

ABSTRACT
Cryptococcus neoformans is a human fungal pathogen and a major cause of fungal meningitis in immunocompromised individuals. Treatment options for cryptococcosis are limited. Of the two major antifungal drug classes, azoles are active against C. neoformans but exert a fungistatic effect, necessitating long treatment regimens and leaving open an avenue for emergence of azole resistance. Drugs of the echinocandin class, which target the glucan synthase and are fungicidal against a number of other fungal pathogens, such as Candida species, are ineffective against C. neoformans. Despite the sensitivity of the target enzyme to the drug, the reasons for the innate resistance of C. neoformans to echinocandins remain unknown. To understand the mechanism of echinocandin resistance in C. neoformans, we screened gene disruption and gene deletion libraries for mutants sensitive to the echinocandin-class drug caspofungin and identified a mutation of CDC50, which encodes the β-subunit of membrane lipid flippase. We found that the Cdc50 protein localized to membranes and that its absence led to plasma membrane defects and enhanced caspofungin penetration into the cell, potentially explaining the increased caspofungin sensitivity. Loss of CDC50 also led to hypersensitivity to the azole-class drug fluconazole. Interestingly, in addition to functioning in drug resistance, CDC50 was also essential for fungal resistance to macrophage killing and for virulence in a murine model of cryptococcosis. Furthermore, the surface of cdc50Δ cells contained increased levels of phosphatidylserine, which has been proposed to act as a macrophage recognition signal. Together, these results reveal a previously unappreciated role of membrane lipid flippase in C. neoformans drug resistance and virulence.
IMPORTANCE
Cryptococcus neoformans is a fungal pathogen that is the most common cause of fungal meningitis, causing over 620,000 deaths annually. The treatment options for cryptococcosis are very limited. The most commonly used drugs are either fungistatic (azoles) or highly toxic (amphotericin B). Echinocandins are the newest fungicidal drug class that works well in treating candidiasis and aspergillosis, yet they are ineffective in treating cryptococcosis. In this study, we showed that the regulatory subunit of the lipid translocase (flippase), a protein that regulates the asymmetrical orientation of membrane lipids, is required for C. neoformans resistance to caspofungin, as well as for virulence during infection. This discovery identifies lipid flippase as a potential C. neoformans drug target, which plays an important role in the innate resistance of C. neoformans to echinocandins and in fungal virulence.
INTRODUCTION
Cryptococcus neoformans is an opportunistic fungal pathogen that can infect the central nervous system (CNS) in immunocompromised individuals to cause life-threatening cryptococcal meningitis (1, 2). C. neoformans expresses several classical virulence factors, including the ability to grow at body temperature and produce melanin and the polysaccharide capsule. These features protect the fungus against the hostile host environment and help it to evade the host immune response (3, 4). In addition, C. neoformans is a facultative intracellular organism that can survive and proliferate inside macrophages (5). The mechanisms underlying C. neoformans-host interactions and its ability to withstand the antimicrobial activity of macrophages are still very incompletely understood.
Treatment options for cryptococcosis are very limited. Currently, an acute infection is treated with amphotericin B or azoles in combination with flucytosine (6). However, both treatment courses have serious drawbacks. Amphotericin B binds not only fungal but also human sterols and thus causes toxic side effects. Azoles, which specifically target ergosterol biosynthesis, are better tolerated by patients but are fungistatic rather than fungicidal and often require a lifetime regimen of the drug. The fungistatic nature and long-term duration of these regimens increase the probability of the development of drug resistance by C. neoformans (7). Thus, new and more efficacious treatments are urgently needed to combat cryptococcosis.
The therapeutic challenge in developing antifungal agents is that both fungi and their mammalian hosts are eukaryotes and therefore contain similar cellular machinery. One major fungus-specific drug target is the cell wall. Echinocandins are the latest-generation antifungal drug class that targets the cell wall with fungicidal activity against several major fungal pathogens, including Candida and Aspergillus species (8, 9). The target of this new drug class is the β-1,3-glucan synthase, the essential enzyme to produce β-1,3-d-glucan, a major cell wall component. β-1,3-Glucan synthase is encoded by the FKS genes, which were first identified in Saccharomyces cerevisiae (10). In C. albicans, there are three copies of the Fks1 homologs (FKS1/GSC1, GSL1, and GSL2), and FKS1 plays a predominant role (11). In Aspergillus fumigatus and Aspergillus nidulans, a single gene, FKS1, encodes the enzyme activity (12, 13). In C. neoformans, there is a single FKS1 homolog (14). Although this gene is essential for survival in C. neoformans and purified β-glucan synthase from this fungus is strongly inhibited by echinocandin drugs in vitro (15), C. neoformans is naturally resistant to echinocandins, and the mechanism of resistance remains unknown.
To investigate the molecular basis of the inherent resistance of C. neoformans to echinocandins, we performed a high-throughput genetic screen for cryptococcal mutants that are sensitive to caspofungin, a drug of the echinocandin class. After screening over 7,000 mutants from a random mutagenesis library and 3,000 mutants from a gene deletion collection (16), we found that the homolog of the S. cerevisiae CDC50 gene is required for echinocandin resistance in C. neoformans. CDC50 encodes a β-subunit of lipid flippase, which is involved in membrane aminophospholipid translocation, cell surface receptor signal transduction, vacuole organization, and maintenance of the asymmetrical distribution of phospholipids on the bilayer lipid membrane (17). We found that in addition to mediating caspofungin resistance, C. neoformans CDC50 was required for maintaining normal stress resistance and normal development of fungal virulence factors, overcoming the antifungal activity of macrophages, and developing cryptococcosis in the mouse model in vivo. Together, these results implicate the Cdc50-mediated lipid trafficking pathway in antifungal drug resistance and fungal pathogenesis in C. neoformans.
RESULTS
Screening for caspofungin-sensitive mutants.
Using the wild-type strain H99, we generated an Agrobacterium-mediated mutagenesis library with ~7,000 transformants. This library was screened for mutants sensitive to caspofungin at 8 µg/ml in 96-well plates on yeast extract-peptone-dextrose (YPD) medium at 30°C. We identified three transformants (3B1, 5A3, and 11B6) that were sensitive to caspofungin but grew normally in its absence (Fig. 1). To confirm that the caspofungin-sensitive phenotype was linked with nourseothricin (NAT) resistance, the insertion marker, we performed a cosegregation assay by back-crossing these three transformants to the wild-type strain KN99a. In every case, caspofungin resistance segregated with NAT resistance. Next, to identify the sequences flanking the NAT marker and thus the disrupted genes, we used an inverse PCR method (18). Isolate 3B1 had a single insertion in gene ZIT1, which encodes an ion transporter homolog, while 5A3 had a single insertion in HOB1, which encodes an actin-associated protein with roles in endocytosis and exocytosis. The insertion in isolate 6B11 disrupted two adjacent genes: CDC50, which encodes a membrane protein homologous to Cdc50/Lem3 in S. cerevisiae, and MPT1, which encodes a mitogen-activated protein (MAP) kinase phosphatase. To determine which of these two disruptions was responsible for the phenotype in 11B6, we generated gene deletions for both CDC50 and MPT1. We found that only the cdc50Δ null mutant, but not the mpt1Δ mutant, was sensitive to caspofungin (Fig. 1).
FIG 1 .

Identification of genes required for resistance to caspofungin. (A) Cultures of mutants sensitive to caspofungin (CSF) were grown overnight in YPD and diluted to an OD600 of 1.0. Tenfold serial dilutions were prepared, and 5 µl of each was spotted on YPD agar supplemented with 0, 8, or 16 µg/ml caspofungin. (B) Genomic location of T-DNA insertion marker in the mutant 11B6. Arrows indicate the direction of gene transcription. (C) Caspofungin sensitivity of deletion mutants of MPT1 and CDC50 that were identified from 11B6 by inverse PCR. (D) Caspofungin-sensitive mutants identified from the C. neoformans deletion library.
Besides the random mutagenesis library, we also screened two C. neoformans gene deletion collections generated by Hiten Madhani’s group at University of California San Francisco (UCSF) (16). A total of ~3,300 mutants were screened on YPD with 8 µg/ml caspofungin as described above. We identified two mutants (5F6 and 6A3) that were sensitive to caspofungin at this concentration (Fig. 1). Both of them are mutants of genes involved in the late steps of the ergosterol biosynthetic pathway: 5F6 is a null mutant of sterol C-24 reductase (Erg4; CNAG_02830), which catalyzes the last step of the ergosterol biosynthetic pathway, while 6A3 is a null mutant of sterol C-5 desaturase (Erg3; CNAG_00519), also involved in the late steps of the ergosterol biosynthesis. Thus, the two screens identified five genes potentially involved in mediating echinocandin resistance: CDC50, ZIT1, HOB1, ERG4, and ERG3. Among them, strains disrupting the ZIT1 or HOB1 gene also showed slow growth on YPD without drug. Because the cdc50Δ mutant exhibited the most pronounced sensitivity to caspofungin, we focused our further study on this gene.
Cdc50 is required for caspofungin resistance.
