

Abstract
Offshore oil production facilities are frequently victims of internal piping corrosion, potentially leading to human and environmental risks and significant economic losses. Microbially influenced corrosion (MIC) is believed to be an important factor in this major problem for the petroleum industry. However, knowledge of the microbial communities and metabolic processes leading to corrosion is still limited. Therefore, the microbial communities from three anaerobic biofilms recovered from the inside of a steel pipe exhibiting high corrosion rates, iron oxide deposits, and substantial amounts of sulfur, which are characteristic of MIC, were analyzed in detail. Bacterial and archaeal community structures were investigated by automated ribosomal intergenic spacer analysis, multigenic (16S rRNA and functional genes) high-throughput Illumina MiSeq sequencing, and quantitative PCR analysis. The microbial community analysis indicated that bacteria, particularly Desulfovibrio species, dominated the biofilm microbial communities. However, other bacteria, such as Pelobacter, Pseudomonas, and Geotoga, as well as various methanogenic archaea, previously detected in oil facilities were also detected. The microbial taxa and functional genes identified suggested that the biofilm communities harbored the potential for a number of different but complementary metabolic processes and that MIC in oil facilities likely involves a range of microbial metabolisms such as sulfate, iron, and elemental sulfur reduction. Furthermore, extreme corrosion leading to leakage and exposure of the biofilms to the external environment modify the microbial community structure by promoting the growth of aerobic hydrocarbon-degrading organisms.
INTRODUCTION
Metal corrosion is a major concern for the oil industry, potentially leading to environmental pollution, safety issues, and major economic losses (1, 2). Offshore oil facilities represent an important biotope for microorganisms with a corrosive metabolism and are thus markedly affected by microbially influenced corrosion (MIC). The anaerobic conditions combined with petroleum hydrocarbons, organosulfur compounds (e.g., benzothiophenes), volatile fatty acids, and other end products of fermentation present in produced oil and water provide significant amounts of carbon substrates for microorganisms within piping and pipeline networks (3). Furthermore, the steel tubes themselves and the seawater initially injected for secondary oil recovery supply abundant electron acceptors like iron and sulfate (4). Moreover, the ambient temperature in oil-handling facilities (15 to 35°C) is also permissive for microbial growth. However, opportunities to study biofilms covering the inner walls of pipe networks of operational oil facilities are scarce; therefore, MIC has been extensively studied in laboratory experiments with metallic coupons or cultures of specific organisms that are potentially involved in MIC (5–8). Sulfidogenic bacteria (reducing sulfate, thiosulfate, and/or sulfur to sulfide) such as some members of the phylum Deltaproteobacteria (5, 6, 9–11), Firmicutes, or Archaeoglobales (12); specific iron-oxidizing microorganisms (5, 6, 13–15); metal-reducing bacteria such as members of the genera Shewanella (16) and Geobacter (17, 18); and acid-producing fermentative organisms have been incriminated as major actors in MIC, and different processes have been described (18, 19). The main mechanisms of MIC are (i) the reduction of iron to iron sulfide through hydrogen sulfide produced by sulfate-reducing bacteria or archaea in a process referred to as chemical MIC (CMIC) (5, 6) and (ii) direct oxidation of iron (Fe0) by specifically adapted lithotrophic microorganisms that withdraw electrons from iron via electroconductive iron sulfide in a process referred to as electrical MIC (EMIC) (6, 20). Because of the frequent detection of hydrogenotrophic bacteria and hydrogenase activity in corrosion samples, cathodic depolarization of the metal surface by hydrogen-scavenging bacteria has also been suggested as an important microbial corrosive process (21–23). However, the validity of this model is controversial. Previous studies highlighted that incubation of hydrogen-scavenging bacteria on steel coupons resulted in nonsignificant corrosion activities (24), and on the basis of thermodynamic and kinetic considerations, cathodic hydrogen consumption conflicts with the rapid corrosion rates observed in situ (6, 20). Furthermore, additional metabolisms, such as fermentation and methanogenesis (9, 19, 25), might indirectly increase corrosion through the production of organic acids or syntrophy with corrosive microorganisms (18). Direct reduction of iron might also increase corrosion by removing the Fe(III) oxide coating from metal surfaces (26, 27). However, corrosion cannot be linked to a single microbial species and laboratory studies typically exhibit less severe corrosion than is reported in the field, where corrosion is associated with multispecies biofilms (28–30). Likewise, there is no single corrosive biochemical reaction in biofilms, as demonstrated by metabolically versatile bacteria such as Desulfovibrio species, which can scavenge hydrogen, reduce sulfate to hydrogen sulfide, and/or reduce iron, depending on the environmental conditions (6, 16). However, the composition and activity of microbial communities from corrosive biofilms appear to depend on various factors like the temperature (12, 30) and the availability of carbon substrates and electron acceptors (26). Additionally, under certain conditions, microbial biofilms may have a positive role. Depending on the microbial community composition, biofilm architecture and environmental conditions, microorganisms may inhibit or protect against corrosion in process referred to as MIC inhibition (18, 29, 31), leading to contradictory results. Therefore, accurate characterization of microbial communities associated with corrosion, as well as their metabolic potential, remains fundamental to understanding, anticipating, and preventing MIC.
This study presents a unique opportunity to study corrosive biofilms covering the inner walls of an oil industry production piping. Bacterial and archaeal community composition and abundance were investigated by complementary molecular approaches coupling quantitative PCR (qPCR), ribosomal intergenic spacer analysis, and multigenic DNA next-generation sequencing. To analyze the microbial functions and actors involved in MIC in detail, bacterial and archaeal 16S rRNA gene diversity was complemented by sequencing of the dsrAB and mcrA genes, which code for key enzymes in sulfate reduction (32) and methanogenesis (33).
MATERIALS AND METHODS
Site description and sampling.
In July 2014, corrosion issues were reported in offshore oil facilities in the Gulf of Mexico. After 2 years of service, a pinhole leak was reported in a topside 10.9-mm-thick vertical steel pipe carrying oily seawater (produced water and crude oil) at 25°C and atmospheric pressure, which corresponds to a high corrosion rate of 5.45 mm · year−1. The leak was plugged with silica-based resin (Fig. 1b), and then the tube section containing the leak site was removed and a microbial biofilm was observed on the inner wall of the pipe (Fig. 1a and b). Approximatively 10 cm2 of biofilm was sampled in duplicate from three locations in the pipe section with sterile swabs presoaked in RNAlater (Life Technologies, Carlsbad, CA, USA), (i) a site located at the bottom (6 o'clock) of the tube, (ii) at a weld, and (iii) at the leaking point. The swabs were then shipped to the laboratory at 4°C for analysis. Microscopic observations, energy-dispersive X-ray microanalysis (EDX), and X-ray diffraction (XRD) analysis of the leaking site were performed by the Southwest Research Institute (San Antonio, TX, USA).
FIG 1.

(a) The topside piping section removed for analysis in this study. (b) Closeup view of the inner wall of the tube with the leak site (sealed with white silica) showing corrosion products. The scale is in centimeters. (c) EDX spectrum of loose deposits present in the biofilm. (d) Optical micrograph of pitting at the leak site showing the scoops-inside-scoops morphology often observed in MIC.
DNA extraction and purification.
