

SUMMARY
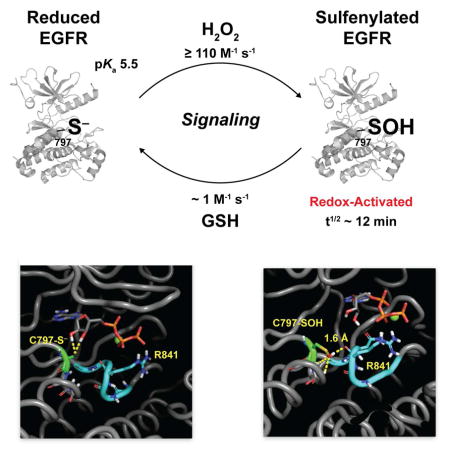
EGFR is a target of signal-derived H2O2 and oxidation of active site cysteine 797 to sulfenic acid enhances kinase activity. Although a major class of covalent drugs targets C797, nothing is known about its catalytic importance or how S-sulfenylation leads to activation. Here, we report the first detailed functional analysis of C797. In contrast to prior assumptions, mutation of C797 diminishes catalytic efficiency in vitro and cells. The experimentally determined pKa and reactivity of C797 towards H2O2 correspondingly distinguish this residue from the bulk of the cysteinome. Molecular dynamic simulation of reduced versus oxidized EGFR, reinforced by experimental testing, indicates that sulfenylation of C797 allows new electrostatic interactions to be formed with the catalytic loop. Finally, we show that chronic oxidative stress yields an EGFR subpopulation that is refractory to the FDA-approved drug, afatinib. Collectively, our data highlight the significance of redox biology to understanding kinase regulation and drug pharmacology.
eTOC BLURB
Truong et al. elucidate the molecular mechanism underlying redox-activation of EGFR. Sulfenylation of C797 activates EGFR kinase through new electrostatic interactions, which may induce a favorable change in catalytic loop dynamics. Furthermore, C797 sulfenylation and H2O2 stress impacts the pharmacology of covalent, thiol-targeted EGFR inhibitors.
INTRODUCTION
Epidermal growth factor receptor (EGFR) is one of the most studied receptor tyrosine kinases (RTK) and serves as a quintessential model for understanding RTKs in physiological signaling (Schlessinger, 2000) and has generated significant interest as a target for cancer therapeutics (Shawver, et al., 2002). EGFR is a member of the ErbB family of growth factor receptors (EGFR, Her2, Her3, and Her4) and at least 10 ligands are known to bind and activate family members (Bogdan and Klambt, 2001). Ligand binding to the extracellular domain results in receptor dimerization with the same or different family members, autophosphorylation, kinase activation, and the generation of binding sites for various intracellular proteins that regulate cellular processes such as proliferation, survival, motility, adhesion, invasion, and angiogenesis. Elevated levels, mutation, and constitutive activation of EGFR are common features of many cancer types and promote tumor development by driving cell proliferation and survival (Blume-Jensen and Hunter, 2001). Consequently, numerous modalities, including antibodies to the extracellular domain of the receptor and small-molecule inhibitors that reversibly or irreversibly bind to the intracellular kinase domain (Hubbard, 2005), have been developed to target EGFR and are currently in various stages of preclinical evaluation and clinical use (Wu, et al., 2015).
Ligand-mediated activation of RTKs, including EGFR, also leads to production of endogenous hydrogen peroxide (H2O2) by membrane-bound NADPH oxidases (NOXs) (Brewer, et al., 2015; Holmstrom and Finkel, 2014). Receptor activation triggers rapid activation of NOX multi-protein complexes that catalyze the reduction of molecular oxygen (O2) using cytosolic NADPH to generate superoxide (O2•−) and its subsequent dismutation product, H2O2 (Dickinson and Chang, 2011). Once formed, H2O2 transmits the redox signal via oxidation of cysteine residues in reactive sensor proteins, with concomitant modulation of biological function (Paulsen and Carroll, 2013; Veal, et al., 2007). In this way, H2O2 serves as a second messenger in growth factor-induced signaling cascades. Reaction of cysteines with H2O2 occurs solely with the thiolate anion (Cys-S−) and direct product of this reaction is the sulfenic acid (Cys-SOH), also referred to as S-sulfenylation (Chouchani, et al., 2016; Dotsey, et al., 2015; Finkel, 2012; Lo Conte and Carroll, 2013). The oxidation and reduction of cysteine residues presents a mechanism to rapidly and reversibly alter activity, stability, localization and protein-protein interactions.
Using a selective, cell-permeable probe for detecting sulfenic acid, known as DYn-2 we have previously demonstrated that EGF-mediated signaling events induce dynamic changes in global protein S-sulfenylation, concurrent with changes in intracellular H2O2 (Paulsen, et al., 2012). Our study also revealed that ligand-induced EGFR activation resulted in NOX2-dependent sulfenylation of a cysteine residue (C797; Figure 1A) at the hinge region of the catalytic ATP-binding pocket and that this modification was associated with enhanced tyrosine kinase activity. Sequence alignment shows that 10 other kinases possess a cysteine at the same structural position as C797 of EGFR (Truong and Carroll, 2012). These kinases include members of the Erb family (Her2, Her4), the Tec family (BMX, BTK, ITK, TEC, and TXK), one Src member (BLK), MKKa7 and JAK3. Proximity to the catalytic pocket and selective oxidation strongly support an important regulatory function for sulfenylation of C797 (Heppner and van der Vliet, 2015; Truong and Carroll, 2013); however, the precise molecular mechanism for redox-based activation of EGFR remains entirely unknown. This question is critical to address both from a fundamental perspective and owing to implications for related kinases.
FIGURE 1.

EGFR catalytic kinase domain and covalent inhibitors. (A) Crystal structure of the EGFR kinase domain (PDB 3VJO) bound to AMP-PNP showing all six conserved cysteine residues. The catalytic loop is highlighted in lime green, T790/L858 residues in magenta, and E746/A750 residues in lilac. (B) Top: Structure of two electrophilic covalent EGFR inhibitors, afatinib and PD168393. The acrylamide functional groups in each inhibitor are highlighted in the gray circle. Bottom: Crystal structure of the EGFR kinase domain (PDB 4G5J) in a covalent thioether bond with afatinib at C797.
EGFR kinase domain mutations have been identified as a cause of non-small cell lung cancer (NSCLC), and the two most frequent include the exon 19 point mutation (L858R) and the exon 19 deletion (Δ746–750) (Johnson and Janne, 2005) (Figure 1A). L858R is the single most common mutation (~40%) and resides as part of the kinase activation loop (A-loop). The L858R mutation causes constitutive activation of EGFR by destabilizing the auto-inhibitory conformation of the A-loop, which is its normal state in the absence of ligand (Yun, et al., 2007; Zhang, et al., 2006). Cells bearing this EGFR mutation exhibit decreased ATP affinity and are generally more sensitive to ATP-competitive inhibitors as compared to cells expressing wild type EGFR (Lynch, et al., 2004). Approximately one-half of lung cancer patients harboring the L858R mutation eventually develop resistance to ATP-competitive EGFR inhibitors due to a second-site mutation (T790M) (Kobayashi, et al., 2005). Residue 790 is located at the posterior of the ATP binding pocket and is important for inhibitor binding and specificity. Although these activating mutations are often observed in human tumors, it is not known whether these kinases undergo cysteine oxidation and, if so, whether they exhibit a different redox sensitivity compared to wild type EGFR.