To confirm the caspofungin sensitivity of the cdc50Δ mutant in C. neoformans, we deleted the entire CDC50 open reading frame (ORF), replaced it with a NEO marker, and tested the resulting mutant as well as the complemented strain for growth in the presence and absence of caspofungin. We found that the cdc50Δ deletion mutant showed the same caspofungin sensitivity as the original insertion mutant and that drug resistance was restored by reintroducing the CDC50 gene (Fig. 2A). Furthermore, we found that the cdc50Δ mutant was hypersensitive to caspofungin both on agar plates and in liquid medium (Fig. 2A and B). MIC testing showed that the MIC90 of the cdc50Δ mutant was >4-fold lower relative to wild-type strain H99 (4 µg/ml compared to over 16 µg/ml [Table 1]) following a 72-h incubation. Interestingly, the same mutant was also sensitive to another glucan synthase inhibitor, MK-3118, an enfumafungin derivative, but not to anidulafungin and micofungin, two other echinocandin-class drugs (Table 1). To determine whether caspofungin kills the cdc50Δ mutant or only inhibits its growth, we measured the viability of strains grown in liquid medium containing 16 µg/ml of caspofungin. Our results showed that fungal CFU of the cdc50Δ mutant decreased after drug treatment, indicating that caspofungin is fungicidal in the absence of CDC50 (Fig. 2C).
FIG 2 .

The cdc50Δ mutant is hypersensitive to caspofungin (CSF). (A) Cultures of indicated strains were grown overnight in YPD and diluted to an OD600 of 1.0. Tenfold serial dilutions were prepared, and 5 µl of each was spotted on YPD agar supplemented with 0, 4, 8, or 16 µg/ml caspofungin. (B) Of each strain, 1 × 105 cells were inoculated into liquid YPD with or without 16 µg/ml caspofungin and incubated for 24 h at 30°C. Cell density was determined every 30 min by measuring OD600. (C) Survival rate of H99 and cdc50Δ strains on YPD with or without 16 µg/ml caspofungin. Yeast CFU were determined at different time points after incubation by plating onto drug-free medium.
TABLE 1 .
MICs on YPD at 30°C
| Drug | MIC (µg/ml) of strain at 72 h on YPD |
MIC range tested (µg/ml) | |
|---|---|---|---|
| H99 | cdc50Δ mutant | ||
| Anidulafungin | >64 | >64 | 64–0 |
| Caspofungin | 16 | 4 | 64–0 |
| Micafungin | >64 | >64 | 64–0 |
| MK-3118 | 4 | 2 | 64–0 |
| Fluconazole | 16 | 1 | 128–0 |
| Voriconazole | 0.25 | 0.01 | 8–0 |
| Amphotericin B | 2 | 0.5 | 8–0 |
Capsule and melanin are two unique features of cryptococci that are required for fungal virulence. To evaluate whether capsule or melanin is required for fungal resistance to echinocandin drugs, we examined mutants lacking these two virulence factors for their response to caspofungin (Fig. 2A). The cap59Δ mutant does not produce capsule (19), and the lac1Δ mutant shows a significant melanin production defect (20). We also used the pka1Δ mutant, which has a defect on both capsule and melanin production (21, 22). Our results showed that all three mutant strains had levels of drug resistance similar to that of the wild-type H99, indicating that neither capsule nor melanin is required for echinocandin resistance.
Cdc50 prevents caspofungin uptake by C. neoformans.
The Cdc50 that we identified in C. neoformans is the only homolog of an S. cerevisiae CDC50 gene family that contains three members (Cdc50, Lem3, and Crf1) (Fig. 3A). These proteins interact with type IV P-type ATPase and regulate its lipid flippase function, which is necessary to create and maintain an asymmetrical distribution of phospholipids in the plasma membrane as well as the membranes of the late secretory pathway and the endocytic compartments (17, 23, 24). Both Cdc50 and Lem3 in S. cerevisiae are localized to the endoplasmic reticulum (ER) and endosomal and plasma membranes to regulate the translocation of aminophospholipids, such as phosphatidylserine (PS). To determine if Cdc50 in C. neoformans also functions in these processes, we first analyzed its subcellular localization by expressing a Cdc50-green fluorescent protein (GFP) fusion protein under the control of the histone H4 promoter in the cdc50Δ mutant background. The GFP-expressing strain fully complemented the drug sensitivity of the cdc50Δ mutant (Fig. 2). We observed that the Cdc50-GFP fluorescent signal was mostly concentrated in membranes and largely colocalized with ER-Tracker staining, indicating that Cdc50 is localized to the ER membrane and also to the plasma membrane (Fig. 3B). We also treated the cells with caspofungin and observed increased plasma membrane localization after drug treatment, which is consistent with our finding that Cdc50 is required for caspofungin drug resistance in C. neoformans (Fig. 3B).
FIG 3 .

Loss of Cdc50 results in increased caspofungin uptake activity. (A) Phylogenetic analysis of Cdc50 homologs in different eukaryotes. The protein sequences were aligned with CLUSTAL X, and a neighbor-joining tree was generated using a Dayhoff model, 1,000 bootstrap replicates, and a pairwise deletion for gaps/missing data. (B) Fluorescent signal generated by Cdc50-GFP in cells grown on YPD or YPD with 8 µg/ml caspofungin (CSF) was colocalized with ER-Tracker Red. Bar, 5 µm. DIC, differential inference contrast. (C) H99 and cdc50Δ cultures were coincubated with 5 µmol BODIPY-labeled caspofungin for 30 min at 30°C. The fluorescent signal of fungal cells was detected by confocal fluorescence microscopy. (D) Fluorescent signal intensity was quantified using flow cytometry for 100,000 yeast cells of H99, the cdc50Δ mutant, and its complemented strain (cdc50Δ+CDC50), as well as S. cerevisiae strain BY4742 and its lem3Δ and cdc50Δ mutants treated with 5 µM BODIPY-caspofungin for 30 min at 30°C. Triplicates were used for each measurement. (E) Representative images of fluorescent signal quantification results using flow cytometry.
Because loss of CDC50 is expected to perturb membrane function, we hypothesized that the cdc50Δ mutant may exhibit enhanced sensitivity to caspofungin because it alters drug uptake into cells. To test this hypothesis, we monitored drug uptake by using boron dipyrromethene difluoride (BODIPY)-labeled caspofungin (25). We observed that while H99 failed to take up significant amounts of fluorescently labeled caspofungin, producing only limited intracellular fluorescence, the cdc50Δ mutant showed a significantly higher level of fluorescent signal, indicative of an increased intracellular drug concentration (Fig. 3C; see also Fig. S1A in the supplemental material). We quantified this fluorescent signal for ~100,000 cells using flow cytometry and found that the cdc50Δ mutant had a significantly increased BODIPY-caspofungin uptake and that this increase was reversed by complementing the mutant with wild-type CDC50 (Fig. 3D and E). Because the cdc50Δ mutant is more sensitive to caspofungin, we checked that the observed phenotype was not due to increased cell death of cdc50Δ cells in the presence of BODIPY-caspofungin. We measured cell viability after a 30-min BODIPY-caspofungin treatment and found that this short treatment did not result in increased killing of the mutant cells compared to the wild-type strain (see Fig. S1B).
We also examined the potential role of Cdc50 homologs in S. cerevisiae in caspofungin internalization. S. cerevisiae wild-type strain BY4741 and its LEM3 and CDC50 deletion mutant strains (sclem3Δ and sccdc50Δ, respectively) were coincubated with BODIPY-caspofungin as described above, and the fluorescent signal was analyzed by flow cytometry and microscopic observation. Our data showed that the overall signal intensities among the three strains were similar: the fluorescence signals for both sclem3Δ and sccdc50Δ mutants were mostly concentrated on the cell surface, similar to that of the wild type (Fig. 3D and E; see also Fig. S1C in the supplemental material).
Together, these results demonstrate that the cdc50Δ mutant significantly increases the internalization of caspofungin in C. neoformans, suggesting that poor drug uptake in the wild-type fungus may be one mechanism of drug resistance in C. neoformans. Because the S. cerevisiae parental strain BY4741 and its sclem3Δ and sccdc50Δ mutants had similar drug binding activities and drug localizations in the cell, the increased internalization of caspofungin in the cdc50Δ mutant in C. neoformans may be specific to this fungus.
Cdc50 is required for azole resistance.
As fluconazole is the first-line drug to treat cryptococcosis, we also investigated the role of C. neoformans CDC50 in resistance to azole drugs at both 30°C and 37°C. While fluconazole is fungistatic in C. neoformans, we used fluconazole concentrations (2 µg/ml and 4 µg/ml) that were not inhibitory to the growth of the wild-type strain on solid medium. The cdc50Δ mutant showed increased sensitivity to fluconazole at 37°C compared to the wild-type H99, while sensitivity was only moderately increased when the strain was incubated at 30°C (Fig. 4A). When the strains were grown in liquid YPD medium at 37°C in the absence or presence of 8 µg/ml fluconazole, the cdc50Δ mutant also showed much greater growth inhibition than did H99 (Fig. 4B and C). These results show that C. neoformans Cdc50 plays a broader role in fungal drug resistance.
FIG 4 .

