

Abstract
Background
Lung cancers express an autocrine cholinergic loop in which secreted acetylcholine can stimulate tumor growth through both nicotinic and muscarinic receptors. Because activation of mAChR and nAChR stimulates growth; tumor growth can be stimulated by both locally synthesized acetylcholine as well as acetylcholine from distal sources and from nicotine in the high percentage of lung cancer patients who are smokers. The stimulation of lung cancer growth by cholinergic agonists offers many potential new targets for lung cancer therapy.
Methods
The potential of cholinergic targets to inhibit lung cancer growth can be evaluated by use of antagonists, agonists, gene silencing and gain of function.
Results
Cholinergic signaling can be targeted at the level of choline transport; acetylcholine synthesis, secretion and degradation; and nicotinic and muscarinic receptors. In addition, the newly describe family of ly-6 allosteric modulators of nicotinic signaling such as lynx1 and lynx2 offers yet another new approach to novel lung cancer therapeutics.
Conclusions
Each of these targets has their potential advantages and disadvantages for the development of new lung cancer therapies which are discussed in this review.
Keywords: cancer, acetylcholine, nicotine, nicotinic receptors, muscarinic receptors, lynx-1
Graphical abstract
1. Introduction
Lung cancer is the number one cause of cancer death in the World, with deaths in 2012 estimated to exceed 1.5 million [1]. Lung cancer is classified into small cell lung carcinoma (SCLC), which accounts for approximately 15–20% of cases and non-small cell lung carcinoma (NSCLC), which accounts for the remaining 80–85%. The two most common forms of NSCLC are squamous cell lung carcinoma (SCC) and lung adenocarcinoma which together represent at least 80% of all NSCLC [2–4]. Based on histology, gene expression and location, SCC are considered to arise from bronchial epithelial cells (BEC) of large airways, and adenocarcinomas from epithelial cells of smaller airways [5] SCLC primarily from pulmonary neuroendocrine cells (PNEC) [6]. Despite improvements in responses to increasingly sophisticated combinations of surgery, radiation and targeted chemotherapy [7], lung cancer survival remains low [8]. Thus, the development of new therapeutic approaches is clearly needed. The ability of cholinergic signaling to modify lung cancer growth offers diverse potential new targets.
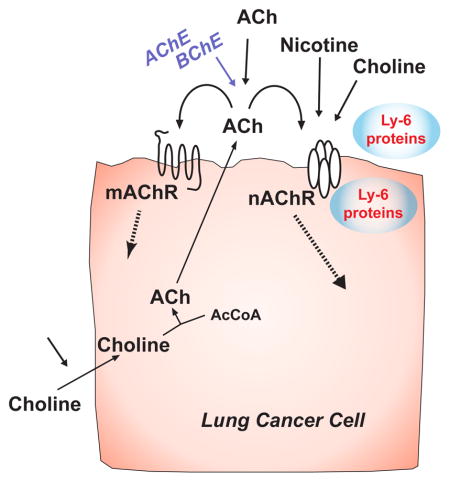
Bronchial epithelial cells and pulmonary neuroendocrine cells express a cholinergic autocrine loop in which the cells synthesize and secrete acetylcholine (ACh) that stimulates the cells to grow through nicotinic acetylcholine receptors (nAChR) and muscarinic acetylcholine (mAChR) receptors (Fig. 1). The secreted ACh can also act as an endocrine or paracrine factor to stimulate local or distant cells. In turn, airway cells that express cholinergic receptors can be stimulated to grow by ACh from autocrine, paracrine, exocrine or neuronal sources. Adding a critically important layer of complexity for consideration of lung cancer growth, cells can also be stimulated by other cholinergic agonists that include nicotine, choline, secreted ly-6 proteins and some classes of tobacco specific nitrosamines. Each of these stimulatory pathways provides targets for potential modulation of lung cancer growth. As shown in figure 1, cholinergic stimulation of lung cancer growth can be modulated beginning at the level of choline transport, then proceeding through ACh synthesis, secretion, degradation, nicotinic signaling and muscarinic signaling.
Figure 1.

Cholinergic signaling pathways in lung cancers. Diagram of pathways for cholinergic signaling and potential points where signaling can be interrupted. Choline is taken into cells; acetylcholine (ACh) synthesized by the action of choline acetyltransferase (ChAT), packaged and secreted to interact with nAChR and mAChR receptors on the same or neighboring cells. Muscarinic and nicotinic receptors can also be activated by ACh from neighboring or distal sources. Nicotinic receptors can be activated by nicotine and up- or down-regulated by both membrane bound ly-6 proteins such as lynx1 or lynx2 or secreted ly-6 proteins such as slurp-1.
2. Evidence that acetylcholine is an autocrine growth factor for lung cancer
In the 90’s, initial reports by Quik et al [9], Maneckjee and Minna [10] and Schuller et al [11] demonstrated that nicotine and ACh could stimulate lung cancer growth through both nicotinic and muscarinic mechanisms. In 2003, our laboratory [12] demonstrated that most SCLC expressed both nAChR and mAChR and also synthesized and secreted ACh that stimulated lung cancers to grow through both nicotinic and muscarinic mechanisms (Fig. 2); thus, establishing ACh as an autocrine growth factor for SCLC. Our laboratory then extended that observation to NSCLC and demonstrated that cholinergic signaling was upregulated in NSCLC as established by increased levels of ACh and increased levels of some nAChR subtypes [13]. Consistent with the ability of ACh and nicotine to stimulate lung cancer cell growth, inhibitors of cholinergic signaling could block lung cancer cell growth thus establishing cholinergic signaling as a potential therapeutic target for lung cancer growth (Fig. 2).
Figure 2.

