

Abstract
Type 1 diabetes is an autoimmune disorder afflicting millions of people worldwide. Once diagnosed, patients require lifelong insulin treatment and can experience numerous disease-associated complications. The last decade has seen tremendous advances in elucidating the causes and treatment of the disease based on extensive research both in rodent models of spontaneous diabetes and in humans. Integrating these advances has led to the recognition that the balance between regulatory and effector T cells determines disease risk, timing of disease activation, and disease tempo. Here we describe current progress, the challenges ahead and the new interventions that are being tested to address the unmet need for preventative or curative therapies.
Type 1 diabetes represents one of more than 80 diseases considered to have an autoimmune aetiology. The disease occurs as a consequence of the organ-specific immune destruction of the insulin-producing β-cells in the islets of Langerhans within the pancreas1. The β-cells are elegant glucose ‘thermostats’, sensing glucose and releasing insulin to maintain physiologic glucose levels within a relatively narrow range. They thus comprise much more than just an insulin factory. Once those cells are destroyed, patients with type 1 diabetes lose blood glucose control, which can result in both acute conditions (for example, ketoacidosis and severe hypoglycaemia)2 and secondary complications (including heart disease, blindness and kidney failure)—even with current insulin replacement therapies3,4. Type 1 diabetes develops as a consequence of a combination of genetic predisposition, largely unknown environmental factors, and stochastic events. For many reasons, postulated to involve population hygiene, sun exposure, and other environmental factors, its incidence has increased dramatically over the last two decades, especially in children less than five years old5. Those under the age of 18 are most often afflicted6, but an equal number of adults over 18 are thought to develop the disease, although incidence in older people receives less media/research attention. In this review, we discuss our current understanding of the cellular/molecular mechanisms of disease aetiology and progression, the usefulness and limitations of rodent models of spontaneous diabetes, the factors that are influencing the current increased incidence and the clinical opportunities for those affected.
Pathophysiology of type 1 diabetes in mouse and human
Although the clinical picture of type 1 diabetes as a progressive loss of β-cell function over a period of years and the requirement for daily insulin treatment for patient survival has been apparent for over a century, the precise immunologic, genetic and physiologic events that control disease initiation and progression continue to be elucidated. During the last 25 years, two key animal models of type 1 diabetes—the inbred BioBreeding (BB) rat7 and non-obese diabetic (NOD) mouse1,8—have been used to study the genetics, pathophysiology and environmental impact on the spontaneous form of this disease. The rodent models have many aspects in common with the human disease, including a number of similarities in genetic loci of susceptibility, influence of the environment and pathogenesis of disease. The studies in NOD mice have demonstrated that the disease occurs as a consequence of a breakdown in immune regulation, resulting in the expansion of autoreactive CD4+ and CD8+ T cells9–11, autoantibody-producing B lymphocytes12–14, and activation of the innate immune system that collaborate to destroy the insulin-producing β-cells15,16. These attributes of the disease are consistent with studies of human type 1 diabetes. We note that of 26 loci identified through the genome-wide association study (GWAS17) of human type 1 diabetes, at least 6 loci are shared between the NOD mouse model and humans at risk for type 1 diabetes, and 19 are associated with immune regulation17,18.
Although the presence of islet tissue-specific autoantibodies in sera from patients with type 1 diabetes was the first diagnostic of autoimmunity (Fig. 1a), there is overwhelming evidence in both the NOD mouse and human disease that autoreactive T cells play a dominant role in disease initiation and progression. CD11c+ dendritic cells and ER-MP23+ macrophages are the first cells to infiltrate the pancreas of NOD mice at approximately three weeks of age. At the same time, or shortly thereafter, potentially pathogenic T cells can be detected surrounding the islets (this is termed peri-insulitis) (Fig. 1b, c)1. These T cells are presumably activated in the pancreatic draining lymph nodes as a result of high turnover of β-cells in the islets leading to antigen presentation19, although the molecular events that initiate the loss of tolerance in this setting are still speculative. Further islet damage leads to the release of self-antigens, leading to epitope spreading (that is, presentation of new autoantigens to the inflamed immune system, leading to newly activated T cells), and amplification by complex islet mononuclear cell infiltrates present at the time of disease onset. Both major histocompatibility complex (MHC) classes I and II restricted islet-antigen-reactive T cells have been identified in NOD mice and in the peripheral blood of type 1 diabetes patients. In many instances, these T cells have been shown to recognize islet autoantigens similar to those seen by autoantibodies (such as insulin, glutamic acid decarboxylase (GAD) and zinc transporter 8 (ZnT8)). The T cells also recognise other islet antigens, such as islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) and chromogranin A, in NOD mice and humans that have particular susceptibility alleles10,20,21. In fact, autoreactive T cells are observed in very young NOD mice and are often found in the blood of susceptible individuals before disease onset. A recent study used an elegant retrogenic mouse T-cell receptor system to demonstrate directly that islet retention of T cells is antigen-specific and cell-intrinsic22. These studies suggest that a limited number of primary islet autoantigens recognized by early-infiltrating T cells may be responsible for disease initiation (Fig. 2). These findings raise the critical question of whether there is a single self-protein responsible for initiating type 1 diabetes.
Figure 1. Markers of diabetes.
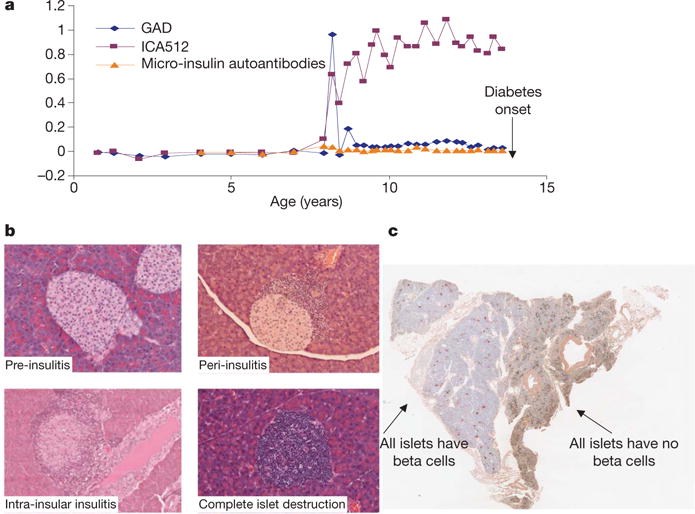
a, Typical child followed from birth until development of diabetes in the DAISY (Diabetes Auto Immunity Study in the Young; http://www.uchsc.edu/misc/diabetes/Teddy/DAISY/DAISY_home.htm) study (M. Rewers, unpublished work), expressing multiple autoantibodies (GAD, the islet cell antibody ICA512 and low-level insulin autoantibodies). b, Islet invasion by lymphocytes of NOD mice is asynchronous during progression to diabetes, often with a mixture of normal islets, peri-insulitis, intra-islet insulitis, and complete β-cell destruction. c, Pathology of the pancreas in a long-term type 1 diabetic (the nPOD program, pancreas 6028, see http://www.jdrfnpod.org/). The section shows lobular areas in which all β-cells (insulin-producing) in all islets have been destroyed (pseudoatrophic islets in which only glucagon, somatostatin, and pancreatic polypeptide cells are present) juxtaposed with regions in which all islets contain insulin-containing β-cells.