Three months after sampling, head swabs were aseptically cut in two for independent duplicate DNA extractions. Nucleic acids were extracted with a FastDNA spin kit for soil (MP Biomedicals, Santa Ana, CA, USA) with modifications (34). Because of the small amounts of biomass recovered on the swabs and the occurrence of enzymatic inhibitors, DNA extraction duplicates were pooled and purified with a QIAamp DNA minikit (Qiagen, Hilden, Germany) before PCR amplification. Procedural blanks were subjected to the same extraction and purification procedures and then analyzed in the same way as samples to identify potential contaminants.
qPCR.
Estimates of the relative abundances of Bacteria and Archaea were obtained by qPCR with the Bact1369f/Bact1492r and Arc787f/Arc1059r primer sets, respectively (Table 1). Amplifications were performed in triplicate with a Rotor-Gene Q system (Qiagen, Hilden, Germany) in a final volume of 25 μl with PerfeCTa SYBR green SuperMix (Quanta Bioscience, Gaithersburg, MD, USA), 0.5 μM each primer, and 0.1 ng of template DNA. The qPCR conditions used were 40 cycles of denaturation at 95°C for 15 s and then annealing and extension at 60°C for 60 s. Standard curves were prepared in triplicate with dilutions ranging from 0.001 to 100 nM DNA extracted from Desulfobulbus propionicus (ATCC 33891) and Methanococcoides methylutens (ATCC 33938). The R2 values of standard curves obtained by qPCR were >0.997, and PCR efficiencies were above 92% and 94% for Archaea and Bacteria, respectively. Samples were diluted until the crossing point decreased log linearly with the sample dilution, indicating absence of PCR inhibition. Because of the partial recovery of the biofilms by the sampling technique and DNA purification step, qPCR results were expressed in terms of numbers of 16S rRNA gene copies per nanogram of genomic DNA.
TABLE 1.
PCR primers used for PCR, ARISA, and real-time qPCR
| Primer | Function | Target group | Sequence (5′–3′) | Amplicon size (bp) | Annealing temp (°C) | Primer concn (μM) | Reference |
|---|---|---|---|---|---|---|---|
| ITSf | ARISA | Bacteria | GTC-GTA-ACA-AGG-TAG-CCG-TA | Variable | 55 | 0.5 | 75 |
| ITSreub | GCC-AAG-GCA-TCC-ACC | ||||||
| 934f | ARISA | Archaea | AGG-AAT-TGG-CGG-GGG-AGC-A | Variable | 55 | 0.5 | 76 |
| 71r | TCG-GYG-CCG-AGC-CGA-GCC-ATC-C | ||||||
| BACT1369F | Q-PCR | Bacteria | CGG-TGA-ATA-CGT-TCY-CGG | 142 | 60 | 0.6 | 77 |
| BACT1492R | GGW-TAC-CTT-GTT-ACG-ACT-T | ||||||
| ARC787F | Q-PCR | Archaea | ATT-AGA-TAC-CCS-BGT-AGT-CC | 273 | 60 | 0.5 | 78 |
| ARC1059R | GCC-ATG-CAC-CWC-CTC-T | ||||||
| S-D-Bact-0516-a-S-18 | Sequencing | Bacteria | TGC-CAG-CAG-CCG-CGG-TAA | 420 | 58 | 0.5 | 38 |
| S-D-Bact-0907-a-A-20 | CCG-TCA-ATT-CMT-TTG-AGT-TT | ||||||
| S-D-Arch-0008-b-S-18 | Sequencing | Archaea | TCY-GGT-TGA-TCC-TGS-CGG | 530 | 58 | 0.5 | 38 |
| S-D-Arch-0519-a-A-19 | GGT-DTT-ACC-GCG-GCK-GCT-G | ||||||
| DSR2060f | Sequencing | DsrAB | CAA-CAT-CGT-YCA-YAC-CCA-GGG | 380 | 50 | 0.5 | 79 |
| Dsr4RdegN | GTR-TAR-CAG-TTD-CCR-CA | ||||||
| MLf | Sequencing | McrA | GGT-GGT-GTM-GGA-TTC-ACA-CAR-TAY-GCW-ACA-GC | 550 | 55 | 0.5 | 33 |
| MLr | TTC-ATT-GCR-TAG-TTW-GGR-TAG-TT | ||||||
| Adaptor F | Sequencing | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG | |||||
| Adaptor R | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG |
ARISA.
Automated ribosomal intergenic spacer analysis (ARISA) was carried out for low-resolution assessment of the structure of biofilm microbial communities as previously described (35, 36). ARISA-PCR was performed with the ITSf/ITSreub and 934f/71r primer sets (Table 1), targeting the bacterial and archaeal 16S-23S RNA intergenic regions, respectively. PCRs were carried out in duplicate with AccuPrime Pfx DNA polymerase (Invitrogen, Carlsbad, CA, USA), 0.5 μM each primer, and 1.5 μl of template DNA in a 25-μl reaction volume. The PCR conditions used were 35 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s and extension for 1 min 20 s at 72°C, followed by a final extension step at 72°C for 12 min. A 1-μl sample of each PCR product was analyzed according to the manufacturer's protocol on a DNA 7500 Chip with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Data were recovered and normalized as previously detailed (36) and then analyzed with PAST software (37).
Illumina MiSeq library preparation and sequencing.
To minimize the effect of primer selectivity and recover a larger proportion of the microbial diversity present, 16S rRNA gene PCRs were carried out with two different primer sets, S-D-Bact-0516-a-S-18/S-D-Bact-0907-a-A-20, targeting the V4-V5 regions of the bacterial 16S rRNA genes, and S-D-Arch-0008-b-S-18/S-D-Arch-0519-a-A-19, targeting the V1-V2-V3 regions of the archaeal 16S RNA genes (Table 1) (38). These primer pairs amplify fragments of approximately 420 and 530 bp, respectively. Additionally, the diversity of sulfate reducers and methanogens was investigated by amplification and sequencing of the dissimilatory (bi)sulfite reductase (dsrAB) metabolic gene with the DSR2060f/Dsr4RdegN primers (39) and the methyl coenzyme M reductase mcrA gene with the MLf/MLr primers (33) (Table 1). These primer sets produce PCR products of 380 and 550 bp, respectively, allowing the generation of contigs from the paired-end sequences. MiSeq adaptors (Table 1) were fused to the 5′ regions of the primers. All PCRs were conducted in triplicate with negative controls and AccuPrime Pfx DNA polymerase (Invitrogen, Carlsbad, CA, USA), 0.5 μM each primer, and 1.5 μl of template DNA in a 25-μl reaction volume. For 16S rRNA genes, the PCR conditions comprised 35 cycles of denaturation at 95°C for 30 s, annealing at 52°C for 30 s, and extension for 30 s at 72°C, followed by a final extension step at 72°C for 5 min. dsrAB and mcrA amplifications used the same PCR parameters, except that the annealing temperature was adjusted to 50°C and 55°C for dsrAB and mcrA, respectively. Replicate amplicons were pooled and purified from agarose gels with a Qiagen MinElute gel purification kit (Qiagen, Hilden, Germany). PCR products were indexed with a Nextera XT kit (Illumina Inc., San Diego, CA, USA) according to manufacturer's recommendations. Indexed amplicons were quantified with a Qubit dsDNA HS assay kit (Life Technologies, Carlsbad, CA, USA) and diluted to give an equimolar mix of products at a final concentration of 4 nM for MiSeq library preparation. The DNA library was diluted at 4 pM, and then paired-end Illumina MiSeq sequencing was performed with the Illumina MiSeq v3 kit (Illumina Inc., San Diego, CA, USA), as recommended by the manufacturer, to obtain two 300-bp paired-end sequences.