Cholinergic modification of lung cancer cell growth. H82 small cell lung carcinoma cells were plated in 96-well culture plates and cell proliferation measured after specified drug treatments. Cell numbers were measured at 0, 6, 12 days using the MTS assay. A. The nicotinic agonist nicotine; B. The muscarinic agonist carbachol; C. The nicotinic antagonist mecamylamine; D. The muscarinic antagonist atropine; E. The choline transport inhibitor hemicholinium-3; F. The vesicular acetylcholine transporter inhibitor (VAChT) vesamicol. * p<0.05 by Neuman-Keuls test following ANOVA. All data are expressed as the mean ± SE of twelve replicates. Drug concentrations as shown in panel A. Modified after Song et al [12].
The expression of the cholinergic autocrine loop in lung cancer does not uniquely occur in lung cancer cells, but is manifest in most epithelial tissues [14,15]. Studies from our laboratory and others [16,17] have established that bronchial epithelial cells and PNEC also express a cholinergic autocrine loop. Therefore, the expression of this loop in lung cancers continues the expression pattern of the non-transformed cells though the levels of cholinergic signaling by lung cancer cells is significantly increased over non-transformed cells [13], further emphasizing its potential as a lung cancer therapeutic target.
One consideration in attempting to target cholinergic signaling by lung cancer cells is the ability to target non-neuronal cholinergic signaling without targeting neuronal signaling, thus lessening potential side effects. As discussed further below some aspects of non-neuronal cholinergic signaling are different than neuronal signaling, thus providing especially intriguing targets.
3. Targeting choline transport needed for ACh synthesis in lung cancer
The first step in cholinergic signaling is transport of choline into the cell so ACh can be synthesized from choline and acetyl-CoA by the action of choline acetyltransferase (ChAT). Because transport of choline is essential to many fundamental cell processes such as phospholipid synthesis, there are a multitude of low, medium and high affinity choline transporters. In neurons there is a specific high affinity choline transporter (CHT1, CHT, SLC5A7) that is only expressed in cholinergic neurons [18]. If that gene is knocked out, cholinergic neurons can no longer synthesize ACh [19]. Thus, CHT1 is specifically coupled to ACh synthesis in neurons. CHT1 plays a role in non-neuronal cell ACh synthesis as it is expressed in trachea and large airway epithelium [17,20].
Notably however, some non-neuronal cell type can synthesize ACh in the absence of CHT1 including colon epithelial cells [21] and lung cancer cells [12,22]. Thus, choline transport in non-neuronal cells cannot be solely dependent on CHT1 and must, by necessity, utilize other choline transporters such as the recently described family of five choline transporters designated as the choline transporter-like proteins 1–5 (CTL1-5) [23,24]. The CTLs are Na+-independent and have an intermediate-affinity for choline and hemicholinium-3 (HC-3) as compared to CHT1 [23,25]. CTL1, in particular, has been shown to transport choline in renal tubule epithelia [26], keratinocytes [27], and lung adenocarcinoma cells [28]. CTL2 has also been shown to transport choline in lung adenocarcinoma cells [28]. In particular, Song et al [29] have demonstrated that knockdown of CTL4 in lung cancer cells decreases ACh synthesis while increasing expression of CTL4 increases choline transport and ACh synthesis. Besides CTL4, the organic cation transporter (OCT) 1 and 3 have also been linked to non-neuronal ACh synthesis, as demonstrated by expression in cell types that synthesize ACh but do not express CHT1 [30,31].
That lung cancer cells rely on a different transporter for ACh synthesis than neurons, suggests that targeting CTL4 would be a way to inhibit ACh synthesis by lung cancers without interfering with neuronal ACh synthesis. Consistent with this, Song et al [29] has demonstrated that knockdown of CTL4 inhibits lung cancer cell growth. The potential effectiveness of targeting choline transport to inhibit lung cancer cell growth is also supported by the fact that increased levels of media choline linearly increase levels of ACh synthesis and secretion by lung cancer cells [29]. The increased ACh secretion in turn stimulates lung cancer cell growth which can be blocked by cholinergic antagonists [29]. Making choline transport an especially intriguing target for lung cancer is lung cancers concentrate choline [32] and that inhibition of choline kinase, the first step in utilization of choline for phospholipid synthesis, also inhibits lung cancer cell growth [33,34]. As shown in figure 2E, inhibition of choline transport by hemicholinium-3 inhibits lung cancer cell growth.
Consistent with the different affinity for hemicholinium-3 for CHT1 and the CTL’s, hemicholinium blocks ACh synthesis in neurons and tissues that rely on CHT1 with a Ki close to 10 nM, while μM concentrations of hemicholinium are required to inhibit CTL activity [35]. This difference in affinity suggests that more specific inhibitors of the CTL’s verus CHT1 could be developed to provide another approach to target non-neuronal ACh synthesis.
4. Targeting ACh synthesis, secretion and degradation in lung cancer
Targeting ACh synthesis, secretion and degradation as a therapeutic approach to lung cancer presents multiple targets and levels, though overlap with neuronal pathways does present, challenges. ACh is synthesized by the same enzyme ChAT in lung cancers as in neurons, so inhibition of ChAT in cancers would also inhibit ChAT in neurons; therefore, targeting this enzyme would require care in pharmacokinetics and target tissues levels. However inhibitors of ChAT synthesis such as bromoacetylcholine have been shown to inhibit cancer cell growth.
One way in which non-neuronal cholinergic signaling dramatically differs from neuronal cholinergic signaling is regulation of ACh secretion. In neurons, ACh is packaged in synaptic vesicles and released as a function of action potentials and closely regulated synaptic signaling. By contrast lung cancer cells are not excitable, do not have synaptic vesicles, and ACh secretion appears less closely regulated. Consistent with this, simply increasing or decreasing levels of choline in media causes linear increases or decreases in ACh synthesis [29]. While lung cancer cells do not package ACh in synaptic vesicles, they do package ACh in related secretory vesicles. This packaging requires the vesicular acetylcholine transporter (VAChT) which is also required for packaging ACh in synaptic vesicles. All cells that synthesize ACh also express VAChT, and the inhibitor vesamicol inhibits ACh secretion in both neuronal systems [36] and lung cancer cells (Fig. 2F) [12]. Thus while vesamicol or other mechanisms of decreasing VAChT activity can inhibit lung cancer cell growth, this will not be specific to lung cancer ACh secretion. Overall, regulation of non-neuronal ACh synthesis remains poorly understood.