Figure 2. Immunologic history of type 1 diabetes.
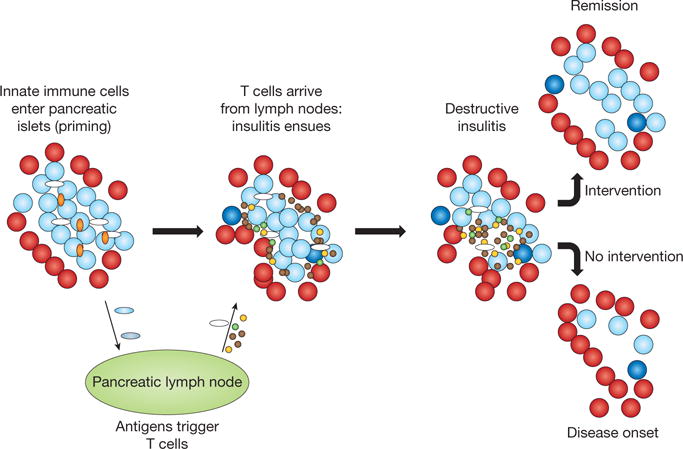
An as-yet-undefined immunologic insult occurs in an individual with genetic predisposition and initiates a chronic low-grade immunologic process (priming). The initiating events involve infiltration of innate immune cells (such as monocytes and natural killer cells with autoreactive B cells) (orange ovals) into the pancreatic islets. The principal site of antigen presentation is thought to be the pancreatic lymph node where islet antigens are presented by antigen-presenting cells (white ovals) to T cells (brown dots). Blue ovals are antigen-presenting cells loaded with islet antigens. B cells (green dots) and dendritic cells may be among the early antigen-presenting cells. The cellular infiltration of islets ensues but the insulitis is uneven. Islets with infiltration may be situated near to islets without cells. The process specifically targets insulin-producing β-cells (light blue circles), while other endocrine cells (red circles) within the islet are spared. In the lymph nodes, the cycle of antigen presentation, activation of adaptive immune cells, licensing of effector T cells and epitope spreading continues with the loss of β-cells over time. There is evidence for a regenerative attempt of β-cells in the midst of the islet inflammation (dark blue circles). Tertiary lymphoid organs are thought to develop within the islets, which may lead to amplification of the adaptive immune response. Regulatory T cells (yellow dots) may arrest this process in its early and late stages but are not able to contain the amplified process in the late stages despite an increase in their numbers. With continued loss of β-cells, hyperglycaemia can be detected. The loss of metabolic function at presentation may be both functional and anatomic, because immune therapies can restore cells that have lost the capacity to produce insulin but have not been destroyed. Without intervention, however, β-cell loss continues.
In this regard, there is strong evidence that defects in thymic T-cell negative selection related to insulin reactivity itself is key to the genetic predisposition towards the disease23,24. First, approximately 20% of individuals with spontaneous mutations of the autoimmune regulator gene AIRE develop type 1 diabetes with other autoimmune diseases, which is thought to reflect their inability to select against islet antigen reactivity during T-cell development25. Insulin is an AIRE-regulated islet protein ectopically expressed in the thymic medullary epithelial cells. In addition, polymorphisms in the insulin promoter that map to a diabetes susceptibility allele control expression of insulin in the thymus, potentially regulating the autoimmune repertoire. Second, a high percentage of pathogenic T cells isolated from early islet infiltrates recognise insulin in the NOD mouse model. In fact, it appears that there is a unique relationship between the expression of certain T-cell receptor Vα regions and the recognition of an insulin B-chain peptide (B9–23)26. However, preclinical evidence supports the notion of a sequential loss of tolerance to multiple epitopes, suggesting that multiple self-antigen specificities are probably involved in disease progression. For example, tolerance to proinsulin prevents induction of IGRP-reactive T cells in NOD mice, whereas diabetes is seen in mice tolerant to IGRP in spite of the absence of IGRP-reactive cells.
Multiple genes within the MHC have been recognized for more than two decades as the dominant loci associated with disease in both the NOD model and human disease. The MHC class I and class II molecules have been suggested to account for both positive and negative selection of autoreactive T cells by virtue of creating the binding groove for antigens presented to T lymphocytes, the final effectors of disease. For instance, the NOD MHC I-Ag7 allele is essential for disease development while alternative MHC alleles (such as MHC I-Ek) act in a dominant fashion to protect NOD mice from disease occurrence. The human MHC is called the human leukocyte antigen (HLA) region. In humans, the presence of susceptibility loci within the HLA complex (the DRB1 0401, DRB1 0402, DRB1 0405, DQA1 0301, DQB1 0302 or DQB1 0201 alleles), which are common in North Americans of European ancestry and Europeans, result in a lod score (the logarithm of the odds of linkage) of 116 (which is 50-fold higher than the next susceptibility locus identified in GWAS studies)17,27. However, strong protection from certain HLA alleles (DRB1 1501, 1401 or 0701 and DQB1 0602, 0503 or 0303 alleles) is dominant, suggesting that the HLA may have a more significant role of protection than predisposition. The key role of HLA genetics in disease susceptibility has proved useful in screening for prevention trials because certain alleles are used to identify patients at high risk of disease, and exclusion of individuals with resistance alleles can reduce the sample size needed for clinical prevention studies. Finally, although T cells reactive with MHC class II may be the driver for disease initiation, CD8+ T cells are likely also to be involved in disease pathogenesis because CD8+ T cells reactive with MHC class I-restricted antigens can be found in the blood and islets of mice and humans with type 1 diabetes.
The mediators of islet destruction have not been precisely defined. In studying cadaver pancreases from humans with type 1 diabetes, it has been rare to observe the degree of insulitis seen in the animal models or evidence of pathogenic T cells28 (see http://www.jdrfnpod.org/). Notably, studies of the pancreas from both new-onset and long-term patients with residual β-cells indicate destruction of β-cells in lobules of the pancreas (likely to underlie slow progression) despite the presence of autoantibodies, typically for years before hyperglycaemia occurs29. Circulating T cells that can be studied in the peripheral blood of patients may be an indirect reflection of the events that are occurring in the pancreas. The lack of easy accessibility to pancreatic tissue from patients except in unusual circumstances28,30 has made analyses of human disease difficult. However, a recent study of cadaver pancreases isolated from patients with new-onset diabetes showed that CD8+ cells and CD68+ appeared to dominate the infiltrates31. Moreover, the creation of a nation-wide network, nPOD (http://www.jdrfnpod.org/), has allowed increased access to potentially informative samples (Fig. 1c).