The cdc50Δ mutant is hypersensitive to fluconazole treatment. (A) Cultures of the (H99) cdc50Δ mutant and its complemented strains, as well as the CDC50 overexpression strain, were incubated overnight in YPD and diluted to an OD600 of 1.0. Tenfold serial dilutions were prepared and plated on YPD containing 0, 2, and 4 µg/ml fluconazole (FLC) and incubated for 48 h at 30°C and 37°C. (B) Of each indicated strain, 1 × 105 cells were inoculated in liquid YPD with or without 8 µg/ml fluconazole and grown for 24 h at 37°C. The OD600 of each culture was measured every 30 min. (C) Fungal survival rate was determined by CFU for H99 and cdc50Δ strains that were grown in liquid YPD with or without 8 µg/ml fluconazole.
Cdc50 is required for membrane integrity.
Next, we determined whether deleting CDC50 affected cellular resistance to other types of stress, including cell wall and membrane stress, as well as oxidative, nitrosative, and osmotic stresses (Fig. 5A). No obvious change in sensitivity was observed in the presence of agents that disturb the cell wall—calcofluor white (CFW) and Congo red. However, the cdc50Δ cells were more sensitive to SDS, a phenotype that is usually associated with altered cellular membrane integrity. Cdc50 was not required for oxidative stress (3 mM H2O2) or nitrosative stress (1 mM NaNO2) resistance in our assays but did show increased sensitivity to osmotic stress (1.5 M KCl) and alkaline pH (Fig. 5A). Together, these data show that C. neoformans CDC50 is required for maintaining membrane integrity.
FIG 5 .

Cdc50 is involved in stress resistance and is required for normal development of classical C. neoformans virulence factors. (A) Cultures of H99, cdc50Δ, cdc50Δ+CDC50, and CDC50 overexpression (cdc50Δ+PHIS-GFP-CDC50) strains were prepared with serial dilutions and were grown on YPD with 0.3% SDS, 250 µg/ml calcofluor white (CFW), 0.5% Congo red, 3 mM H2O2, 1 mM nitric oxide (NaNO2), or 1.5 M KCl. Cells were also grown on l-DOPA or YPD (pH 8.0) buffered with HEPES. The results were photographed after a 48-h incubation. Their growth on YPD or l-DOPA at 37°C was also determined by serial dilutions. (B) Capsule sizes of the above strains were determined by India ink staining after incubation on DME medium for 3 days at 37°C. Relative capsule size was determined by measuring over 50 cells for each strain. Bar, 10 µm; *, P < 0.01. (C) Electromobility of secreted GXM from H99 and the cdc50Δ mutant was detected in a Western blot assay as described in reference 50. Culture supernatants were collected at 0, 24, 48, and 72 h after inoculation and loaded on an 0.6% certified megabase agarose gel. The gel was transferred to a polyvinylidene difluoride membrane, and the GXM signal was detected using the GXM monoclonal antibody 18B7. (D) Quantification of secreted GXM from H99 and the cdc50Δ mutant. Secreted total polysaccharides were purified from a 500-ml YPD culture of each strain using the CTAB precipitation method as described previously (51). The amount of total GXM was determined by the phenol sulfuric method. Triplicates were used for each experiment. *, P < 0.01.
Cdc50 is required for the development of fungal virulence factors.
To further probe the potential roles of Cdc50 in the pathogenesis of C. neoformans, we investigated whether its major virulence factors—ability to grow at 37°C and production of capsule and melanin—were affected in the cdc50Δ mutant. The cdc50Δ mutant grew normally at 30°C and exhibited a growth defect relative to wild-type and complemented strains at 37°C (Fig. 5A). The mutant also showed normal melanin production at both 30°C and 37°C. Interestingly, deletion of CDC50 resulted in a significantly increased capsule size in cells grown on Dulbecco modified Eagle (DME) medium for 3 days (Fig. 5B). This capsule enlargement in the mutant cells was not observed when fungal cells were grown on noninducing media, such as YPD and yeast nitrogen base (YNB), suggesting that this phenotype is specific to capsule-inducible conditions (see Fig. S2 in the supplemental material). We also measured glucuronoxylomannan (GXM) secretion using two approaches. Using a Western blot assay with the GXM monoclonal antibody 18B7, we detected GXM components secreted into the medium after culturing cells for 0, 24, 48, and 72 h. Our results showed that the wild type secreted more GXM than the mutant, with a somewhat higher average molecular weight (Fig. 5C). We also quantified the amount of GXM secreted by purifying the secreted GXM in the medium supernatants after a 24-h incubation. We also found that the wild type secreted more GXM than the cdc50Δ mutant (Fig. 5D). Together, we conclude that the cdc50Δ mutant exhibited increased capsule size but reduced GXM secretion.
Cdc50 is essential for C. neoformans to overcome the antifungal activity of macrophages.
As it is a facultative intracellular pathogen, a key characteristic of C. neoformans is its ability to survive and replicate within the phagolysosome of macrophages (1, 26, 27). We investigated the potential role of Cdc50 in survival in macrophages. After coincubation of C. neoformans with macrophage cell line J774.16 for 1.5 h, macrophages were fixed with methanol and stained with Giemsa stain and the phagocytosis rate was determined by the number of yeast cells inside each macrophage. Our results showed that when cocultured with the wild-type strain H99, ~80% of macrophages contained yeast cells, with ~1.8 yeast cells on average inside each macrophage. These numbers increased to ~90% and ~4.3, respectively, when the cdc50Δ mutant was coincubated with macrophages, suggesting that Cdc50 helps cell resistance to phagocytosis (Fig. 6A to C). To further test macrophage killing of the fungal cells, C. neoformans and macrophages were coincubated for 2, 4, and 24 h; fungal cells were collected from macrophage cultures; and yeast survival rate was determined by CFU. Our results showed that the cdc50Δ mutant was hypersensitive to macrophage killing (Fig. 6D). To determine whether the killing of the cdc50Δ mutant cells was truly related to the presence of macrophages and not the medium, we cocultured fungal strains with YPD, fresh DME, fresh DME with 10% heat-killed fetal bovine serum (FBS), and the spent medium of J774.16 macrophage culture. Our results clearly showed that none of these culture conditions caused a significant CFU reduction in the cdc50Δ mutant, indicating that the killing of mutant cells is independent of the medium (see Fig. S3 in the supplemental material). These results demonstrate that CDC50 is essential for fungal survival in macrophages.
FIG 6 .

Cdc50 is required for C. neoformans to overcome the antifungal activity of macrophages. (A) The phagocytosis rate of C. neoformans in the J774 macrophage cell line was measured in 48-well plates. PBS-washed C. neoformans H99, cdc50Δ, and cdc50Δ+CDC50 strains were added to the activated macrophages and incubated for 1.5 h at 37°C with 5% CO2. Cells were fixed with methanol and stained with Giemsa stain before counting. The percentage of macrophages containing internalized yeast cells was measured from over 100 macrophages. (B) In the same assay, the number of yeast cells in each macrophage was also calculated under an inverted microscope. The data were generated from counting over 500 macrophages. *, P < 0.01. (C) Typical field views of phagocytosis for each sample. (D) Intracellular proliferation of C. neoformans in the macrophage cell line. Cryptococcal strains were added to the activated macrophages and incubated for 2 h at 37°C with 5% CO2. Nonadherent extracellular yeast cells were then removed by washing with fresh DME medium. Numbers of fungal CFU from macrophage cultures after an additional 0, 2, or 22 h of incubation were used to determine intracellular proliferation and macrophage killing. The error bars indicate standard deviations for three experiments. *, P < 0.01. (E) RT-PCR analysis of CDC50 gene expression under different culture conditions, including growth on YNB, YPD, YPD containing 8 µg/ml caspofungin (CSF), or YPD with 4 µg/ml fluconazole (FLC) and coculture with macrophages. The GAPDH gene was used as a reference. AmB, amphotericin B.
CDC50 is highly expressed during interaction with macrophages.
To determine the expression pattern of the CDC50 gene, we prepared C. neoformans RNAs from a range of culture conditions and measured gene expression using the quantitative real-time reverse transcription-PCR (qRT-PCR) method. Our data showed that the CDC50 gene was highly induced when cocultured with the J774.16 macrophage cell line but that its expression was unaffected under other culture conditions, including treatment with caspofungin, fluconazole, or amphotericin B for 30 min, or under a variety of stress conditions, including SDS treatment and salt stress (Fig. 6E). Thus, CDC50 expression is specifically induced during interaction with macrophages.
Cdc50 regulates phosphatidylserine abundance on the cell surface.
In S. cerevisiae, CDC50 encodes the β-subunit of the lipid flippase that directs the translocation of aminophospholipids, such as phosphatidylserine (PS), to maintain the asymmetry of the bilayer membrane structure (24). Our protein subcellular localization assay showed that C. neoformans Cdc50 localization is similar to that of Cdc50 proteins in other organisms (23, 24) (Fig. 3B), suggesting that Cdc50 in C. neoformans may also play a role in lipid translocation.
The mutants of the Cdc50 homolog in mammalian cells have been reported to exhibit an aberrant exposure of endogenous aminophospholipids, including PS, on the cell surface (28). It has been suggested that PS exposure on the cell surface may serve as an “eat-me” signal for macrophages to recognize and internalize such cells (28). Therefore, the hypersensitivity of the cdc50Δ mutant to macrophage killing may be due not only to its defect in membrane integrity but also to increased exposure of PS on the cell surface. To test this hypothesis, we analyzed PS trafficking using 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD)-labeled PS (NBD-PS). While wild-type H99 cells efficiently took up NBD-labeled PS, the cdc50Δ mutant displayed very weak NBD-PS binding and uptake, indicating a blockage of PS translocation, which is consistent with the function of Cdc50 as a subunit of the lipid flippase (Fig. 7A).