There is also increased levels of ACh in lung cancer cells compared to normal lung [13], suggesting that ACh synthesis could be targeted on the basis that lung cancer cells will be more sensitive than non-transformed cells, as underlies many cancer chemotherapies. The increased levels of ACh in lung cancer come not just from increased ChAT activity but also from decreased levels of acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) the enzymes that metabolize ACh [13]. Notably levels of cholinesterase activity have been linked to prognosis and patients with lower cholinesterase levels tend to have a worse prognosis [37,38]. This would suggest that restoration of normal cholinesterase activities in lung cancers might provide a new therapeutic approach. This approach also has challenges since little is known about regulation of cholinesterase activities in lung cancers, and cholinesterase activity is also a critical target for neurologic disorders such as Alzheimer’s disease and myasthenia gravis.
Levels of ACh in normal and neoplastic tissues represent a balance between synthesis and degradation. While ACh has a short half-life in serum [39], there is enough synthesis from multiple epithelial and endothelial sources that ACh can be readily detected in blood. Thus alterations in levels of ChAT or cholinesterase activities can be expected to increase blood and tissue levels of ACh to have either a local or distal effect.
5. Nicotinic signaling in lung cancer
The most straightforward approach to targeting cholinergic stimulation of lung cancer growth is by blocking nAChR and mAChR. Blockade of nicotinic signaling is discussed in this section, muscarinic signaling in the section below. Multiple studies have shown that the interaction of nicotine, ACh and nicotinic agonists stimulates lung cancer cell growth and that the stimulation can be blocked by nAChR antagonists [9,12,40–44]. The nAChR are ligand gated ion channels such that the interaction of agonist with the receptor allows influx of Na+ and Ca++ into the cell thus triggering downstream events such as cell proliferation. In non-neuronal cells, nAChR also have a metabotropic function activating downstream kinases that lead to increased cell growth [45,46] that still remains poorly characterized. Fourteen genes that code for neuronal nicotinic subunits have been identified to date; four β subunits and ten α subunits. nAChR can be heteromers composed of both α and β subunits, or homomers composed of one type of α subunit. Agonist, antagonist and nicotine sensitivity varies depending on subunit composition.
As shown in figure 2, nicotine stimulates growth of lung cancer cells and the effect can be blocked by mecamylamine. Surprisingly classic α7 nAChR antagonist such as α-bungarotoxin (αBGT) or methyllycaconitine (MLA) do not block lung cancer cell growth (Fig. 3A). This suggests that non-alpha7 containing nAChR mediate the effect of nicotine on lung cancer cell growth. The finding of lack of effect of αBGT on lung cancer cell growth has been reported by several laboratories as well as our own [47]. However as shown in figure 3B & C, the role of α7 nAChR in lung cancer cell growth is more complicated, since knockdown of α7 decreases lung cancer cell growth. The ability of α7 knockdown to decrease cell growth while classic α7 antagonists do not, supports that non-ion channel signaling of α7 may play a role in stimulation of lung cancer growth. This is an important finding as it suggests new classes of nAChR modifiers that act as negative modulators [48,49] or target non-ion channel aspects of α7 signaling [50,51], may prove to be effective inhibitors of lung cancer growth. In particular, in a series of compounds reported by Millar et al [48,49], 2,6MP-TQS (cis-trans-4-(2,6-dimethylphenyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-8-sulfonamide) acts as a negative allosteric modulator and 2,4,6MP-TQS blocks the effects of other positive allosteric modulators. As reported by Papke et al, NS6740 acts by targeting non-conducting states of α7 [51]. As signaling in this pathway has still not been clearly delineated, screening for these compounds is still difficult.
Figure 3.

α7 nAChR and lung cancer cell growth. A. The α7 nAChR antagonist methyllycaconitine (MLA) has no effect on cell growth of A549 lung adenocarcinoma cells. Similar results were seen with α-bungarotoxin and in the H82 and H520 cell lines (data not shown). B. siRNA knockdown of α7 decreases cell growth in H520 squamous cell lung carcinoma cells compared to cells transfected with control siRNA. C. siRNA knockdown of α7 decreases cell growth in A549 lung adenocarcinoma cells compared to cells transfected with control siRNA.
Studies by Gardner et al also suggest a role for α3, α4 and α5 containing heteromeric receptors as shown by antagonist and shRNA studies [42]. Implication of a role for α5 receptors in lung cancer growth is also consistent with the reports that polymorphisms in the α3, α5, β4 nAChR gene locus are associated with increased risk of lung cancer [52]. These polymorphisms are also associated with increased smoking so it is still not clear if these polymorphisms are associated with increased risk of lung cancer because of a direct role of the nAChR in the lung cancer or because increase smoking or depth of smoking leads to increased exposure to carcinogens in tobacco [52]. Schuller et al have also shown tobacco specific nitrosamines such as NNK specifically interact with α7 nAChR to stimulate lung cancer growth so antagonists that block interaction of tobacco-specific nitrosamines with nAChR could have therapeutic benefit [53,54].
In discussing the connection between cholinergic signaling and lung cancer, it must be remembered that lung cancer is fundamentally caused by nicotine addiction. Therefore the most effective anticholinergic approach to lung cancer would be the use of nicotinic partial agonists for smoking cessation such as varenicline which is a partial agonist for α4β2 and a full agonist for α7 nAChR [55,56] and cytisine which is a partial agonist for α4β2 and a full agonist for α3β4 [56,57]. The ability of nicotine to stimulate lung cancer growth also raises safety issues for the use e-cigarettes and there is little data yet whether there will be a link between long term use of e-cigarettes and lung cancer.
