft L. V., Urquhart A. J., Bolderson E., Zhang S.-D., O'Byrne K. J. and Richard D. J. (2014). Néstor-Guillermo Progeria Syndrome: a biochemical insight into Barrier-to-Autointegration Factor 1, alanine 12 threonine mutation. BMC Mol. Biol. 15, 27 10.1186/s12867-014-0027-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendas A. M., Zhou Z., Cadinanos J., Freije J. M., Wang J., Hultenby K., Astudillo A., Wernerson A., Rodriguez F., Tryggvason K. et al. (2002). Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 31, 94-99. 10.1038/ng871 [DOI] [PubMed] [Google Scholar]
- Pennarun G., Hoffschir F., Revaud D., Granotier C., Gauthier L. R., Mailliet P., Biard D. S. and Boussin F. D. (2010). ATR contributes to telomere maintenance in human cells. Nucleic Acids Res. 38, 2955-2963. 10.1093/nar/gkp1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitrez P. R., Rosa S. C., Praca C. and Ferreira L. (2016). Vascular disease modeling using induced pluripotent stem cells: focus in Hutchinson-Gilford Progeria Syndrome. Biochem. Biophys. Res. Commun. 473, 710-718. 10.1016/j.bbrc.2015.10.014 [DOI] [PubMed] [Google Scholar]
- Poitelon Y., Kozlov S., Devaux J., Vallat J.-M., Jamon M., Roubertoux P., Rabarimeriarijaona S., Baudot C., Hamadouche T., Stewart C. L. et al. (2012). Behavioral and molecular exploration of the AR-CMT2A mouse model Lmna (R298C/R298C). Neuromol. Med. 14, 40-52. 10.1007/s12017-012-8168-z [DOI] [PubMed] [Google Scholar]
- Pollina E. A. and Brunet A. (2011). Epigenetic regulation of aging stem cells. Oncogene 30, 3105-3126. 10.1038/onc.2011.45 [DOI] [PubMed] [Google Scholar]
- Puente X. S., Quesada V., Osorio F. G., Cabanillas R., Cadiñanos J., Fraile J. M., Ordóñez G. R., Puente D. A., Gutiérrez-Fernández A., Fanjul-Fernández M. et al. (2011). Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am. J. Hum. Genet. 88, 650-656. 10.1016/j.ajhg.2011.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzianowska-Kuznicka M. and Kuznicki J. (2005). Genetic alterations in accelerated ageing syndromes: do they play a role in natural ageing? Int. J. Biochem. Cell Biol. 37, 947-960. 10.1016/j.biocel.2004.10.011 [DOI] [PubMed] [Google Scholar]
- Rando T. A. and Chang H. Y. (2012). Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell 148, 46-57. 10.1016/j.cell.2012.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raya A., Rodríguez-Pizà I., Guenechea G., Vassena R., Navarro S., Barrero M. J., Consiglio A., Castellà M., Rio P., Sleep E. et al. (2009). Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 460, 53-59. 10.1038/nature08129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Righolt C. H., van ‘t Hoff M. L. R., Vermolen B. J., Young I. T. and Raz V. (2011). Robust nuclear lamina-based cell classification of aging and senescent cells. Aging 3, 1192-1201. 10.18632/aging.100414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Torres J., Acín-Perez R., Cabezas-Sánchez P., Osorio F. G., Gonzalez-Gómez C., Megias D., Cámara C., López-Otín C., Enríquez J. A., Luque-Garcia J. L. et al. (2013). Identification of mitochondrial dysfunction in Hutchinson-Gilford progeria syndrome through use of stable isotope labeling with amino acids in cell culture. J. Proteomics 91, 466-477. 10.1016/j.jprot.2013.08.008 [DOI] [PubMed] [Google Scholar]
- Roos C. M., Zhang B., Palmer A. K., Ogrodnik M. B., Pirtskhalava T., Thalji N. M., Hagler M., Jurk D., Smith L. A., Casaclang-Verzosa G. et al. (2016). Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. [Epub ahead of print] 10.1111/acel.12458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengardten Y., McKenna T., Grochová D. and Eriksson M. (2011). Stem cell depletion in Hutchinson-Gilford progeria syndrome. Aging Cell 10, 1011-1020. 10.1111/j.1474-9726.2011.00743.x [DOI] [PubMed] [Google Scholar]
