

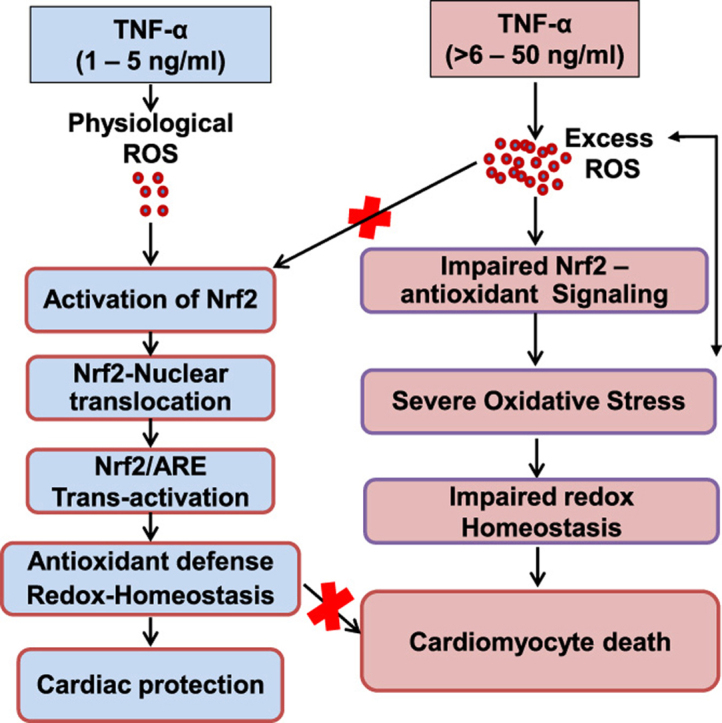
Abstract
Antagonizing TNF-α signaling attenuates chronic inflammatory disease, but is associated with adverse effects on the cardiovascular system. Therefore the impact of TNF-α on basal control of redox signaling events needs to be understand in more depth. This is particularly important for the Keap1/Nrf2 pathway in the heart and in the present study we hypothesized that inhibition of a low level of TNF-α signaling attenuates the TNF-α dependent activation of this cytoprotective pathway. HL-1 cardiomyocytes and TNF receptor1/2 (TNFR1/2) double knockout mice (DKO) were used as experimental models. TNF-α (2–5 ng/ml, for 2 h) evoked significant nuclear translocation of Nrf2 with increased DNA/promoter binding and transactivation of Nrf2 targets. Additionally, this was associated with a 1.5 fold increase in intracellular glutathione (GSH). Higher concentrations of TNF-α (>10–50 ng/ml) were markedly suppressive of the Keap1/Nrf2 response and associated with cardiomyocyte death marked by an increase in cleavage of caspase-3 and PARP. In vivo experiments with TNFR1/2-DKO demonstrates that the expression of Nrf2-regulated proteins (NQO1, HO-1, G6PD) were significantly downregulated in hearts of the DKO when compared to WT mice indicating a weakened antioxidant system under basal conditions. Overall, these results indicate that TNF-α exposure has a bimodal effect on the Keap1/Nrf2 system and while an intense inflammatory activation suppresses expression of antioxidant proteins a low level appears to be protective.
Key words: Nrf2 signaling, TNF-α, Antioxidants, Oxidative stress, Apoptosis
Graphical abstract
Highlights
-
•
TNF-α promotes oxidative stress in a dose dependent manner in HL-1 cardiomyocytes.
-
•
Lower concentration of TNF-α evoked nuclear translocation of Nrf2.
-
•
TNF-α induced Nrf2 is functionally active in regulating antioxidant response.
-
•
Abrogation of TNF-α signaling selectively impairs Nrf2-dependent antioxidant regulation in double receptor knockout mice.
1. Introduction
The family of redox regulated transcription factors including p53, NF-κB, AP-1, Nrf2 and Keap1 and their partner proteins have critical cysteine residues in the DNA binding domain which form the basis of their redox sensitive transcriptional regulation [1], [2], [3], [4]. Of these, one of the best studied is Nrf2 which is a redox-sensitive basic leucine zipper transcription factor and a master regulator of the electrophile response element (EpRe or ARE) dependent cellular defense system [5], [6]. Using Nrf2 knockout in vivo and in vitro models, several studies reveal important roles for this transcription factor in regulating antioxidant and cytoprotective gene expression in various pathological conditions including cancer, inflammation and neurodegenerative diseases [5], [7], [8], [9]. Under basal conditions, Nrf2 is sequestered by the Kelch like ECH associated protein (Keap1), a BTB-Kelch substrate adapter protein, in the cytoplasm by binding to its Neh2 domain [10]. This promotes the rapid ubiquitin-dependent ligation to the cullin-3 ubiquitin ligase leading to proteasomal degradation of Nrf2 thus preventing its nuclear entry [11]. Modification of critical thiol residues on Keap1 by electrophiles or other oxidants leads to the release of Nrf2 to the nucleus [12], [13]. Therefore, the Keap1/Nrf2 pathway is considered as one of the primary regulatory nodal points for the cellular oxidative/electrophilic stress response.
Studies with TNF-α [14], [15], [16] have shown an important role in a number of pathologies associated with oxidative stress including diabetes, cancer, cardiac hypertrophy and cardiomyopathy [17], [18], [19], [20]. Consistent with these reports, genetic ablation of TNF-α and its resultant signaling enabled increased but short-term protection against fibro-proliferative effects of asbestos inhalation and dextran sodium sulfate-induced colitis in mouse models [21], [22]. Subsequently, there are several clinical studies demonstrating that TNF antagonists can be an effective class of anti-inflammatory, with an associated antioxidant effect [23], [24], [25]. Over 5 anti-TNF drugs are currently being used therapeutically and typically function as neutralizing antibodies or a soluble TNF receptor. Notably, chronic use of TNF-α blockers and anti-TNF α therapies have shown to be deleterious in various cells and organ systems and is associated with risk of developing cancer, demyelinating disorders and cardiovascular complications [15], [26], [27], [28], [29], [30]. In addition, prolonged TNF-α signaling deficiency cause wide range of immune system defects starting from asymptomatic immunological abnormalities to life-challenging autoimmune diseases [28], [31], [32]. At present the mechanisms which underlie these adverse effects due to TNF-α inhibition are not clear. One possibility is that prolonged TNF-α blockade may suppress ROS generation below the threshold which is required for physiological regulation of the Keap1/Nrf2 pathway. In support of this concept recent studies suggest that basal levels of TNF-α may have beneficial effects in acute heart ischemia [33], [34].