FIG 7 .
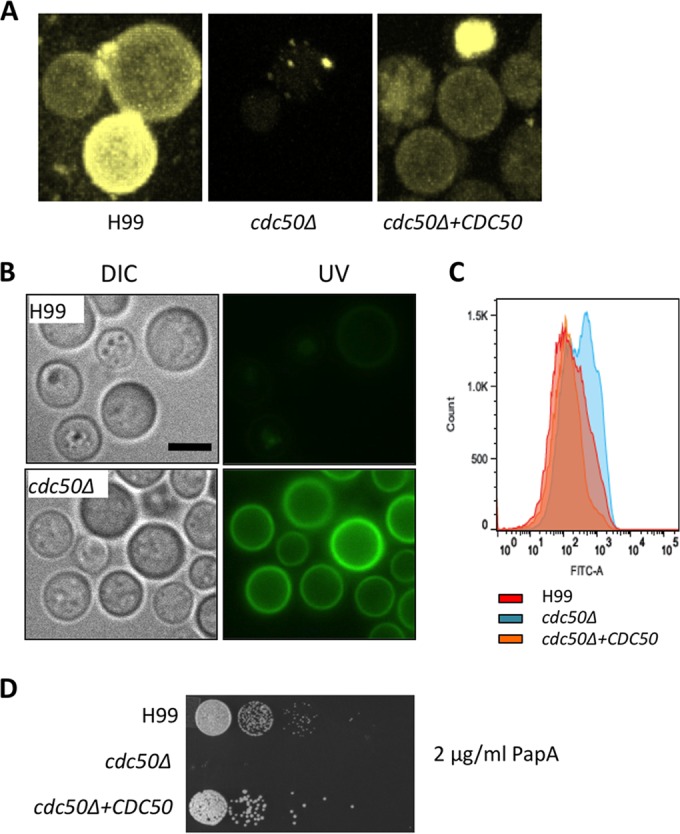
Loss of Cdc50 causes reduced phosphatidylserine (PS) uptake and increased PS levels on plasma membrane. (A) The PS uptake assay for H99, cdc50Δ, and cdc50Δ+CDC50 strains was performed by coincubating fungal cells with 1 µmol NBD-PS. The fluorescent signal was photographed after a 30-min incubation at 30°C. Bar, 5 µm. (B) The fungal strains were coincubated with FITC-conjugated annexin V for 30 min before being fixed and observed by fluorescence microscopy. Bar, 5 µm. DIC, differential inference contrast. (C) The fluorescent signal of stained H99 and cdc50Δ cells was also determined by flow cytometry. One hundred thousand cells were counted for each sample. (D) The above strains were inoculated on YPD with 2 µg/ml papuamide A (PapA) and incubated at 30°C for 48 h.
We also measured cell surface PS levels using fluorescein isothiocyanate (FITC)-conjugated annexin V staining (FITC-annexin), which efficiently binds to PS on the cell surface. Annexin V staining is commonly used to measure apoptosis because apoptotic cells have more PS on the outer plasma membrane. Our results showed that the FITC signal was much stronger in the mutant than in the wild-type strain (Fig. 7B). Signaling intensity quantification using flow cytometry confirmed the visual observations (Fig. 7C). In addition, we found that the cdc50Δ mutant is hypersensitive to papuamide A, a cytotoxic lipopeptide that kills fungal cells by binding to PS (Fig. 7D) (29, 30). These results indicate that the cdc50Δ cells have higher levels of PS exposure on the outer plasma membrane, which is consistent with observations in other eukaryotes (28). Because it has been reported that PS exposure causes macrophage recognition and macrophage killing in mammalian cells, this result may indicate that the increased sensitivity to killing by macrophages of the C. neoformans cdc50Δ mutant may be at least partially due to the high PS exposure in this mutant.
Cdc50 is essential for fungal virulence.
Our results showing increased stress sensitivity, killing by macrophages, increased PS cell surface exposure, and altered virulence factor development in the cdc50Δ strain suggested that this mutant may affect C. neoformans virulence in vivo. To test this hypothesis, we used the wild type, the cdc50Δ mutant, and the complemented strain in a murine inhalation model of cryptococcosis. We observed that while mice infected by the wild-type strain died at around 20 days, as expected, mice infected by the cdc50Δ mutant remained healthy after 60 days postinoculation (Fig. 8A). Analysis of the fungal burden in the lungs and brains of cdc50Δ mutant-infected mice after 7 days postinfection showed that fungal cells were completely cleared in the lung (Fig. 8B). No fungal cells were observed in the sections of lungs infected by the mutant using either hematoxylin and eosin (H&E) or Grocott’s methenamine silver (GMS) staining, in contrast to the presence of abundant cryptococcal cells in the lung infected by the wild-type cells (Fig. 8C). The dramatic virulence attenuation and clearance of fungal cells from the lungs of mice infected by the cdc50Δ strain showed that the Cdc50 lipid flippase is essential for cryptococcal virulence in vivo.
FIG 8 .

Cdc50 is required for virulence in vivo. (A) Survival curves for infected mice in a murine nasal inhalation model of systemic cryptococcosis. Female A/Jcr mice were inoculated via nasal inhalation with the (H99) cdc50Δ and cdc50Δ+CDC50 strains. Mice were infected with 1 × 105 yeast cells of each strain. (B) Fungal burden in infected lungs and brains was determined by CFU 7 days postinfection. (C) H&E- and GMS-stained slides were prepared from cross sections of infected lung at 7 days postinfection and visualized by light microscopy. The cryptococci are indicated by arrows.
DISCUSSION
In this study, we showed that loss of lipid flippase component Cdc50 function leads to enhanced echinocandin and fluconazole sensitivity and increases C. neoformans sensitivity to several types of stress. Moreover, we found that the CDC50 expression was specifically regulated by macrophage interaction and that the cdc50Δ mutant was highly sensitive to macrophage killing in vitro. Finally, Cdc50 was essential for fungal virulence in a murine model of systemic cryptococcosis. The detection of increased caspofungin uptake in the cdc50Δ mutant and increased PS exposure on the mutant cell surface help provide a mechanistic understanding of Cdc50 function in echinocandin resistance and fungal virulence.
Although β-1,3-glucan synthase is both functional and essential in C. neoformans, this fungus is highly resistant to echinocandin treatment (14). It is not currently understood why the efficacy of echinocandin therapy in C. neoformans is limited. Echinocandin drug resistance in Candida species is mostly due to the point mutations in β-1,3-glucan synthase subunits (9, 31). Because purified cryptococcal β-1,3-glucan synthase is sensitive to echinocandin in vitro (15), it is unlikely that this enzyme in C. neoformans naturally exists in an echinocandin-resistant mutant form, suggesting instead that the drug is not able to efficiently interact with the enzyme or disrupt the enzyme production in vivo. This conclusion is supported by our observation that a mutant that allows increased echinocandin entry into C. neoformans cells shows elevated echinocandin sensitivity.
Cdc50 homologs are best studied in S. cerevisiae, which contains three Cdc50 family members: Cdc50, Lem3, and Crf1. Cdc50 and Lem3 interact with P4-type ATPases Drs2 and Dnf1 to produce Cdc50-Drs2 and Lem3-Dnf1 complexes, respectively, which function as lipid flippases (17, 23, 32). Cdc50-like proteins contain two transmembrane domains and a large luminal loop (23), which is required for interacting with the transported phospholipids to translocate them through the membrane, thus maintaining the asymmetry of the bilayer lipid membrane (33). In Cryptococcus, Cdc50 is the only homolog of the Cdc50 gene family, and our study shows that it shares several properties with Cdc50 and Lem3 in S. cerevisiae, including its membrane localization and its role in aminophospholipid translocation. These similarities, together with the drug-sensitive and virulence-defective phenotype of the cdc50Δ mutant, suggest that cryptococcal Cdc50 also functions as part of a lipid flippase and that the function of this protein complex is necessary for both normal virulence and drug resistance.
The involvement of Cdc50 homologs in drug uptake has been reported previously. For instance, Cdc50A in human cells has been reported to play a role in the uptake of the anticancer drug perifosine (34). Also, Lem3 in S. cerevisiae has been shown to play an essential role in the uptake and potency of the alkylphosphocholine drugs edelfosine and miltefosine, which have been studied for treatment of protozoal and fungal diseases (35). However, in both cases, in contrast to C. neoformans cdc50Δ cells, the mutants lacking either CDC50 or LEM3 showed increased drug resistance due to blockage of drug uptake and translocation. In fact, S. cerevisiae diploids homozygous for lem3Δ have a higher caspofungin MIC than do the wild-type strains (36). Therefore, the drug-sensitive phenotype of the C. neoformans cdc50Δ mutant is unusual and may indicate a unique role of the lipid flippase in drug transport in this organism.