Data sets were split into reads from individual indexed amplicons in silico by MiSeq reporter software. Reads were assembled into single paired-end sequences, which were curated by Qiime (40). Sequences with low-quality scores or flagged as chimeras by UChime were removed. Alignment and determination of the taxonomic affiliation of the reads were carried out with the Silva release 119 (41), dsrAB (39), and mcrA (42) sequence databases as references.
Nucleotide sequence accession number.
Raw sequences were deposited in the NCBI database under BioProject accession no. PRJNA302156.
RESULTS
Corrosion observations.
After sampling, the surface of the tube section was observed with a light microscope to examine the corrosion morphology and by EDX and XRD analyses. Corrosion morphologies known as “scoops inside scoops” were observed (Fig. 1d), and EDX profiles of cross sections of the deposits indicated substantial quantities of iron (47.18 weight percent), sulfur (16.29 weight percent), chloride (9.33 weight percent), and barium (17.76 weight percent) along the tube inner wall (Fig. 1c). The quantity of sulfur identified by EDX analysis far exceeds the stoichiometric ratio of sulfur present as barium sulfate, indicating that most of the sulfur present occurs as iron sulfide. XRD analysis of the same deposits confirmed the presence of barium sulfate, iron hydroxide/chloride, and iron oxides, which is characteristic of MIC.
Relative abundance of bacterial and archaeal 16S rRNA genes.
The relative abundances of bacteria and archaea in the biofilms were estimated by qPCR. Overall, bacterial 16S rRNA genes were predominant within the biofilms, encompassing 90% of the total 16S rRNA genes at the weld (5.69 × 105 16S rRNA gene copies/ng of DNA) and bottom (1.13 × 106 16S rRNA gene copies/ng of DNA) of the pipe, as well as up to 96% at the leak site (1.51 × 105 16S rRNA gene copies/ng of DNA) (Fig. 2). In contrast, the archaeal abundances were around 1.47 × 104 and 3.43 × 104 16S rRNA gene copies/ng of DNA at the weld and bottom of the tube section and close to 5.73 × 103 16S rRNA gene copies/ng of DNA at the leak site (Fig. 2). Therefore, microbial abundance was 7.5 times lower at the leak site than at the bottom of the pipe. No archaea were detected in the DNA extraction control (blank), while around 5 × 103 bacterial 16S rRNA gene copies/ng were quantified in the blank, indicating a potential overestimation of bacterial abundance of only 0.05 to 2% due to contaminating DNA in DNA extraction kit components.
FIG 2.
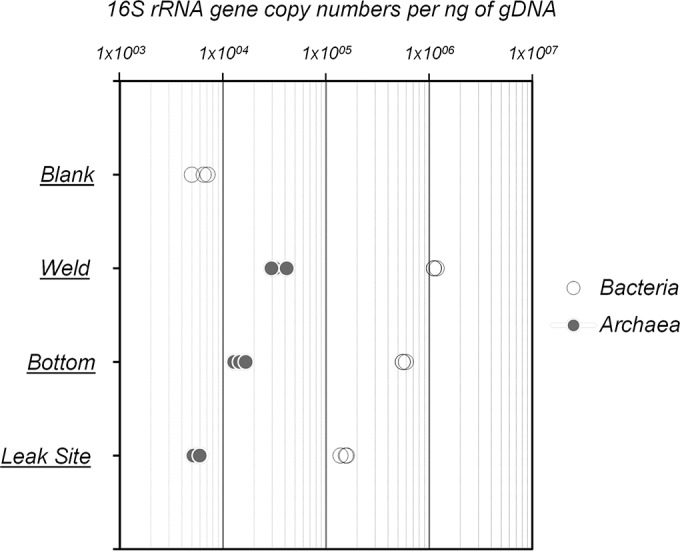
rRNA gene abundances per nanogram of genomic DNA of Bacteria and Archaea from biofilms sampled at the leak site, the bottom, and a weld of the tube. Blank corresponds to the DNA extraction and purification negative control.
Biofilm microbial community composition.
Microbial community composition and diversity within the biofilm were investigated by ARISA fingerprinting and 16S rRNA, mcrA, and dsrAB gene sequencing. No archaeal dsrAB or mcrA sequences were detected in the procedural blank, while the bacterial 16S rRNA sequences detected in the procedural blank were dominated by Burkholderiales-related sequences (90% of the sequences), resulting in a microbial community structure that was extremely different from that recovered from the biofilm samples (Fig. 3a; Tables 2 and 3). Further, sequences detected in the procedural blank (Burkholderia, Enterobacteriaceae) have previously been identified as recurring contamination from DNA extraction kits and are often observed in analysis of samples where DNA concentrations are low (43). Congruently with this, the sample where the highest proportion of potential contaminant sequences was detected had the lowest bacterial and archaeal abundances based on qPCR analysis of 16S rRNA genes. These contaminants represented <2% of the sequences in data generated from the weld and bottom samples and 6% of those from the leak site, indicating that >94% of the sequences originated from the biofilms. Overall, ARISA fingerprinting and 16S rRNA gene sequencing indicated relatively low biodiversity within the bottom and weld samples (Simpson's 1 − D = 0.74; Table 3) and a slightly higher diversity at the leak site (Simpson's 1 − D = 0.82; Table 3). Similarity analyses of both ARISA and 16S rRNA sequence data sets indicated comparable microbial communities in the biofilms sampled at the weld and at the bottom of the tube (Bray-Curtis similarity indices of >0.73; Table 2). In contrast, the microbial profile of the sample from the leak site exhibited a slightly different pattern (Bray-Curtis similarity indices of <0.663; Table 2).
FIG 3.

(a) Phylogenetic affiliations of bacterial 16S rRNA and dsrAB genes at the leak site, the bottom, and a weld of the tube. Shades of blue and green denote gammaproteobacterial and deltaproteobacterial lineages, respectively. (b) Phylogenetic affiliations of archaeal 16S rRNA and mcrA genes at the leak site, the bottom, and a weld of the tube section. Shades of orange denote lineages from the order Methanosarcinales. Blank represents sequences amplified from the DNA extraction and purification negative control. The amount of PCR product obtained from mcrA gene amplification of material from the leak site sample was too small for sequencing.
TABLE 2.
Bray-Curtis similarity indices based on pairwise comparisons of ARISA profiles (lightface) and 16S rRNA gene taxonomic affiliations (boldface)
| 16S rRNA gene | ARISAa |
|||
|---|---|---|---|---|
| Blank | Weld | Bottom | Leak site | |
| Blank | 1 | 0.051 | 0.0438 | 0.147 |
| Weld | 0.01 | 1 | 0.879 | 0.405 |
| Bottom | 0.024 | 0.73 | 1 | 0.429 |
| Leak site | 0.079 | 0.593 | 0.663 | 1 |
Maximum divergence between samples is represented by 0, and perfect similarity is represented by 1.
TABLE 3.
Diversity indices based on 16S rRNA gene taxonomic affiliations based on pairwise comparisons of ARISA profiles and 16S rRNA gene taxonomic affiliations
| Index | Blank | Weld | Bottom | Leak site |
|---|---|---|---|---|
| Simpson's 1 − D | 0.17 | 0.78 | 0.74 | 0.82 |
| Shannon's H | 0.45 | 1.85 | 1.73 | 2.17 |
| EHb | 0.13 | 0.27 | 0.32 | 0.51 |
Maximum divergence between samples is represented by 0, and perfect similarity is represented by 1.