6. Muscarinic signaling in lung cancer
A particularly attractive target in the cholinergic pathway to block lung cancer growth are mAChR. The mAChR are G-protein coupled receptors and there are 5 subtypes; M1, M3 and M5 are coupled to Gq and M2 and M4 are coupled to Gi. The majority of reports link the Gq coupled receptors to proliferation and evidence is particularly strong for a proliferative role of the M3 receptor in lung cancer and colon cancer [58–60]. M3 receptors are a particularly intriguing target since M3 muscarinic receptor antagonists are in wide clinical use for overactive bladder [61] (eg, darifenacin and solifenacin) and COPD [62] (tiotropium, aclidinium, glycopyrronium) and are generally well tolerated. As shown in figure 4, M3 antagonists block the ability of ACh to increase intracellular calcium. This in turn translates to blocking activation of Akt and MAPK and inhibition of cell growth (Fig. 4). This then translates into inhibition of growth in tumors by M3 antagonists in nude mice in vivo (Fig. 4). Thus M3 receptor antagonists taken either systemically as for darifenacin or solifenacin or inhaled as for tiotropium or aclidinium may have potential as lung cancer therapeutics.
Figure 4.

Muscarinic modulation of lung cancer cell growth. A. The M3 antagonist darifenacin blocks ACh-induced increase in intracellular Ca++ in H82 SCLC cells. B. The M3 mAChR antagonist 4-DAMP inhibited H82 cell proliferation in a concentration-dependent manner. * p < 0.001 and † p < 0.05 compared to control at 9 days by Tukey-Kramer multiple comparison test after Two-way ANOVA. C. Effect of the M3-antagonist darifenacin on growth of H82 tumor xenografts in nude mice. Tumor volume. * p < 0.05 compared to control at same time point by Tukey-Kramer multiple comparison test after repeated measures ANOVA. D. Effect of darifenacin on MAPK and Akt phosphorylation in the tumor xenografts. Ratio of density of phosphorylated to unphosphorylated Akt and MAPK in H82 tumor xenografts is shown along with representative bands from western blots for each treatment. Modified after Song et al [58].
7. Nicotinic allosteric modulators and lung cancer
Perhaps the most intriguing target for cholinergic therapy of lung cancer are the ly-6 allosteric modulators of lung cancer. The ly-6 proteins are a large family of small proteins related to snake α-neurotoxins [63,64] including the α7 nAChR antagonist α-bungarotoxin. Ly-6 proteins are also described as 3 finger proteins because of their conserved structure of 3 fingers created by multiple cysteine residues. In humans more than 25 genes encoding ly-6 proteins have been identified, many with multiple forms produced by alternate splicing [64–66]. Most of the ly-6 proteins are membrane bound thorough a GPI linkage; however some of family members are secreted.
Consistent with the similarity of structure of the mammalian ly-6 proteins to α-bungarotoxin, many of the ly-6 proteins modulate nAChR signaling. Miwa et al [67,68] used similarity to α-bungarotoxin to identify a mammalian α-bungarotoxin-like protein in mouse brain which they named lynx1 and then demonstrated it was a negative allosteric regulator of α4β2 and α7 nAChR receptors. In addition to lynx1, Miwa and co-workers also identified a second ly-6 protein in brain, lynx2 and showed it too was a negative regulator of α7 nAChR [69].
Lynx1 and lynx2 are not the only ly-6 proteins that regulate nAChR. The ly-6 protein, slurp-1, was initially characterized for its role in the skin disease Mal de Maleda [70,71] and is a positive regulator of α7 nAChR [72,73]. Slurp-1 lacks a GPI linkage and is one of the secreted ly-6 proteins. Slurp-2 is encoded by an alternate transcript of the LYNX1 gene and has been reported to be a negative nAChR regulator [74]. Prostate stem cell antigen (PSCA) has been shown by Nishi and co-workers to be yet another ly-6 protein that negatively regulates α7 nAChR [75]. Thus lynx1, lynx2, Slurp-1, Slurp-2 and PSCA are ly-6 proteins that have been demonstrated to modulate nAChR activity but it is likely that other members of the ly-6 family will also turn out to modulate nicotinic signaling. Alignment of lynx1, lynx2 and PSCA is shown in figure 5 and the conserved cysteine residues that define the family can be seen.
Figure 5. Alignment of Lynx1, lynx2 and PSCA ly-6 proteins.

Proteins were aligned using the Clustal Omega alignment tool. The conserved cysteine residues that define the family are shown by a triangle. The putative GPI cleavage site as predicted by PredGPI is boxed [78]. * indicates fully conserved amino acids other than Cysteine, : indicates conservation of strongly similar amino acids, . indicates conservation of weakly similar amino acids. The secreted ly-6 proteins slurp-1 and slurp-2 are not included in the alignment, but if they were they would lack the C-terminal hydrophobic residues and the GPI cleavage site, but would still maintain the conserved cysteine residues.
The potential role for lynx1 in regulating lung cancer cell growth is also supported by our previous report that levels of lynx1 are decreased in most lung cancers as compared to adjacent normal tissue [13]. As shown in figure 6A, levels of lynx1 are significantly decreased in lung cancer compared to normal lung and the magnitude of this decrease increases as the tumors become less differentiated. This raises the key question of whether levels of lynx1 can modulate growth of lung cancer cells? As shown in figure 6B, siRNA knockdown of lynx1 significantly increased growth of lung cancer cells. This suggests that the decreased levels of lynx1 in lung cancers shown in figure 6 may have significance in terms of rate of growth of lung cancers. Conversely, over expression of lynx1 significantly decreased growth of the A549 cells compared to cells infected with a control lentivirus (Fig. 6C) [76]. This confirms the role of lynx1 as a modulator of lung cancer cell growth and suggests that small molecule mimetics of lynx1 could have therapeutic potential in lung cancer.