The detailed mechanisms of Cdc50-mediated caspofungin resistance will require further investigation. Studies in Candida glabrata have revealed a sphingolipid-dependent drug resistance mechanism where changes in membrane lipid composition lead to a tighter or looser interaction between various echinocandin-class drugs and their target, β-1,3-glucan synthase (36). Thus, it is possible that a change in membrane lipid distribution in the cdc50Δ mutant may alter the interaction of β-1,3-glucan synthase with caspofungin, resulting in improved drug efficacy. Another hypothesis is that caspofungin can be efficiently removed from the outer leaflet of the membrane, where it appears to act on glucan synthase in the wild-type C. neoformans cells by a lipid flippase-dependent process, and that this process is impaired in the mutant strain. Indeed, our caspofungin uptake assay using the BODIPY-labeled caspofungin showed a significantly increased intracellular caspofungin level in the mutant cells, suggesting that the mechanism of resistance is related to the caspofungin binding and internal caspofungin concentrations. In fact, lipid flippase is involved in the function of the ER and Golgi apparatus and has been reported to regulate the endocytic and exocytic recycling in S. cerevisiae (23, 32). Therefore, it is possible that the lipid flippase may contribute to echinocandin resistance in C. neoformans by either blocking drug penetration and interacting with the drug target or enhancing the drug exocytosis, leading to low intracellular drug concentrations.
We observed that Cdc50 also plays a role in the sensitivity of C. neoformans to azole drugs, such as fluconazole. Previous studies in S. cerevisiae have shown that Cdc50-mediated lipid flippase regulates the ergosterol distribution and trafficking in the Golgi network (37). Therefore, disruption of Cdc50 function could lead to altered ergosterol distribution on the membrane to cause lethality when treated with fluconazole. In agreement with this argument, it has been reported in S. cerevisiae that disruption of both CDC50 and genes acting at late steps of the ergosterol biosynthesis pathway (ERG2 and ERG6) results in synthetic lethality (38). Our screens also identified disruptions of three other genes encoding membrane proteins that have an elevated sensitivity to caspofungin. Two of them, ERG3 and ERG4, encode enzymes involved in the ergosterol biosynthetic pathway. This discovery is consistent with our observation that many of our caspofungin-sensitive mutants are also sensitive to fluconazole.
Besides its role in drug resistance, our results have revealed the requirement of C. neoformans Cdc50 for fungal virulence both in vitro and in vivo. The cdc50Δ mutant showed a reduced growth rate at 37°C under some growth conditions in vitro, which may contribute to the virulence attenuation. Interestingly, cdc50Δ cells produced even larger capsules than did the wild type and caused no obvious difference in melanin production. Thus, it is not clear whether the virulence defect of the cdc50Δ mutant stems from altered production of these known cryptococcal virulence factors. Our results support another, non-mutually exclusive hypothesis for the role of Cdc50 in virulence: that Cdc50 activity is required for counteracting the antimicrobial activities of host macrophages. This hypothesis is supported by our findings that CDC50 expression is specifically induced during interaction with macrophages, that cdc50Δ cells are engulfed by macrophages at a higher rate than the wild type and are more sensitive to macrophage killing in vitro, and that cdc50Δ mutants display higher levels of PS on the outer membrane, which may serve as a macrophage recognition signal, as demonstrated in other systems (28).
It is not clear whether the interaction of macrophages with PS exposed on the surface of cryptococcal cells is necessarily direct, as the presence of the large polysaccharide capsule in C. neoformans may interfere with this process. While we have no direct experimental evidence to address this question, studies in mammalian cells have shown that a direct interaction between macrophages and PS may be not required to trigger phagocytosis. In apoptotic cells where PS exposure on the cell surface acts as an “eat-me” signal to trigger phagocytosis, several macrophage-secreted proteins required for the recognition have been reported, such as milk fat-globule epidermal growth factor 8 (MFG-8), growth arrest-specific 6 (Gas6), and protein S (PROS), as reviewed in reference 39. These secreted proteins bind to PS to form a bridge between PS and macrophage cells during the recognition process. Thus, it is possible that macrophages are able to sense PS levels on the C. neoformans surface despite the presence of the capsule. One way to address this issue is to examine the phagocytosis of the cdc50Δ mutant in wild-type macrophages and macrophages lacking PS receptors. Furthermore, besides functioning as an “eat-me” signal, PS has recently been identified as a global immunosuppressive signal in efferocytosis, infectious disease, and cancer, as summarized in reference 40. Therefore, PS exposure on the cell surface may have multiple effects on C. neoformans-host interaction during infection, which will be interesting to investigate in the future.
In summary, we identified Cdc50, the only β-subunit homolog of lipid flippases in C. neoformans, as essential both for resistance to antifungal drugs in this organism and for virulence in a murine model of infection. These results reveal phospholipid translocation and trafficking as a new potential target for antifungal drug development.
MATERIALS AND METHODS
Strains and media.
An Agrobacterium-mediated mutagenesis library was created from Cryptococcus neoformans var. grubii (serotype A) strain H99. Two Cryptococcus neoformans gene deletion libraries were generated by Hiten Madhani at UCSF and purchased from the American Type Culture Collection (ATCC) and the Fungal Genetic Stock Center (FGSC), respectively. Other C. neoformans and Saccharomyces cerevisiae strains used in this study are listed in Table 2. In all growth assays, transformants were grown in nutrient-rich yeast extract-peptone-dextrose (YPD) medium at 30°C. After measurements were taken, cultures were stored at 4°C. Papuamide A was kindly provided by Kirk Gustafson at the National Cancer Institute. The GXM monoclonal antibody 18B7 was kindly provided by Arturo Casadevall at Johns Hopkins University. BODIPY-labeled caspofungin was synthesized in-house, while NBD-PS was purchased from Avanti. Caspofungin was provided by Merck. Other common medium preparation and growth conditions followed the instructions described previously (41, 42).
TABLE 2 .
Strains used in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| C. neoformans | ||
| H99 | MATα | 56 |
| KN99a | MATa | 57 |
| CUX178 | Caspofungin-sensitive strain 11B6 from ATMT library | This study |
| CUX195 | MATα mpt1Δ::NEO | This study |
| CUX196 | MATα cdc50Δ::NEO | This study |
| CUX202 | MATα cdc50Δ::NEO pHIS-GFP-CDC50-NAT | This study |
| CUX208 | MATα cdc50Δ::NEO CDC50-NAT | This study |
| S. cerevisiae | ||
| BY4741 | MATa his3Δ1 leu2Δ0 ura3Δ0 | ATCC deletion collection |
| YUX87 | MATa his3Δ1 leu2Δ0 ura3Δ0 lem3Δ::KanMX | ATCC deletion collection |
| YUX89 | MATa his3Δ1 leu2Δ0 ura3Δ0 cdc50Δ::KanMX | ATCC deletion collection |
Generation and screen of Agrobacterium-mediated mutagenesis library.
A random mutagenesis library was created using strains C. neoformans var. grubii H99 and Agrobacterium tumefaciens EHA105 as previously described (18). Briefly, cells were grown in various fungal-to-bacterial cell ratios. After 48 h, cultures were transferred to YPD with NAT and cefotaxime to select for C. neoformans strains with transfer DNA (T-DNA) insertions. Cells that grew on the selective medium were transferred to 96-well plates and grown to saturation for further screening.
To screen for mutants sensitive to caspofungin, mutagenesis libraries were activated by inoculating 5 µl stock cells to 96-well plates containing 150 µl YPD per well and incubated at 30°C. Once cultures were saturated, transformants were screened for sensitivity to 8 µg/ml of caspofungin. Ninety-six-well plates were prepared with 150 µl YPD plus 8 µg/ml of caspofungin. Five microliters of saturated cultures was inoculated into the corresponding wells. The optical density at 600 nm (OD600) was measured every 24 h, and growth in the presence of drug was compared to growth in YPD only. Transformants identified as having an increased sensitivity to drug compared to the wild type were selected for rescreening. After repeated screenings, sensitive mutants were selected for further analysis. Overnight cultures of transformants identified as sensitive to caspofungin were washed and diluted to a standardized OD600 of 1.0. Tenfold serial dilutions were prepared, and cultures were inoculated at different concentrations of caspofungin or fluconazole on YPD agar plates.
Inverse PCR and gene identification.
Isolates identified as sensitive to caspofungin were crossed with strain KN99a. Cells from each culture were mixed and inoculated on MS and V8 mating media. Plates were incubated at 22.5°C in the dark for 10 days. Spores were dissected and inoculated on YPD-plus-NAT plates and incubated at 30°C for 3 days. Progenies were crossed with wild-type strain H99α to determine the mating type. Progenies with the a mating type were selected for phenotypic and genotypic analysis to determine whether the drug-sensitive phenotype of a progeny is cosegregated with the NAT marker.
For the mutants sensitive to caspofungin due to T-DNA insertion confirmed by cosegregation assay, their target genes were identified using the inverse PCR method as previously described (18) with minor adjustment. Briefly, genomic DNA was prepared and digested with ClaI, NcoI, NdeI, BglII, and XhoI for a total of five reactions. Digested fragments were self-ligated with T4 DNA ligase, the flanking regions of the NAT insert were amplified, and their sequences were determined. The location of the T-DNA insertion and subsequent gene disruption was determined using BLAST analysis of sequencing results with the C. neoformans genome database of the Broad Institute.
Cryptococcus-macrophage interaction assay.