Since the early 1990s, EGFR C797 has been targeted for therapeutic intervention with the objective to develop covalent irreversible inhibitors for the treatment of cancer (Liu, et al., 2013). These small-molecule inhibitors generally incorporate a Michael acceptor (electrophile warhead) to covalently modify EGFR C797 (nucleophile target) and a binding element specific for the kinase active site. Examples include FDA-approved, afatinib (Figure 1B) as well as neratinib, dacomitinib, and rociletinib currently in Phase III clinical trials. In addition to modulation of EGFR kinase activity, sulfenylation of C797 has the potential to impact drug potency in two significant ways. Most obviously, variation in the redox state of Cys797 (e.g., –SOH, –SO2H, –SSG) can give rise to local conformational changes, which can impact the reversible binding affinity of EGFR inhibitors. Not quite so obvious, but equally important, is that oxidation of the cysteine γ-sulfur blunts its nucleophilic reactivity (Gupta and Carroll, 2014). The above-mentioned inhibitors all incorporate an acrylamide electrophile that undergoes facile reaction with the nucleophilic cysteine thiol group. The same reaction, however, does not take place with cysteine sulfenic acid, where the electrophilic reactivity of the γ-sulfur is dominant in the aqueous physiological environment. S-sulfenylation is a dynamic and reversible process, involving the glutathione (GSH) and/or thioredoxin (TRX) systems. Even so, chronic H2O2 stress, such as that encountered in cultured malignant cells and solid tumors, would be expected to increase the steady-state population of oxidized cysteine residues. A key question, therefore, is whether chronic oxidative stress yields a population of EGFR that is refractory to C797-targeted irreversible drugs.
RESULTS
H2O2 Activates Native EGFR Through a Direct and Reversible Mechanism
Our work and that of others has shown that moderate exogenous or endogenous H2O2 elevation can lead to a net increase of EGFR autophosphorylation in cells [reviewed in (Corcoran and Cotter, 2013; Heppner and van der Vliet, 2015; Truong and Carroll, 2012)]. Using recombinant GST-tagged EGFR kinase domain with a peptide-based assay, we have demonstrated that sulfenylation of C797 enhances the intrinsic tyrosine kinase activity (Paulsen, et al., 2012). However, the impact of sulfenylation on the autophosphorylation activity of purified full-length EGFR has not been investigated. Thus, we isolated native EGFR from A431 cells by immunoprecipitation and assayed for its ability to autophosphorylate tyrosine residues within its cytoplasmic domain (Figure 2A, B). Control experiments indicated that EGFR kinase activity increased nearly twofold after EGF treatment, congruent with findings from other groups (Cohen, et al., 1980). Next, we investigated whether H2O2 treatment of native EGFR would also lead to an increase in receptor autophosphorylation. From 1–10 μM of H2O2, a dose-dependent increase in EGFR kinase activity was observed, with a maximal stimulation of three-fold. Importantly, reduction with dithiothreitol (DTT) totally abrogated the enhancing effect of H2O2 on kinase activity, consistent with sulfenic acid formation (Figure 2A, B). Site-specific generation of cysteine sulfenic acid under these conditions was further supported by observation of dimedone-labeled C797 by peptide mapping using LC-MS/MS (Figure S1). Indeed, tandem MS analysis including all oxidizable cysteine residues in the kinase domain (~95% overall amino acid sequence coverage) verified that oxidation occurs exclusively at C797, consistent with our earlier findings (Paulsen, et al., 2012) and as confirmed by Schwartz et al. (Schwartz, et al., 2014). At H2O2 levels greater than 25 μM, the enzymatic activity of EGFR began to decrease, likely through formation of sulfinic acid (–SO2−), which is known to reduce the fraction of functional active sites in EGFR (Schwartz, et al., 2014). Control experiments confirmed that pre-treatment of native EGFR with the irreversible inhibitor, afatinib completely inhibited autophosphorylation, as expected (Figure 2A, B). Coupled with our earlier findings that sulfenylation of C797 enhances catalytic activity with GST-tagged EGFR kinase domain in a peptide assay, these new data show that exposure to physiological concentrations of H2O2 enhances autophosphorylation of native EGFR by reversible sulfenylation.
FIGURE 2.

H2O2 stimulation of EGFR autophosphorylation and biochemical properties of C797. (A) Full length native EGFR was treated with EGF (3.3 ng/μl) or H2O2 (0–100 μM). Western blot shows autophosphorylated (p) and total EGFR. (B) Densitometric quantification of A. Data are normalized to untreated control and are representative of three independent readings (mean ± s.e.m). *p<0.05, **p<0.01, ***p< 0.001 compared to untreated control. (C) Bromobimane fluorescence labeling of recombinant EGFR over a pH range (2–6.5) yields a single pKa value for C797 of 5.5 ± 0.1. Data are representative of two independent readings. (D) Plotting the rate of sulfenic acid formation over a range of H2O2 concentrations (10, 20, 40, 80 μM) yields a second-order rate constant for the H2O2 reactivity of C797 of ≥110 M−1s−1. Data represent the average of two independent readings (mean ± s.e.m.).
The Physical Properties of EGFR C797
Solvent accessibility and cysteine reactivity (modulated by pKa and microenvironment) are generally considered as the most important factors underlying thiol redox reactions. Consistent with this hypothesis, C797 has the greatest solvent accessible surface area of any cysteine in the EGFR kinase domain [as calculated by GETAREA (Fraczkiewicz and Braun, 1998) four are buried: C781, C818, C929, and C950; C775 is somewhat solvent exposed, but with ~10-fold less accessible area than C797; Figure 1A]. In addition, C797 is distinguished from all other cysteine residues in this domain by virtue of its location at the N-terminus of an α-helix (referred to the Ncap position). Owing to the α-helix dipole, pKa perturbations significantly below the intrinsic thiol pKa (~8.3) are well documented at the Ncap position (Anderson and Sauer, 2003; Miranda, 2003). Since thiolates are significantly stronger nuclephiles than thiol groups and C797 preferentially reacts with H2O2, we hypothesized that this residue should exhibit a pKa value lower than the unperturbed value of 8.5. To test this proposal, we determined the pKa of C797 by its reactivity with the fluorescent thiol alkylating agent, monobromobimane, over a range of pH values. Using this method, the pKa value of C797 was determined to be 5.5 ± 0.1 (Figure 2C), approximately three orders of magnitude lower than the pKa of free sulfhydryl in solution. Pretreatment with the irreversible C797-targeted inhibitor, PD168393 (Fry, et al., 1998), suppressed fluorescence labeling as expected. Finally, we measured the reactivity of EGFR C797 with H2O2. Since a thiol or thiolate is unreactive with sulfenic acid probe, DYn-2, the increase in reaction rate with this reagent as H2O2 increases reports on reactivity of the target residue. Using this technique, a lower limit (imposed by further oxidation to sulfinic acid) for the second-order rate constant of C797 with H2O2 was determined to be ≥110 M−1 s−1 at pH 7.4 (Figure 2D). Together, these data indicate that EGFR C797 is differentiated from the bulk of the cysteine proteome with respect to solvent accessibility, pKa, and H2O2 reactivity.