Equitability (evenness).
A total of 140,588 bacterial 16S rRNA gene sequences were obtained from the biofilm samples (see Table S1 in the supplemental material). Bacterial 16S rRNA gene surveys of all of our biofilm samples were dominated mainly by deltaproteobacterial lineages (Desulfovibrio and Pelobacter species; up to 88% of the sequences at the weld) (Fig. 3a). Clostridiales (Acetobacterium and Syntrophomonas), Thermotogales (Geotoga), Gammaproteobacteria (Pseudomonas), and Alphaproteobacteria (Rhodospirillaceae) lineages were also detected as a minority of the sequences in all of the samples. Sequences from organisms related to known aerobic hydrocarbon-degrading gammaproteobacterial lineages (Oceanospirillaceae and Marinobacter, 8.1% of the sequences) were detected only in the leak site sample (Fig. 3a).
A total of 135,341 archaeal 16S rRNA gene sequences were identified (see Table S1 in the supplemental material). The archaeal communities in all of the biofilm samples were composed of various methanogens affiliated with five different orders (Methanobacteriales, Methanococcales, Methanomicrobiales, Methanosarcinales, and Methanomassiliicoccales) (Fig. 3b). 16S rRNA gene sequences indicated that Methanobacterium (∼70%) and Methanolobus (∼20%) species were predominant at the weld and at the bottom of the tube, while sequences from Methanosarcina and Methanomassiliicoccales represented <10% of the total. In contrast, at the leak site, the archaeal community was dominated by Methanobacterium (∼35%), Methanomassiliicoccales (∼35%), and archaea related to Methanosarcina (∼20%) (Fig. 3b).
Functional gene survey.
The 16S rRNA gene inventory was complemented by analysis of dsrAB genes, coding for the dissimilatory (bi)sulfite reductase involved in sulfate reduction, and mcrA genes, coding for a methyl coenzyme M reductase involved in methanogenesis pathways. A total of 396,292 dsrAB sequences were obtained for the biofilm samples (see Table S1 in the supplemental material). Four different (93% sequence identity) dsrAB gene sequences related to dsrAB from Desulfovibrio species were detected, confirming the occurrence of this sulfate-reducing lineage in the biofilms. Sequences from the two dominant dsrAB operational taxonomic units (OTUs) were affiliated with the Desulfovibrio desulfuricans lineage, while sequences from the minority OTUs were distantly related to an uncharacterized Desulfovibrio group. However, no dsrAB sequences affiliated with Desulfobacteraceae and Desulfonauticus were identified, despite their detection in a 16S RNA gene survey of the sample from the leak site (Fig. 3a), probably because of primer specificity (Desulfobacteraceae and Desulfonauticus dsrAB sequences have more than three mismatches with database sequences at the primer target sites).
Consistent with the archaeal 16S rRNA gene survey, mcrA genes were detected by PCR in the three biofilm samples. However, probably because of the lowest abundance of archaea in the leak site sample, amplification of mcrA DNA from this part of the biofilm was too weak to allow sequencing of the amplicons. A total of 74,378 mcrA sequences were recovered from the bottom and weld samples (see Table S1 in the supplemental material). The mcrA sequences recovered were affiliated mainly with Methanolobus vulcani and Methanosarcina barkeri (Fig. 3b). Methanococcales-, Methanomicrobiales-, and Methanomassiliicoccales-related sequences were also identified but in much lower relative abundances (<1%). No Methanobacterium-related sequence was detected because of the poor coverage of the Methanobacteriales lineage by the ML primers (more than seven mismatches with database sequences).
DISCUSSION
In this study, we identified various microbial populations, previously detected in oil facilities (4, 44) and corrosive biofilms (25, 45), potentially involved in MIC.
The microorganisms detected were mainly mesophilic, corresponding to the temperature at these surface facilities (∼25°C) and were previously detected in these environments (44, 46, 47). Dissolved hydrocarbons in this stream and the continuous recycling of the seawater from the sump to the production system might have enriched some of these microorganisms at these locations. ARISA and multigenic sequencing highlighted the similar microbial community compositions of biofilms sampled at the weld and at the bottom of the tube, indicating microbial homogeneity in all parts of the piping section. However, differences in microbial abundance and community composition were observed at the point of leakage. At the leak site, microbial abundance decreased 7.5-fold, whereas bacterial diversity increased (Simpson's 1 − D = 0.82; Table 3). This is probably a consequence of ingress of oxygen to the biofilm at the leak site by connection with the external environment. Indeed, the presence of oxygen would be toxic for most of the anaerobic lineages detected elsewhere in the other biofilms, leading to a decrease in microbial abundance. Oxygen inputs would also explain the higher diversity detected since lineages identified specifically at the leak site by 16S rRNA gene sequencing (Marinobacter, Oceanospirillaceae) are affiliated with gammaproteobacterial aerobic hydrocarbon degraders (48, 49). Finally, differential sensitivity to oxygen might also explain the modification of the methanogenic archaeal community. The opening to the external environment offered by the leak site appeared to change the microbial community composition by modifying the availability of electron acceptors and promoting aerobic metabolism. Intrusion of oxygen might also exacerbate the corrosion due to the chemical attack of oxygen on the steel (29, 50), potentially leading to the iron oxides detected by EDX and secondary oxidation of iron sulfides to highly corrosive elemental sulfur (51).
Potentially corrosive microorganisms.
Since they have been detected in association with highly corrosive biofilms, all of the microorganisms found in this study might play a direct or indirect a role in corrosion. The detection of various iron oxides and corrosion products (9, 20) and the high corrosion rate observed in the piping section (5.45 mm · year−1), which far exceeds the highest reported in vitro corrosion rates reported for single bacterial strains (up to 0.88 mm · year−1 for Desulfobulbus sp. strain IS6 [52]), suggest that the mechanisms underlying MIC are diverse and may interact (Fig. 4). In these biofilms, Bacteria, representing more than 98% of the microbial population estimated by qPCR, particularly Desulfovibrio species, detected as highly predominant on the basis of both bacterial 16S rRNA and dsrAB gene analyses, were likely the main culprits of MIC, as has been proposed previously (16, 53, 54). Desulfovibrio species are metabolically diverse and often versatile, coupling hydrogen or volatile fatty acid consumption with sulfate, iron, or nitrate reduction according to electron acceptor availability or alcohol fermentation in the absence of electron acceptors (55). Different mechanisms explaining the corrosion activity of Desulfovibrio spp. have been suggested. Production of corrosive phosphorus compounds through phosphate reduction by Desulfovibrio desulfuricans (56), which is the closest cultivated relative of the organisms identified in the biofilms studied here, was suspected. However, iron phosphide was not detected in the system, and this corrosion process is controversial (9, 57). Desulfovibrio ferrophilus strain IS5 can also exploit iron directly as an electron donor, coupling sulfate reduction to iron oxidation in the EMIC process (6). However, this metabolic capacity is not shared by all Desulfovibrio lineages and appears to be restricted to specific strains. Further, no genetic marker is available to identify this metabolic feature; thus, we cannot exclude the possibility that the Desulfovibrio species detected in our study contains this pathway and some corrosion of the piping may be a result of direct iron oxidation by Desulfovibrio species. Corrosiveness of Desulfovibrio by cathodic hydrogen scavenging was previously investigated in single-species biofilm, but no increase in corrosion was detected in those studies (20, 24). Therefore, since sulfate was present in the piping environment and iron sulfide was detected in the biofilm, the corrosiveness of Desulfovibrio species seems more likely to be due to chemical MIC and production of corrosive sulfide (Fig. 4). Furthermore, in laboratory experiments when H2 concentrations are not limiting, Desulfovibrio desulfuricans can reduce sulfate and iron simultaneously (16). This might suggest that, in proximity to hydrogen-producing fermenters, detected in our 16S rRNA gene survey, some Desulfovibrio bacteria might potentially catalyze the reduction of both sulfate and iron oxide at the same time. This metabolism would remove any iron oxide coating and reexpose steel to corrosive products like H2S, which is simultaneously produced, likely resulting in a significant part of the high corrosion rates observed (Fig. 4).