Yet so far, most of the information we have on disease pathogenesis has been from the NOD mouse studies. Although T helper 1 cells, producing interferon gamma (IFNγ) can transfer disease, especially in CD4+ T-cell-receptor transgenic models, the role of IFNγ in the spontaneous disease remains controversial because IFNγ-deficient NOD mice develop the disease similarly to wild-type NOD mice. Interestingly, disruption of Tbx21, the gene encoding the transcription factor Tbet that controls IFNγ production, ameliorates disease; however, this may be due to the role Tbet plays in T helper 1 cell trafficking and dendritic cell function32. Similar discrepancies have also been observed in studies disrupting the gene encoding the cytokine interleukin (IL)-12, which is key to the T helper 1 cell lineage. In contrast, there is good evidence that a variety of cytotoxicity-inducing molecules (for example, the tumour necrosis factor TNFα, perforin and granzyme B) are critical in disease pathogenesis mediated by both the CD4+ and CD8+ T cells1. The identification of IL-17-producing T helper 17 cells raised the possibility that this population (and the associated IL-23 cytokine) may be involved in the disease. However, recent studies have largely discounted this possibility33.
Other cell types are also involved in disease pathogenesis in NOD mice and are present in human insulitis. This includes natural killer cells, monocytes and potentially other cells of the innate immune system34,35 (modelled in Fig. 2). In the case of B cells, their precise role is less clear. Although they may have a role in autoantibody production, the antigen-specific B cells are very efficient in presenting antigens and produce cytokines that can either promote or suppress immunity36,37. Additionally, the innate immune cells also contribute to the inflammatory milieu, promoting islet cell destruction and immune activation. It is becoming increasingly clear that type 1 diabetes in NOD mice, and possibly in humans, is amplified by immune components of tissue inflammation, analogous to that described for type 2 diabetes, Alzheimer’s, atherosclerosis and other diseases38,39.
Although there are a number of GWAS-identified susceptibility alleles that may contribute to T-cell-mediated islet cell destruction, these immune-response genes have not helped us to identify monogenic pathways that directly lead to disease. Rather, it appears that a global problem of immune regulation may underlie disease susceptibility. For example, mutations of genes encoded in several of the susceptibility loci—including Il2, Il2ra (CD25), Ctla4, PTPN22, and Pdca1 (PD-1)—in multiple animal models lead to the development of a diverse array of autoimmune diseases, including type 1 diabetes. The proteins encoded by these genes have been implicated in maintaining immune homeostasis either by directly tuning the signal strength of T and B cell receptor complex or indirectly through the control of regulatory cell populations critical to controlling autoimmunity.
Most prominent is the potential role of these pathways in the control of Foxp3+ regulatory T (Treg) cells, which are essential suppressors of unwanted autoimmune responses40,41. Treg cells, whose T-cell-receptor repertoire is skewed towards self-protein recognition, are thought to modulate T-cell activation and promote tolerance by suppressing adaptive immunity through direct cell–cell interactions and production of immune modulatory cytokines such as the transforming growth factor TGF-β and IL-10 (ref. 42). However, other proposed Treg cell functions target more generalized mechanisms of inflammation, such as the redox reaction, ATP utilization, tryptophan metabolism and the nitric oxide pathways. These results suggest that Treg cells may play a more generalized role in β-cell survival and function as ‘innate’ regulators of immune-mediated tissue damage. Treg cells exist in fat and control obesity-related inflammatory responses that alter insulin resistance43,44. In fact, Treg cells may affect tissue repair, linking elements of type 1 diabetes and β-cell stress45. Mutations of Foxp3 result in the immune polyendocrinopathy enteropathy X-linked (IPEX) syndrome, which includes autoimmune diabetes within the first years of life. However, polymorphisms in Foxp3 alleles have not been found among the susceptibility loci for type 1 diabetes, so the manifestations of Treg cell alterations controlled by the susceptibility alleles must be subtle.
Nonetheless, the critical importance of Treg cells in the autoimmune setting has been documented. Several studies have shown that Treg cell numbers and functions are altered during disease activity and the subset has been shown to be unstable in some settings, such as the inflamed pancreas of autoimmune mice46. At the time of disease onset in NOD mice, the number and function of the Treg cells, especially the stable CD25+ subset in the pancreas, is significantly reduced in the inflamed islet tissue, most probably owing to defects in IL-2 production47. In fact, a subset of the Treg cells loses its Foxp3 expression, turns on IFNγ, and when adoptively transferred can become pathogenic in its own right46. A similar observation has been made in humans with type 1 diabetes, demonstrating a significant increase in the number of IFNγ-producing Foxp3+ cells in the circulation of new-onset type 1 diabetes patients, concomitant with slightly reduced Foxp3 expression in the circulating Treg cell subset (S. A. McClymont, A. L. Putnam, M. R. Lee, J. H. Esensten, W. Liu, U. Baron, S. Olek, J.A.B. and T. M. Brusko, unpublished work). Coupling these observations with the findings in both the NOD mouse and humans with type 1 diabetes that T effector cells change over time to become resistant to Treg-cell-mediated suppression48,49, we are left with a model that suggests that ultimate disease progression is a direct consequence of the imbalance of Treg cell to effector T cells42 (Fig. 3).
Figure 3. Immune system balance is key to disease pathogenesis.
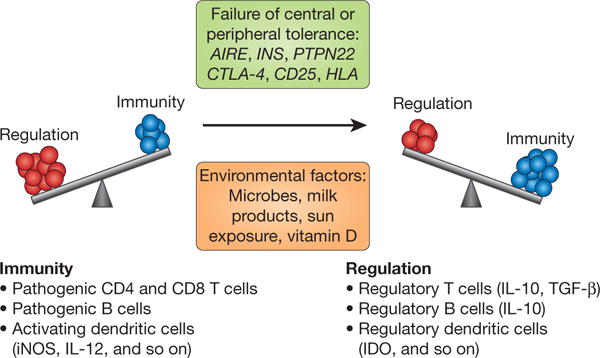
This schematic illustrates the fine balance of immune regulation versus pathogenesis, highlighting a number of genes that are likely to influence the balance through effects on central and peripheral tolerance and the environmental factors that control immunity. The key cell types that affect the balance locally during immune responses are listed (with the regulatory cytokines and proteins given in parentheses). iNOS, inducible nitric oxide synthase. IDO, indoleamine-2,3 dioxygenase.