Activity of the C797S and C797A Mutants in vitro and in Cells
EGFR C797 is the target for a major class of covalent drugs, yet its functional importance in catalysis still remains unknown. Because C797 is located in the hinge region of the ATP-binding pocket, we first characterized the activity of recombinant serine (C797S) or alanine (C797A) EGFR kinase domain (residues 696–1022). The kinetic parameters for ATP and a peptide substrate were determined using a well-established continuous in vitro kinase assay (Yun, et al., 2007) and are summarized in Table 1 (rate plots are presented in Figure S2). The catalytic efficiency (kcat/Km) of the wild type and mutant kinases is also plotted in Figure 3A, B. Overall, the kcat/Km of C797S and C797A was 4.4 to 6.7-fold lower than that of wild type EGFR. The decreases in kcat/Km were attributable both to lower kcat and higher Km values, with the C797A mutant more affected than C797S. By comparison, the value of kcat/Km for the activating L858R mutant has been reported to be ~20-fold higher than that for the wild type enzyme (Zhang, et al., 2006). In contrast to wild type EGFR, C797S/A mutants did not exhibit any discernable increase in kinase activity following H2O2 treatment (Figure S2).
TABLE 1.
Kinetic Parameters of recombinant wild type, C797S, and C797A EGFR.
| ATPa | Peptideb | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Km (μM) | kcat (min−1) | kcat/Km (μM−1 min−1 × 102) | Fold Decreasec | Km (μM) | kcat (min−1) | kcat/Km (μM−1 min−1 × 103) | Fold Decreasec | |
| WT | 7.5 ± 0.4 | 0.33 ± 0.02 | 4.4 | (1) | 906 ± 48 | 1.1 ± 0.02 | 1.2 | (1) |
| C797S | 19.6 ± 0.8 | 0.19 ± 0.01 | 0.97 | 4.5 | 1911 ± 119 | 0.51 ± 0.04 | 0.27 | 4.4 |
| C797A | 21.7 ± 1.1 | 0.17 ± 0.06 | 0.78 | 5.6 | 2134 ± 129 | 0.38 ± 0.01 | 0.18 | 6.7 |
Parameters for ATP were determined using a consensus peptide with the sequence RAHEEIYHFFFAKKK.
Parameters for peptide were determined using poly [Glu4Tyr1] as the peptide substrate.
Fold decrease was calculated by determining the catalytic efficiency (kcat/Km) for each protein and dividing by kcat/Km for WT enzyme.
FIGURE 3.

Effect of C797 mutation on recombinant and cellular EGFR kinase activity. (A) Catalytic efficiency (kcat/Km) with respect to ATP. Relative efficiency was calculated by determining kcat/Km for each protein with varying ATP and saturating peptide substrate (RAHEEIYHFFFAKKK) and dividing by the kcat/Km of wild type enzyme. (B) Catalytic efficiency (kcat/Km) with respect to peptide substrate. Relative efficiency was calculated by determining kcat/Km for each protein with varying peptide substrate (poly[Glu4Tyr1]) and saturating ATP and dividing by the kcat/Km of wild type enzyme. (C, E) EGF and H2O2-mediated activation of C797 mutants. Wild type (WT), C797S, and C797A EGFR were transfected into HeLa cells and stimulated with EGF or H2O2 for 5 min. (C, E) Western blot shows phosphorylated (p) and total EGFR. Mutants exhibit decreased autophosphorylation levels compared to WT. (D, F) Densitometric quantification of C and E respectively. At higher H2O2 concentrations (i.e., 500 μM), C797S phosphorylation levels (##) increase, which can be attributed to PTP inhibition. Data are normalized to untreated wild type (WT) control and are representative of three independent readings (mean ± s.e.m). *p<0.05, **p<0.01, ***p<0.001 compared to untreated wild type control.
To place our biochemical findings into a cellular context, plasmids expressing full-length wild type, C797S or C797A EGFR were transfected into HeLa cells with low endogenous levels of receptor expression, treated with EGF or H2O2, followed by Western blot and analysis of autophosphorylation (pEGFR) levels. In the absence of EGF, cells transfected by wild type EGFR displayed a markedly higher level of basal pEGFR compared to C797S/A mutants (Figure 3C, D). In the presence of EGF, each receptor showed a dose-dependent increase autophosphorylation; however, both mutations diminished the response to EGF with the alanine mutant most severely affected, corroborating our earlier biochemical findings. Additionally, H2O2-activation of EGFR was blunted in C797S/A mutants relative to wild type (Figure 3E, F and Figure S3). Both C797S/A mutants showed little detectable increase in receptor activation compared to corresponding basal levels when cells were exposed to H2O2 at doses of 50, 100 or 250 μM [note that intracellular concentrations of H2O2 are ~7–10-fold less than the externally applied concentration (Antunes and Cadenas, 2000)]. In contrast to lower exogenous H2O2 doses, treatment of C797S-expressing cells with H2O2 at 500 μM abruptly increased pEGFR levels (Figure 3E, F). These findings are consistent with other studies showing that exogenous H2O2 concentrations ≥500 μM broadly inhibit protein tyrosine phosphatases (PTPs), thereby increasing the net level of pEGFR (Garcia and Carroll, 2014; Haque, et al., 2011; Leonard, et al., 2011; Meng, et al., 2002). Collectively, these data show that C797 is integral for cellular EGFR activity and H2O2-mediated activation.
Modeling Predicts that C797 Sulfenylation Alters Catalytic Loop Dynamics
To examine possible mechanisms through which sulfenylation of C797 could enhance EGFR kinase activity, we generated a molecular model of sulfenyl C797 (Cys-SOH) based on the wild type EGFR kinase domain structure. Molecular dynamic (MD) simulations were subsequently performed for reduced and oxidized C797 (Movies S1–4). Although C797 predominantly exists as the thiolate anion (Figure 2C) or sulfenic acid [pKa ~7 in proteins (Gupta and Carroll, 2016a)] under physiological conditions, calculations were obtained for neutral and deprotonated states for reduced (cysteine – SH and cysteine thiolate –S−) and oxidized (sulfenic acid –SOH and sulfenate –SO−) C797 so as to comprehensively explore potential differences between functional group interactions. The MM-PBSA end-state method was employed to calculate binding free energies of simulated EGFR-ATP complexes. In the reduced (C797-SH and C797-S−) and oxidized (C797-SOH and C797-SO−) forms, EGFR kinase gave similar estimated interaction energetics with ATP, suggesting that sulfenyl modification does not have a direct impact on ATP binding affinity (Table S1). Among neutral and deprotonated C797 in reduced and oxidized states, the deprotonated forms of C797 (Cys-S− and Cys-SO−) showed weaker estimated electrostatic interaction with ATP than the neutral states (Cys-SH and Cys-SOH), likely due to close proximity of the negatively charged oxygen atoms of the phosphate tail of ATP (Table S2).