FIG 4.

Conceptual model (not to scale) of microbial communities and processes involved in MIC of the tube section. Each microbial group was characterized by their potential metabolic functions: fermentation, iron oxidation, iron reduction, sulfate reduction, elemental sulfur reduction, and biofouling.
Desulfovibrio species were not the only hydrogen sulfide-producing bacteria detected in the 16S rRNA gene survey. Indeed, sequences affiliated with Geotoga, a mesophilic genus of the order Thermotogales previously isolated from oil reservoirs (58), were also detected. Geotoga species are fermentative bacteria capable of reducing elemental sulfur to hydrogen sulfide as a “hydrogen sink” (58). Elemental sulfur was detected at the site of corrosion, providing a potential niche for Geotoga in the corroding system. Thereby, like Desulfovibrio species, Geotoga species might contribute to chemical MIC (Fig. 4).
Sequences affiliated with Pelobacter bacteria represented around 10% of the reads in all of the samples, suggesting a potentially important role within the corrosive biofilm. Pelobacter bacteria are commonly present in marine sediments (59), oil reservoirs (12, 46), and microbial fuel cells (60). Cultivated lineages of Pelobacter bacteria are able to grow by fermentation of various hydrocarbon-derived substrates (acetylene, polyethylene glycol, trihydroxybenzenes), generating hydrogen that might be consumed in syntrophic metabolism with hydrogen scavengers such as methanogens or Acetobacterium bacteria (61, 62), both also detected in the corrosion biofilm. However, in the presence of iron oxides, sulfide, and/or elemental sulfur, as detected by EDX microanalysis of the biofilms, Pelobacter bacteria grow by indirect reduction of iron via a cryptic sulfur reduction/oxidation cycle, with Fe(III) reduction being coupled to the chemical oxidation of sulfide to S0 (63, 64). This metabolism might have substantial consequences for steel corrosion. Similarly to Desulfovibrio sulfate and iron coreduction, dissolution of ferric iron from metal surfaces could reexpose the underlying metal to corrosive products such as the simultaneously produced S0, leading to potentially continuous high corrosion rates (Fig. 4). Pseudomonas species were detected as a significant proportion of the bacterial community in all of our samples (15% of the sequences at the bottom of the pipe). Under anaerobic conditions, some Pseudomonas species can reduce iron oxides directly with H2 by using a metabolic pathway involving various cytochromes (65). Therefore, like Pelobacter species, Pseudomonas species might reexpose the piping metal to corrosive products, enhancing corrosion (Fig. 4) (16, 17).
Organic acid-producing members of the order Clostridiales and the genera Acetobacterium and Syntrophomonas together represented 5% of the bacterial 16S rRNA gene sequences recovered. Cultivated Acetobacterium species are chemolithotrophs, scavenging hydrogen in a syntrophic relationship with methanogens such as members of the order Methanomicrobiales or Methanosarcina barkeri (66), both detected in 16S rRNA and mcrA gene surveys, whereas Syntrophomonas bacteria produce organic acids and H2 from the fermentation of long-chain fatty acids (67). Organic acids produced from fermentation, such as lactate, would subsequently supply Desulfovibrio species, which are known to use lactate as an electron donor, leading to sulfide production and corrosion. Alternatively, organic acids might also dissolve iron and therefore enhance corrosion rates (Fig. 4) (18, 19).
Finally, other proteobacterial lineages were detected in all of our biofilms, but their role in corrosion remains unclear. Members of the family Rhodospirillaceae within the class Alphaproteobacteria were previously found in oil facilities and biofilms (15, 68), and like Pseudomonas and Desulfovibrio species (18, 53, 69), these Alphaproteobacteria could contribute to biofilm formation by polysaccharide production, for example (Fig. 4) (68). By creating a confined environment, these bacteria might enhance interspecies interactions leading to corrosion (29).
On the basis of qPCR analysis, archaea represented a minority of the total microbial community. However, they may play an important role in the ecosystem (19, 30). All of the archaea detected in 16S rRNA gene surveys were related to methanogenic lineages belonging to five different orders. Methane is chemically inert toward iron; thus, potential MIC by methanogens relies on different mechanisms. Although Methanobacterium lineages were not detected by mcrA sequencing because of the lack of coverage of the genus Methanobacterium by the mcrA primers, they represented 70% of the archaeal community detected by 16S rRNA gene sequencing. Methanobacterium spp. are hydrogen scavengers and have been frequently detected in corrosive biofilms (6, 25) and in syntrophic consortia with fermentative bacteria (25) (e.g., Pelobacter and Syntrophomonas, also identified in these biofilms) (Fig. 4). However, direct utilization of iron by specific Methanobacterium species has been reported, with corrosion rates of up to 0.37 mm · year−1 (6, 13). This suggests that the predominant archaeal lineage present in the biofilms could contribute to corrosion by EMIC and partially explain the presence of iron oxides subsequently used by the iron reducers detected in the biofilm (Fig. 4).
Organisms related to Methanosarcina barkeri and Methanolobus vulcani were both detected in our 16S rRNA and mcrA gene surveys. M. barkeri is a metabolically versatile methanogen and can grow on hydrogen, methylated compounds, methanol, or acetate (70, 71), while M. vulcani uses only methanol as a growth substrate (72). However, in the presence of elemental sulfur, as detected at the biofilm-metal interface in the corroding piping section, these methanogens, as well as Methanobacterium species, can switch their metabolism to dissimilatory elemental sulfur reduction and produce high concentrations of hydrogen sulfide (73), potentially contributing to CMIC in this way (Fig. 4).
Additionally, sequences related to organisms of the order Methanomassiliicoccales were detected by 16S rRNA and mcrA gene sequencing. Members of this order were previously identified in the digestive tracts of various animals and are known to grow on methanol with hydrogen (74). To our knowledge, this is the first report of Methanomassiliicoccales from an oil-related environment. As a consequence of the moderate temperature, various carbon substrates and hydrogen availability, as well as low levels of oxygen, oil processing and transport facilities might represent a new biotope for some members of the order Methanomassiliicoccales. However, the involvement of these methanogens in EMIC or CMIC remains to be determined.
Conclusion.
Previous investigations of MIC focused on single-species biofilms (6) or incubation of metal coupons (5, 25), and opportunities to study biofilms covering the inner walls of piping and pipelines from operational oil facilities are limited. In this study, the microbial community structure and abundance of steel-corrosive biofilms were investigated in detail, revealing various microbial lineages with different but complementary metabolisms (fermentation and hydrogen consumption) potentially involved in metal corrosion. Sulfate-, elemental sulfur-, and iron-reducing microorganisms were predominant in the biofilms and are likely to have made an important contribution to the high corrosion rate and corrosive products measured in this particular piping section. However, other organisms, by their potential fermentative metabolism or production of exopolysaccharides, might provide essential substrates (hydrogen, volatile fatty acids) and a favorable environment for microbial growth and corrosive activities. However, since most of the microbial lineages identified are metabolically versatile, an integrated study of corrosion chemistry and kinetics by metatranscriptomic analysis might lead to a better understanding of the interaction between microbial activities and redox chemistry involved in MIC.