Finally, the environment is likely to have a strong effect on the development and progression of type 1 diabetes. In the case of NOD mice, the incidence of diabetes dramatically decreases when mice are exposed to microbial stimuli, by injection with mycobacteria, or through contact with various microbial products50,51. Similar observations have been noted in humans, with the development of the so-called “hygiene hypothesis”, which postulates that the increase in type 1 diabetes incidence is most notable in industrialized societies with reduced exposure to parasites52. In this regard, recent studies have shown that the type of “commensal” bacteria can protect NOD mice from developing type 1 diabetes51. Specifically, disruption of the Myd88 gene, encoding an adaptor for multiple innate immune receptors that recognize microbial stimuli, protects NOD mice from developing type 1 diabetes by altering the composition of gut microbiota51. Other environmental factors may also influence disease incidence, ranging from cows’ milk/bovine serum albumin, meat preservatives/N-nitroso compounds, and so on52. There is evidence of a role for vitamin D and its analogues, omega-3 fatty acids, and environmental stress and toxins53–56. Finally, ongoing epidemiological and laboratory research efforts support a potential role for viruses in the disease pathogenesis, either owing to antigenic mimicry or to the possibility that certain viral infections break self-tolerance by activating innate immunity, potentially in concert with risk alleles influencing innate immune function57,58. Each of Coxsackie B4, the seasonal incidence of rhinovirus and influenza has been implicated56. It has been difficult to identify the most significant environmental factors involved because of the multitude of suggested causes. The TEDDY (The Environmental Determinants of Diabetes in the Young; http:teddy.epi.usf.edu/) study is prospectively analysing environmental factors that may be responsible for modifying the incidence of this disease.
Opportunities for preventing and treating type 1 diabetes
As highlighted above, our increased understanding of the pathogenesis of the disease and the identification of genes and environmental factors that control disease incidence have provided a wealth of potential targets for disease intervention (Fig. 4).
Figure 4. Targets of immune intervention in type 1 diabetes.
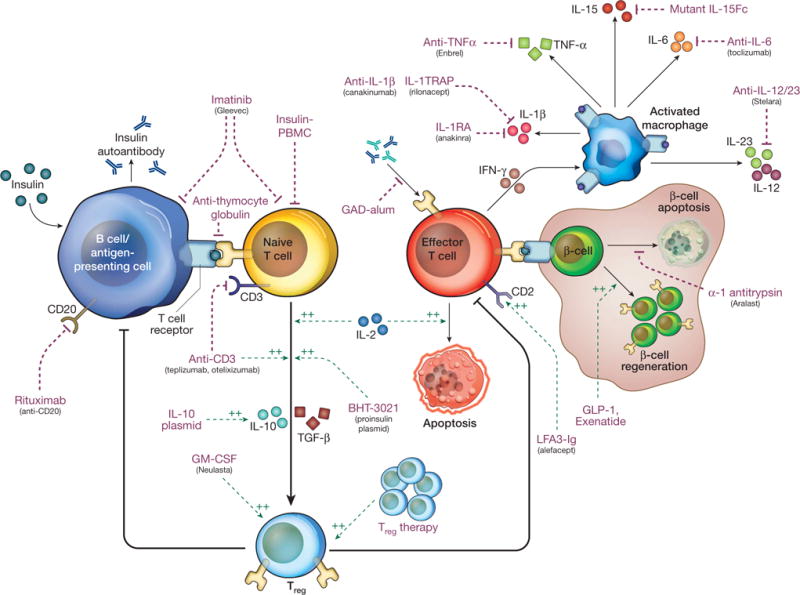
This schematic provides an overview of the pathogenesis of type 1 diabetes, highlighting a number of key pathways that are being targeted by current therapeutics. Although not exhaustive (see Supplementary Table 1), this figure shows that both non-specific and antigen-specific therapies are being tested, which inhibit effector cells and antigen presentation as well as boost regulatory pathways. Purple and green dotted arrows indicate the therapeutics, black arrows are immune and metabolic pathways; a green dotted arrow indicates a positive effect and a purple dotted arrow indicates a negative effect. In addition, these immunotherapies are being combined with drugs that promote β-cell survival to potentially replenish insulin-producing β-cells. The figure has been redrawn after ref. 87, with permission.
Clinical trials for new-onset disease are feasible and have been conducted over the past 20 years but have the following limitations. First, given the reduced level of β-cell numbers at the time of diagnosis, the goal of most clinical trials in type 1 diabetes is to improve functional residual β-cell mass, optimally through induction of immunologic tolerance, while preserving protective immune responses. By definition, this will rarely “cure” the disease because of the significant β-cell destruction that preceded the treatment. Second, since there are no biomarkers of the disease process that are reliably correlated with the pathogenic process, endpoints for studies have been limited to measures of β-cell function and clinical parameters after one or two years, which by definition does not “read-out” the immune pathogenesis but rather the metabolic consequences of the disease. Third, timing may be important. Metabolic and immunologic data from the Diabetes Prevention Trial-1 (DPT-1; (http://www.diabetestrialnet.org/publications/publications.htm#dpt1) and the European Nicotinamide Diabetes Intervention Trial (ENDIT; http://www.bristol.ac.uk/clinicalsciencenorth/diabetes/ri/endit.html), and studies after clinical presentation have been consistent with the model of a chronic autoimmune process that relentlessly leads to β-cell destruction. Human data are consistent with this notion, because the risk of diabetes exceeds 90% in first-degree relatives of patients who are positive for at least two biochemical autoantibodies, whereas it is less than 20% in relatives who are positive for only one autoantibody. On the basis of this model, therefore, antigen-specific tolerance would be predicted to have greatest success in the earliest stages of disease, whereas a broader approach would be needed at the time of diagnosis when there is a polyclonal autoreactive repertoire.
Indeed, prevention trials, which enroll individuals at risk, identified after the development of autoantibodies have been uniformly unsuccessful. Prevention trials involving relatives of people with type 1 diabetes require the screening of a large number of individuals in order to identify a study group because the frequency of autoantibody positivity within this population is only approximately 3.5%. In the DPT-1 trial, parenteral insulin was administered to individuals with positive autoantibody levels and impaired insulin responses to intravenous glucose; this approach was based on studies in the NOD mouse and pilot human studies that showed this treatment to prevent disease progression (http://www.diabetestrialnet.org/index.htm)88. However, the trial failed to show a significant effect of insulin administration on the progression of disease. The ENDIT trial, which proposed to arrest early stages of β-cell injury, enrolled subjects with positive autoantibodies and administered nicotinamide, but likewise showed no beneficial effects. The negative outcomes of these large trials have led to speculation regarding differences between the animal model (the NOD mouse) that is used for preclinical studies and human disease. However, a number of therapies described throughout this review have been translated to humans. So the lack of success in these trials may be due to the use of the wrong dose of the interventional agent or the timing of the intervention, given that the trials involved individuals in whom autoimmunity and β-cell destruction was already initiated.
The most extensive clinical interventions have been performed at the time of disease onset, when nearly all individuals have ‘clinically significant’ levels of C-peptide production (that is, at least 0.2 pmol ml−1 following metabolic stimulation). Some 15 years after completion of the Diabetes Control and Complications Trial (DCCT; http://diabetes.niddk.nih.gov/dm/pubs/control/) there is still compelling evidence that improving short-term control of blood sugars, which can be achieved by preserving endogenous β-cell function, can lead to long-term reductions in diabetes complications, through a process referred to as “metabolic memory”4.