We also performed hydrogen-bond analysis on the EGFR simulations with various C797 oxidation states. Two cutoff distances (3.0 and 3.5 Å) were compared (Table S3). Deprotonated C797 in reduced and oxidized states (Cys-S− and Cys-SO−) showed more extensive hydrogen bonding with the ribose hydroxyl hydrogen than their corresponding neutral states; however, this extensive interaction does not result in strong affinity with ATP (Table S1) and is therefore not likely to account for increased kinase activity. Interestingly, only the sulfenic acid (Cys-SOH) state of C797 was predicted to form new electrostatic interactions with the active site of EGFR. Simulations indicated extensive interaction with the carbonyl backbone oxygen of arginine 841 (R841) via the sulfenyl hydrogen in nearly 50% of trajectories with an average distance of 2.8 Å (Figure 4A, B and Movies S1–2). R841 resides in the catalytic loop, a conserved motif (HRDLXXXN) in protein kinases found in the subdomain VIb, which positions substrates and conducts catalytic phosphotransfer of ATP to its peptide substrate. Reduced C797 thiol hydrogen interacted only sparsely (<5%) with the backbone carbonyl oxygen of R841 and occurred at distances >3.0 Å (Figure 4A, B). Stronger electrostatic interactions were predicted between C797 sulfenic acid (Cys-SOH) and R841 (−3.5 kcal/mol) than with reduced neutral C797 (−1.4 kcal/mol) and deprotonated C797 (2.4 kcal/mol) (Table S2). Other intermolecular interactions (e.g., van der Waals) did not exhibit any significant changes. Finally, when compared to reduced EGFR, the atomic fluctuations of several key regions were affected by oxidation of EGFR C797 to sulfenic acid (Figure S4), including R841 in the catalytic segment, the glycine-rich phosphate-binding loop (P-loop) and the activation loop (A-loop), which harbors Tyr845, a phosphorylation site. Collectively, our simulations predict that oxidation of EGFR affords new hydrogen bond and electrostatic interactions between the C797 sulfenyl hydrogen and R841, which may influence the structural dynamics of several critical regions surrounding the kinase active site.
FIGURE 4.

MD simulation of (A) reduced (–S−) and (B) sulfenylated (–SOH) EGFR. Dashed yellow lines indicate interactions predicted to occur between the C797 thiolate (A) or sulfenic acid (B), ATP substrate and/or the backbone carbonyl of R841. Figure depicts movie frame depicting the shortest distance (1.6 Å) for sulfenic acid hydrogen bonding with the R841 carbonyl. (C, E) EGF and H2O2-mediated activation of R841 mutants. WT, R841K, and R841A EGFR were transfected into HeLa cells and stimulated with EGF or H2O2 for 5 min. Western blot shows phosphorylated (p) and total EGFR. (D, F) Densitometric quantification of C and E respectively. Data are normalized to untreated wild type (WT) control and are representative of three independent readings (mean ± s.e.m). *p<0.05, **p<0.01, ***p<0.001 compared to untreated wild type control.
Catalytic Loop Residue R841 is Critical for EGFR Function and C797 Sulfenyation
To follow up on our computational studies and probe the functional importance of R841 in redox-based EGFR activation, we replaced this residue with lysine (R841K) or alanine (R841A). Then, plasmids expressing wild type, R841K, or R841A EGFR were transfected into HeLa cells, treated with EGF or H2O2, followed by Western blot and analysis of pEGFR levels. In the absence of EGF, cells transfected by wild type EGFR gave higher levels of basal pEGFR compared to R841K/A mutants (Figure 4C, D). Wild type and R841K/A mutants exhibited EGF-dependent increases in receptor autophosphorylation; however, the pEGFR level for R841K/A mutant kinases was reduced approximately 3- to 5-fold, with the alanine variant being only slightly more affected than the lysine variant. Although autophosphorylation was reduced several fold in R841K/A mutants, H2O2-mediated activation of these variants was virtually abolished (Figure 4E, F). Consistent with the loss of redox activation, sulfenylation of C797 was essentially nonexistent in both R841 mutants compared to the response observed for wild type EGFR (Figure S3). Together with our MD simulations, our functional studies identify R841 as important for kinase activity, and that this residue may facilitate C797 sulfenylation or help to stabilize the sulfenic acid modification at this site.
Oncogenic EGFR Mutants Exhibit Different Redox Sensitivity
Given the profound effect that protein environment can have on cysteine oxidation we next wondered whether C797 shows altered redox sensitivity in frequently occurring oncogenic EGFR mutants (L858R/T790M and Δ746–750). To examine this question, we stimulated adenocarcinomic human alveolar basal epithelial cell lines A549 (wild type EGFR), NCI-H1875 (L858R/T790M EGFR) and HCC827 (Δ746–750 EGFR) with EGF and probed for kinase sulfenylation. EGF-mediated sulfenylation was detectable in both oncogenic mutants as well as wild type, although basal oxidation levels (i.e., minus EGF) differed among these kinases (Figure 5A–C). Of note, the dose-dependent change in sulfenylation of EGFR was greatest in the L858R/T790M mutant compared to wild type and the deletion mutant, Δ746–750 (Figure 5D). In the case of wild type and Δ746–750 EGFR, peak sulfenylation was observed at 4 ng/ml EGF, while oxidation of L858R/T790M was greatest at five-fold higher growth factor (i.e., 20 ng/ml). Given the location of L858R/T790M with respect to C797, these two point mutations may alter structural and electrostatic features of the kinase active site pocket, leading to different redox sensitivity. Overall, these data suggest that clinically relevant oncogenic EGFR mutants can exhibit unique cysteine sulfenylation profiles.
FIGURE 5.

Sulfenylation profiles of oncogenic EGFR mutations. (A–C) Western blots show sulfenylated and total EGFR. Lung cancer cells expressing wild type (A) A549 (WT) or EGFR mutants (B) NCI-H1975 (L858R/T790M) and (C) HCC827 (ΔE746-A750) were stimulated with EGF or vehicle for 2 min and sulfenic acids were detected by Strep-HRP western blot. (D) Densitometric quantification of A–C. Data are normalized to untreated control for each respective cell line and representative of three independent readings (mean ± s.e.m). *p<0.05, **p <0.01, ***p<0.001 compared to the untreated control for each cell line.
Chronic H2O2 Stress Alters the Pharmacology of Irreversible EGFR Inhibitors
Covalent EGFR inhibitors with acrylamide “warheads” irreversibly bind to C797 through Michael addition to the reduced form of the kinase (Singh, et al., 2010). Of these, the most well known are based on the 4-anilinoquinazoline scaffold, which targets this inhibitor class to the EGFR active site for covalent reaction with the C797 thiol (Singh, et al., 1997). Due to the nature of this chemical reaction, oxidation of C797 to any redox form (e.g., –SOH and/or –SO2H and/or –SSG) is expected to impact the pharmacology of such acrylamide-based inhibitors. To investigate the effect of chronic oxidative stress on inhibitor potency in a cellular context, we used two 4-anilinoquinazoline inhibitors: afatinib (Li, et al., 2008) and PD168393 (Fry, et al., 1998) (Figure 1B). For simulation of chronic oxidative stress, glucose oxidase (GO) was added to the medium at a non-toxic dosage (2 U/mL) to generate an intracellular steady-state level of H2O2 of 1~2 μM, as previously described (Askoxylakis, et al., 2011; Long, et al., 2012; Millonig, et al., 2009; Mueller, et al., 2009). Following a 3 h exposure to H2O2 (via the GO system) and 1 h inhibitor treatment, cells were stimulated with EGF and analyzed for changes in EGFR autophosphorylation. In the absence of oxidative stress, EGFR was inhibited by >95% at 1 μM afatinib or PD169383 (Figure 6A, B; Western blots depicted in Figure S5). In the presence of H2O2, potency was demonstrably reduced at inhibitor concentrations of 1 μM and above, with an apparent leveling off of the dose-response at ~25% pEGFR. At dosages ≥10 μM, incomplete inhibition by either compound (Figure 6A, B) reflects a population of EGFR, which has been rendered resistant to irreversible targeting.
FIGURE 6.

The effect of chronic oxidative stress on irreversible EGFR inhibitors and model for redox regulation of EGFR in cells. A431 cells were incubated with glucose oxidase (2 u/ml) for 3 h to simulate chronic H2O2 stress, treated with (A) afatinib or (B) PD168393 for 1 h, and subsequently treated with EGF (100 ng/ml) for 5 min. Plots represent densitometric quantification of Western blotting for phosphorylated (p) and total EGFR. Data are normalized to untreated control for each respective condition and representative of two independent readings (mean ± s.e.m). *p<0.05, **p<0.01, ***p<0.001 compared to untreated control for each respective condition. (C) Model for H2O2 modification of EGFR C797 in cells.