Supplementary Material
ACKNOWLEDGMENTS
We thank people on the offshore facility for the sampling effort and Shell Global Solutions for logistic support.
This work was supported by Shell Global Solutions.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03842-15.
REFERENCES
- 1.Schmitt G. 2009. Global needs for knowledge dissemination, research, and development in materials deterioration and corrosion control. World Corrosion Organization, New York, NY: http://www.corrosion.org/wco_media/whitepaper.pdf. [Google Scholar]
- 2.Koch GH, Brongers MP, Thompson NG, Virmani YP, Payer JH. 2002. Corrosion cost and preventive strategies in the United States. US Federal Highway Administration, Washington, DC: https://www.nace.org/uploadedFiles/Publications/ccsupp.pdf. [Google Scholar]
- 3.Larter SR, Head IM, Huang H, Bennett B, Jones M, Aplin AC, Murray A, Erdmann M, Wilhelms A, Di Primio R. 2005. Biodegradation, gas destruction and methane generation in deep subsurface petroleum reservoirs: an overview. Pet Geol Conf Ser 6:633–639. [Google Scholar]
- 4.Youssef N, Elshahed MS, McInerney MJ. 2009. Microbial processes in oil fields: culprits, problems, and opportunities. Adv Appl Microbiol 66:141–251. doi: 10.1016/S0065-2164(08)00806-X. [DOI] [PubMed] [Google Scholar]
- 5.Venzlaff H, Enning D, Srinivasan J, Mayrhofer KJJ, Hassel AW, Widdel F, Stratmann M. 2013. Accelerated cathodic reaction in microbial corrosion of iron due to direct electron uptake by sulfate-reducing bacteria. Corros Sci 66:88–96. doi: 10.1016/j.corsci.2012.09.006. [DOI] [Google Scholar]
- 6.Dinh HT, Kuever J, Muszmann M, Hassel AW, Stratmann M, Widdel F. 2004. Iron corrosion by novel anaerobic microorganisms. Nature 427:829–832. doi: 10.1038/nature02321. [DOI] [PubMed] [Google Scholar]
- 7.Pankhania IP, Moosavi AN, Hamilton WA. 1986. Utilization of cathodic hydrogen by Desulfovibrio vulgaris (Hildenborough). J Gen Microbiol 132:3357–3365. [Google Scholar]
- 8.Beese P, Venzlaff H, Srinivasan J, Garrelfs J, Stratmann M, Mayrhofer KJJ. 2013. Monitoring of anaerobic microbially influenced corrosion via electrochemical frequency modulation. Electrochim Acta 105:239–247. doi: 10.1016/j.electacta.2013.04.144. [DOI] [Google Scholar]
- 9.Enning D, Garrelfs J. 2014. Corrosion of iron by sulfate-reducing bacteria: new views of an old problem. Appl Environ Microbiol 80:1226–1236. doi: 10.1128/AEM.02848-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsen J, Rasmussen K, Pedersen H, Sørensen K, Lundgaard T, Skovhus TL. Consortia of MIC bacteria and archaea causing pitting corrosion in top side oil production facilities. NACE International Foundation, Houston, TX. [Google Scholar]
- 11.Almahamedh HH, Williamson C, Spear JR, Mishra B, Olson DL. Identification of microorganisms and their effects on corrosion of carbon steels [sic] pipelines. NACE International Foundation, Houston, TX. [Google Scholar]
- 12.Duncan KE, Gieg LM, Parisi VA, Tanner RS, Tringe SG, Bristow J, Suflita JM. 2009. Biocorrosive thermophilic microbial communities in Alaskan North Slope oil facilities. Environ Sci Technol 43:7977–7984. doi: 10.1021/es9013932. [DOI] [PubMed] [Google Scholar]
- 13.Daniels L, Belay N, Rajagopal BS, Weimer PJ. 1987. Bacterial methanogenesis and growth from CO2 with elemental iron as the sole source of electrons. Science 237:509–511. doi: 10.1126/science.237.4814.509. [DOI] [PubMed] [Google Scholar]
- 14.Kato S, Yumoto I, Kamagata Y. 2015. Isolation of acetogenic bacteria that induce biocorrosion by utilizing metallic iron as the sole electron donor. Appl Environ Microbiol 81:67–73. doi: 10.1128/AEM.02767-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uchiyama T, Ito K, Mori K, Tsurumaru H, Harayama S. 2010. Iron-corroding methanogen isolated from a crude-oil storage tank. Appl Environ Microbiol 76:1783–1788. doi: 10.1128/AEM.00668-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovley DR, Roden EE, Phillips EJP, Woodward JC. 1993. Enzymatic iron and uranium reduction by sulfate-reducing bacteria. Mar Geol 113:41–53. doi: 10.1016/0025-3227(93)90148-O. [DOI] [Google Scholar]
- 17.Lovley DR, Phillips EJP. 1988. Novel mode of microbial energy metabolism: organic carbon oxidation coupled to dissimilatory reduction of iron or manganese. Appl Environ Microbiol 54:1472–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kip N, van Veen JA. 2015. The dual role of microbes in corrosion. ISME J 9:542–551. doi: 10.1038/ismej.2014.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Usher KM, Kaksonen AH, MacLeod ID. 2014. Marine rust tubercles harbour iron corroding archaea and sulphate reducing bacteria. Corros Sci 83:189–197. doi: 10.1016/j.corsci.2014.02.014. [DOI] [Google Scholar]
- 20.Enning D, Venzlaff H, Garrelfs J, Dinh HT, Meyer V, Mayrhofer K, Hassel AW, Stratmann M, Widdel F. 2012. Marine sulfate-reducing bacteria cause serious corrosion of iron under electroconductive biogenic mineral crust. Environ Microbiol 14:1772–1787. doi: 10.1111/j.1462-2920.2012.02778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Booth GH, Tiller AK. 1968. Cathodic characteristics of mild steel in suspensions of sulphate-reducing bacteria. Corros Sci 8:583–600. doi: 10.1016/S0010-938X(68)80094-0. [DOI] [Google Scholar]
- 22.Bryant RD, Jansen W, Boivin J, Laishley EJ, Costerton JW. 1991. Effect of hydrogenase and mixed sulfate-reducing bacterial populations on the corrosion of steel. Appl Environ Microbiol 57:2804–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Windt W, Boon N, Siciliano SD, Verstraete W. 2003. Cell density related H2 consumption in relation to anoxic Fe(0) corrosion and precipitation of corrosion products by Shewanella oneidensis MR-1. Environ Microbiol 5:1192–1202. doi: 10.1046/j.1462-2920.2003.00527.x. [DOI] [PubMed] [Google Scholar]
- 24.Mori K, Tsurumaru H, Harayama S. 2010. Iron corrosion activity of anaerobic hydrogen-consuming microorganisms isolated from oil facilities. J Biosci Bioeng 110:426–430. doi: 10.1016/j.jbiosc.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 25.Zhang T, Fang HHP, Ko BCB. 2003. Methanogen population in a marine biofilm corrosive to mild steel. Appl Microbiol Biotechnol 63:101–106. doi: 10.1007/s00253-003-1396-2. [DOI] [PubMed] [Google Scholar]
- 26.Herrera LK, Videla HA. 2009. Role of iron-reducing bacteria in corrosion and protection of carbon steel. Int Biodeterior Biodegradation 63:891–895. doi: 10.1016/j.ibiod.2009.06.003. [DOI] [Google Scholar]