The macrophage-Cryptococcus interaction assay was done as previously described (43). Macrophage-like cell line J774 cells were cultured in DME medium with 10% heat-inactivated FBS at 37°C with 5% CO2. J774 cells (5 × 104) in 0.5 ml fresh DME medium were added into each well of a 48-well culture plate and incubated at 37°C in 5% CO2 overnight. To activate macrophage cells, 50 units/ml gamma interferon (IFN-γ; Invitrogen) and 1 µg/ml lipopolysaccharide (LPS; Sigma) were added into each well. C. neoformans overnight cultures were washed with phosphate-buffered saline (PBS) twice and opsonized with 20% mouse complement. Cryptococcus cells (2 × 105) were added into each well (yeast/J774 ratio, 4:1). To assess the phagocytosis rate, the cells were washed with PBS after a 1.5-h coincubation and fixed with methanol for 30 min. Giemsa stain was added to the wells at a 1:10 dilution, and the plates were incubated overnight at 4°C. Cells were washed once with PBS and analyzed using an inverted microscope. To assess intracellular proliferation of C. neoformans, nonadherent extracellular yeast cells were removed by washing with fresh DME medium after a 2-h coincubation and cultures were incubated for another 0, 2, and 22 h. At indicated time points, the medium in each well was replaced with distilled water (dH2O) to lyse macrophage cells for 30 min at room temperature. The lysate was spread on YPD plates, and CFU were counted to determine intracellular proliferation.
qRT-PCR.
Cryptococcus cells from overnight culture or mating cultures were collected. Total RNA extraction and first-strand cDNA synthesis were followed with the protocol as described previously (42). Expression of CDC50 and GAPDH was analyzed using SYBR Advantage QPCR premix reagents (Clontech). Gene expression levels were normalized using the endogenous control gene GAPDH, and the relative levels were determined using the comparative threshold cycle (CT) method (44). Real-time PCRs were performed using an Mx4000 QPCR system (Stratagene) as previously described (45).
Generation of cdc50Δ mutant and its complemented strain.
The cdc50Δ mutants were generated by overlap PCR as previously described (46). The 5′ and 3′ regions of each CDC50 gene were amplified from H99 genomic DNA with primers CX614/CX615 and CX616/CX617, respectively (see Table S1 in the supplemental material for primer sequences). The dominant selectable markers (Neor) were amplified with the M13 primers (M13F and M13R) from plasmid pJAF1 (47). Each target gene replacement cassette was generated by overlap PCR with primers CX614/CX617 (see Table S1). Purified overlap PCR products were precipitated onto 10 µl gold microcarrier beads (0.6 µm; Bio-Rad), and H99 was biolistically transformed as described previously (48). Stable transformants were selected on YPD medium containing G418 (200 mg/liter). To screen for mutants of the CDC50 gene, diagnostic PCR was performed by analyzing the 5′ junction of the disrupted mutant alleles with primers CX620 and JH8994 (see Table S1). Positive transformants identified by PCR screening were further confirmed by Southern blot analysis.
To generate complemented strains of cdc50 mutants, a genomic DNA fragment that contains a 1.5-kb upstream promoter region, the CDC50 ORF, and its 500-bp downstream region was amplified in a PCR using primers CX625/CX626. This PCR fragment was cloned into the plasmid pJAF13, which contains a NAT marker, by Infusion cloning (Clontech). The linearized plasmid was biolistically transformed in a cdc50Δ mutant strain to generate the complemented strain CUX208.
To generate a GFP-tagged strain, the CDC50 full-length cDNA was amplified with primers CX647/CX648 and cloned into the BamHI/NotI sites of a vector containing the Cryptococcus actin promoter (49) and a GFP epitope, to generate a plasmid that contains the CDC50-GFP fusion. The above plasmids were biolistically transformed into the wild-type H99 strain to generate strain CUX202, which would express Cdc50-GFP.
Assays for melanin, capsule production, and stress response.
Melanin production was assayed by inoculating C. neoformans strains into 2 ml YPD liquid medium and incubating them overnight at 30°C. Five microliters of each overnight culture with serial dilutions was placed on l-3,4-dihydroxyphenylalanine (l-DOPA) agar medium. The agar plates were incubated at 30°C or 37°C for 2 days, and pigmentation of fungal colonies was assessed. To examine capsule production, yeast cells were inoculated on Dulbecco modified Eagle (DME) medium. The DME medium plates were incubated at 37°C for 3 days before capsule size was visualized by adding a drop of India ink to the cell suspensions and observed on an Olympus AX70 microscope (Melville, NY). The relative capsule size was calculated by dividing capsule size with the whole-cell size (capsule and cell size). The average and standard deviation from at least 50 cells were calculated for each condition tested. Electromobility of secreted GXM from H99 and the cdc50Δ mutant was detected in a Western blot assay as described in reference 50. Secreted total polysaccharides were purified from a 500-ml YPD culture of each strain using the cetyltrimethylammonium bromide (CTAB) precipitation method as described previously (51). The amount of the total GXM was determined by the phenol sulfuric method. The two-pair t test method was used to determine the statistical significance of the difference between samples.
To assay for stress responses, yeast cells from overnight cultures were washed, resuspended, and serially diluted (1:10) in dH2O and spotted (5 µl) on YPD agar plates containing 1.0 M KCl for osmotic shock, 2.5 mM H2O2 for oxidative stress, and 1 mM NaNO2 for nitrosative stress as previously described (43, 52). To test cell integrity, cells were also spotted on YPD agar plates containing 0.03% SDS, 0.5% Congo red, or 250 µg/ml calcofluor white (CFW) and incubated for 2 days at 30°C.
Cellular staining assays. (i) BODIPY-caspofungin localization assay.
C. neoformans and S. cerevisiae overnight cultures were reinoculated in YPD and cultured for 4 h. Fresh cultures were washed with PBS, and BODIPY-fluorescently labeled caspofungin was added at a final concentration of 5 µM and incubated for 30 min. Cell suspensions were then observed under a fluorescence microscope for cellular localization of the fluorescent signal. Fluorescent signal intensity for 100,000 cells was also quantified by flow cytometry.
(ii) Annexin V assay.
Cryptococcus strains were cultured in YPD for 48 h. Cells were harvested and washed in binding buffer (10 mM HEPES-NaOH, pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2) and resuspended in 1 ml binding buffer containing 5 µl FITC-conjugated annexin V (Life Technologies, Inc.) as described previously (53). After being incubated for 1 h at 30°C with shaking, cells were fixed by 3.7% formaldehyde for 10 min at 37°C and then washed in PBS containing 1% formaldehyde before being observed under fluorescence microscopy. Fluorescent signal intensity was also quantified by flow cytometry.
(iii) NBD-PS uptake assay.
C. neoformans cells from overnight cultures were washed in NaCl buffer (0.7 M NaCl, 0.5 mM MgCl2, 35 mM potassium phosphate, pH 6.8) twice and treated with 15 U/ml lyticase (Sigma) for 2 h at 30°C as previously described (54). Cells were incubated in NaCl buffer with 100 µmol NBD-PS for 30 min at 30°C and then washed 3 times with NaCl buffer containing 4% bovine serum albumin (BSA) before being fixed with 3.7% formaldehyde for 10 min at 37°C. Fixed cells were washed with NaCl buffer and observed under fluorescence microscopy or quantified using flow cytometry.
(iv) ER-Tracker Red staining.
C. neoformans overnight cultures were washed in Hanks balanced salt solution (HBSS) buffer (1.26 mM CaCl2, 5.33 mM KCl, 0.44 mM KH2PO4, 0.5 mM MgCl2⋅6H2O, 0.44 mM MgSO4⋅7H2O, 138 mM NaCl, 4 mM NaHCO3, 0.3 mM Na2HPO4, and 5.6 mM d-glucose) twice before being fixed with 3.7% formaldehyde for 2 to 5 min at 30°C. Fixed cells were washed with HBSS buffer 3 times and stained with 1 µmol ER-Tracker (Life Technologies, Inc.) for 30 min at 30°C before being observed by fluorescence microscopy (Nikon).
Virulence studies in vivo and fungal burden in infected organs.
Groups of 10 female A/Jcr mice (NCI-Frederick) were intranasally infected with 105 yeast cells of each strain as previously described (41, 55). Over the course of the experiments, animals that appeared moribund or in pain were sacrificed by CO2 inhalation. Survival data from the murine experiments were statistically analyzed between paired groups using the log rank test of the PRISM program 4.0 (GraphPad Software, San Diego, CA). P values of <0.001 were considered significant.
Infected animals were sacrificed at the endpoint of the experiment according to the Rutgers IACUC-approved animal protocol. For mice infected by the cdc50Δ mutant strain, the experiment was terminated 60 days postinfection. To compare fungal burdens, lungs and brains from mice infected by H99, the cdc50Δ mutant, or its complemented strain were isolated 7 days postinfection. Infected lungs and brains were isolated and homogenized in 1× PBS buffer using a homogenizer. Resuspensions were diluted, and 100 µl of each dilution was spread on YPD medium; fungal colonies were counted after 3 days of incubation at 30°C. The isolated animal tissues were also fixed in 10% formalin solution and sent to the Rutgers histopathological core facility for section preparation and staining with hematoxylin and eosin (H&E) and Grocott’s methenamine silver (GMS) stain.