Previous studies have shown that TNF-α activates Nrf2 in human monocytes [35], but in this cell type this is also associated with a change to metabolically glycolytic phenotype and this response could therefore underlie this response [36]. The effect of TNF-α-dependent activation of the Keap1/Nrf2 pathway in the cardiomyocyte are unknown and this is important since cardiomyocytes rely more on oxidative capacity and change to glycolysis is not possible to meet the energetic demands of the heart [37]. A range of concentrations of TNF-α (1–100 ng/ml) have been commonly used in different cellular models and elicit diverse cellular responses ranging from sub-inflammatory to inflammatory and apoptotic effects [38], [39]. These responses are partly due to the inherent differences in the levels of TNFR1 and TNFR2 expression and the ability of TNF-α to activate these receptors in the context of the differential redox status that exists in different cell types [35], [40], [41], [42], [43], [44]. Moreover, sustained TNF-α signaling and the adverse effects of its over-activation have received wide attention because of their effects on cell death and inflammatory pathways in cardiomyocytes [38]. The physiologically relevant and/or sub-inflammatory effects of TNF-α in regulating cytoprotective mechanisms (involving Nrf2) in cardiomyocytes are poorly understood. Given that (i) a broad spectrum of biological effects of TNF-α depends on the type and growth state of the target cell [45], (ii) its signaling is not constitutive within the heart but rather is temporally coupled to the response to metabolic or oxidative stress [46], [47], [48], it is important to note that the response to cytokines will be cell specific. Here, the concentrations of TNF-alpha (1–50 ng/ml) used encompass for the first time the range from sub-inflammatory to inflammatory with the impact on the Keap1/Nrf2 signaling pathway in cardiomyocytes. Further, we have tested the hypothesis, that TNF-α dependent activation is essential for redox regulation of Keap1/Nrf2 using HL-1 cardiomyocytes and TNFR1/2 DKO mice.
2. Methods
2.1. Reagents and antibodies
TNF–α, Claycomb medium, Fetal Bovine Serum, norepinephrine, gelatin and fibronectin were purchased from Sigma (St. Louis, MO, USA). Trypsin, PBS, Penicillin/streptomycin, l-glutamine etc., were obtained from Life technologies (Carlsbad, CA, USA). Reagents for RNA extraction and real-time RT-PCR quantification were purchased from Qiagen (Valencia, CA, USA). The primers used in this study were obtained from IDT technologies, (Coralville, IA, USA). The fluorescent probes Annexin V-FITC, Propidium Iodide (PI), 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) were from Molecular Probes/Invitrogen Corp., (Carlsbad, CA, USA). TransAM kit for Nrf2 was purchased from Active motif (Carlsbad, CA, USA). Glutathione kit was from Cayman (Ann Arbor, MI, USA). Protein assay reagent was from Biorad (Hercules, CA, USA). PVDF membrane was obtained from Millipore (Billerica, MA, USA). Antibodies were obtained from different sources and they are as follows: Catalase (219,010, Calbiochem, Merck KGaA, Germany), the following from Abcam (Cambridge, MA, USA): glutathione peroxidase (GPX1; ab22604), NQO1 (ab34173), SOD1 (ab13498), SOD2 (ab13534), GCLC (ab41463), GCLM (ab81445), GAPDH (ab9485) and G6PD (NB100-236, Novus Biologicals, CO, USA). Secondary rabbit and mouse antibodies conjugated with horseradish peroxidase IgG were purchased from Vector Laboratories (Burlingame, CA, USA). ECL kit was obtained from Thermo Fisher Scientific (Waltham, MA, USA).
2.2. Cell culture and treatments
HL-1 mouse cardiomyocytes were cultured in 0.02% gelatin and fibronectin pre-coated cell culture grade plates/flasks using Claycomb medium supplemented with 10% fetal bovine serum, 1% penicillin/ streptomycin, 1% l-glutamine and Norepinephrine at 37 °C in 5% CO2−95% air. Cells were cultured in 6 well, T-25 or 60 mm culture plates. All the TNF-α treatment was carried out in serum free media. Cells were treated with different concentrations of TNF-α (0, 1, 2, 5, 10 and 50 ng/ml) for 2 h and 24 h and washed with ice cold phosphate buffered saline (PBS) and used for further analysis.
2.3. Annexin-V Staining and FACS analysis
Annexin V binding was used to measure the apoptosis in cells. After TNF-α treatment, untreated and treated cells were washed with PBS. The harvested cells were centrifuged and the pellets were suspended in buffer containing annexin V-FITC, PI and were incubated in the dark for 15 min in room temperature. Untreated single cell events were gated and used as a control for the analysis. At least 10,000 cells were counted for each measurement. The stained cells were tested using flow cytometer (Becton Dickinson) and analyzed using Cell Quest (BD Biosciences) software.
2.4. Measurement of DCFDH-reactive signals
HL-1 cardiomyocytes were grown in 6 well plates treated with TNF-α at indicated concentrations. The treated cells were washed with PBS followed by H2DCFDA (5 µM) incubation for 30 min in the dark. The unbound H2DCFDA was removed by washing with PBS and H2DCFDA fluorescence was visualized and images were captured using fluorescence microscopy. The fluorescence intensity was measured using Image J and graphs were made using Graph pad Prism software.
2.5. Immunofluorescence determination of Nrf2 nuclear translocation
Nuclear translocation of Nrf2 induced by TNF-α was determined by immunofluorescence and TransAM Nrf2 binding assay. At the end of TNF-α treatment, cells were fixed with 4% paraformaldehyde at 37 °C for 15 min and rinsed with PBS containing 2% BSA. Cells were then permeabilized with 1% Triton X-100 25 °C for 30 min. Followed by rinsing with PBS, cells were incubated with anti-Nrf2 antibody (1:500) for 2 h and incubated with red-fluorescent Alexa Flour 594 rabbit anti-mouse IgG antibody for 1 h. DAPI (Abcam) was used to stain the nuclei. The cells were imaged using a Nikon Eclipse A1 Microscopy System and the intensity was measured using Image J software.
2.6. ELISA based measurement of Nrf2 activity
Briefly, 10 µg of nuclear extracts from each sample in triplicate were incubated in a 96-well plate that was coated with oligonucleotide containing a consensus binding site for Nrf2. For competitive binding experiments, which measure the specificity of the assay, 10 μg of nuclear extract from the TNF-α-treated cells were assayed in the presence of wild-type or mutated competitor oligonucleotides. After 1 h of incubation, the wells were washed and incubated with 100 μl of a 1:1000 dilution of rabbit polyclonal antibody against Nrf2. Incubation with normal rabbit polyclonal IgG was also performed separately to determine the specificity of the Nrf2 antibody. The wells were then washed, followed by incubation with 100 μl of a 1:1000 dilution of horseradish peroxidase-conjugated, anti-rabbit secondary antibody at room temperature. The wells were developed using 100 μl of TMB substrate for 15 min before addition of 100 μl of stop solution. Absorbance was read at 450 nm with a reference wavelength of 650 nm using a plate reader from Bio-Rad Laboratories. All the values for the TNF-α treated samples (for each concentration) were divided by the control values and expressed as a fold expression.