- 27.Lee A, Newman D. 2003. Microbial iron respiration: impacts on corrosion processes. Appl Microbiol Biotechnol 62:134–139. doi: 10.1007/s00253-003-1314-7. [DOI] [PubMed] [Google Scholar]
- 28.Kan J, Chellamuthu P, Obraztsova A, Moore JE, Nealson KH. 2011. Diverse bacterial groups are associated with corrosive lesions at a Granite Mountain Record Vault (GMRV). J Appl Microbiol 111:329–337. doi: 10.1111/j.1365-2672.2011.05055.x. [DOI] [PubMed] [Google Scholar]
- 29.Videla HA, Herrera LK. 2005. Microbiologically influenced corrosion: looking to the future. Int Microbiol 8:169–180. [PubMed] [Google Scholar]
- 30.Davidova IA, Duncan KE, Perez-Ibarra BM, Suflita JM. 2012. Involvement of thermophilic archaea in the biocorrosion of oil pipelines. Environ Microbiol 14:1762–1771. doi: 10.1111/j.1462-2920.2012.02721.x. [DOI] [PubMed] [Google Scholar]
- 31.Zuo R. 2007. Biofilms: strategies for metal corrosion inhibition employing microorganisms. Appl Microbiol Biotechnol 76:1245–1253. doi: 10.1007/s00253-007-1130-6. [DOI] [PubMed] [Google Scholar]
- 32.Wagner M, Loy A, Klein M, Lee N, Ramsing NB, Stahl DA, Friedrich MW. 2005. Functional marker genes for identification of sulfate-reducing prokaryotes. Methods Enzymol 397:469–489. doi: 10.1016/S0076-6879(05)97029-8. [DOI] [PubMed] [Google Scholar]
- 33.Luton PE, Wayne JM, Sharp RJ, Riley PW. 2002. The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148:3521–3530. doi: 10.1099/00221287-148-11-3521. [DOI] [PubMed] [Google Scholar]
- 34.Webster G, Parkes RJ, Cragg BA, Newberry CJ, Weightman AJ, Fry JC. 2006. Prokaryotic community composition and biogeochemical processes in deep subseafloor sediments from the Peru Margin. FEMS Microbiol Ecol 58:65–85. doi: 10.1111/j.1574-6941.2006.00147.x. [DOI] [PubMed] [Google Scholar]
- 35.Vigneron A, Cruaud P, Pignet P, Caprais J-C, Cambon-Bonavita M-A, Godfroy A, Toffin L. 2013. Archaeal and anaerobic methane oxidizer communities in the Sonora Margin cold seeps, Guaymas Basin (Gulf of California). ISME J 7:1595–1608. doi: 10.1038/ismej.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vigneron A, Cruaud P, Roussel EG, Pignet P, Caprais J-C, Callac N, Ciobanu M-C, Godfroy A, Cragg BA, Parkes JR, Van Nostrand JD, He Z, Zhou J, Toffin L. 2014. Phylogenetic and functional diversity of microbial communities associated with subsurface sediments of the Sonora Margin, Guaymas Basin. PLoS One 9:e104427. doi: 10.1371/journal.pone.0104427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammer Ø, Harper DAT, Ryan PD. 2001. PAST: palaeontological statistics software package for education and data analysis. Palaeontological Association, Oxford, United Kingdom: http://palaeo-electronica.org/2001_1/past/issue1_01.htm. [Google Scholar]
- 38.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. 2013. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Müller AL, Kjeldsen KU, Rattei T, Pester M, Loy A. 2015. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J 9:1152–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41(Database issue):D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang S, Liebner S, Alawi M, Ebenhöh O, Wagner D. 2014. Taxonomic database and cut-off value for processing mcrA gene 454 pyrosequencing data by MOTHUR. J Microbiol Methods 103:3–5. doi: 10.1016/j.mimet.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 43.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW. 2014. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gieg L, Jack T, Foght J. 2011. Biological souring and mitigation in oil reservoirs. Appl Microbiol Biotechnol 92:263–282. doi: 10.1007/s00253-011-3542-6. [DOI] [PubMed] [Google Scholar]
- 45.Zhang T, Fang H. 2001. Phylogenetic diversity of a SRB-rich marine biofilm. Appl Microbiol Biotechnol 57:437–440. doi: 10.1007/s002530100770. [DOI] [PubMed] [Google Scholar]
- 46.Hubert CRJ, Oldenburg TBP, Fustic M, Gray ND, Larter SR, Penn K, Rowan AK, Seshadri R, Sherry A, Swainsbury R, Voordouw G, Voordouw JK, Head IM. 2012. Massive dominance of Epsilonproteobacteria in formation waters from a Canadian oil sands reservoir containing severely biodegraded oil. Environ Microbiol 14:387–404. doi: 10.1111/j.1462-2920.2011.02521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gittel A, Sørensen KB, Skovhus TL, Ingvorsen K, Schramm A. 2009. Prokaryotic community structure and sulfate reducer activity in water from high-temperature oil reservoirs with and without nitrate treatment. Appl Environ Microbiol 75:7086–7096. doi: 10.1128/AEM.01123-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berlendis S, Cayol J-L, Verhé F, Laveau S, Tholozan J-L, Ollivier B, Auria R. 2010. First evidence of aerobic biodegradation of BTEX compounds by pure cultures of Marinobacter. Appl Biochem Biotechnol 160:1992–1999. doi: 10.1007/s12010-009-8746-1. [DOI] [PubMed] [Google Scholar]
- 49.Satomi M, Fujii T. 2014. The family Oceanospirillaceae, p 491–527. In Rosenberg E, DeLong E, Lory S, Stackebrandt E, Thompson F (ed), The prokaryotes. Springer, Berlin, Germany. [Google Scholar]
- 50.Huang YH, Zhang TC. 2005. Effects of dissolved oxygen on formation of corrosion products and concomitant oxygen and nitrate reduction in zero-valent iron systems with or without aqueous Fe2+. Water Res 39:1751–1760. doi: 10.1016/j.watres.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 51.Lee W, Lewandowski Z, Nielsen PH, Hamilton WA. 1995. Role of sulfate-reducing bacteria in corrosion of mild steel: a review. Biofouling 8:165–194. doi: 10.1080/08927019509378271. [DOI] [Google Scholar]
- 52.Enning D. 2012. Bioelectrical corrosion of iron by lithotrophic sulfate-reducing bacteria. Ph.D. dissertation. University of Bremen, Bremen, Germany: http://elib.suub.uni-bremen.de/edocs/00102721-1.pdf. [Google Scholar]
- 53.Ilhan-Sungur E, Cansever N, Cotuk A. 2007. Microbial corrosion of galvanized steel by a freshwater strain of sulphate reducing bacteria (Desulfovibrio sp.). Corros Sci 49:1097–1109. doi: 10.1016/j.corsci.2006.05.050. [DOI] [Google Scholar]
- 54.Miranda E, Bethencourt M, Botana FJ, Cano MJ, Sánchez-Amaya JM, Corzo A, de Lomas JG, Fardeau ML, Ollivier B. 2006. Biocorrosion of carbon steel alloys by an hydrogenotrophic sulfate-reducing bacterium Desulfovibrio capillatus isolated from a Mexican oil field separator. Corros Sci 48:2417–2431. doi: 10.1016/j.corsci.2005.09.005. [DOI] [Google Scholar]