- Saadatzadeh M. R., Bijangi-Vishehsaraei K., Kapur R. and Haneline L. S. (2009). Distinct roles of stress-activated protein kinases in Fanconi anemia-type C-deficient hematopoiesis. Blood 113, 2655-2660. 10.1182/blood-2008-09-181420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin E. and DePinho R. A. (2010). Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464, 520-528. 10.1038/nature08982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A., Kaarniranta K. and Kauppinen A. (2012). Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging 4, 166-175. 10.18632/aging.100444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P. and Misteli T. (2005). Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 11, 440-445. 10.1038/nm1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P. and Misteli T. (2006). Lamin A-dependent nuclear defects in human aging. Science 312, 1059-1063. 10.1126/science.1127168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P. and Misteli T. (2008). Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 10, 452-459. 10.1038/ncb1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye-Knudsen M., Croteau D. L. and Bohr V. A. (2013). Mitochondrial deficiency in Cockayne syndrome. Mech. Ageing Dev. 134, 275-283. 10.1016/j.mad.2013.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye-Knudsen M., Mitchell S. J., Fang E. F., Iyama T., Ward T., Wang J., Dunn C. A., Singh N., Veith S., Hasan-Olive M. M. et al. (2014). A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840-855. 10.1016/j.cmet.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B., Hoeijmakers J. H. and Garinis G. A. (2009). Sealing the gap between nuclear DNA damage and longevity. Mol. Cell. Endocrinol. 299, 112-117. 10.1016/j.mce.2008.10.031 [DOI] [PubMed] [Google Scholar]
- Seemanova E., Sperling K., Neitzel H., Varon R., Hadac J., Butova O., Schrock E., Seeman P. and Digweed M. (2006). Nijmegen breakage syndrome (NBS) with neurological abnormalities and without chromosomal instability. J. Med. Genet. 43, 218-224. 10.1136/jmg.2005.035287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanske A., Caride D. G., Menasse-Palmer L., Bogdanow A. and Marion R. W. (1997). Central nervous system anomalies in Seckel syndrome: report of a new family and review of the literature. Am. J. Med. Genet. 70, 155-158. [DOI] [PubMed] [Google Scholar]
- Sharma G. G., So S., Gupta A., Kumar R., Cayrou C., Avvakumov N., Bhadra U., Pandita R. K., Porteus M. H., Chen D. J. et al. (2010). MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell. Biol. 30, 3582-3595. 10.1128/MCB.01476-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto A., Kagawa H., Zensho K., Sera Y., Kazuki Y., Osaki M., Oshimura M., Ishigaki Y., Hamasaki K., Kodama Y. et al. (2014). Reprogramming suppresses premature senescence phenotypes of Werner syndrome cells and maintains chromosomal stability over long-term culture. PLoS ONE 9, e112900 10.1371/journal.pone.0112900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumaker D. K., Dechat T., Kohlmaier A., Adam S. A., Bozovsky M. R., Erdos M. R., Eriksson M., Goldman A. E., Khuon S., Collins F. S. et al. (2006). Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 103, 8703-8708. 10.1073/pnas.0602569103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh L., Brennan T. A., Kim J.-H., Egan K. P., McMillan E. A., Chen Q., Hankenson K. D., Zhang Y., Emerson S. G., Johnson F. B. et al. (2013). Long-term functional engraftment of mesenchymal progenitor cells in a mouse model of accelerated aging. Stem Cells 31, 607-611. 10.1002/stem.1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria-Valles C., Osorio F. G., Gutierrez-Fernandez A., De Los Angeles A., Bueno C., Menendez P., Martin-Subero J. I., Daley G. Q., Freije J. M. P. and Lopez-Otin C. (2015a). NF-kappaB activation impairs somatic cell reprogramming in ageing. Nat. Cell Biol. 17, 1004-1013. 10.1038/ncb3207 [DOI] [PubMed] [Google Scholar]
- Soria-Valles C., Osorio F. G. and López-Otín C. (2015b). Reprogramming aging through DOT1L inhibition. Cell Cycle 14, 3345-3346. 10.1080/15384101.2015.1093443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. E. and Armanios M. (2015). The short and long telomere syndromes: paired paradigms for molecular medicine. Curr. Opin. Genet. Dev. 33, 1-9. 10.1016/j.gde.2015.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockklausner C., Raffel S., Klermund J., Bandapalli O. R., Beier F., Brümmendorf T. H., Bürger F., Sauer S. W., Hoffmann G. F., Lorenz H. et al. (2015). A novel autosomal recessive TERT T1129P mutation in a dyskeratosis congenita family leads to cellular senescence and loss of CD34+ hematopoietic stem cells not reversible by mTOR-inhibition. Aging 7, 911-927. 10.18632/aging.100835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strandgren C., Nasser H. A., McKenna T., Koskela A., Tuukkanen J., Ohlsson C., Rozell B. and Eriksson M. (2015). Transgene silencing of the Hutchinson-Gilford progeria syndrome mutation results in a reversible bone phenotype, whereas resveratrol treatment does not show overall beneficial effects. FASEB J. 29, 3193-3205. 10.1096/fj.14-269217 [DOI] [PubMed] [Google Scholar]