2.7. RNA isolation, reverse transcription, and gene expression using qPCR analysis
Total RNA was isolated from control and TNF-α treated HL-1 cells or TNFαR1/R2-DKO mouse heart using Qiagen RNeasy Mini kit and 1.25 µg RNA was processed for cDNA synthesis using Qiagen reverse transcription kit (205,311) as per the supplier instructions. An aliquot of cDNA template (25–50 ng), 10 μl of QuantiFast SYBR green master mix (204,054), and appropriate primers were used for quantitative real-time RT-PCR (qPCR) analysis and analyzed in a Light Cycler (Roche Bio) [49]. Fold changes of mRNA expression of different targets were quantified using Ct values, and the relative mRNA expression levels for all samples were obtained by normalizing to the level of the housekeeping gene Arbp1 or Gapdh mRNA expression. All the primers used for qPCR were given in Table 1. Semi-quantitative PCR was also carried out for TNFR1, TNFR2 and GAPDH using gene specific primer by following PCR conditions; an initial denaturation at 95 °C for 3 min followed by 95 °C for 30 s, 60 °C for 30 s, 72 °C for 45 s, and finally an extension at 72 °C for 5 min. The PCR products were run in 2% agarose gel and the products were seen at −100 bp sizes by ethidium bromide staining [50] using Amersham Imager 600 RGB.
Table 1.
Primer list used for the semi-quantitative and quantitative PCR.
| Genes | Sequences (5′….3′) |
|---|---|
| mNrf2 F | CTGAACTCCTGGACGGGACTA |
| mNrf2 R | CGGTGGGTCTCCGTAAATGG |
| mKeap1 F | TGCCCCTGTGGTCAAAGTG |
| mKeap1 R | GGTTCGGTTACCGTCCTGC |
| mCat F | GGAGGCGGGAACCCAATAG |
| mCat R | GTGTGCCATCTCGTCAGTGAA |
| mGpx 1 F | CCACCGTGTATGCCTTCTCC |
| mGpx 1 R | AGAGAGACGCGACATTCTCAAT |
| mNqo1 F | AGGATGGGAGGTACTCGAATC |
| mNqo1R | TGCTAGAGATGACTCGGAAGG |
| mSod-1 F | AACCAGTTGTGTTGTCAGGAC |
| mSod-1 R | CCACCATGTTTCTTAGAGTGAGG |
| mSod2 F | TGGACAAACCTGAGCCCTAAG |
| mSod2 R | CCCAAAGTCACGCTTGATAGC |
| mGclc F | GGACAAACCCCAACCATCC |
| mGclc R | GTTGAACTCAGACATCGTTCCT |
| mGclm F | CTTCGCCTCCGATTGAAGATG |
| mGclm R | AAAGGCAGTCAAATCTGGTGG |
| mGapdh F | TGACCTCAACTACATGGTCTACA |
| mGapdh R | CTTCCCATTCTCGGCCTTG |
| mG6pd F | TCAGACAGGCTTTAACCGCAT |
| mG6pd R | CCATTCCAGATAGGGCCAAAGA |
| mTnfr1 F | CCGGGAGAAGAGGGATAGCTT |
| mTnfr1 R | TCGGACAGTCACTCACCAAGT |
| mTnfr2 F | GCCCAGGTTGTCTTGACACC |
| mTnfr2 R | CACAGCACATCTGAGCCTTCC |
2.8. Preparation of cell lysates, heart homogenates, SDS-PAGE, and immunoblotting
Cells lysates were prepared from control and TNF-α treated HL-1 cardiomyocytes using lysis buffer (20 mM HEPES, 400 mM NaCl, 1.5 mM EDTA, 1.5 mM MgCl2, with freshly prepared 0.1 mM phenyl methylsulfonyl fluoride (PMSF), 1 mM dithiothreitol, 1% Triton X-100, pH 7.9), and centrifuged at 6000 rpm for 10 min. Heart tissues from WT and TNFR DKO mice were harvested and flash-frozen in liquid nitrogen. Tissue homogenates were prepared using cytosolic extraction buffer (10 mM HEPES, 60 mM KCl, 1 mM EDTA with freshly prepared 0.1 mM phenyl methylsulfonyl fluoride (PMSF), 1 mM dithiothreitol and 1% Triton X-100), and centrifuged at 5000 rpm for 6 min. Equal amount (25 µg) of proteins were resolved by electrophoresis on 10% polyacrylamide gels and electrotransferred to PVDF membrane and blocked using 5% milk. After incubation with specific primary antibodies against NQO1, HO-1, G6PD, GCLC, GCLM, GPX1, SOD1, SOD2 and CAT, the membranes were washed with PBS-Tween 20 and incubated with horseradish peroxidase conjugated with anti-rabbit or anti-mouse IgG for 1 h. The antigen-antibody complex was detected using an ECL chemiluminescence kit. The same membranes were stripped and reprobed with Gapdh antibodies. Quantification of specific protein signals were performed using ImageJ software and were normalized to the signal intensity of GAPDH [51].
2.9. Measurement of glutathione levels
Intracellular levels of reduced GSH and GSSG were assessed by a GSH detection kit from Cayman (Ann Arbor, MI, USA). In brief, MES buffer was used to prepare the cell extracts and centrifuged at 500 rpm for 5 min at 4 °C. An aliquot of the supernatant was taken for protein determination and equal amount of 10% MPA (meta-phosphoric acid, Cat. No. 239,275, Sigma) was added to the remaining samples to precipitate the proteins. 100 μl of the MPA extracts were treated with TEAM (triethanolamine) reagent and additionally GSH and GSSG standards were prepared and processed similarly to prepare a standard graph. After deproteination, 10 μl of 1 M 2-vinylpyridine was added and then the assay was performed as per the manufacture's instruction using a plate reader (Bio-Tek; epoch) [49].
2.10. Animals
C57BL/6×129 S background breeding pairs of male and female WT and TNF-α receptors knockout (TNFR-DKO, Cat #003243) mice were obtained from Jackson Laboratory. All the mice were maintained in pathogen free facility with 12 h day/night cycle and the mice were fed with standard rodent diet and water ad libitum and humanely treated accordance with Guidelines for the Care and Use of Laboratory Animals from the National Institutes of Health. The mice were genotyped using PCR primers and protocols recommended by Jackson Laboratories. All the animal experiments were approved by institutional animal Care and Use Committee (IACUC), University of Utah, Utah, USA. Age-matched WT and TNFR-DKO mice were used to study the effect of TNFR-DKO on Nrf2 dependent antioxidant protein expression.