SUPPLEMENTAL MATERIAL
Cdc50 in C. neoformans, but not in S. cerevisiae, plays a negative role in caspofungin internalization. (A) Cultures of C. neoformans H99, cdc50Δ, and cdc50Δ+CDC50 strains were coincubated with 5 µmol BODIPY-labeled caspofungin for 30 min at 30°C. The fluorescent signal of fungal cells was detected by fluorescence microscopy. (B) C. neoformans survival rate was determined by CFU for the above strains treated with 5 µmol BODIPY-caspofungin for 30 min at 30°C. (C) Cultures of S. cerevisiae strain BY4742 and its lem3Δ and cdc50Δ mutants were coincubated with 5 µmol BODIPY-labeled caspofungin for 30 min at 30°C. The fluorescent signal of fungal cells was detected by fluorescence microscopy. Download
The C. neoformans cells lacking CDC50 produced normal capsule under noninducing conditions for capsule. C. neoformans H99, cdc50Δ, and cdc50Δ+CDC50 strains and the CDC50 overexpression strain (PHIS-GFP-CDC50) were cultured on YNB and YPD for 3 days, respectively. Capsule production of these cells on YNB (A) and YPD (B) was visualized by India ink staining. (C) Capsule sizes were measured for the above strains cultured in either YNB or YPD medium from over 100 cells for each condition. Download
The C. neoformans cells lacking CDC50 had normal growth rates on different media. C. neoformans H99, cdc50Δ, and cdc50Δ+CDC50 strains were cultured on YPD, DME, or DME with 10% FBS or on macrophage spent medium. Numbers of CFU were used to determine live cell numbers after incubation for 2, 4, and 24 h in different media. Download
Primers used in this study.
ACKNOWLEDGMENTS
We thank Erika Shor for critical reading and editing of the manuscript and valuable comments for the study. We thank Tong-Bao Liu, Peter Nakagami, and Delfin Balili for technical assistance on the project. We thank Eliseo Eugenin for support on confocal microscopy and Riccardo Arrigucci for assistance on flow cytometry. We thank Merck for providing caspofungin, Kirk Gustafson for providing papuamide A, and Arturo Casadevall for providing GXM antibody for this study. We also acknowledge use of the C. neoformans genome sequences at the Broad Institute and use of the C. neoformans deletion collections generated by Hiten Madhani with funding support from the NIH (R01AI00272).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Huang W, Liao G, Baker GM, Wang Y, Lau R, Paderu P, Perlin DS, Xue C. 2016. Lipid flippase subunit Cdc50 mediates drug resistance and virulence in Cryptococcus neoformans. mBio 7(3):e00478-16. doi:10.1128/mBio.00478-16.
REFERENCES
- 1.Casadevall A, Perfect JR. 1998. Cryptococcus neoformans. ASM Press, Washington, DC. [Google Scholar]
- 2.Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23:525–530. doi: 10.1097/QAD.0b013e328322ffac. [DOI] [PubMed] [Google Scholar]
- 3.O’Meara TR, Alspaugh JA. 2012. The Cryptococcus neoformans capsule: a sword and a shield. Clin Microbiol Rev 25:387–408. doi: 10.1128/CMR.00001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doering TL, Nosanchuk JD, Roberts WK, Casadevall A. 1999. Melanin as a potential cryptococcal defence against microbicidal proteins. Med Mycol 37:175–181. doi: 10.1080/j.1365-280X.1999.00218.x. [DOI] [PubMed] [Google Scholar]
- 5.Feldmesser M, Kress Y, Novikoff P, Casadevall A. 2000. Cryptococcus neoformans is a facultative intracellular pathogen in murine pulmonary infection. Infect Immun 68:4225–4237. doi: 10.1128/IAI.68.7.4225-4237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perfect JR, Dismukes WE, Dromer F, Goldman DL, Graybill JR, Hamill RJ, Harrison TS, Larsen RA, Lortholary O, Nguyen MH, Pappas PG, Powderly WG, Singh N, Sobel JD, Sorrell TC. 2010. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis 50:291–322. doi: 10.1086/649858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loyse A, Dromer F, Day J, Lortholary O, Harrison TS. 2013. Flucytosine and cryptococcosis: time to urgently address the worldwide accessibility of a 50-year-old antifungal. J Antimicrob Chemother 68:2435–2444. doi: 10.1093/jac/dkt221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denning DW. 2003. Echinocandin antifungal drugs. Lancet 362:1142–1151. doi: 10.1016/S0140-6736(03)14472-8. [DOI] [PubMed] [Google Scholar]
- 9.Perlin DS. 2011. Current perspectives on echinocandin class drugs. Future Microbiol 6:441–457. doi: 10.2217/fmb.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazur P, Morin N, Baginsky W, el-Sherbeini M, Clemas JA, Nielsen JB, Foor F. 1995. Differential expression and function of two homologous subunits of yeast 1,3-beta-d-glucan synthase. Mol Cell Biol 15:5671–5681. doi: 10.1128/MCB.15.10.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douglas CM, D’Ippolito JA, Shei GJ, Meinz M, Onishi J, Marrinan JA, Li W, Abruzzo GK, Flattery A, Bartizal K, Mitchell A, Kurtz MB. 1997. Identification of the FKS1 gene of Candida albicans as the essential target of 1,3-beta-d-glucan synthase inhibitors. Antimicrob Agents Chemother 41:2471–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beauvais A, Bruneau JM, Mol PC, Buitrago MJ, Legrand R, Latgé JP. 2001. Glucan synthase complex of Aspergillus fumigatus. J Bacteriol 183:2273–2279. doi: 10.1128/JB.183.7.2273-2279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly R, Register E, Hsu MJ, Kurtz M, Nielsen J. 1996. Isolation of a gene involved in 1,3-beta-glucan synthesis in Aspergillus nidulans and purification of the corresponding protein. J Bacteriol 178:4381–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson JR, Douglas CM, Li W, Jue CK, Pramanik B, Yuan X, Rude TH, Toffaletti DL, Perfect JR, Kurtz M. 1999. A glucan synthase FKS1 homolog in Cryptococcus neoformans is single copy and encodes an essential function. J Bacteriol 181:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maligie MA, Selitrennikoff CP. 2005. Cryptococcus neoformans resistance to echinocandins: (1,3)beta-glucan synthase activity is sensitive to echinocandins. Antimicrob Agents Chemother 49:2851–2856. doi: 10.1128/AAC.49.7.2851-2856.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu OW, Chun CD, Chow ED, Chen C, Madhani HD, Noble SM. 2008. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 135:174–188. doi: 10.1016/j.cell.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lenoir G, Williamson P, Puts CF, Holthuis JC. 2009. Cdc50p plays a vital role in the ATPase reaction cycle of the putative aminophospholipid transporter Drs2p. J Biol Chem 284:17956–17967. doi: 10.1074/jbc.M109.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Idnurm A, Reedy JL, Nussbaum JC, Heitman J. 2004. Cryptococcus neoformans virulence gene discovery through insertional mutagenesis. Eukaryot Cell 3:420–429. doi: 10.1128/EC.3.2.420-429.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang YC, Kwon-Chung KJ. 1994. Complementation of a capsule-deficient mutation of Cryptococcus neoformans restores its virulence. Mol Cell Biol 14:4912–4919. doi: 10.1128/MCB.14.7.4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu X, Williamson PR. 2004. Role of laccase in the biology and virulence of Cryptococcus neoformans. FEMS Yeast Res 5:1–10. doi: 10.1016/j.femsyr.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 21.Hu G, Steen BR, Lian T, Sham AP, Tam N, Tangen KL, Kronstad JW. 2007. Transcriptional regulation by protein kinase A in Cryptococcus neoformans. PLoS Pathog 3:e42. doi: 10.1371/journal.ppat.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D’Souza CA, Alspaugh JA, Yue C, Harashima T, Cox GM, Perfect JR, Heitman J. 2001. Cyclic AMP-dependent protein kinase controls virulence of the fungal pathogen Cryptococcus neoformans. Mol Cell Biol 21:3179–3191. doi: 10.1128/MCB.21.9.3179-3191.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saito K, Fujimura-Kamada K, Furuta N, Kato U, Umeda M, Tanaka K. 2004. Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol Biol Cell 15:3418–3432. doi: 10.1091/mbc.E03-11-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka K, Fujimura-Kamada K, Yamamoto T. 2011. Functions of phospholipid flippases. J Biochem 149:131–143. doi: 10.1093/jb/mvq140. [DOI] [PubMed] [Google Scholar]
- 25.Pratt A, Garcia-Effron G, Zhao Y, Park S, Mustaev A, Pillai S, Perlin DS. 2013. Evaluation of fungal-specific fluorescent labeled echinocandin probes as diagnostic adjuncts. Med Mycol 51:103–107. doi: 10.3109/13693786.2012.685767. [DOI] [PubMed] [Google Scholar]
- 26.Alvarez M, Casadevall A. 2006. Phagosome extrusion and host-cell survival after Cryptococcus neoformans phagocytosis by macrophages. Curr Biol 16:2161–2165. doi: 10.1016/j.cub.2006.09.061. [DOI] [PubMed] [Google Scholar]