DISCUSSION
Cysteine oxidation modulates kinase activity during intracellular signal transduction (Truong and Carroll, 2012; Truong and Carroll, 2013). Coupled with our earlier findings that sulfenylation of C797 enhances catalytic activity with GST-tagged EGFR kinase domain in a peptide assay, our findings using native EGFR demonstrates that exposure to physiological concentrations of H2O2 leads to sulfenylation of C797 and enhanced receptor autophosphorylation in cells (Figure 1). The location of C797 with respect to the C-helix and catalytic loop raises the possibility that new interactions made possible through C797 sulfenylation may lead to transition-state stabilization or destabilization of its auto-inhibitory conformation (Zhang, et al., 2006).
EGFR C797 is located at the amino-terminus of an α-helix, also referred to as the Ncap position. Cysteines are the most sparsely occurring Ncap residue, comprising <1% of this position (Penel, et al., 1999). Even so, Ncap cysteines are enriched among redox-active proteins, like human thioredoxin. The alignment of peptide bond dipole moments within an α-helix generates a macroscopic dipole along the helical axis and negatively charged residues are often found at the amino termini where they have a stabilizing interaction with the positive charge of the helix dipole. At this unique structural position, electrostatic interactions of the thiol with the helix dipole can lower its pKa and increase the susceptibility of this residue to oxidation (Anderson and Sauer, 2003; Miranda, 2003). Consistent with this prospect, we determined that EGFR C797 has a pKa of 5.5 ± 0.1 (nearly three orders of magnitude lower than the pKa of free cysteine sulfhydryl in solution) and a lower-limit for H2O2 reactivity of ≥110 M−1 s−1 [more than two-orders of magnitude higher than GSH and at least five-fold more reactive than the protein tyrosine phosphatase, PTP1B (Gupta and Carroll, 2014)].
Using selective cell-permeable chemical probes, direct evidence for H2O2 generation with EGFR stimulation in A431 cells was first provided by Miller et al. (Miller, et al., 2007). In A431 cells, the average number of EGFRs at the cell surface corresponds to ~640/μm2 or approximately 1.2 × 106 receptors per cell (Zhang, et al., 2015). Given the inhomogenous and two-dimensional nature of the plasma membrane, extrapolating these values to reflect a three-dimensional environment is difficult. With this caveat in mind, a back-of-the-envelope-calculation using these above values suggests a lower limit for receptor concentration of around 1 μM. With this estimated concentration and known second-order rate constants, the ratio (r = k1[substrate 1]/k2[substrate 2]) of H2O2 reacting with GSH relative to EGFR would be ~10:1 (Concentrations and rate constants: EGFR 1 μM and 110 M−1s−1; GSH, 1 mM and 0.89 M−1s−1). This ratio may underestimate H2O2 selectivity, due to a lower limit value for receptor reactivity with H2O2 (≥110 M−1 s−1) and existence of receptors within networks of high-density membrane patches (i.e., ≥1 μM). Once the C797 thiolate is sulfenylated by H2O2, with GSH supplying the reducing equivalents, at steady-state, the cellular half-life of EGFR C797–SOH is estimated to be ~12 minutes (Figure 6C). This figure correlates well with our observation that global protein sulfenylation peaks ~5 min after EGF addition to cells, with a subsequent decay over 30 min (Paulsen, et al., 2012). As an alternative to GSH restoring sulfenylated receptor back to its reduced state, EGFR C797-SOH can be oxidized further to sulfinic acid (–SO2H), which is generally an irreversible modification in biological systems (Lo Conte and Carroll, 2012; Lo Conte and Carroll, 2013; Lo Conte, et al., 2015). Although not explicitly measured in this work for EGFR, rates of sulfinic acid formation are typically 10- to 1000-fold slower than for the equivalent thiolate owing to reduced nucleophilic character of sulfur in the sulfenic acid state (Gupta and Carroll, 2014; Lo Conte and Carroll, 2013; Paulsen and Carroll, 2013). With this approximation, the rate constant for sulfinylation of EGFR is anticipated to be ~0.1–10 M−1s−1 (Figure 6C). For this reason, it is unlikely that any significant amount of over-oxidized EGFR C797 is generated in a normal cellular environment. In short, our observation of EGF-dependent sulfenylation of EGFR in cells is fully compatible with the physical and reactivity properties of C797 observed in our biochemical experiments.
C797 mutants exhibit altered autophosphorylation and sulfenylation properties upon cell stimulation (Figure 3) and have lowered catalytic efficiency (kcat/Km) compared to wild type EGFR (Table 1). The behavior of C797S/A mutants in biochemical and cellular studies supports a model wherein C797 functions as a redox-switch to stimulate EGFR autophosphorylation. Using computational modeling, we identified an arginine residue, R841 that is predicted to have enhanced intermolecular interactions due to hydrogen bonding with the sulfenic acid form of C797 (Tables S1–3 and Figure 4). R841 is a highly conserved residue found in the catalytic loop of the tyrosine kinase family that plays a role in coordinating the phosphate tail and associated Mg2+ ions (Gajiwala, et al., 2013). Therefore, oxidation of C797 has the potential to influence catalytic loop dynamics (Figure 4, Figure S4 and Movie S1). Hydrogen bonding between sulfenyl C797 and the backbone carbonyl of R841 could stabilize the greater catalytic loop, facilitating metal ion coordination and overall interactions with the ATP phosphate tail, ultimately stimulating kinase activity. From a more granular molecular perspective, new electrostatic interactions between R841 and ATP substrate might position the positively charged guanidinium group near the bridging oxygen between the β- and γ-phosphates to stabilize the negative charge that builds up on this oxygen as the nucleophile attacks. Our simulations are in good agreement with the determined kinetic parameters of the C797 mutants, where ATP binding affinity (Km) is modestly affected, while turnover (kcat) is affected to a greater extent (Table 1). Interestingly, sequence alignment reveals that some kinases, like c-Src, b-Raf contain a serine at the position corresponding to EGFR C797. From this standpoint, the hydroxyl side chain of serine may participate in interactions with R841, analogous to C797–SOH, and could account for the increased reactivity of EGFR C797S compared to C797A that observed in cells. The competence of EGFR C797S, relative to C797A, is also underscored by the recent findings that this mutation is acquired in NSCLC patients who developed resistance to the irreversible inhibitor, AZD9291 (Thress, et al., 2015).
We also determined whether commonly occurring oncogenic EGFR mutations (L858R/T790M and Δ746–750) affected their propensity to undergo sulfenylation. Given their relative proximity to the active site and C797, the L858R/T790M mutant was of particular interest, whereas Δ746–750 is more distal and might be expected to have little to no effect on sulfenylation (Figure 1A). Our findings indicate that the L858R/T790M mutations correlate with an increased propensity to undergo sulfenylation, compared to the Δ746–750 mutant (Figure 5). In this context, structural rearrangements and increased positive electrostatic potential stemming from the L858R mutation may facilitate reaction of the C797 thiolate with H2O2. In addition, the T90M mutation causes an increase in the frequency of active site opening (i.e., movement between the N- and C-lobes), which would increase the solvent accessibility of the active site and C797. Another interesting finding from these experiments is that basal levels of sulfenylation were consistently higher in oncogenic mutants, relative to wild type EGFR. An increase in the steady-state population of sulfenylated C797 is relevant to the overall kinase activity of EGFR and may also impact the pharmacology of irreversible inhibitors designed to target these resistance mutations in EGFR-driven cancer cells.