2.11. Statistical analysis
Data are expressed as mean±SD. “Mann Whitney test” was used to determine significant differences between control and TNF-α treated groups and all the statistical comparisons were made between control and TNF-α treated groups. p Values smaller than 0.05 were considered statistically significant.
3. Results
3.1. Dose dependent effects of TNF-α on the survival of HL1 cardiomyocytes
First, the survival of HL-1 cardiomyocytes were assessed following exposure to TNF-α (1 ng, 2 ng, 5 ng, 10 ng and 50 ng/ml) for 24 h followed by staining with annexin V and propidium iodide (PI). Low concentrations of TNF-α (1 ng, 2 ng and 5 ng) did not cuase significant apoptosis and cell death, while 10 ng/ml reduced survival by approximately 20% which was further decreased to 72% at 50 ng/ml (Fig. 1A–C). We next determined if TNF-α induced apoptosis in HL-1 cardiomyocytes was associated with caspase-3 activation and PARP-1 cleavage, two hallmarks of apoptosis. As illustrated in Fig. 1D, there was negligible detection of cleaved caspase fragment (p17) in at the low TNF-α concentration (1–5 ng/ml) while at concentrations of 10 ng and 50 ng/ml proteolytic activation of pro-caspase 3 was detected. Since PARP-1 cleavage is widely recognized as characteristic of the execution phase of cell death, we next determined PARP-1 degradation. Consistent with the previous data at doses ranging from 10 ng and 50 ng/ml, the extent of PARP cleavage was increased (Fig. 1D) and, taken together, these data show that the higher doses of TNF-α induce apoptosis.
Fig. 1.

TNF-α treatment in cell viability and apoptosis in HL-1 cardiomyocytes. HL-1 cells were treated with TNF-α 1, 2, 5, 10, 50 ng/ml for 24 h. (A) At the end of experiment, cells were stained with annexin-V/FITC and read immediately by flow cytometry to measure the extent of apoptosis (n=3). Quantification of percentage live cells at each concentration of TNF-α (B) and percentage dead cells (C) relative to that of control. (D) HL-1 cells treated with TNF-α as in Panel A and protein expression for cleaved fragments of caspase-3 and PARP was determined by immunoblotting. '+' indicates Staurosporine, a positive control. Equal loading was analyzed by anti-GAPDH. Statistical significance was calculated by Mann Whitney test, where *p<0.05 vs untreated control.
3.2. Dose dependent effects of TNFα on the generation of ROS in HL1 cardiomyocytes
It has been shown that TNF-α induces a pro-oxidant environment in the cell as measured by a number of methods including a range of structurally diverse fluorescent dyes, lipid and protein oxidation [52], [53], [54]. In the present study we used DCFDH to detect treatment change in the intracellular oxidative milieu [55], [56], [57]. Untreated control cells displayed very low levels of DCFDH-dependent fluorescence. While 2–5 ng/ml TNF-α treatment of HL-1 cells for 2 h showed a moderate, yet significant DCF- fluorescence (Fig. 2A and B), that was further intensified at 10 ng/ml–50 ng/ml consistent with an increased oxidative intracellular milieu. These levels of DCF fluorescence were moderately sustained at 24 h (data not shown).
Fig. 2.

TNF-α induced ROS levels in HL-1 cardiomyocytes by H2DCFDA. (A) HL-1 cells were treated with indicated concentrations of TNF-α (as in panel 1 A) and immunofluorescence analysis was performed using H2DCFDA (green fluorescence, 5 µM). The cells were visualized using fluorescence microscopy and images captured using 20× magnification. (B) Three different fields were randomly counted for green positive cells using Image J and the average fluorescence intensity of each concentration of TNF-α relative to that of control was depicted. Statistical significance was calculated by Mann Whitney test, where *p<0.05 vs control. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
3.3. TNF-α, at low concentrations, induces nuclear translocation of Nrf2 along with enhanced Nrf2 transcript expression and antioxidant proteins in HL1 cardiomyocytes
Since an increased pro-oxidant environment can promote nuclear accumulation of Nrf2 protein and activate Nrf2/EpRE signaling [11], [49], we next determined if TNF-α exposure resulted in modulation of Nrf2-dependent antioxidant expression in HL-1 cardiomyocytes. HL-1 cardiomyocytes were treated with TNF-α (1–50 ng/ml, 2 and 24 h) and immunofluorescence analysis was used to detect the localization of the endogenous nuclear Nrf2 protein. In control cells, a fraction of the Nrf2 was found in the nucleus (Fig. 3A and B control −2 h) which was substantially increased by treatment with 2 or 5 ng/ml TNF-α (Fig. 3A and B). In contrast, 10 ng and 50 ng/ml TNF-α did not result in significant translocation of Nrf2 to the nucleus (Fig. 3A and B). TNF-α exposure at 24 h gave essentially similar results consistent with a sustained translocation of Nrf2 to the nucleus (Fig. 3A and C). We next determined if the nuclear translocated Nrf2 was capable of binding to the EpRE following TNF-α exposure. An ELISA based Nrf2 transcription factor assay was performed on the nuclear extracts treated with and without TNF-α. A 2 fold increase in the Nrf2 binding was observed in HL-1 cardiomyocytes stimulated with 2 ng/ml TNF-α between 30 min and 2 h which again was not evident at the higher concentrations (Fig. 4A and B). Taken together, these results demonstrate that TNF-α at the lower concentrations induces nuclear translocation of Nrf2 and leads to increased DNA binding activity. Next, we assessed if the effect of TNF-α signaling on increased Nrf2 nuclear levels is due to an overall increase in Nrf2 transcription by quantitative real time PCR analysis. A 3 fold stimulation of Nrf2 gene expression was noted as early as 2 h at 5 ng/ml but decreased at the 10 ng and 50 ng/ml concentrations (Fig. 4C). After 24 h TNF-α stimulation resulted in a significant increase in Nrf2 gene expression even at the lower concentrations of 2 ng/ml (p<0.05, Fig. 4D). Similar to 2 h treatment, the Nrf2 gene expression was found to approach the baseline levels at 24 h when treated with higher concentration of TNF-α (10 ng and 50 ng/ml). These data indicate that lower doses of TNF-α activated Nrf2 gene expression while higher doses are repressive. As shown in Fig. 5, we observed a robust differential regulation of Nrf2 target genes 24 h after TNF-α treatment consistent with translocation to the nucleus which also showed a biphasic response with respect to concentration.
Fig. 3.