Figure 6. Lynx1 expression and function in lung cancers cells.

A. Relative RNA levels of Lynx1 in squamous cell carcinomas compared to normal and also plotted according to degree of differentiation. †p < 0.03 by t-test, *p < 0.05 compared to normal tissue by Tukey-Kramer after One-way ANOVA. Number of samples of each grade is shown in figure legend in parentheses. Figure modified after Song et al [13]. B. siRNA knockdown of lynx1 increased cell growth of A549 lung adenocarcinoma cells. Levels of lynx1 were decreased by ~70% by the siRNA knockdown (data not shown). Data are mean ± SD of 20 replicates in two separate experiments. * p < 0.05 versus control siRNA. C. Effect of increased lynx1 expression in A549 cells. A lentivirus expressing lynx1 was prepared and A549 cells transduced with the lynx-1 lentiviral vector or a control lentivirus. Lynx1 protein expression was highly expressed after transduction (data not shown). Cell growth is shown relative to day 0. * p < 0.05 compared to controls. From Fu et al [76].
While we have shown data for lynx1 here, there is also evidence supporting roles of other nAChR-modifying ly-6 proteins in cancer. PSCA was identified in part due to its ability to modify neuroblastoma growth [75] and polymorphisms in PSCA are associated with clinical course of gastric carcinomas [77]. Similarly, the ability of lynx2 to modulate alpha7 signaling suggests that it may similarly modulate lung cancer cell growth.
8. Conclusions
As discussed above, there are multiple potential lung cancer therapeutic targets in the cholinergic proliferative pathway in lung cancer. Considering potential side effects, delivery of drug to target and druggability of target the most appealing targets are modulators of choline transport, nicotinic antagonists that target metabotropic functions of the nAChR, muscarinic antagonists, and ly-6 protein mimetics. Muscarinic antagonists are already in clinical use so would appear to be particularly appealing. Small molecule ly-6 mimetics are still relatively rare, but given the success in developing other allosteric modulators of nAChR [48–50] this is a particularly intriguing target.
Acknowledgments
This research was supported by NIH grants CA151601, HL087710 and OD011092
References
- 1.Islami F, Torre LA, Jemal A. Global trends of lung cancer mortality and smoking prevalence. Transl Lung Cancer Res. 2015;4:327–38. doi: 10.3978/j.issn.2218-6751.2015.08.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wahbah M, Boroumand N, Castro C, El-Zeky F, Eltorky M. Changing trends in the distribution of the histologic types of lung cancer: a review of 4,439 cases. Ann Diagn Pathol. 2007;11:89–96. doi: 10.1016/j.anndiagpath.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Gabrielson E. Worldwide trends in lung cancer pathology. Respirology. 2006;11:533–8. doi: 10.1111/j.1440-1843.2006.00909.x. [DOI] [PubMed] [Google Scholar]
- 4.Devesa SS, Bray F, Vizcaino AP, Parkin DM. International lung cancer trends by histologic type: male:female differences diminishing and adenocarcinoma rates rising. Int J Cancer. 2005;117:294–9. doi: 10.1002/ijc.21183. [DOI] [PubMed] [Google Scholar]
- 5.Kumar V, Abbas AK, Fausto N, Aster JC. Robbins and Cotran Patholologic Basis of Disease. 8. Philadelphia, PA: W.B. Saunders Company; 2009. [Google Scholar]
- 6.Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011;19:754–64. doi: 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 7.Ginsburgh PJ, Voles EE, Rosenzweig K. Non-small cell lung cancer. In: DeVita VT, Hellman S, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. Philadelphia: Lippincott, Williams and Wilkins; 2005. pp. 925–83. [Google Scholar]
- 8.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 9.Quik M, Chan J, Patrick J. alpha-Bungarotoxin blocks the nicotinic receptor mediated increase in cell number in a neuroendocrine cell line. Brain Res. 1994;655:161–7. doi: 10.1016/0006-8993(94)91610-1. [DOI] [PubMed] [Google Scholar]
- 10.Maneckjee R, Minna JD. Opioids induce while nicotine suppresses apoptosis in human lung cancer cells. Cell Growth Differ. 1994;5:1033–40. [PubMed] [Google Scholar]
- 11.Schuller HM, Nylen ES, Park P, Becker KL. Nicotine, acetylcholine and bombesin are trophic growth factors in neuroendocrine cell lines derived from experimental hamster lung tumors. Life Sci. 1990;47:571–8. doi: 10.1016/0024-3205(90)90618-2. [DOI] [PubMed] [Google Scholar]
- 12.Song P, Sekhon HS, Jia Y, et al. Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer Res. 2003;63:214–21. [PubMed] [Google Scholar]
- 13.Song P, Sekhon HS, Fu XW, et al. Activated cholinergic signaling provides a target in squamous cell lung carcinoma. Cancer Res. 2008;68:4693–700. doi: 10.1158/0008-5472.CAN-08-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klapproth H, Reinheimer T, Metzen J, et al. Non-neuronal acetylcholine, a signalling molecule synthezised by surface cells of rat and man. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:515–23. doi: 10.1007/pl00004977. [DOI] [PubMed] [Google Scholar]