The concentration of H2O2 in transformed cells is dependent upon the cancer cell type, oxygen tension, and expression of scavenger enzymes. Notwithstanding this variation, the steady-state level of H2O2 in cancer cells has been shown to increase by as much as 40-fold compared to their non-transformed counterparts (Spitz, et al., 2012). Given an intracellular H2O2 concentration of ~10–50 nM in normal tissue (Sies, 2014; Winterbourn, 2013), this would translate into ~0.4–2 μM in cancer cells. Another study in cancer cells across a range of tumor types indicates that rates of H2O2 production are comparable in magnitude to the respiratory burst in human neutrophils (Szatrowski and Nathan, 1991), corresponding to an intracellular steady-state H2O2 concentration of ~1–4 μM. While these concentrations assume a homogenous distribution of H2O2 across the cell, it is increasingly appreciated that diffusion of H2O2 across the cytoplasm is limited (Dickinson and Chang, 2011; Mishina, et al., 2011). Instead, H2O2 localizes to microdomains near the sites of its generation during RTK signaling (Holmstrom and Finkel, 2014). Thus, the H2O2 concentration near the signalsome formed by activated EGFR may be higher than the abovementioned values. At steady-state, we estimate that the fraction of EGFR C797 in the sulfenic acid form in cancer cells could reach ~0.2 or 20% (Concentrations and rate constants: EGFR 1 μM and 110 M−1s−1; GSH, 1 mM and 0.89 M−1s−1; H2O2 2 μM). Note that our estimate infers site-localized oxidation of EGFR at the plasma membrane where reactive peroxidases, such as peroxiredoxins, are either absent or have been inactivated through hyperoxidation (Wood, et al., 2003) or phosphorylation (Woo, et al., 2010).
As discussed above, deregulation of EGFR signaling is prevalent in human cancers and has motivated the development of inhibitors that target EGFR C797 (Liu, et al., 2013). Under chronic H2O2-induced oxidative stress (1–2 μM intracellular H2O2), we observed a leveling off of the dose-response around ~25% pEGFR for both afatinib and PD168393 (Figure 6 A, B). Incomplete inhibition by these inhibitors is indicative of an oxidized population of EGFR, which does not undergo covalent adduction by thiol-targeted electrophiles. Along these lines, a separate study has demonstrated that glutathionylation of C797 can also diminish the potency of covalent EGFR inhibitors (Schwartz, et al., 2014). Although the authors of this study generated glutathionylated EGFR in vitro through incubation of the C797 thiolate with oxidized GSH (GSSG), the kinetically competent and more probable pathway for glutationylation in vivo would be attack of GSH on the C797-SOH oxoform (Figure 6C). Irrespective, from our collective studies, it is clear that cysteine oxidation can alter kinase active site topography, affect the activity of reversible and irreversible covalent inhibitors designed to target wild type EGFR, and likely contribute to mechanisms of drug resistance.
In patients with advanced solid tumors, the recommended daily dose of afatinib (40 mg orally once daily) gives a gMean maximum plasma concentration (Cmax) value at steady state of 38.0 ng/mL or ~80 nM (Wind, et al., 2013). The effectiveness of afatinib against EGFR has also been reported (Schwartz, et al., 2014): kinact/K*i ~2 × 105 M−1s−1 (Kinetic parameters: kinact ~2 × 10−3 s−1 and K*i ~4.4 nM). This second-order rate constant is ~2,000-fold higher than the reactivity of H2O2 with EGFR C797. Then again, the plasma concentration of afatinib is ~25-fold less than the level of H2O2 estimated in cancer cells (~2 μM) and ostensibly lower at the site of action, leading to a reactivity differential of 75-fold or less. If new EGFR is resynthesized in the presence of afatinib and H2O2 [depending upon cell type, the half-life of EGFR is 8–24 h (Sorkin and Duex, 2010)] it is expected that alkyation of C797 would predominate. However, this analysis does not take into account the low specific chemical reactivity between afatinib and C797. In other words, afatinib may bind to EGFR with high reversible binding affinity, but the ensuing Michael reaction is extremely slow. In this scenario, H2O2 could oxidize C797 in the noncovalent EGFR•drug complex and the competition between afatinib and H2O2 modification could be less than or equal to 10:1 (kinact ~2 × 10−3 s−1 versus kox ~2 × 10−4 s−1). Interestingly, afatinib has a long terminal elimination half-life (37.2 h at steady-state), supporting a once-daily dose regimen (Wind, et al., 2013). Higher doses are often associated with adverse events, such as diarrhea and rash. Therefore, it is unlikely that the efficacy of afatinib could be increased significantly either through increased dosage or altered frequency.
The susceptibility of EGFR C797 to oxidation and its mutation to serine in NSCLC patients treated with AZD9291 highlight the ongoing need to develop novel strategies and combination therapies to inhibit EGFR signaling. Toward this end, the sulfenic acid modification is a unique functional group in the cellular milieu and may afford a new opportunity in inhibitor design, where nucleophilic “warheads” (Gupta and Carroll, 2016a; Gupta and Carroll, 2016b) are appended to existing or novel inhibitor scaffolds and to target the sulfur atom in its oxidized state. In this approach, the propensity of H2O2-sensitive cysteines in kinases and other proteins of therapeutic interest, like tyrosine phosphatases, can be exploited to develop new classes of inhibitors akin to proof-of-concept studies reported earlier by our group (Garcia and Carroll, 2014; Leonard, et al., 2011).
Finally, kinases containing a cysteine that is structurally homologous to C797, including Her2 and BTK, represent another critical area of future research. Additionally, more than 150 kinases contain a cysteine in or around the nucleotide-binding site, where oxidation has the potential to impact both function and drug pharmacology (Zhang, et al., 2009). Of these 150 kinases, in recent proteomic studies, we have mapped redox-active cysteine residues in GAK, MAP2K1,3,4, PBK, PRK, ZAK (Yang, et al., 2014) and FAK, LIM, and NEK (Gould, et al., 2015). Given the findings of this work, our recent proteomic analyses, and the clinical importance of this class of enzymes, it remains a high priority to delineate the full scope of the redox-modified and redox-regulated kinomes.
SIGNIFICANCE
Hydrogen peroxide is generated as a signaling molecule in a wide variety of signal transduction pathways and is intimately involved in cell fate decisions. The mechanism of “redox signaling” includes H2O2-mediated oxidation of reactive cysteines to rapidly and reversibly alter protein activity, stability, localization and interactions. This study provides, for the first time, a molecular understanding of how cysteine sulfenylation (Cys-SOH) can augment protein kinase activity, as well as impact inhibitor pharamacology in cells. We expect that this work will lay the foundation to further elucidate redox-regulatory mechanisms in the kineome, and to develop a new class of therapeutics that target cysteine residues in their sulfenylated form.