TNF-α treatment on Nrf2 Nuclear translocation in HL-1 cardiomyocytes. (A) Representative immunofluorescence photomicrograph (original magnification, 20×) from control and 2 h and 24 h TNFα-treated HL-1 cells (2, 5, 10, 50 ng/ml) showing Nrf2 nuclear localization. DAPI was used as a nuclear counterstain. Data are representative of 3 independent experiments. Relative fluorescence intensity was calculated for Nrf2 nuclear translocation for (B) 2 h and (C) 24 h. Statistical significance was determined by Mann Whitney test, where *p<0.05 compared with untreated control. For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.
Fig. 4.

Nrf2 binding activity and mRNA expression in HL-1 cardiomyocytes by TNF-α treatment. Nrf2- DNA binding activity was measured using Trans-AM Nrf2 kit in control and TNF-α treated HL-1 cardiomyocytes (n=3). (D) 2 h and (E) 24 h. Nrf2 gene expression determined in TNF-α treated HL-1 samples for (F) 2 h and (G) 24 h by qPCR. The relative gene expression was calculated by normalizing the mRNA levels of Nrf2 with the levels of GAPDH. In panels, d-G, statistical significance was determined by Mann Whitney test, where *p<0.05 compared with untreated control.
Fig. 5.

Increased Nrf2 regulated antioxidant genes in HL-1 cardiomyocytes by 24 h of TNF-α treatment. Nrf2 regulated antioxidant Gene expression Profiles (Gclc, Gclm, G6pd, Gpx1, Cat, Nqo1, Sod1 and Sod2) in TNF-α treated HL-1 cell samples for 24 h were analyzed by quantitative PCR and gene levels were normalized with Gapdh (n=3). Mann Whitney test was used to establish statistical significance, where *p<0.05 vs control.
Next, we determined whether the Nrf2 target gene expression is reflected at the protein level and thus we performed immunoblotting analysis for these Nrf2 targets at 2 and 24 h. Increases in the protein expression, of GCLC, GPX1, CAT, NQO1 were significantly increased at the lowest TNF-α concentration (Fig. 6, Fig. 7) which was further enhanced at 2 ng/ml. The protein levels of GPX1, CAT and NQO1 attained a maximal level at 2 ng/ml that paralleled with the corresponding transcript expression (compare 2 ng/ml 3 vs 5A, 5B). The biphasic effect evident in the mRNA data was paralleled in the protein data for GCLC, SOD2 and NQO1 but not for GCLM, CAT, SOD1 or GPX1 suggesting selective differential turnover of these antioxidant proteins with increasing concentrations of TNF-α. Next, we measured the cellular GSH level to determine whether the Nrf2 regulated antioxidant protein expression is reflected in cellular GSH synthesis. Interestingly, we observed ~1.5 and ~2.0 fold increase in the levels of reduced glutathione (GSH) and a corresponding change in the GSH/GSSG ratio at 2 and 5 ng/ml of TNF-α treatment, respectively, at 24 h. However, the GSH levels declined and GSH/GSSG ratio shifted to a more oxidized state in the 10 ng. Consistent with the message and protein data, the GSH levels and GSH/GSSG ratio were significantly decreased with exposure to 50 ng/ml TNF- α (Fig. 8).
Fig. 6.

Lower concentration of TNF-α treatment for 2 h increases Nrf2 regulated antioxidant proteins in HL-1 cardiomyocytes. (A) Immunoblot analyses of Nrf2 regulated antioxidant proteins (GCLC, GCLM, G6PD, GPX1, CAT, NQO1, SOD1 and SOD2) in 2 h of TNFα treated HL-1 cells (n=3). (B) Relative intensity of the protein signal was calculated using Image-J software and normalized to GAPDH. Statistical significance was calculated by Mann Whitney test, where *p<0.05 vs control.
Fig. 7.

TNF-α treatment for 24 h increases Nrf2 regulated antioxidant proteins in HL-1 cardiomyocytes. (A) Immunoblots showing protein expression of antioxidant enzymes in TNF-α treated HL-1 cells (GCLC, GCLM, G6PD, GPX1, CAT, NQO1, SOD1 and SOD2) for 24 h (n=3). (B) Densitometry quantification for appropriate immunoblots normalized with GAPDH intensity. Statistical significance among different treatments were analyzed by Mann Whitney test, where *p<0.05 vs control.
Fig. 8.

TNF-α induces GSH synthesis in HL-1 cardiomyocytes. (A) HL-1 cells were treated with 2, 5, 10, 50 ng/ml of TNF-α and after 24 h, the cells were processed for quantification of GSH levels (n=3), (B) The GSH/GSSG ratio was calculated from GSH, GSSG values and represented as a bar graph (n=3). Mann Whitney test was performed to establish statistical significance, where *p<0.05 vs control.
3.4. Abrogation of TNF-αR1/R2 impairs Nrf2-dependent antioxidant signaling in the mouse (double knockout) heart
The previous data with the HL-1 cells suggested to us that there is a basal requirement of TNF-α signaling for maintaining the Nrf2 response in cardiomyocytes. Considering the fact that TNF-α mediates its effect on target cells by binding to one of its two specific cell surface membrane receptors (TNFR1 and TNFR2) [18], we extend the relevance of in vitro observations to in vivo setting and assessed the requirement of basal TNFα signaling in regulating Nrf2/ARE pathway using TNFR1 and TNFR2 double knockout mice (TNFR-DKO). Semi quantitative and real time PCR analysis for TNFα receptors in TNFR-DKO mice hearts confirmed the knockdown of receptors (Fig. 9A and B). Real time PCR analysis for Nrf2 and Keap1 indicated that there was no change in the transcript levels of both genes (Fig. 9C). Interestingly, the Nrf2 regulated protein levels of NQO1, HO-1, G6PD were found to be significantly decreased (p<0.05) in the TNFR-DKO mice compared to the WT (Fig. 9D and E). The protein expression of other Nrf2 targets such as SOD1, SOD2 and CAT was unaffected (Fig. 9D and E).
Fig. 9.

TNF-α receptors knockout animals showed decreased basal antioxidant gene/proteins in the heart. (A) One step RT-PCR analysis of TNF-α receptors (TNFR1 and TNFR2) and housekeeping gene Gapdh were determined in WT and TNFR DKO heart samples. (B) Quantitative real time PCR analysis of TNFα receptors (TNFR1 and TNFR2) and housekeeping gene Gapdh were determined in WT and TNFR DKO heart samples (n=4). (C) Nrf2 and Keap1 gene expression profiles were measured by qPCR. Relative gene expression was calculated by normalizing the mRNA levels of gene of interest with Gapdh. (D) Nrf2 regulated antioxidant proteins (NQO1, HO-1, G6PD, SOD1, SOD2 and CAT) were determined by Immunoblot analysis in WT and DKO heart tissue homogenates (n=4). (E) Densitometric analysis of proteins normalized with GAPDH intensity using Image J. Statistical differences were determined by Mann Whitney test, where *p<0.05 vs control.