- 27.Ma H, Croudace JE, Lammas DA, May RC. 2006. Expulsion of live pathogenic yeast by macrophages. Curr Biol 16:2156–2160. doi: 10.1016/j.cub.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 28.Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. 2014. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344:1164–1168. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- 29.Chen YL, Montedonico AE, Kauffman S, Dunlap JR, Menn FM, Reynolds TB. 2010. Phosphatidylserine synthase and phosphatidylserine decarboxylase are essential for cell wall integrity and virulence in Candida albicans. Mol Microbiol 75:1112–1132. doi: 10.1111/j.1365-2958.2009.07018.x. [DOI] [PubMed] [Google Scholar]
- 30.Ford PW, Gustafson KR, McKee TC, Shigematsu N, Maurizi LK, Pannell LK, Williams DE, Dilip de Silva E, Lassota P, Allen TM, Van Soest R, Andersen RJ, Boyd MR. 1999. Papuamides A-D, HIV-inhibitory and cytotoxic depsipeptides from the sponges Theonella mirabilis and Theonella swinhoei collected in Papua New Guinea. J Am Chem Soc 121:5899–5909. doi: 10.1021/ja990582o. [DOI] [Google Scholar]
- 31.Perlin DS. 2007. Resistance to echinocandin-class antifungal drugs. Drug Resist Updat 10:121–130. doi: 10.1016/j.drup.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furuta N, Fujimura-Kamada K, Saito K, Yamamoto T, Tanaka K. 2007. Endocytic recycling in yeast is regulated by putative phospholipid translocases and the Ypt31p/32p-Rcy1p pathway. Mol Biol Cell 18:295–312. doi: 10.1091/mbc.E06-05-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bryde S, Hennrich H, Verhulst PM, Devaux PF, Lenoir G, Holthuis JC. 2010. CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J Biol Chem 285:40562–40572. doi: 10.1074/jbc.M110.139543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muñoz-Martínez F, Torres C, Castanys S, Gamarro F. 2010. CDC50A plays a key role in the uptake of the anticancer drug perifosine in human carcinoma cells. Biochem Pharmacol 80:793–800. doi: 10.1016/j.bcp.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 35.Hanson PK, Malone L, Birchmore JL, Nichols JW. 2003. Lem3p is essential for the uptake and potency of alkylphosphocholine drugs, edelfosine and miltefosine. J Biol Chem 278:36041–36050. doi: 10.1074/jbc.M305263200. [DOI] [PubMed] [Google Scholar]
- 36.Healey KR, Katiyar SK, Raj S, Edlind TD. 2012. CRS-MIS in Candida glabrata: sphingolipids modulate echinocandin-Fks interaction. Mol Microbiol 86:303–313. doi: 10.1111/j.1365-2958.2012.08194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hankins HM, Sere YY, Diab NS, Menon AK, Graham TR. 2015. Phosphatidylserine translocation at the yeast trans-Golgi network regulates protein sorting into exocytic vesicles. Mol Biol Cell 26:4674–4685. doi: 10.1091/mbc.E15-07-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kishimoto T, Yamamoto T, Tanaka K. 2005. Defects in structural integrity of ergosterol and the Cdc50p-Drs2p putative phospholipid translocase cause accumulation of endocytic membranes, onto which actin patches are assembled in yeast. Mol Biol Cell 16:5592–5609. doi: 10.1091/mbc.E05-05-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Segawa K, Nagata S. 2015. An apoptotic “eat me” signal: phosphatidylserine exposure. Trends Cell Biol 25:639–650. doi: 10.1016/j.tcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, Barcinski M, Brekken RA, Huang X, Hutchins JT, Freimark B, Empig C, Mercer J, Schroit AJ, Schett G, Herrmann M. 2016. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ doi: 10.1038/cdd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Liu TB, Delmas G, Park S, Perlin D, Xue C. 2011. Two major inositol transporters and their role in cryptococcal virulence. Eukaryot Cell 10:618–628. doi: 10.1128/EC.00327-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu TB, Wang Y, Stukes S, Chen Q, Casadevall A, Xue C. 2011. The F-Box protein Fbp1 regulates sexual reproduction and virulence in Cryptococcus neoformans. Eukaryot Cell 10:791–802. doi: 10.1128/EC.00004-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu TB, Xue C. 2014. Fbp1-mediated ubiquitin-proteasome pathway controls Cryptococcus neoformans virulence by regulating fungal intracellular growth in macrophages. Infect Immun 82:557–568. doi: 10.1128/IAI.00994-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 45.Xue C, Liu T, Chen L, Li W, Liu I, Kronstad JW, Seyfang A, Heitman J. 2010. Role of an expanded inositol transporter repertoire in Cryptococcus neoformans sexual reproduction and virulence. mBio 1:e00478-16. doi: 10.1128/mBio.00084-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davidson RC, Blankenship JR, Kraus PR, de Jesus Berrios M, Hull CM, D’Souza C, Wang P, Heitman J. 2002. A PCR-based strategy to generate integrative targeting alleles with large regions of homology. Microbiology 148:2607–2615. doi: 10.1099/00221287-148-8-2607. [DOI] [PubMed] [Google Scholar]
- 47.Fraser JA, Subaran RL, Nichols CB, Heitman J. 2003. Recapitulation of the sexual cycle of the primary fungal pathogen Cryptococcus neoformans var. gattii: implications for an outbreak on Vancouver Island, Canada. Eukaryot Cell 2:1036–1045. doi: 10.1128/EC.2.5.1036-1045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davidson RC, Cruz MC, Sia RA, Allen B, Alspaugh JA, Heitman J. 2000. Gene disruption by biolistic transformation in serotype D strains of Cryptococcus neoformans. Fungal Genet Biol 29:38–48. doi: 10.1006/fgbi.1999.1180. [DOI] [PubMed] [Google Scholar]
- 49.Bahn YS, Kojima K, Cox GM, Heitman J. 2005. Specialization of the HOG pathway and its impact on differentiation and virulence of Cryptococcus neoformans. Mol Biol Cell 16:2285–2300. doi: 10.1091/mbc.E04-11-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoneda A, Doering TL. 2008. Regulation of Cryptococcus neoformans capsule size is mediated at the polymer level. Eukaryot Cell 7:546–549. doi: 10.1128/EC.00437-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wozniak KL, Levitz SM. 2009. Isolation and purification of antigenic components of Cryptococcus. Methods Mol Biol 470:71–83. doi: 10.1007/978-1-59745-204-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shea JM, Kechichian TB, Luberto C, Del Poeta M. 2006. The cryptococcal enzyme inositol phosphosphingolipid-phospholipase C confers resistance to the antifungal effects of macrophages and promotes fungal dissemination to the central nervous system. Infect Immun 74:5977–5988. doi: 10.1128/IAI.00768-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen S, Wang J, Muthusamy BP, Liu K, Zare S, Andersen RJ, Graham TR. 2006. Roles for the Drs2p-Cdc50p complex in protein transport and phosphatidylserine asymmetry of the yeast plasma membrane. Traffic 7:1503–1517. doi: 10.1111/j.1600-0854.2006.00485.x. [DOI] [PubMed] [Google Scholar]
- 54.Madeo F, Fröhlich E, Fröhlich KU. 1997. A yeast mutant showing diagnostic markers of early and late apoptosis. J Cell Biol 139:729–734. doi: 10.1083/jcb.139.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cox GM, Mukherjee J, Cole GT, Casadevall A, Perfect JR. 2000. Urease as a virulence factor in experimental cryptococcosis. Infect Immun 68:443–448. doi: 10.1128/IAI.68.2.443-448.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perfect JR, Schell WA, Rinaldi MG. 1993. Uncommon invasive fungal pathogens in the acquired immunodeficiency syndrome. J Med Vet Mycol 31:175–179. doi: 10.1080/02681219380000211. [DOI] [PubMed] [Google Scholar]
- 57.Nielsen K, Cox GM, Wang P, Toffaletti DL, Perfect JR, Heitman J. 2003. Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and α isolates. Infect Immun 71:4831–4841. doi: 10.1128/IAI.71.9.4831-4841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cdc50 in C. neoformans, but not in S. cerevisiae, plays a negative role in caspofungin internalization. (A) Cultures of C. neoformans H99, cdc50Δ, and cdc50Δ+CDC50 strains were coincubated with 5 µmol BODIPY-labeled caspofungin for 30 min at 30°C. The fluorescent signal of fungal cells was detected by fluorescence microscopy. (B) C. neoformans survival rate was determined by CFU for the above strains treated with 5 µmol BODIPY-caspofungin for 30 min at 30°C. (C) Cultures of S. cerevisiae strain BY4742 and its lem3Δ and cdc50Δ mutants were coincubated with 5 µmol BODIPY-labeled caspofungin for 30 min at 30°C. The fluorescent signal of fungal cells was detected by fluorescence microscopy. Download
The C. neoformans cells lacking CDC50 produced normal capsule under noninducing conditions for capsule. C. neoformans H99, cdc50Δ, and cdc50Δ+CDC50 strains and the CDC50 overexpression strain (PHIS-GFP-CDC50) were cultured on YNB and YPD for 3 days, respectively. Capsule production of these cells on YNB (A) and YPD (B) was visualized by India ink staining. (C) Capsule sizes were measured for the above strains cultured in either YNB or YPD medium from over 100 cells for each condition. Download
The C. neoformans cells lacking CDC50 had normal growth rates on different media. C. neoformans H99, cdc50Δ, and cdc50Δ+CDC50 strains were cultured on YPD, DME, or DME with 10% FBS or on macrophage spent medium. Numbers of CFU were used to determine live cell numbers after incubation for 2, 4, and 24 h in different media. Download
Primers used in this study.