- 55.Bryant MP, Campbell LL, Reddy CA, Crabill MR. 1977. Growth of Desulfovibrio in lactate or ethanol media low in sulfate in association with H2-utilizing methanogenic bacteria. Appl Environ Microbiol 33:1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iverson WP. 1968. Corrosion of iron and formation of iron phosphide by Desulfovibrio desulfuricans. Nature 217:1265–1267. doi: 10.1038/2171265a0. [DOI] [PubMed] [Google Scholar]
- 57.Beech IB, Sunner JA. 2007. Sulphate-reducing bacteria and their role in corrosion of ferrous materials, p 459–482. In Barton LL, Hamilton WA (), Sulphate-reducing bacteria. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 58.Davey ME, Wood WA, Key R, Nakamura K, Stahl DA. 1993. Isolation of three species of Geotoga and Petrotoga: two new genera, representing a new lineage in the bacterial line of descent distantly related to the “Thermotogales.” Syst Appl Microbiol 16:191–200. [Google Scholar]
- 59.Bowman JP, Rea SM, McCammon SA, McMeekin TA. 2000. Diversity and community structure within anoxic sediment from marine salinity meromictic lakes and a coastal meromictic marine basin, Vestfold Hills, Eastern Antarctica. Environ Microbiol 2:227–237. doi: 10.1046/j.1462-2920.2000.00097.x. [DOI] [PubMed] [Google Scholar]
- 60.Richter H, Lanthier M, Nevin KP, Lovley DR. 2007. Lack of electricity production by Pelobacter carbinolicus indicates that the capacity for Fe(III) oxide reduction does not necessarily confer electron transfer ability to fuel cell anodes. Appl Environ Microbiol 73:5347–5353. doi: 10.1128/AEM.00804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schink B, Pfennig N. 1982. Fermentation of trihydroxybenzenes by Pelobacter acidigallici gen. nov. sp. nov., a new strictly anaerobic, non-sporeforming bacterium. Arch Microbiol 133:195–201. doi: 10.1007/BF00415000. [DOI] [Google Scholar]
- 62.Stackebrandt E, Wehmeyer U, Schink B. 1989. The phylogenetic status of Pelobacter acidigallici, Pelobacter venetianus, and Pelobacter carbinolicus. Syst Appl Microbiol 11:257–260. doi: 10.1016/S0723-2020(89)80022-0. [DOI] [Google Scholar]
- 63.Lovley DR, Phillips EJ, Lonergan DJ, Widman PK. 1995. Fe(III) and S0 reduction by Pelobacter carbinolicus. Appl Environ Microbiol 61:2132–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haveman SA, DiDonato RJ, Villanueva L, Shelobolina ES, Postier BL, Xu B, Liu A, Lovley DR. 2008. Genome-wide gene expression patterns and growth requirements suggest that Pelobacter carbinolicus reduces Fe(III) indirectly via sulfide production. Appl Environ Microbiol 74:4277–4284. doi: 10.1128/AEM.02901-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Obuekwe CO, Westlake DWS. 1982. Effects of medium composition on cell pigmentation, cytochrome content, and ferric iron reduction in a Pseudomonas sp. isolated from crude oil. Can J Microbiol 28:989–992. doi: 10.1139/m82-148. [DOI] [PubMed] [Google Scholar]
- 66.Winter J, Wolfe R. 1980. Methane formation from fructose by syntrophic associations of Acetobacterium woodii and different strains of methanogens. Arch Microbiol 124:73–79. doi: 10.1007/BF00407031. [DOI] [PubMed] [Google Scholar]
- 67.McInerney MJ, Bryant MP, Hespell RB, Costerton JW. 1981. Syntrophomonas wolfei gen. nov., sp. nov., an anaerobic, syntrophic, fatty acid-oxidizing bacterium. Appl Environ Microbiol 41:1029–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elifantz H, Horn G, Ayon M, Cohen Y, Minz D. 2013. Rhodobacteraceae are the key members of the microbial community of the initial biofilm formed in eastern Mediterranean coastal seawater. FEMS Microbiol Ecol 85:348–357. doi: 10.1111/1574-6941.12122. [DOI] [PubMed] [Google Scholar]
- 69.Stadler R, Wei L, Fürbeth W, Grooters M, Kuklinski A. 2010. Influence of bacterial exopolymers on cell adhesion of Desulfovibrio vulgaris on high alloyed steel: corrosion inhibition by extracellular polymeric substances (EPS). Mater Corros 61:1008–1016. doi: 10.1002/maco.201005819. [DOI] [Google Scholar]
- 70.Krzycki JA, Kenealy WR, DeNiro MJ, Zeikus JG. 1987. Stable carbon isotope fractionation by Methanosarcina barkeri during methanogenesis from acetate, methanol, or carbon dioxide-hydrogen. Appl Environ Microbiol 53:2597–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hippe H, Caspari D, Fiebig K, Gottschalk G. 1979. Utilization of trimethylamine and other N-methyl compounds for growth and methane formation by Methanosarcina barkeri. Proc Natl Acad Sci U S A 76:494–498. doi: 10.1073/pnas.76.1.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kadam PC, Boone DR. 1995. Physiological characterization and emended description of Methanolobus vulcani. Int J Syst Bacteriol 45:400–402. doi: 10.1099/00207713-45-2-400. [DOI] [Google Scholar]
- 73.Stetter KO, Gaag G. 1983. Reduction of molecular sulphur by methanogenic bacteria. Nature 305:309–311. doi: 10.1038/305309a0. [DOI] [Google Scholar]
- 74.Dridi B, Fardeau M-L, Ollivier B, Raoult D, Drancourt M. 2012. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int J Syst Evol Microbiol 62:1902–1907. doi: 10.1099/ijs.0.033712-0. [DOI] [PubMed] [Google Scholar]
- 75.Cardinale M, Brusetti L, Quatrini P, Borin S, Puglia AM, Rizzi A, Zanardini E, Sorlini C, Corselli C, Daffonchio D. 2004. Comparison of different primer sets for use in automated ribosomal intergenic spacer analysis of complex bacterial communities. Appl Environ Microbiol 70:6147–6156. doi: 10.1128/AEM.70.10.6147-6156.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Casamayor EO, Massana R, Benlloch S, Ovreas L, Diez B, Goddard VJ, Gasol JM, Joint I, Rodriguez-Valera F, Pedros-Alio C. 2002. Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ Microbiol 4:338–348. doi: 10.1046/j.1462-2920.2002.00297.x. [DOI] [PubMed] [Google Scholar]
- 77.Suzuki MT, Taylor LT, DeLong EF. 2000. Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl Environ Microbiol 66:4605–4614. doi: 10.1128/AEM.66.11.4605-4614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu Y, Lee C, Kim J, Hwang S. 2005. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng 89:670–679. doi: 10.1002/bit.20347. [DOI] [PubMed] [Google Scholar]
- 79.Perry V. 2014. Metabolic activities and diversity of microbial communities associated with anaerobic degradation. Ph.D. dissertation. Georgia State University, Atlanta, GA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.