4. Discussion
Heart is an organ with the highest O2 consumption among all body organs that can increase consumption eightfold or more under maximal workload conditions and Nrf2 signaling is a key pathway that is sensitive to redox changes. Maintaining functional redox signaling is crucial for living cells to protect them from various pathological insults such as inflammation, apoptosis, oxidative stress, toxic chemical exposure and genetic mutations. Although TNF-α is well known to induce oxidative stress, mitochondrial dysfunction and associated with or causally linked to a number of pathophysiological conditions, its primary role, at lower concentrations is less well understood. In this context numerous studies have highlighted the necessity of a basal physiological maintenance of oxidants to activate and sustain the regulation of transcription factors such as Nrf2 for control of redox modulators necessary for cell signaling. The basal mechanisms which control Nrf2 signaling remain elusive. The results from the present study suggest that sub-inflammatory levels of TNF-α are necessary and sufficient to (i) promote Nrf2 signaling and (ii) maintain basal transcription and translation of antioxidants in the HL-1 cardiomyocytes/mouse myocardium.
Interestingly, certain Nrf2 targets were activated at lower concentrations of TNF-α such as Gclc, Gpx1, Catalase and Nqo1, whereas others (i.e. Gclm, G6pd, Sod1, and Sod2) only at higher concentration of TNF-α. This could be attributed to the fact that there might be certain ‘early-onset’ genes that are required for the initial regulatory redox cascade and those are under the primary control of Nrf2. In contrast, the ‘second wave genes’ that might be involved in a delayed later maintenance cascade and those may be under secondary control of Nrf2 that requires the interplay of other factors (activation or repression) for subsequent gene regulation. In addition, we observed a bimodal concentration dependent effect of TNF-α with respect to expression of many of the Nrf2 target genes/proteins. Overall, our results indicate that TNF-α stimulation resulted in a general pattern of Nrf2 dependent target activation that is consistent with the increased nuclear Nrf2 levels and binding activity in HL-1 cardiomyocytes. Further, the TNF-α induced increase of GCLC, NQO1, GPX1 and Catalase in combination with increased glutathione (GSH) indicates a potential requirement for a low-dose of TNF-α to maintain Nrf2 signaling under basal conditions.
Our findings also show that exposure of TNF-α to cells at concentrations well below the threshold associated with sub-inflammation significantly increased Nrf2 activity and its nuclear translocation [35], [42]. The transcriptional induction of Nrf2 and its subsequent targets occurs in response to a low-dose (>2–<10 ng/ml) of TNF-α, while concentrations >10 ng/ml were markedly suppressive and associated with cell death. In support of a critical role for sub-inflammatory effects of TNFα, other studies have shown that a complete neutralization and/or chronic TNF-α blockade in the elderly resulted in impaired immune function, increased risk of cancer, and cardiovascular complications [27], [58].
Finally, the present study with the TNFR1/2 double knockout mice and HL-1 cardiomyocytes makes a first step forward in understanding the bimodal effects of the cytokine, TNF-α in regulating the redox-sensitive Keap1/Nrf2 antioxidant pathway. This study has potential importance in the field of cardiovascular signaling because the TNF-α induced biphasic regulation of the Nrf2 pathway suggest that a certain threshold of TNF/ROS signaling is essential to prime and activate the Nrf2 protective signaling pathway. This data also implies that abrogation of TNF-α signaling similar to those that can occur in chronic anti-TNF therapy may disrupt the sub-inflammatory basal signaling in cardiomyocytes leading to toxicity and organ malfunction.
Conflicts of interest
The authors declare that they have no competing interests.
Author contributors
N.S.R, G.S and N.M designed the study, involved in interpretation of the data and wrote the manuscript; G.S, S.R and R.K.R performed the experiments, analyzed data; P.S provided resources and interpreted the immunofluorescence data, C.D, JRH and VDU contributed in critical discussion and interpretation of the data. All the authors approved the final version of the manuscript.
Acknowledgments
This study was supported by funding from NHLBI (HL118067), NIA (AG042860), the AHA (BGIA 0865015F), University of Utah center for Aging (Pilot grant#2009), the Division of Cardiovascular Medicine/Department of Medicine, University of Utah and the start-up funds (3115851.000.213115851.392300000.0000 for NSR) by Department of Pathology, the University of Alabama at Birmingham, AL and UABAMC21 reload multi-investigator grant by the University of Alabama at Birmingham, AL (NSR and VDU).
Authors' acknowledge Mrs. Jennifer Schroff and Mrs. Jessica Keiper for their editorial assistance with the manuscript. Also, thank Gayatri Khanderao for technical assistance with HL-1 cell cultures and treatment.
References
- 1.Haddad J.J. Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell Signal. 2002;14:879–897. doi: 10.1016/s0898-6568(02)00053-0. [DOI] [PubMed] [Google Scholar]
- 2.Korashy H.M., El-Kadi A.O. The role of redox-sensitive transcription factors NF-kappaB and AP-1 in the modulation of the Cyp1a1 gene by mercury, lead, and copper. Free Radic. Biol. Med. 2008;44:795–806. doi: 10.1016/j.freeradbiomed.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Rahman I., Adcock I.M. Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 2006;28:219–242. doi: 10.1183/09031936.06.00053805. [DOI] [PubMed] [Google Scholar]
- 4.Maillet A., Pervaiz S. Redox regulation of p53, redox effectors regulated by p53: a subtle balance. Antioxid. Redox Signal. 2012;16:1285–1294. doi: 10.1089/ars.2011.4434. [DOI] [PubMed] [Google Scholar]
- 5.Lee J.M., Li J., Johnson D.A., Stein T.D., Kraft A.D., Calkins M.J., Jakel R.J., Johnson J.A. Nrf2, a multi-organ protector? FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2005;19:1061–1066. doi: 10.1096/fj.04-2591hyp. [DOI] [PubMed] [Google Scholar]