The first successful immunosuppressive agent used in a placebo-controlled, double-blind clinical trial for type 1 diabetes was cyclosporine A59,60. Unfortunately, although β-cell function appeared to be preserved (on the basis of reduced insulin usage), its excessive nephrotoxicity forced termination of therapy in those studies after one year. Moreover, the protective effect was not extended beyond treatment cessation61. In the mid-1980s, a commercial anti-thymocyte globulin in conjunction with prednisone was shown to reduce insulin requirements in a small number of new-onset patients at a time when many were still questioning the autoimmune aetiology of type 1 diabetes62. Finally, based on the hygiene hypothesis and related studies described above, there have been attempts to use a variety of adjuvants, including the bacillus Calmette-Guerin, without success63.
In 2002, there was a theoretical paradigm shift when Herold et al.64 reported that treatment of newly diagnosed patients with type 1 diabetes with the anti-T-cell humanized anti-CD3 monoclonal antibody, teplizimab (mutated to prevent binding to Fc receptors) led to a sustained preservation of C-peptide (insulin) production. A second European report showed similar efficacy using a second anti-CD3 monoclonal antibody otelixizumab (similarly Fc-mutated)65. In an example of mouse models of type 1 diabetes equating with the human disease, a short (that is, two weeks or less) course of an anti-CD3 monoclonal antibody drug (145-2C11) revealed that diabetes was permanently reversed in NOD mice, whereas in humans, C-peptide levels were sustained for at least one to two years in most of the patients and in some cases five years or more, suggesting that short-term therapy can have a long-term effect (of at least two years)66,89. Further studies in mice and patients have suggested that by virtue of its function as a partial T-cell-receptor agonist, Treg cells are selectively preserved by treatment with the anti-CD3 monoclonal antibody. In mice, these cells are localized to the pancreatic lymph nodes and require TGF-β, whereas in humans, both CD4+ and CD8+ Treg cells have been described67,68. The probable mechanism of anti-CD3 monoclonal antibody therefore involves the preservation of Treg cells that may regulate antigen-specific responses involved in the disease. The duration of this effect is still unknown, as is whether the Treg preservation can be boosted by repeated drug administration. These questions are being addressed in clinical trials. In addition, an effect of anti-CD3 monoclonal antibody on the ability of residual effector T cells to be regulated has not been studied.
Initial preclinical studies that showed transfer of diabetes by purified T cells, independently of B lymphocytes, cast doubt on a role of B cells late in the disease course. It was shown, however, that membrane-bound immunoglobulin could partially restore disease to mice deficient in secreted immunoglobulin, suggesting that although there was not an essential role of soluble immunoglobulins (that is, autoantibodies), B cells might be important for antigen presentation or other functions90. Hu et al. used mice expressing the human CD20 transgene to demonstrate that depletion of cells could prevent, and even reverse, diabetes when the cells were depleted at diagnosis69. Preclinical studies suggested that B cells with regulatory function were induced with anti-CD20 monoclonal antibody treatment69,70. More recently, in a blinded placebo-controlled clinical trial, the anti-human CD20 antibody, rituximab, given in four weekly doses was shown to improve provoked C-peptide responses three months after diagnosis71. Subtle differences in B cell subsets were identified in the clinical trial and correlated with clinical responses. The long-term effects of rituximab treatment have not been reported, but a number of observations are of interest. The greatest difference in responses between drug- and placebo-treated subjects occurred three months after entry into the study (which occurred within three months of diagnosis), and the decline in C-peptide production by six months after the treatment began and thereafter was similar in drug and placebo groups. In addition, the duration of immune effects could be problematic, because immunoglobulin M levels remained depressed long-term. The application of this therapy to clinical practice, therefore, requires careful weighing and further study because, in general, long-term immune suppression is not acceptable as a therapeutic option.
The safety concerns and adverse effects of antigen non-specific interventions, as well as the lack of permanent remission of disease with any agent tested to date have heightened interest in antigen-specific interventions that might modulate the disease. Even a partial response, requiring repeated administration of a drug, might be preferable to a broad immune-suppressive agent in this patient population. The polyclonal nature of the autoimmune response, reflected by the presence of multiple autoantibodies and multiple T-cell epitopes that are recognized by peripheral blood cells, might suggest that this goal cannot be achieved, but immunologic tolerance mechanisms, even by antigen-specific cells, are not restricted to a single antigen. The notion of “bystander suppression” mediated by soluble products or cell–cell contact explains how T cells with regulatory properties could modulate the function of T cells specific for other antigens72–74. Cytokines such as IL-10, TGF-β and even IL-4, produced by T cells in response to antigen or antigen-presenting cells, can modulate the function of effector T cells in the vicinity of the antigen. In this way, activated Treg cells exert their function by entering the site of the incipient immune response and need not directly recognize the antigen(s) recognized by effector T cells. By modulating the differentiation of other antigen-specific T cells, pathogenicity could be modified permanently in cells otherwise destined to become effector T cells (“infectious tolerance”)75,76. In addition, CD4+CD25+FoxP3+ and other regulatory T cells are able to modulate the function of effector T cells through contact-dependent mechanisms. We have shown that polyclonal diabetogenic T cells may be inhibited by antigen-specific Treg cells in vitro and in an adoptive transfer model of diabetes77.
Hence, two trials, the oral insulin arm of the DPT-1 and the GAD65 immunization trial in patients with new-onset disease (http://clinicaltrials.gov/ct2/show/NCT00435981?term=GAD65&rank=4), support the concept that an antigen-specific intervention may have broad immunologic effects, even late in the immune progression, provided the antigen selected is directly involved in the disease pathogenesis and/or regulation. A second arm of the DPT-1 study enrolled patients who had positive insulin autoantibodies but did not show the same impairment in insulin responses as those enrolled in the parenteral arm, and administered oral insulin based on the hypothesis that an oral antigen would induce a tolerogenic (producing immunological tolerance) response. The oral insulin administration yielded intriguing results in a subset of patients with high levels of insulin autoantibodies, a finding now being studied in detail by Trialnet78. Immunization with alum GAD65 was also shown to attenuate the loss of C-peptide in individuals treated within six months of diagnosis79. These studies are a prelude to a number of additional antigen-specific therapies that are in late pre-clinical or clinical development. This includes a number of additional insulin-specific therapies that target tolerogenic pathways, other potential regulatory autoantigens and combination therapies that include autoantigens as part of the tolerogenic cocktail (see Fig. 4 and Supplementary Tables 1 and 2). By more directly targeting the pathogenic response with regulatory cells, combined with adoptive immune therapy, we might find an approach, which, if found to be safe, could be repeated on a regular basis without the need to treat patients with broadly active immunosuppressive drugs and put them at risk of treatment-associated adverse events. This strategy is now being tested in type 1 diabetes using CD4+CD25+Foxp3+ Treg cells as well as T regulatory 1 cells IL-10+) adaptive cells80.