EXPERIMENTAL PROCEDURES
General Reagents and Methods
EGF (BD Biosciences) and H2O2 (Sigma) were prepared in ddH2O. DYn-2 (≥99% purity) was synthesized as previously described and prepared in DMSO (250 mM) (Paulsen, et al., 2012). Dimedone (250 mM, Sigma) was prepared in 70% DMSO/30% Bis-Tris (500 mM, pH 7.4). Catalase (20,000 U/ml, Sigma) and glucose oxidase (500 U/ml, Sigma) were prepared fresh in 50 mM Tris-HCl pH 7.4. Azide biotin (5 mM, Invitrogen) and TAMRA azide (5 mM, Invitrogen) were prepared in DMSO. BTTP (100 mM, Kerafast) was prepared in DMSO, and stock dilutions were prepared with ddH2O. Sodium L-ascorbate (Sigma) and CuSO4 (Sigma) were prepared fresh in ddH2O. Afatinib (10 mM, ChemieTek, ≥99% purity) and PD168393 (10 mM, Santa Cruz Biotechnology, ≥95% purity) stocks were prepared in DMSO.
Cell Culture
Cell lines (American Type Culture Collection) were maintained at 37 °C in a 5% CO2 humidified atmosphere. A431, and HeLa cells were cultured in DMEM (Invitrogen) containing 10% FBS (Invitrogen), 1% GlutaMax (Invitrogen), 1% MEM nonessential amino acids (Invitrogen), and 1% penicillin-streptomycin (Invitrogen). A549, NCI-H1975, and HCC827 cells were cultured in RPMI 1640 with glutamine (Invitrogen) containing 10% FBS, 1% MEM nonessential amino acids, and 1% penicillin-streptomycin. For EGF and H2O2 treatment, cells were washed with PBS and placed in culture media without serum (serum-starved) for 16 h. Afterwards, cells were treated with EGF or H2O2. Treatment was stopped by removing the media and washing with PBS. For glucose oxidase treatment, cells were washed with PBS and serum-starved for 16 h. Afterwards, cells were treated with glucose oxidase to simulate chronic H2O2 stress. Subsequently, cells were pre-incubated with inhibitor, washed with PBS, and stimulated with EGF. For expression of EGFR or mutant kinases, HeLa cells were transfected with EGFR constructs or an empty vector using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. For EGF and H2O2 treatment, cells were washed with PBS after transfection and serum-starved for 2 h. Cells were treated with EGF or H2O2 as described previously.
In Vitro EGFR Autophosphorylation Assay
A431 cells were serum-starved for 2 h, washed with cold PBS (2X) and lysed in RIPA B buffer as described below. Native full-length EGFR was immunoprecipitated from 1000 μg (1 mg/ml) with goat anti-EGFR conjugated agarose (40 μl) for 30 min at 4 °C with rocking. Resin was collected by centrifugation, washed with cold RIPA B buffer (3X), and resuspended in 40 mM HEPES, pH 7.4 (100 μl). EGFR autophosphorylation was assayed from 50 μg lysate in 10 μl reactions containing 20 mM HEPES pH 7.4, 6 mM MnCl2, 100 μM NaVO4 (New England Biolabs), 1 μM ATP (Promega) and supplemented with EGF (3.3 ng/μl) or H2O2 (0–100 μM). EGF-treated reactions were initiated by the addition of immunoprecipitated EGFR and incubated on ice for 5 min. For H2O2-treated reactions, immunoprecipitated EGFR was pre-incubated with H2O2 for 5 min on ice, prior to addition of ATP and an additional incubation on ice for 5 min. To assess the reversibility of H2O2-induced EGFR kinase activation, immunoprecipitates were pre-incubated +/− H2O2 as previously stated followed by addition of DTT (1 mM) simultaneous with ATP and EGF (as appropriate) for an additional 5 min on ice. To verify autophosphorylation was due to enhanced kinase activity, immunoprecipitates were pre-incubated with afatinib (0 or 1 μM) on ice for 5 min prior to additional incubation with ATP and EGF for 5 min on ice. In all cases, reactions were quenched with 5 μl LDS sample buffer and bound proteins were eluted by boiling, resolved by SDS-PAGE, and analyzed by Western blot.
Supplementary Material
HIGHLIGHTS.
The EGFR active-site cysteine (C797) is a target of NADPH oxidase-derived H2O2.
Unique biochemical properties distinguish C797 within the cysteine proteome.
C797 sulfenylation allows new electrostatic interactions with the catalytic loop.
Chronic oxidative stress yields an EGFR population that is refractory to afatinib.
Acknowledgments
We thank Crystal Yan for technical assistance. This work was supported by the National Institute of Health grants GM102187 (KSC), CA174986 (KSC), and R01 GM108911 (AS and PMUU) and in part through computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai (PMUU and AS).
Footnotes
AUTHOR CONTRIBUTIONS
THT performed cell culture experiments. THT and CEP performed kinetic assays. PBP performed the pKa determination and H2O2 reactivity experiments. PMUU performed molecular modeling and MD simulations. All authors designed experimental strategies. THT and KSC wrote the paper with input from all co-authors.
Supplemental information including additional experimental procedures, five figures and three tables and can be found with article online at http://di.doi.org.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson TA, Sauer RT. Role of an N(cap) residue in determining the stability and operator-binding affinity of Arc repressor. Biophys Chem. 2003;100:341–350. doi: 10.1016/s0301-4622(02)00291-0. [DOI] [PubMed] [Google Scholar]
- Antunes F, Cadenas E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000;475:121–126. doi: 10.1016/s0014-5793(00)01638-0. [DOI] [PubMed] [Google Scholar]
- Askoxylakis V, Millonig G, Wirkner U, Schwager C, Rana S, Altmann A, Haberkorn U, Debus J, Mueller S, Huber PE. Investigation of tumor hypoxia using a two-enzyme system for in vitro generation of oxygen deficiency. Radiat Oncol. 2011;6:35. doi: 10.1186/1748-717X-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Bogdan S, Klambt C. Epidermal growth factor receptor signaling. Curr Biol. 2001;11:R292–295. doi: 10.1016/s0960-9822(01)00167-1. [DOI] [PubMed] [Google Scholar]
- Brewer TF, Garcia FJ, Onak CS, Carroll KS, Chang CJ. Chemical Approaches to Discovery and Study of Sources and Targets of Hydrogen Peroxide Redox Signaling Through NADPH Oxidase Proteins. Ann Rev Biochem. 2015;84:765–790. doi: 10.1146/annurev-biochem-060614-034018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, Pierce KA, Laznik-Bogoslavski D, Vetrivelan R, Clish CB, et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature. 2016 doi: 10.1038/nature17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Carpenter G, King L., Jr Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. J Biol Chem. 1980;255:4834–4842. [PubMed] [Google Scholar]
- Corcoran A, Cotter TG. Redox regulation of protein kinases. FEBS J. 2013;280:1944–1965. doi: 10.1111/febs.12224. [DOI] [PubMed] [Google Scholar]
- Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotsey EY, Jung KM, Basit A, Wei D, Daglian J, Vacondio F, Armirotti A, Mor M, Piomelli D. Peroxide-Dependent MGL Sulfenylation Regulates 2-AG-Mediated Endocannabinoid Signaling in Brain Neurons. Chem Biol. 2015;22:619–628. doi: 10.1016/j.chembiol.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. From sulfenylation to sulfhydration: what a thiolate needs to tolerate. Sci Signal. 2012;5:pe10. doi: 10.1126/scisignal.2002943. [DOI] [PubMed] [Google Scholar]
- Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comp Chem. 1998;19:319–333. [Google Scholar]
- Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc Natl Acad Sci U S A. 1998;95:12022–12027. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajiwala KS, Feng J, Ferre R, Ryan K, Brodsky O, Weinrich S, Kath JC, Stewart A. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure. 2013;21:209–219. doi: 10.1016/j.str.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Garcia FJ, Carroll KS. Redox-based probes as tools to monitor oxidized protein tyrosine phosphatases in living cells. Eur J Med Chem. 2014;88:28–33. doi: 10.1016/j.ejmech.2014.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould NS, Evans P, Martinez-Acedo P, Marino SM, Gladyshev VN, Carroll KS, Ischiropoulos H. Site-Specific Proteomic Mapping Identifies Selectively Modified Regulatory Cysteine Residues in Functionally Distinct Protein Networks. Chem Biol. 2015;22:965–975. doi: 10.1016/j.chembiol.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Carroll KS. Profiling the Reactivity of Cyclic C-Nucleophiles towards Electrophilic Sulfur in Cysteine Sulfenic Acid Chem. Sci. 2016a;7:400–415. doi: 10.1039/c5sc02569a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Carroll KS. Rational design of reversible and irreversible cysteine sulfenic acid-targeted linear C-nucleophiles. Chem Commun (Camb) 2016b;52:3414–3417. doi: 10.1039/c6cc00228e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta V, Carroll KS. Sulfenic acid chemistry, detection and cellular lifetime. Biochim Biophys Acta. 2014;1840:847–875. doi: 10.1016/j.bbagen.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK. Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell. 2011;147:185–198. doi: 10.1016/j.cell.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner DE, van der Vliet A. Redox-dependent regulation of epidermal growth factor receptor signaling. Redox Biol. 2015;8:24–27. doi: 10.1016/j.redox.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- Hubbard SR. EGF receptor inhibition: attacks on multiple fronts. Cancer cell. 2005;7:287–288. doi: 10.1016/j.ccr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Johnson BE, Janne PA. Epidermal growth factor receptor mutations in patients with non-small cell lung cancer. Cancer Res. 2005;65:7525–7529. doi: 10.1158/0008-5472.CAN-05-1257. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Leonard SE, Garcia FJ, Goodsell DS, Carroll KS. Redox-based probes for protein tyrosine phosphatases. Angew Chem Int Ed Engl. 2011;50:4423–4427. doi: 10.1002/anie.201007871. [DOI] [PubMed] [Google Scholar]
- Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Conte M, Carroll KS. Chemoselective ligation of sulfinic acids with aryl-nitroso compounds. Angew Chem Int Ed Engl. 2012;51:6502–6505. doi: 10.1002/anie.201201812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Conte M, Carroll KS. The redox biochemistry of protein sulfenylation and sulfinylation. J Biol Chem. 2013;288:26480–26488. doi: 10.1074/jbc.R113.467738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Conte M, Lin J, Wilson MA, Carroll KS. A Chemical Approach for the Detection of Protein Sulfinylation. ACS Chem Biol. 2015;10:1825–1830. doi: 10.1021/acschembio.5b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MJ, Tucci MA, Benghuzzi HA. Human gingival fibroblast cell response to gox/cat. Biomed Sci Instrum. 2012;48:268–274. [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- Miller EW, Tulyathan O, Isacoff EY, Chang CJ. Molecular imaging of hydrogen peroxide produced for cell signaling. Nat Chem Biol. 2007;3:263–267. doi: 10.1038/nchembio871. [DOI] [PubMed] [Google Scholar]
- Millonig G, Hegedusch S, Becker L, Seitz HK, Schuppan D, Mueller S. Hypoxia-inducible factor 1 alpha under rapid enzymatic hypoxia: cells sense decrements of oxygen but not hypoxia per se. Free Radic Biol Med. 2009;46:182–191. doi: 10.1016/j.freeradbiomed.2008.09.043. [DOI] [PubMed] [Google Scholar]
- Miranda JJ. Position-dependent interactions between cysteine residues and the helix dipole. Protein Sci. 2003;12:73–81. doi: 10.1110/ps.0224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina NM, Tyurin-Kuzmin PA, Markvicheva KN, Vorotnikov AV, Tkachuk VA, Laketa V, Schultz C, Lukyanov S, Belousov VV. Does cellular hydrogen peroxide diffuse or act locally? Antioxid. Redox Signal. 2011;14:1–7. doi: 10.1089/ars.2010.3539. [DOI] [PubMed] [Google Scholar]
- Mueller S, Millonig G, Waite GN. The GOX/CAT system: a novel enzymatic method to independently control hydrogen peroxide and hypoxia in cell culture. Adv Med Sci. 2009;54:121–135. doi: 10.2478/v10039-009-0042-3. [DOI] [PubMed] [Google Scholar]
- Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2012;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penel S, Hughes E, Doig AJ. Side-chain structures in the first turn of the alpha-helix. J Mol Biol. 1999;287:127–143. doi: 10.1006/jmbi.1998.2549. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Schwartz PA, Kuzmic P, Solowiej J, Bergqvist S, Bolanos B, Almaden C, Nagata A, Ryan K, Feng J, Dalvie D, et al. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc Natl Acad Sci U S A. 2014;111:173–178. doi: 10.1073/pnas.1313733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawver LK, Slamon D, Ullrich A. Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer cell. 2002;1:117–123. doi: 10.1016/s1535-6108(02)00039-9. [DOI] [PubMed] [Google Scholar]
- Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem. 2014;289:8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Dobrusin EM, Fry DW, Haske T, Whitty A, McNamara DJ. Structure-based design of a potent, selective, and irreversible inhibitor of the catalytic domain of the erbB receptor subfamily of protein tyrosine kinases. J Med Chem. 1997;40:1130–1135. doi: 10.1021/jm960380s. [DOI] [PubMed] [Google Scholar]
- Singh J, Petter RC, Kluge AF. Targeted covalent drugs of the kinase family. Curr Opin Chem Biol. 2010;14:475–480. doi: 10.1016/j.cbpa.2010.06.168. [DOI] [PubMed] [Google Scholar]
- Sorkin A, Duex JE. Quantitative analysis of endocytosis and turnover of epidermal growth factor (EGF) and EGF receptor. Curr Protoc Cell Biol. 2010;Chapter 15(Unit 15):14. doi: 10.1002/0471143030.cb1514s46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz DR, Dornfeld KJ, Krishnan K, Gius D SpringerLink (Online service) Oxidative Stress in Applied Basic Research and Clinical Practice. Springer New York; New York, NY: 2012. Oxidative Stress in Cancer Biology and Therapy. [Google Scholar]
- Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nature medicine. 2015;21:560–562. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong TH, Carroll KS. Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry. 2012;51:9954–9965. doi: 10.1021/bi301441e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong TH, Carroll KS. Redox regulation of protein kinases. Crit Rev Biochem Mol Biol. 2013;48:332–356. doi: 10.3109/10409238.2013.790873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Wind S, Schmid M, Erhardt J, Goeldner RG, Stopfer P. Pharmacokinetics of afatinib, a selective irreversible ErbB family blocker, in patients with advanced solid tumours. Clin Pharmacokinet. 2013;52:1101–1109. doi: 10.1007/s40262-013-0091-4. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013;528:3–25. doi: 10.1016/B978-0-12-405881-1.00001-X. [DOI] [PubMed] [Google Scholar]
- Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422–439. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Yang J, Gupta V, Carroll KS, Liebler DC. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nature Comm. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang S, Yin L, Yang Y, Guan Y, Wang W, Xu H, Tao N. Quantification of epidermal growth factor receptor expression level and binding kinetics on cell surfaces by surface plasmon resonance imaging. Anal Chem. 2015;87:9960–9965. doi: 10.1021/acs.analchem.5b02572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.