- 15.Wessler I, Kirkpatrick CJ, Racke K. Non-neuronal acetylcholine, a locally acting molecule, widely distributed in biological systems: expression and function in humans. Pharmacol Ther. 1998;77:59–79. doi: 10.1016/s0163-7258(97)00085-5. [DOI] [PubMed] [Google Scholar]
- 16.Reinheimer T, Bernedo P, Klapproth H, et al. Acetylcholine in isolated airways of rat, guinea pig, and human: species differences in role of airway mucosa. Am J Physiol. 1996;270:L722–L728. doi: 10.1152/ajplung.1996.270.5.L722. [DOI] [PubMed] [Google Scholar]
- 17.Proskocil BJ, Sekhon HS, Jia Y, et al. Acetylcholine Is an Autocrine or Paracrine Hormone Synthesized and Secreted by Airway Bronchial Epithelial Cells. Endocrinology. 2004;145:2498–506. doi: 10.1210/en.2003-1728. [DOI] [PubMed] [Google Scholar]
- 18.Kus L, Borys E, Ping CY, et al. Distribution of high affinity choline transporter immunoreactivity in the primate central nervous system. J Comp Neurol. 2003;463:341–57. doi: 10.1002/cne.10759. [DOI] [PubMed] [Google Scholar]
- 19.Ferguson SM, Bazalakova M, Savchenko V, Tapia JC, Wright J, Blakely RD. Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc Natl Acad Sci U S A. 2004;101:8762–7. doi: 10.1073/pnas.0401667101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeil U, Lips KS, Eberling L, Grau V, Haberberger RV, Kummer W. Expression of the high-affinity choline transporter, CHT1, in the rat trachea. Am J Respir Cell Mol Biol. 2003;28:473–7. doi: 10.1165/rcmb.2002-0190OC. [DOI] [PubMed] [Google Scholar]
- 21.Yajima T, Inoue R, Matsumoto M, Yajima M. Non-neuronal release of ACh plays a key role in secretory response to luminal propionate in rat colon. J Physiol. 2011;589:953–62. doi: 10.1113/jphysiol.2010.199976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song P, Spindel ER. Basic and clinical aspects of non-neuronal acetylcholine: expression of non-neuronal acetylcholine in lung cancer provides a new target for cancer therapy. J Pharmacol Sci. 2008;106:180–5. doi: 10.1254/jphs.fm0070091. [DOI] [PubMed] [Google Scholar]
- 23.Traiffort E, Ruat M, O’Regan S, Meunier FM. Molecular characterization of the family of choline transporter-like proteins and their splice variants. J Neurochem. 2005;92:1116–25. doi: 10.1111/j.1471-4159.2004.02962.x. [DOI] [PubMed] [Google Scholar]
- 24.O’Regan S, Traiffort E, Ruat M, Cha N, Compaore D, Meunier FM. An electric lobe suppressor for a yeast choline transport mutation belongs to a new family of transporter-like proteins. Proc Natl Acad Sci U S A. 2000;97:1835–40. doi: 10.1073/pnas.030339697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inazu M, Takeda H, Matsumiya T. Molecular and functional characterization of an Na+-independent choline transporter in rat astrocytes. J Neurochem. 2005;94:1427–37. doi: 10.1111/j.1471-4159.2005.03299.x. [DOI] [PubMed] [Google Scholar]
- 26.Yabuki M, Inazu M, Yamada T, Tajima H, Matsumiya T. Molecular and functional characterization of choline transporter in rat renal tubule epithelial NRK-52E cells. Arch Biochem Biophys. 2009;485:88–96. doi: 10.1016/j.abb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Uchida Y, Inazu M, Takeda H, Yamada T, Tajima H, Matsumiya T. Expression and functional characterization of choline transporter in human keratinocytes. J Pharmacol Sci. 2009;109:102–9. doi: 10.1254/jphs.08291fp. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura T, Fujiwara R, Ishiguro N, et al. Involvement of choline transporter-like proteins, CTL1 and CTL2, in glucocorticoid-induced acceleration of phosphatidylcholine synthesis via increased choline uptake. Biol Pharm Bull. 2010;33:691–6. doi: 10.1248/bpb.33.691. [DOI] [PubMed] [Google Scholar]
- 29.Song P, Rekow SS, Singleton CA, et al. Choline transporter-like protein 4 (CTL4) links to non-neuronal acetylcholine synthesis. J Neurochem. 2013;126:451–61. doi: 10.1111/jnc.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beckmann J, Schubert J, Morhenn HG, Grau V, Schnettler R, Lips KS. Expression of choline and acetylcholine transporters in synovial tissue and cartilage of patients with rheumatoid arthritis and osteoarthritis. Cell Tissue Res. 2015;359:465–77. doi: 10.1007/s00441-014-2036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lips KS, Wunsch J, Zarghooni S, et al. Acetylcholine and molecular components of its synthesis and release machinery in the urothelium. Eur Urol. 2007;51:1042–53. doi: 10.1016/j.eururo.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 32.Yokota H, Guo J, Matoba M, Higashi K, Tonami H, Nagao Y. Lactate, choline, and creatine levels measured by vitro 1H-MRS as prognostic parameters in patients with non-small-cell lung cancer. J Magn Reson Imaging. 2007;25:992–9. doi: 10.1002/jmri.20902. [DOI] [PubMed] [Google Scholar]
- 33.Glunde K, Ackerstaff E, Mori N, Jacobs MA, Bhujwalla ZM. Choline phospholipid metabolism in cancer: consequences for molecular pharmaceutical interventions. Mol Pharm. 2006;3:496–506. doi: 10.1021/mp060067e. [DOI] [PubMed] [Google Scholar]
- 34.Janardhan S, Srivani P, Sastry GN. Choline kinase: an important target for cancer. Curr Med Chem. 2006;13:1169–86. doi: 10.2174/092986706776360923. [DOI] [PubMed] [Google Scholar]
- 35.Tomi M, Arai K, Tachikawa M, Hosoya KI. Na(+)-Independent Choline Transport in Rat Retinal Capillary Endothelial Cells. Neurochem Res. 2007 doi: 10.1007/s11064-007-9367-0. [DOI] [PubMed] [Google Scholar]