Lastly, it has been established that persistent dysregulated inflammation contributes to most chronic diseases, including vascular diseases, cancer, neurodegenerative diseases, metabolic diseases such as type 2 diabetes and autoimmune diseases such as type 1 diabetes. Importantly, it is this dysregulated inflammation that is critical in breaking immune tolerance by disabling key regulatory pathways, such as Treg cells and tolerogenic dendritic cells. Thus, it is not surprising that drugs that target the inflammatory responses are actively being tested in the clinic on the basis of strong preclinical data showing an ability to moderate the progression of type 1 diabetes81. Current trials using anti-cytokine drugs such as IL-1 and IL-15 antagonists, as well as anti-chemokine antagonists, are aimed at reducing inflammation in an attempt to allow re-establishment of immune homeostasis. In fact, these anti-inflammatory drugs have been used preclinically, in combination with other pro-tolerogenic therapies. In this regard, several Food and Drug Administration (FDA)-approved small molecules and biologics have shown very promising results in reversing and inducing long-lasting remission of diabetes in the NOD mouse. Aralast NP (an a1 anti-trypsin used to treat the genetic deficiency)82 and imatinib (Gleevec)83, a cancer drug that inhibits a variety of receptor tyrosine kinases such as c-abl, PDGF and VEGF, have been shown to target the inflammatory macrophages and other innate immune cells that are critical for disease pathogenesis. Both of these drugs are about to enter clinical trials to examine their effect on prolonging residual β-cells in recent-onset diabetics.
Next steps
There are still many unanswered questions that need to be addressed before we are likely to develop an effective and acceptably safe cure for type 1 diabetes. First, to target the T cells involved in the development and progression of the disease, it is essential that we determine the precise specificity of the primary pathogenic T cells. This prerequisite is so that the disease-causing cells can be monitored and the various interventions and complementary strategies can be implemented to eliminate pathogenic cells while enhancing immune regulation. In this regard, recent efforts to access tissues from cadavers of patients with type 1 diabetes may provide a source of antigen-specific T cells at the site of disease activity. Second, linking genotypes with phenotypes and the impact of environment factors will be valuable in determining the best diagnostics and treatments. At present, we have only scratched the surface of understanding the biological pathways of the genes that have been shown to modulate disease activity. Finally, there is a need to broaden our study of disease pathogenesis beyond the T cells. In particular, a better understanding of the role of inflammation and the innate immune response will undoubtedly be important in any comprehensive approach to disease treatment. The recent links between inflammation and multiple diseases ranging from autoimmunity to heart disease, Alzheimer’s and type 2 diabetes suggest that core immune system parameters, including natural killer cells, macrophages and even Treg cells, may act to control tissue damage as a means of modulating the autoimmune phenotype driven by the antigen-specific T cells and perhaps B cells.
It is likely, that, like most autoimmune diseases, combination therapies that take advantage of targeting different arms of the immune system will be most effective at ameliorating disease. A provocative report84 showed that CD34+ stem cell transplantation following myeloablation with cyclophosphamide and conditioning with anti-thymocyte globulin and cyclophosphamide resulted in non-insulin-requiring remissions in 14 of 15 subjects for an average of 16 months—a response that has not been achieved with any single immune modulator. Although the approach is controversial owing to the potentially highly toxic nature of the therapy, its outcome suggests that an aggressive approach may be able to reverse the disease and that certain elements of the protocol (for example, anti-thymocyte globulin and granulocyte/macrophage-colony stimulating factor (GM-CSF) conditioning) may be useful alone or in combination with other agents. More acceptable alternatives may be to combine more generalized immunosuppressives (for example, anti-CD3, anti-B-lymphocytes, and anti-inflammatory drugs) with antigen-specific DNA vaccines and antigenic peptides administered via the mucosa85. A suggested mechanism of this combination involves induction of antigen-specific Treg cells when the antigen is administered under the umbrella of the immune modulator. Moreover, efforts to combine the immunomodulatory therapies with islet-preserving drugs, such as glucagon-like peptide-1 (GLP-1) agonists or islet growth factors, may further enhance therapeutic efficacy86.
Conclusion
The last decade has seen extraordinary progress in our understanding and treatment of type 1 diabetes. The pathophysiology and the genetics are increasingly clear. There are over a dozen novel therapies being tested in individuals with this disease at all stages, including pre-clinically. A better understanding of the mechanisms of disease, combined with a more complete set of results from the clinical trials currently under way, may help to isolate the active components in the disease and eliminate the need for broad immunosuppressive treatment. We are confident that the next decade will bring new insights into the linkages between genotype and phenotype, new sources of islets that can be used to replace the destroyed β-cells, and combination therapies that will selectively target antigen-driven pathways to re-reestablish tolerance in this and other autoimmune settings.
Supplementary Material
Acknowledgments
We thank the many people in our laboratories and the community who have made significant contributions to the research highlighted in this review. We owe special thanks to L. Lanier, M. Anderson and M. Atkinson as well as the members of the Brehm Coalition for many discussions and critiques of the manuscript. Additionally, we thank J. Matthews for putting together Fig. 4. We acknowledge support from NIAID, NIDDK, JDRF, CDF and the Brehm Coalition.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions
Each co-author (G.E., K.H. and J.A.B.) contributed experimental results, data analysis, writing and creative contributions to this work.