- 6.Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 7.Clements C.M., McNally R.S., Conti B.J., Mak T.W., Ting J.P. DJ-1, a cancer- and Parkinson's disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA. 2006;103:15091–15096. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayes J.D., McMahon M. The double-edged sword of Nrf2: subversion of redox homeostasis during the evolution of cancer. Mol. Cell. 2006;21:732–734. doi: 10.1016/j.molcel.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Rangasamy T., Cho C.Y., Thimmulappa R.K., Zhen L., Srisuma S.S., Kensler T.W., Yamamoto M., Petrache I., Tuder R.M., Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bryan H.K., Olayanju A., Goldring C.E., Park B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013;85:705–717. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 11.Itoh K., Wakabayashi N., Katoh Y., Ishii T., O’Connor T., Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells.: Devoted Mol. Cell. Mech. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 12.Higdon A., Diers A.R., Oh J.Y., Landar A., Darley-Usmar V.M. Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochem. J. 2012;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dodson M., Redmann M., Rajasekaran N.S., Darley-Usmar V., Zhang J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem. J. 2015;469:347–355. doi: 10.1042/BJ20150568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calabrese F., Carturan E., Chimenti C., Pieroni M., Agostini C., Angelini A., Crosato M., Valente M., Boffa G.M., Frustaci A., Thiene G. Overexpression of tumor necrosis factor (TNF)alpha and TNFalpha receptor I in human viral myocarditis: clinicopathologic correlations. Mod. Pathol.: Off. J. U.S. Can. Acad. Pathol. Inc. 2004;17:1108–1118. doi: 10.1038/modpathol.3800158. [DOI] [PubMed] [Google Scholar]
- 15.Vattemi G., Marini M., Ferreri N.R., Hao S., Malatesta M., Meneguzzi A., Guglielmi V., Fava C., Minuz P., Tomelleri G. Overexpression of TNF-alpha in mitochondrial diseases caused by mutations in mtDNA: evidence for signaling through its receptors on mitochondria. Free Radic. Biol. Med. 2013;63:108–114. doi: 10.1016/j.freeradbiomed.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 16.Li X., Moody M.R., Engel D., Walker S., Clubb F.J., Jr, Sivasubramanian N., Mann D.L., Reid M.B. Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation. 2000;102:1690–1696. doi: 10.1161/01.cir.102.14.1690. [DOI] [PubMed] [Google Scholar]
- 17.Kim Y.S., Morgan M.J., Choksi S., Liu Z.G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell. 2007;26:675–687. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 18.Wajant H., Pfizenmaier K., Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 19.Swaroop J.J., Rajarajeswari D., Naidu J.N. Association of TNF-α with insulin resistance in type 2 diabetes mellitus. Indian J. Med. Res. 2012;135:127–130. doi: 10.4103/0971-5916.93435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun M., Chen M., Dawood F., Zurawska U., Li J.Y., Parker T., Kassiri Z., Kirshenbaum L.A., Arnold M., Khokha R., Liu P.P. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115:1398–1407. doi: 10.1161/CIRCULATIONAHA.106.643585. [DOI] [PubMed] [Google Scholar]
- 21.Liu J.Y., Brass D.M., Hoyle G.W., Brody A.R. TNF-alpha receptor knockout mice are protected from the fibroproliferative effects of inhaled asbestos fibers. Am. J. Pathol. 1998;153:1839–1847. doi: 10.1016/s0002-9440(10)65698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amini-Shirazi N., Hoseini A., Ranjbar A., Mohammadirad A., Khoshakhlagh P., Yasa N., Abdollahi M. Inhibition of tumor necrosis factor and nitrosative/oxidative stresses by Ziziphora clinopoides (Kahlioti); a molecular mechanism of protection against dextran sodium sulfate-induced colitis in mice. Toxicol. Mech. Methods. 2009;19:183–189. doi: 10.1080/15376510701533996. [DOI] [PubMed] [Google Scholar]
- 23.Kageyama Y., Takahashi M., Ichikawa T., Torikai E., Nagano A. Reduction of oxidative stress marker levels by anti-TNF-alpha antibody, infliximab, in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2008;26:73–80. [PubMed] [Google Scholar]
- 24.Kageyama Y., Takahashi M., Nagafusa T., Torikai E., Nagano A. Etanercept reduces the oxidative stress marker levels in patients with rheumatoid arthritis. Rheumatol. Int. 2008;28:245–251. doi: 10.1007/s00296-007-0419-1. [DOI] [PubMed] [Google Scholar]
- 25.Quan L.-d, Thiele G.M., Tian J., Wang D. The development of novel therapies for rheumatoid arthritis. Expert Opin. Ther. Pat. 2008;18:723–738. doi: 10.1517/13543776.18.7.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saad A.A., Symmons D.P., Noyce P.R., Ashcroft D.M. Risks and benefits of tumor necrosis factor-alpha inhibitors in the management of psoriatic arthritis: systematic review and metaanalysis of randomized controlled trials. J. Rheumatol. 2008;35:883–890. [PubMed] [Google Scholar]
- 27.Bilate A.M., Salemi V.M., Ramires F.J., de Brito T., Russo M., Fonseca S.G., Fae K.C., Martins D.G., Silva A.M., Mady C., Kalil J., Cunha-Neto E. TNF blockade aggravates experimental chronic Chagas disease cardiomyopathy. Microbes Infect./Inst. Pasteur. 2007;9:1104–1113. doi: 10.1016/j.micinf.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 28.Sethi G., Sung B., Kunnumakkara A.B., Aggarwal B.B. Targeting TNF for Treatment of Cancer and Autoimmunity. Adv. Exp. Med. Biol. 2009;647:37–51. doi: 10.1007/978-0-387-89520-8_3. [DOI] [PubMed] [Google Scholar]
- 29.Inanc N., Direskeneli H. Serious infections under treatment with TNF-alpha antagonists compared to traditional DMARDs in patients with rheumatoid arthritis. Rheumatol. Int. 2006;27:67–71. doi: 10.1007/s00296-006-0165-9. [DOI] [PubMed] [Google Scholar]
- 30.Oberstein E.M., Kromo O., Tozman E.C. Type I reaction of Hansen's disease with exposure to adalimumab: a case report. Arthr. Rheum. 2008;59:1040–1043. doi: 10.1002/art.23815. [DOI] [PubMed] [Google Scholar]
- 31.Janssen R., Van Wengen A., Verhard E., De Boer T., Zomerdijk T., Ottenhoff T.H., Van Dissel J.T. Divergent role for TNF-alpha in IFN-gamma-induced killing of Toxoplasma gondii and Salmonella typhimurium contributes to selective susceptibility of patients with partial IFN-gamma receptor 1 deficiency. J. Immunol. (Baltim. Md.: 1950) 2002;169:3900–3907. doi: 10.4049/jimmunol.169.7.3900. [DOI] [PubMed] [Google Scholar]
- 32.Khanna D., McMahon M., Furst D.E. Anti-tumor necrosis factor alpha therapy and heart failure: what have we learned and where do we go from here? Arthr. Rheum. 2004;50:1040–1050. doi: 10.1002/art.20164. [DOI] [PubMed] [Google Scholar]