- Studer L., Vera E. and Cornacchia D. (2015). Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell 16, 591-600. 10.1016/j.stem.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N. M., Niwa A., Yabe M., Hira A., Okada C., Amano N., Watanabe A., Watanabe K.-I., Heike T., Takata M. et al. (2015). Pluripotent cell models of fanconi anemia identify the early pathological defect in human hemoangiogenic progenitors. Stem Cells Transl. Med. 4, 333-338. 10.5966/sctm.2013-0172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W.-H., Baris H., Robson C. D. and Kimonis V. E. (2005). Cockayne syndrome: the developing phenotype. Am. J. Med. Genet. A 135A, 214-216. 10.1002/ajmg.a.30731 [DOI] [PubMed] [Google Scholar]
- Tang S., Xie M., Cao N. and Ding S. (2016). Patient-specific induced pluripotent stem cells for disease modeling and phenotypic drug discovery. J. Med. Chem. 59, 2-15. 10.1021/acs.jmedchem.5b00789 [DOI] [PubMed] [Google Scholar]
- Thill M., Nguyen T. D., Wehnert M., Fischer D., Hausser I., Braun S. and Jackisch C. (2008). Restrictive dermopathy: a rare laminopathy. Arch. Gynecol. Obstet. 278, 201-208. 10.1007/s00404-008-0676-6 [DOI] [PubMed] [Google Scholar]
- Tilstra J. S., Robinson A. R., Wang J., Gregg S. Q., Clauson C. L., Reay D. P., Nasto L. A., St Croix C. M., Usas A., Vo N. et al. (2012). NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Invest. 122, 2601-2612. 10.1172/JCI45785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivey H. S. E., Rokicki M. J., Barnacle J. R., Rogers M. J., Bagley M. C., Kipling D. and Davis T. (2013). Small molecule inhibition of p38 MAP kinase extends the replicative life span of human ATR-Seckel syndrome fibroblasts. J. Gerontol. A Biol. Sci. Med. Sci. 68, 1001-1009. 10.1093/gerona/gls336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugalde A. P., Ramsay A. J., de la Rosa J., Varela I., Mariño G., Cadiñanos J., Lu J., Freije J. M. P. and López-Otín C. (2011). Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. 30, 2219-2232. 10.1038/emboj.2011.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel O., Reshef H., Ravid A., Fabian I., Halperin D., Ram R., Bakhanashvili M., Nordenberg J. and Lahav M. (2008). Oxidative stress causes telomere damage in Fanconi anaemia cells - a possible predisposition for malignant transformation. Br. J. Haematol. 142, 82-93. 10.1111/j.1365-2141.2008.07137.x [DOI] [PubMed] [Google Scholar]
- van der Pluijm I., Garinis G. A., Brandt R. M. C., Gorgels T. G. M. F., Wijnhoven S. W., Diderich K. E. M., de Wit J., Mitchell J. R., van Oostrom C., Beems R. et al. (2007). Impaired genome maintenance suppresses the growth hormone--insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 5, e2 10.1371/journal.pbio.0050002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen J. M. (2014). The role of senescent cells in ageing. Nature 509, 439-446. 10.1038/nature13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhooren V. and Libert C. (2013). The mouse as a model organism in aging research: usefulness, pitfalls and possibilities. Ageing Res. Rev. 12, 8-21. 10.1016/j.arr.2012.03.010 [DOI] [PubMed] [Google Scholar]
- Varela I., Cadiñanos J., Pendás A. M., Gutiérrez-Fernández A., Folgueras A. R., Sánchez L. M., Zhou Z., Rodríguez F. J., Stewart C. L., Vega J. A. et al. (2005). Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 437, 564-568. 10.1038/nature04019 [DOI] [PubMed] [Google Scholar]
- Varela I., Pereira S., Ugalde A. P., Navarro C. L., Suárez M. F., Cau P., Cadiñanos J., Osorio F. G., Foray N., Cobo J. et al. (2008). Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 14, 767-772. 10.1038/nm1786 [DOI] [PubMed] [Google Scholar]