References
- 1.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 2.Vauzelle-Kervroedan F, et al. Analysis of mortality in French diabetic patients from death certificates: a comparative study. Diabete Metab. 1999;25:404–411. [PubMed] [Google Scholar]
- 3.Maahs DM, Rewers M. Editorial: mortality and renal disease in type 1 diabetes mellitus—progress made, more to be done. J Clin Endocrinol Metab. 2006;91:3757–3759. doi: 10.1210/jc.2006-1730. [DOI] [PubMed] [Google Scholar]
- 4.Steffes MW, et al. (EDIC Research Group). Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: the Epidemiology of Diabetes Interventions and Complications (EDIC) study. J Am Med Assoc. 2003;290:2159–2167. doi: 10.1001/jama.290.16.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harjutsalo V, Sjoberg L, Tuomilehto J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet. 2008;371:1777–1782. doi: 10.1016/S0140-6736(08)60765-5. [DOI] [PubMed] [Google Scholar]
- 6.Lorenzen T, Pociot F, Hougaard P, Nerup J. Long-term risk of IDDM in first-degree relatives of patients with IDDM. Diabetology. 1994;37:321–327. doi: 10.1007/BF00398061. [DOI] [PubMed] [Google Scholar]
- 7.Mordes JP, et al. The BB/Wor rat and the balance hypothesis of autoimmunity. Diabetes Metab Rev. 1996;12:103–109. doi: 10.1002/(SICI)1099-0895(199607)12:2<103::AID-DMR161>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 8.Makino S, et al. Breeding of a non-obese, diabetic strain of mice. Exp Anim. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 9.DiLorenzo TP, Serreze DV. The good turned ugly:immunopathogenic basis for diabetogenic CD8+ T cells in NOD mice. Immunol Rev. 2005;204:250–263. doi: 10.1111/j.0105-2896.2005.00244.x. [DOI] [PubMed] [Google Scholar]
- 10.Burton AR, et al. On the pathogenicity of autoantigen-specific T-cell receptors. Diabetes. 2008;57:1321–1330. doi: 10.2337/db07-1129. [DOI] [PubMed] [Google Scholar]
- 11.Han B, et al. Developmental control of CD8 T cell-avidity maturation in autoimmune diabetes. J Clin Invest. 2005;115:1879–1887. doi: 10.1172/JCI24219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serreze DV, et al. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161:3912–3918. [PubMed] [Google Scholar]
- 13.Greeley SA, et al. Elimination of maternally transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nature Med. 2002;8:399–402. doi: 10.1038/nm0402-399. [DOI] [PubMed] [Google Scholar]
- 14.Hu CY, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zipris D, et al. TLR9-signaling pathways are involved in Kilham rat virus-induced autoimmune diabetes in the biobreeding diabetes-resistant rat. J Immunol. 2007;178:693–701. doi: 10.4049/jimmunol.178.2.693. [DOI] [PubMed] [Google Scholar]
- 16.Devendra D, et al. Interferon-α as a mediator of polyinosinic:polycytidylic acid-induced type 1 diabetes. Diabetes. 2005;54:2549–2556. doi: 10.2337/diabetes.54.9.2549. [DOI] [PubMed] [Google Scholar]
- 17.Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med. 2009;360:1646–1654. doi: 10.1056/NEJMra0808284. [DOI] [PubMed] [Google Scholar]
- 18.Wicker LS, et al. Type 1 diabetes genes and pathways shared by humans and NOD mice. J Autoimmun. 2005;25(Suppl):29–33. doi: 10.1016/j.jaut.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Turley S, et al. Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med. 2003;198:1527–1537. doi: 10.1084/jem.20030966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lieberman SM, DiLorenzo TP. A comprehensive guide to antibody and T-cell responses in type 1 diabetes. Tissue Antigens. 2003;62:359–377. doi: 10.1034/j.1399-0039.2003.00152.x. [DOI] [PubMed] [Google Scholar]
- 21.Medarova Z, et al. In vivo imaging of a diabetogenic CD8+ T cell response during type 1 diabetes progression. Magn Reson Med. 2008;59:712–720. doi: 10.1002/mrm.21494. [DOI] [PubMed] [Google Scholar]
- 22.Lennon GP, et al. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity. 2009;31:643–653. doi: 10.1016/j.immuni.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vafiadis P, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nature Genet. 1997;15:289–292. doi: 10.1038/ng0397-289. [DOI] [PubMed] [Google Scholar]
- 24.Pugliese A, et al. The insulin gene is transcribed in the human thymus and transcription levels correlate with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type I diabetes. Nature Genet. 1997;15:293–297. doi: 10.1038/ng0397-293. [DOI] [PubMed] [Google Scholar]
- 25.Gardner JM, Fletcher AL, Anderson MS, Turley SJ. AIRE in the thymus and beyond. Curr Opin Immunol. 2009;21:582–589. doi: 10.1016/j.coi.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi M, et al. Conserved T cell receptor alpha-chain induces insulin autoantibodies. Proc Natl Acad Sci USA. 2008;105:10090–10094. doi: 10.1073/pnas.0801648105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erlich H, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the Type 1 Diabetes Genetics Consortium families. Diabetes. 2008;57:1084–1092. doi: 10.2337/db07-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dotta F, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA. 2007;104:5115–5120. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gianani R, et al. Dimorphic histopathology of long-standing childhood-onset diabetes. Diabetologia. 2010;53(4):690–698. doi: 10.1007/s00125-009-1642-y. [DOI] [PubMed] [Google Scholar]
- 30.Itoh N, et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92:2313–2322. doi: 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willcox A, et al. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155:173–181. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Esensten JH, Lee MR, Glimcher LH, Bluestone JA. T-bet-deficient NOD mice are protected from diabetes due to defects in both T cell and innate immune system function. J Immunol. 2009;183:75–82. doi: 10.4049/jimmunol.0804154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bending D, et al. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest. 2009;119(3):565–572. doi: 10.1172/JCI37865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogasawara K, et al. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18:41–51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- 35.Feuerer M, et al. How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity. 2009;31:654–664. doi: 10.1016/j.immuni.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong FS, et al. Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes. 2004;53:2581–2587. doi: 10.2337/diabetes.53.10.2581. [DOI] [PubMed] [Google Scholar]
- 37.Xiu Y, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol. 2008;180:2863–2875. doi: 10.4049/jimmunol.180.5.2863. [DOI] [PubMed] [Google Scholar]
- 38.Boni-Schnetzler M, et al. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J Clin Endocrinol Metab. 2008;93:4065–4074. doi: 10.1210/jc.2008-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koulmanda M, et al. Modification of adverse inflammation is required to cure new-onset type 1 diabetic hosts. Proc Natl Acad Sci USA. 2007;104:13074–13079. doi: 10.1073/pnas.0705863104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feuerer M, Hill JA, Mathis D, Benoist C. Foxp31 regulatory T cells: differentiation, specification, subphenotypes. Nature Immunol. 2009;10:689–695. doi: 10.1038/ni.1760. [DOI] [PubMed] [Google Scholar]