- 36.Auld DS, Day JC, Mennicken F, Quirion R. Pharmacological characterization of endogenous acetylcholine release from primary septal cultures. J Pharmacol Exp Ther. 2000;292:692–7. [PubMed] [Google Scholar]
- 37.Martinez-Moreno P, Nieto-Ceron S, Torres-Lanzas J, et al. Cholinesterase activity of human lung tumours varies according to their histological classification. Carcinogenesis. 2006;27:429–36. doi: 10.1093/carcin/bgi250. [DOI] [PubMed] [Google Scholar]
- 38.de Martinez-Lopez CA, Nieto-Ceron S, Pons-Castillo A, et al. Cancer-associated differences in the acetylcholinesterase activity in bronchial aspirates of lung cancer patients. Clin Sci (Lond) 2008 doi: 10.1042/CS20070393. [DOI] [PubMed] [Google Scholar]
- 39.Fujii T, Yamada S, Yamaguchi N, Fujimoto K, Suzuki T, Kawashima K. Species differences in the concentration of acetylcholine, a neurotransmitter, in whole blood and plasma. Neurosci Lett. 1995;201:207–10. doi: 10.1016/0304-3940(95)12180-3. [DOI] [PubMed] [Google Scholar]
- 40.Rayford W, Noble MJ, Austenfeld MA, Weigel J, Mebust WK, Shah GV. Muscarinic cholinergic receptors promote growth of human prostate cancer cells. Prostate. 1997;30:160–6. doi: 10.1002/(sici)1097-0045(19970215)30:3<160::aid-pros3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 41.Dasgupta P, Rastogi S, Pillai S, et al. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J Clin Invest. 2006;116:2208–17. doi: 10.1172/JCI28164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Improgo MR, Soll LG, Tapper AR, Gardner PD. Nicotinic acetylcholine receptors mediate lung cancer growth. Front Physiol. 2013;4:251. doi: 10.3389/fphys.2013.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng Y, Ritzenthaler JD, Roman J, Han S. Nicotine Stimulates Human Lung Cancer Cell Growth by Inducing Fibronectin Expression. Am J Respir Cell Mol Biol. 2007;37:681–90. doi: 10.1165/rcmb.2007-0051OC. [DOI] [PubMed] [Google Scholar]
- 44.Grando SA. Connections of nicotine to cancer. Nat Rev Cancer. 2014;14:419–29. doi: 10.1038/nrc3725. [DOI] [PubMed] [Google Scholar]
- 45.Arredondo J, Chernyavsky AI, Jolkovsky DL, Pinkerton KE, Grando SA. Receptor-mediated tobacco toxicity: cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. FASEB J. 2006;20:2093–101. doi: 10.1096/fj.06-6191com. [DOI] [PubMed] [Google Scholar]
- 46.Galitovskiy V, Chernyavsky AI, Edwards RA, Grando SA. Muscle sarcomas and alopecia in A/J mice chronically treated with nicotine. Life Sci. 2012 doi: 10.1016/j.lfs.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alama A, Bruzzo C, Cavalieri Z, et al. Inhibition of the nicotinic acetylcholine receptors by cobra venom alpha-neurotoxins: is there a perspective in lung cancer treatment? PLoS ONE. 2011;6:e20695. doi: 10.1371/journal.pone.0020695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gill-Thind JK, Dhankher P, D’Oyley JM, Sheppard TD, Millar NS. Structurally similar allosteric modulators of alpha7 nicotinic acetylcholine receptors exhibit five distinct pharmacological effects. J Biol Chem. 2014;290:3552–62. doi: 10.1074/jbc.M114.619221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gill JK, Dhankher P, Sheppard TD, Sher E, Millar NS. A series of alpha7 nicotinic acetylcholine receptor allosteric modulators with close chemical similarity but diverse pharmacological properties. Mol Pharmacol. 2012;81:710–8. doi: 10.1124/mol.111.076026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Papke RL, Chojnacka K, Horenstein NA. The minimal pharmacophore for silent agonism of alpha7 nAChR. J Pharmacol Exp Ther. 2014 doi: 10.1124/jpet.114.215236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papke RL, Bagdas D, Kulkarni AR, et al. The analgesic-like properties of the alpha7 nAChR silent agonist NS6740 is associated with non-conducting conformations of the receptor. Neuropharmacology. 2015;91:34–42. doi: 10.1016/j.neuropharm.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bierut LJ. Convergence of genetic findings for nicotine dependence and smoking related diseases with chromosome 15q24–25. Trends Pharmacol Sci. 2010;31:46–51. doi: 10.1016/j.tips.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat Rev Cancer. 2009 doi: 10.1038/nrc2590. [DOI] [PubMed] [Google Scholar]
- 54.Schuller HM, Orloff M. Tobacco-specific carcinogenic nitrosamines. Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem Pharmacol. 1998;55:1377–84. doi: 10.1016/s0006-2952(97)00651-5. [DOI] [PubMed] [Google Scholar]
- 55.Mihalak KB, Carroll FI, Luetje CW. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol Pharmacol. 2006;70:801–5. doi: 10.1124/mol.106.025130. [DOI] [PubMed] [Google Scholar]
- 56.Cahill K, Stevens S, Perera R, Lancaster T. Pharmacological interventions for smoking cessation: an overview and network meta-analysis. Cochrane Database Syst Rev. 2013;5:CD009329. doi: 10.1002/14651858.CD009329.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bohonus VL, Doolittle RF, Pontes M, Strong DD. Complementary DNA sequence of lamprey fibrinogen beta chain. Biochemistry. 1986;25:6512–6. doi: 10.1021/bi00369a026. [DOI] [PubMed] [Google Scholar]