- 33.Kurrelmeyer K.M., Michael L.H., Baumgarten G., Taffet G.E., Peschon J.J., Sivasubramanian N., Entman M.L., Mann D.L. Endogenous tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc. Natl. Acad. Sci. USA. 2000;97:5456–5461. doi: 10.1073/pnas.070036297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El-Ani D., Philipchik I., Stav H., Levi M., Zerbib J., Shainberg A. Tumor necrosis factor alpha protects heart cultures against hypoxic damage via activation of PKA and phospholamban to prevent calcium overload. Can. J. Physiol. Pharmacol. 2014;92:917–925. doi: 10.1139/cjpp-2014-0092. [DOI] [PubMed] [Google Scholar]
- 35.Rushworth S.A., Shah S., MacEwan D.J. TNF mediates the sustained activation of Nrf2 in human monocytes. J. Immunol. (Baltim. Md.: 1950) 2011;187:702–707. doi: 10.4049/jimmunol.1004117. [DOI] [PubMed] [Google Scholar]
- 36.Ravi S., Mitchell T., Kramer P.A., Chacko B., Darley-Usmar V.M. Mitochondria in monocytes and macrophages-implications for translational and basic research. Int. J. Biochem. Cell Biol. 2014;53:202–207. doi: 10.1016/j.biocel.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopaschuk G.D., Jaswal J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010;56:130–140. doi: 10.1097/FJC.0b013e3181e74a14. [DOI] [PubMed] [Google Scholar]
- 38.Haudek S.B., Taffet G.E., Schneider M.D., Mann D.L. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J. Clin. Investig. 2007;117:2692–2701. doi: 10.1172/JCI29134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krown K.A., Page M.T., Nguyen C., Zechner D., Gutierrez V., Comstock K.L., Glembotski C.C., Quintana P.J., Sabbadini R.A. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J. Clin. Investig. 1996;98:2854–2865. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Machuca C., Mendoza-Milla C., Cordova E., Mejia S., Covarrubias L., Ventura J., Zentella A. Dexamethasone protection from TNF-alpha-induced cell death in MCF-7 cells requires NF-kappaB and is independent from AKT. BMC Cell Biol. 2006;7:9. doi: 10.1186/1471-2121-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murakami A., Kawabata K., Koshiba T., Gao G., Nakamura Y., Koshimizu K., Ohigashi H. Nitric oxide synthase is induced in tumor promoter-sensitive, but not tumor promoter-resistant, JB6 mouse epidermal cells cocultured with interferon-gamma-stimulated RAW 264.7 cells: the role of tumor necrosis factor-alpha. Cancer Res. 2000;60:6326–6331. [PubMed] [Google Scholar]
- 42.Jia G., Cheng G., Gangahar D.M., Agrawal D.K. Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol. Cell Biol. 2006;84:448–454. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 43.Marques-Fernandez F., Planells-Ferrer L., Gozzelino R., Galenkamp K.M., Reix S., Llecha-Cano N., Lopez-Soriano J., Yuste V.J., Moubarak R.S., Comella J.X. TNFalpha induces survival through the FLIP-L-dependent activation of the MAPK/ERK pathway. Cell Death Dis. 2013;4:e493. doi: 10.1038/cddis.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anwar A., Zahid A.A., Scheidegger K.J., Brink M., Delafontaine P. Tumor necrosis factor-alpha regulates insulin-like growth factor-1 and insulin-like growth factor binding protein-3 expression in vascular smooth muscle. Circulation. 2002;105:1220–1225. doi: 10.1161/hc1002.105187. [DOI] [PubMed] [Google Scholar]
- 45.Fiers W., Beyaert R., Boone E., Cornelis S., Declercq W., Decoster E., Denecker G., Depuydt B., De Valck D., De Wilde G., Goossens V., Grooten J., Haegeman G., Heyninck K., Penning L., Plaisance S., Vancompernolle K., Van Criekinge W., Vandenabeele P., Vanden Berghe W., Van de Craen M., Vandevoorde V., Vercammen D. TNF-induced intracellular signaling leading to gene induction or to cytotoxicity by necrosis or by apoptosis. J. Inflamm. 1995;47:67–75. [PubMed] [Google Scholar]
- 46.Bader T., Weitzerbin J. Nuclear accumulation of interferon gamma. Proc. Natl. Acad. Sci. USA. 1994;91:11831–11835. doi: 10.1073/pnas.91.25.11831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giroir B.P., Horton J.W., White D.J., McIntyre K.L., Lin C.Q. Inhibition of tumor necrosis factor prevents myocardial dysfunction during burn shock. Am. J. Physiol. 1994;267:H118–H124. doi: 10.1152/ajpheart.1994.267.1.H118. [DOI] [PubMed] [Google Scholar]
- 48.Giroir B.P., Johnson J.H., Brown T., Allen G.L., Beutler B. The tissue distribution of tumor necrosis factor biosynthesis during endotoxemia. J. Clin. Invest. 1992;90:693–698. doi: 10.1172/JCI115939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muthusamy V.R., Kannan S., Sadhaasivam K., Gounder S.S., Davidson C.J., Boeheme C., Hoidal J.R., Wang L., Rajasekaran N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 2012;52:366–376. doi: 10.1016/j.freeradbiomed.2011.10.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Narasimhan M., Mahimainathan L., Rathinam M.L., Riar A.K., Henderson G.I. Overexpression of Nrf2 protects cerebral cortical neurons from ethanol-induced apoptotic death. Mol. Pharmacol. 2011;80:988–999. doi: 10.1124/mol.111.073262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rajasekaran N.S., Connell P., Christians E.S., Yan L.J., Taylor R.P., Orosz A., Zhang X.Q., Stevenson T.J., Peshock R.M., Leopold J.A., Barry W.H., Loscalzo J., Odelberg S.J., Benjamin I.J. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Houstis N., Rosen E.D., Lander E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 53.Vemula V., Ni Z., Fedorova M. Fluorescence labeling of carbonylated lipids and proteins in cells using coumarin-hydrazide. Redox Biol. 2015;5:195–204. doi: 10.1016/j.redox.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pastukhov A.V., Ropson I.J. Fluorescent dyes as probes to study lipid-binding proteins. Proteins. 2003;53:607–615. doi: 10.1002/prot.10401. [DOI] [PubMed] [Google Scholar]
- 55.Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic. Biol. Med. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 56.Kalyanaraman B., Darley-Usmar V., Davies K.J., Dennery P.A., Forman H.J., Grisham M.B., Mann G.E., Moore K., Roberts L.J., 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forman H.J., Augusto O., Brigelius-Flohe R., Dennery P.A., Kalyanaraman B., Ischiropoulos H., Mann G.E., Radi R., Roberts L.J., 2nd, Vina J., Davies K.J. Even free radicals should follow some rules: a guide to free radical research terminology and methodology. Free Radic. Biol. Med. 2015;78:233–235. doi: 10.1016/j.freeradbiomed.2014.10.504. [DOI] [PubMed] [Google Scholar]
- 58.Chung E.S., Packer M., Lo K.H., Fasanmade A.A., Willerson J.T. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–3140. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]