- 41.Bacchetta R, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest. 2006;116:1713–1722. doi: 10.1172/JCI25112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Q, Bluestone JA. The Foxp31 regulatory T cell: a jack of all trades, master of regulation. Nature Immunol. 2008;9:339–344. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winer S, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nature Med. 2009;15:921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feuerer M, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nature Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan Z, Garg SK, Kipnis J, Banerjee R. Extracellular redox modulation by regulatory T cells. Nature Chem Biol. 2009;5:721–723. doi: 10.1038/nchembio.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang Q, et al. Central role of a defective IL-2 production in triggering islet autoimmune destruction. Immunity. 2008;28:687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou X, et al. Foxp3 instability leads to the generation of pathogenic memory T cells in vivo. Nat Immun. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider A, et al. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol. 2008;181:7350–7355. doi: 10.4049/jimmunol.181.10.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.D’Alise AM, et al. The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proc Natl Acad Sci USA. 2008;105:19857–19862. doi: 10.1073/pnas.0810713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qin HY, Singh B. BCG vaccination prevents insulin-dependent diabetes mellitus (IDDM) in NOD mice after disease acceleration with cyclophosphamide. J Autoimmun. 1997;10:271–278. doi: 10.1006/jaut.1997.0136. [DOI] [PubMed] [Google Scholar]
- 51.Wen L, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weintrob N, et al. Type 1 diabetes environmental factors and correspondence analysis of HLA class II genes in the Yemenite Jewish community in Israel. Diabetes Care. 2001;24:650–653. doi: 10.2337/diacare.24.4.650. [DOI] [PubMed] [Google Scholar]
- 53.Virtanen SM, et al. Age at introduction of new foods and advanced beta cell autoimmunity in young children with HLA-conferred susceptibility to type 1 diabetes. Diabetologia. 2006;49:1512–1521. doi: 10.1007/s00125-006-0236-1. [DOI] [PubMed] [Google Scholar]
- 54.Akerblom HK, et al. Dietary manipulation of beta cell autoimmunity in infants at increased risk of type 1 diabetes: a pilot study. Diabetologia. 2005;48:829–837. doi: 10.1007/s00125-005-1733-3. [DOI] [PubMed] [Google Scholar]
- 55.Norris JM, et al. Omega-3 polyunsaturated fatty acid intake and islet autoimmunity in children at increased risk for type 1 diabetes. J Am Med Assoc. 2007;298:1420–1428. doi: 10.1001/jama.298.12.1420. [DOI] [PubMed] [Google Scholar]
- 56.Jun HS, Yoon JW. A new look at viruses in type 1 diabetes. ILAR J. 2004;45:349–374. doi: 10.1093/ilar.45.3.343. [DOI] [PubMed] [Google Scholar]
- 57.Richardson SJ, et al. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52:1143–1151. doi: 10.1007/s00125-009-1276-0. [DOI] [PubMed] [Google Scholar]
- 58.Nejentsev S, et al. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bougneres PF, et al. Factors associated with early remission of type I diabetes in children treated with cyclosporine. N Engl J Med. 1988;318:663–670. doi: 10.1056/NEJM198803173181103. [DOI] [PubMed] [Google Scholar]
- 60.Stiller CR, et al. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science. 1984;223:1362–1367. doi: 10.1126/science.6367043. [DOI] [PubMed] [Google Scholar]
- 61.Feutren G, et al. Cyclosporin increases the rate and length of remissions in insulin dependent diabetes of recent onset. Results of a multicentre double-blind trial. Lancet. 1986;2:119–124. doi: 10.1016/s0140-6736(86)91943-4. [DOI] [PubMed] [Google Scholar]
- 62.Eisenbarth GS, et al. Anti-thymocyte globulin and prednisone immunotherapy of recent onset type I diabetes mellitus. Diabetes Res. 1985;2:271–276. [PubMed] [Google Scholar]
- 63.Allen HF, et al. Effect of BCG vaccination on new-onset insulin-dependent diabetes mellitus: a randomized clinical study. Diabetes Care. 1998;22:1703–1707. doi: 10.2337/diacare.22.10.1703. [DOI] [PubMed] [Google Scholar]
- 64.Herold KC, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 65.Keymeulen B. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 66.Herold KC, et al. Treatment of patients with new onset type 1 diabetes with a single course of anti-CD3 mAb teplizumab preserves insulin production for up to 5 years. Clin Immunol. 2009;132:166–173. doi: 10.1016/j.clim.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nature Rev Immunol. 2007;7:622–632. doi: 10.1038/nri2134. [DOI] [PubMed] [Google Scholar]
- 68.Bisikirska B, et al. TCR stimulation with modified anti-CD3 mAb expands CD8 T cell population and induces CD8 CD25 Tregs. J Clin Invest. 2005;115:2904–2913. doi: 10.1172/JCI23961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu CY, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev. 2008;224:201–214. doi: 10.1111/j.1600-065X.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- 71.Pescovitz MD, et al. Rituxan, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wise M, et al. CD4 T cells can reject major histocompatibility complex class I-incompatible skin grafts. Eur J Immunol. 1999;29:156–167. doi: 10.1002/(SICI)1521-4141(199901)29:01<156::AID-IMMU156>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 73.Homann D, et al. Autoreactive CD4+ T cells protect from autoimmune diabetes via bystander suppression using the IL-4/Stat6 pathway. Immunity. 1999;11:463–472. doi: 10.1016/s1074-7613(00)80121-1. [DOI] [PubMed] [Google Scholar]
- 74.Masteller EL, et al. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol. 2005;175:3053–3059. doi: 10.4049/jimmunol.175.5.3053. [DOI] [PubMed] [Google Scholar]
- 75.Qin S, et al. ‘Infectious’ transplantation tolerance. Science. 1993;259:974–977. doi: 10.1126/science.8094901. [DOI] [PubMed] [Google Scholar]
- 76.Waldmann H, Cobbold S. How do monoclonal antibodies induce tolerance? A role for infectious tolerance? Annu Rev Immunol. 1998;16:619–644. doi: 10.1146/annurev.immunol.16.1.619. [DOI] [PubMed] [Google Scholar]
- 77.Tang Q, et al. In vitro expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Skyler JS, et al. Effects of oral insulin in relatives of patients with type 1 diabetes: the Diabetes Prevention Trial—Type 1. Diabetes Care. 2005;28:1068–1076. doi: 10.2337/diacare.28.5.1068. [DOI] [PubMed] [Google Scholar]
- 79.Ludvigsson J, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359:1909–1920. doi: 10.1056/NEJMoa0804328. [DOI] [PubMed] [Google Scholar]
- 80.Levings MK, Sangregorio R, Roncarolo MG. Human CD25(+)CD4(+) T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001;193:1295–1302. doi: 10.1084/jem.193.11.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Strom TB, Koulmanda M. Cytokine related therapies for autoimmune disease. Curr Opin Immunol. 2008;20:676–681. doi: 10.1016/j.coi.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Koulmanda M, et al. Curative and beta cell regenerative effects of α1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc Natl Acad Sci USA. 2008;105:16242–16247. doi: 10.1073/pnas.0808031105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Louvet C, et al. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA. 2008;105:18895–18900. doi: 10.1073/pnas.0810246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Couri CE, Voltarelli JC. Stem cell therapy for type 1 diabetes mellitus: a review of recent clinical trials. Diabetol Metabol Syndr. 2009;1:1–19. doi: 10.1186/1758-5996-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bresson D, et al. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116:1371–1381. doi: 10.1172/JCI27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suarez-Pinzon WL, et al. Combination therapy with epidermal growth factor and gastrin increases beta-cell mass and reverses hyperglycemia in diabetic NOD mice. Diabetes. 2005;54:2596–2601. doi: 10.2337/diabetes.54.9.2596. [DOI] [PubMed] [Google Scholar]
- 87.Matthews JB, et al. Developing combination immunotherapies for type 1 diabetes: recommendations from the ITN-JDRF Type 1 Diabetes Combination Therapy Assessment Group. Clin Exp Immunol. doi: 10.1111/j.1365-2249.2010.04153.x. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Skyler JS, et al. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial–Type 1. Diabetes Care. 2005;28:1068–1075. doi: 10.2337/diacare.28.5.1068. [DOI] [PubMed] [Google Scholar]
- 89.Herold KC, et al. A single course of anti-CD3 monoclonal antibody hOKT3γ1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of Type 1 diabetes. Diabetes. 2005;54:1763–1769. doi: 10.2337/diabetes.54.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wong FS, et al. Investigation of the role of β-cells in type 1 diabetes in the NOD mouse. Diabetes. 2004;53:2581–2587. doi: 10.2337/diabetes.53.10.2581. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.