- 58.Song P, Sekhon HS, Lu A, et al. M3 muscarinic receptor antagonists inhibit small cell lung carcinoma growth and mitogen-activated protein kinase phosphorylation induced by acetylcholine secretion. Cancer Res. 2007;67:3936–44. doi: 10.1158/0008-5472.CAN-06-2484. [DOI] [PubMed] [Google Scholar]
- 59.Raufman JP, Samimi R, Shah N, et al. Genetic ablation of M3 muscarinic receptors attenuates murine colon epithelial cell proliferation and neoplasia. Cancer Res. 2008;68:3573–8. doi: 10.1158/0008-5472.CAN-07-6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spindel ER. Muscarinic receptor agonists and antagonists: effects on cancer. Handb Exp Pharmacol. 2012;208:451–68. doi: 10.1007/978-3-642-23274-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maman K, Aballea S, Nazir J, et al. Comparative efficacy and safety of medical treatments for the management of overactive bladder: a systematic literature review and mixed treatment comparison. Eur Urol. 2014;65:755–65. doi: 10.1016/j.eururo.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 62.Alagha K, Palot A, Sofalvi T, et al. Long-acting muscarinic receptor antagonists for the treatment of chronic airway diseases. Ther Adv Chronic Dis. 2014;5:85–98. doi: 10.1177/2040622313518227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsetlin V. Snake venom alpha-neurotoxins and other ‘three-finger’ proteins. Eur J Biochem. 1999;264:281–6. doi: 10.1046/j.1432-1327.1999.00623.x. [DOI] [PubMed] [Google Scholar]
- 64.Tsetlin VI. Three-finger snake neurotoxins and Ly6 proteins targeting nicotinic acetylcholine receptors: pharmacological tools and endogenous modulators. Trends Pharmacol Sci. 2015;36:109–23. doi: 10.1016/j.tips.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 65.Galat A, Gross G, Drevet P, Sato A, Menez A. Conserved structural determinants in three-fingered protein domains. FEBS J. 2008;275:3207–25. doi: 10.1111/j.1742-4658.2008.06473.x. [DOI] [PubMed] [Google Scholar]
- 66.Miwa JM, Lester HA, Walz A. Optimizing cholinergic tone through lynx modulators of nicotinic receptors: implications for plasticity and nicotine addiction. Physiology (Bethesda) 2012;27:187–99. doi: 10.1152/physiol.00002.2012. [DOI] [PubMed] [Google Scholar]
- 67.Ibanez-Tallon I, Miwa JM, Wang HL, et al. Novel modulation of neuronal nicotinic acetylcholine receptors by association with the endogenous prototoxin lynx1. Neuron. 2002;33:893–903. doi: 10.1016/s0896-6273(02)00632-3. [DOI] [PubMed] [Google Scholar]
- 68.Miwa JM, Ibanez-Tallon I, Crabtree GW, et al. Lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron. 1999;23:105–14. doi: 10.1016/s0896-6273(00)80757-6. [DOI] [PubMed] [Google Scholar]
- 69.Tekinay AB, Nong Y, Miwa JM, et al. A role for LYNX2 in anxiety-related behavior. Proc Natl Acad Sci U S A. 2009;106:4477–82. doi: 10.1073/pnas.0813109106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fischer J, Bouadjar B, Heilig R, et al. Mutations in the gene encoding SLURP-1 in Mal de Meleda. Hum Mol Genet. 2001;10:875–80. doi: 10.1093/hmg/10.8.875. [DOI] [PubMed] [Google Scholar]
- 71.Chimienti F, Hogg RC, Plantard L, et al. Identification of SLURP-1 as an epidermal neuromodulator explains the clinical phenotype of Mal de Meleda. Hum Mol Genet. 2003;12:3017–24. doi: 10.1093/hmg/ddg320. [DOI] [PubMed] [Google Scholar]
- 72.Horiguchi K, Horiguchi S, Yamashita N, et al. Expression of SLURP-1, an endogenous alpha7 nicotinic acetylcholine receptor allosteric ligand, in murine bronchial epithelial cells. J Neurosci Res. 2009;87:2740–7. doi: 10.1002/jnr.22102. [DOI] [PubMed] [Google Scholar]
- 73.Narumoto O, Niikura Y, Ishii S, et al. Effect of secreted lymphocyte antigen-6/urokinase-type plasminogen activator receptor-related peptide-1 (SLURP-1) on airway epithelial cells. Biochem Biophys Res Commun. 2013;438:175–9. doi: 10.1016/j.bbrc.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 74.Arredondo J, Chernyavsky AI, Jolkovsky DL, Webber RJ, Grando SA. SLURP-2: A novel cholinergic signaling peptide in human mucocutaneous epithelium. J Cell Physiol. 2006;208:238–45. doi: 10.1002/jcp.20661. [DOI] [PubMed] [Google Scholar]
- 75.Hruska M, Keefe J, Wert D, et al. Prostate stem cell antigen is an endogenous lynx1-like prototoxin that antagonizes alpha7-containing nicotinic receptors and prevents programmed cell death of parasympathetic neurons. J Neurosci. 2009;29:14847–54. doi: 10.1523/JNEUROSCI.2271-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu XW, Song PF, Spindel ER. Role of Lynx1 and related Ly6 proteins as modulators of cholinergic signaling in normal and neoplastic bronchial epithelium. Int Immunopharmacol. 2015;29:93–8. doi: 10.1016/j.intimp.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu Y, Chen J, Ding Y, et al. Genetic variation of PSCA gene is associated with the risk of both diffuse- and intestinal-type gastric cancer in a Chinese population. Int J Cancer. 2010;127:2183–9. doi: 10.1002/ijc.25228. [DOI] [PubMed] [Google Scholar]
- 78.Pierleoni A, Martelli PL, Casadio R. PredGPI: a GPI-anchor predictor. BMC Bioinformatics. 2008;9:392. doi: 10.1186/1471-2105-9-392. [DOI] [PMC free article] [PubMed] [Google Scholar]