- Varga R., Eriksson M., Erdos M. R., Olive M., Harten I., Kolodgie F., Capell B. C., Cheng J., Faddah D., Perkins S. et al. (2006). Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 103, 3250-3255. 10.1073/pnas.0600012103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez-Cruz R., Zadorin A. S., Coin F. and Egly J.-M. (2013). Sirt1 suppresses RNA synthesis after UV irradiation in combined xeroderma pigmentosum group D/Cockayne syndrome (XP-D/CS) cells. Proc. Natl. Acad. Sci. USA 110, E212-E220. 10.1073/pnas.1213076110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij W. P., Hoeijmakers J. H. J. and Pothof J. (2016). Genome integrity in aging: human syndromes, mouse models, and therapeutic options. Annu. Rev. Pharmacol. Toxicol. 56, 427-445. 10.1146/annurev-pharmtox-010814-124316 [DOI] [PubMed] [Google Scholar]
- Vijg J. and Suh Y. (2013). Genome instability and aging. Annu. Rev. Physiol. 75, 645-668. 10.1146/annurev-physiol-030212-183715 [DOI] [PubMed] [Google Scholar]
- Villa-Bellosta R., Rivera-Torres J., Osorio F. G., Acin-Perez R., Enriquez J. A., Lopez-Otin C. and Andres V. (2013). Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation 127, 2442-2451. 10.1161/CIRCULATIONAHA.112.000571 [DOI] [PubMed] [Google Scholar]
- Walne A. J., Vulliamy T., Kirwan M., Plagnol V. and Dokal I. (2013). Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am. J. Hum. Genet. 92, 448-453. 10.1016/j.ajhg.2013.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S. R., Prahalad A. K., Yang J. and Hock J. M. (2006). RECQL4-deficient cells are hypersensitive to oxidative stress/damage: insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochem. Biophys. Res. Commun. 345, 403-409. 10.1016/j.bbrc.2006.04.093 [DOI] [PubMed] [Google Scholar]
- Wood A. M., Rendtlew Danielsen J. M., Lucas C. A., Rice E. L., Scalzo D., Shimi T., Goldman R. D., Smith E. D., Le Beau M. M. and Kosak S. T. (2014). TRF2 and lamin A/C interact to facilitate the functional organization of chromosome ends. Nat. Commun. 5, 5467 10.1038/ncomms6467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z.-M., LaDana C., Wu D. and Cao K. (2013). An inhibitory role of progerin in the gene induction network of adipocyte differentiation from iPS cells. Aging 5, 288-303. 10.18632/aging.100550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z.-M., Choi J. Y., Wang K., Zhang H., Tariq Z., Wu D., Ko E., LaDana C., Sesaki H. and Cao K. (2016). Methylene blue alleviates nuclear and mitochondrial abnormalities in progeria. Aging Cell 15, 279-290. 10.1111/acel.12434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon A., Peng G., Brandenburger Y., Zollo O., Xu W., Rego E. and Ruggero D. (2006). Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 312, 902-906. 10.1126/science.1123835 [DOI] [PubMed] [Google Scholar]
- Yu C.-E., Oshima J., Fu Y.-H., Wijsman E. M., Hisama F., Alisch R., Matthews S., Nakura J., Miki T., Ouais S. et al. (1996). Positional cloning of the Werner's syndrome gene. Science 272, 258-262. 10.1126/science.272.5259.258 [DOI] [PubMed] [Google Scholar]
- Zhang X., Sejas D. P., Qiu Y., Williams D. A. and Pang Q. (2007). Inflammatory ROS promote and cooperate with the Fanconi anemia mutation for hematopoietic senescence. J. Cell Sci. 120, 1572-1583. 10.1242/jcs.003152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.-S., Marquez-Loza L., Eaton L., Duncan A. W., Goldman D. C., Anur P., Watanabe-Smith K., Rathbun R. K., Fleming W. H., Bagby G. C. et al. (2010). Fancd2-/- mice have hematopoietic defects that can be partially corrected by resveratrol. Blood 116, 5140-5148. 10.1182/blood-2010-04-278226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Lian Q., Zhu G., Zhou F., Sui L., Tan C., Mutalif R. A., Navasankari R., Zhang Y., Tse H.-F. et al. (2011). A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 8, 31-45. 10.1016/j.stem.2010.12.002 [DOI] [PubMed] [Google Scholar]
- Zhang H., Xiong Z.-M. and Cao K. (2014). Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1. Proc. Natl. Acad. Sci. USA 111, E2261-E2270. 10.1073/pnas.1320843111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Li J., Suzuki K., Qu J., Wang P., Zhou J., Liu X., Ren R., Xu X., Ocampo A. et al. (2015). Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348, 1160-1163. 10.1126/science.aaa1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlateska B., Ciccolini A. and Dror Y. (2015). Treatment of dyskeratosis congenita-associated pulmonary fibrosis with danazol. Pediatr. Pulmonol. 50, E48-E51. 10.1002/ppul.23235 [DOI] [PubMed] [Google Scholar]