

Abstract
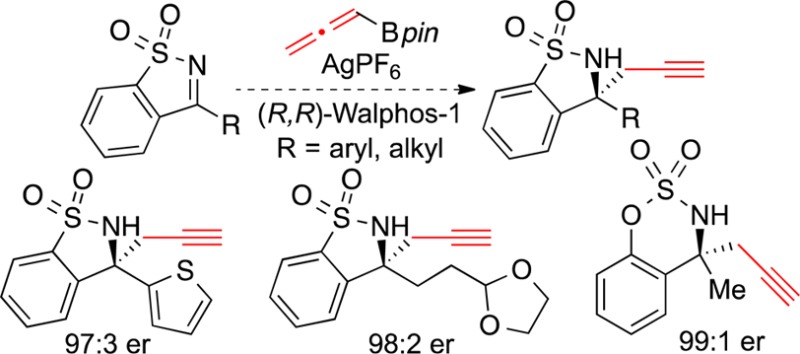
The enantioselective silver-catalyzed propargylation of N-sulfonylketimines is described. This reaction proceeds in high yield and excellent enantiomeric ratio and is compatible with a wide variety of diaryl- and alkylketimines. Synthetic transformations of homopropargylic products via enyne ring-closing metathesis, Sonogashira cross-coupling, and reduction reactions proceed with high stereochemical fidelity. Both allenyl and propargyl borolane reagents can be used to obtain homopropargylic products, a distribution most consistent with a mechanism involving transmetalation of the silver catalyst with the borolane reagent.
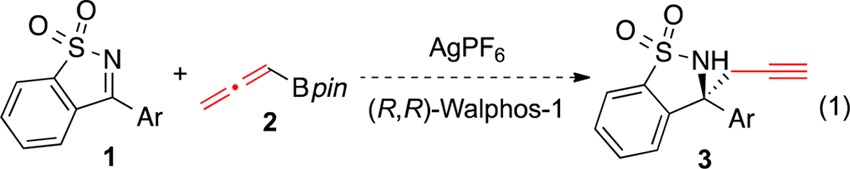
Nearly half of the top 200 pharmaceuticals in 2012 contain functional groups that can be prepared from α-chiral amines.1 To access this moiety, numerous enantioselective methods for the synthesis of chiral amines have been developed, many of which involve addition of organometallic nucleophiles to aldimines.2 Additions to ketimines pose specific challenges. For example, mixtures of E and Z isomers can lead to low levels of enantioinduction.3 These obstacles have inspired creative approaches4 including use of cyclic N-sulfonylketimines (e.g., 1), which do not undergo E/Z isomerization and are synthesized in one step from saccharin.5,6 Hayashi and co-workers have pioneered the rhodium-catalyzed enantioselective arylation reactions of N-sulfonylketimines; other elegant examples of arylation, allylation, and alkenylation reactions have also been reported.5c,7 An enantioselective propargylation reaction would afford a chiral sultam with a pendant terminal alkyne, a valuable functional group handle that can be easily derivatized for further synthetic elaboration.8 In this paper, we report the first enantioselective propargylation reaction of ketimines (eq 1).
 |
1 |
Building on early advances in enantioselective propargylation reactions of aldehydes, in the past five years there has been rapid development of enantioselective propargylation reactions of ketones and aldimines.9−11 Our laboratory has reported the silver-catalyzed enantioselective propargylation reactions of aldimines and diaryl ketones.10b,12,13 Using AgF and chiral phosphine ligands from the Walphos family provided a variety of homopropargylic amines and alcohols in good yield and high enantiomeric excess (ee). We reasoned that a Ag/Walphos catalyst would be able to differentiate between the Re and Si faces of diarylketimines, based on their structural similarity to diaryl ketones. Indeed, we found that employing AgPF6 with Walphos-1 resulted in formation of homopropargylic amine 3a in 99:1 enantiomeric ratio (er) and modest yield (Table 1, entry 1). In comparison, preforming the catalyst in methanol and running the reaction in THF gave high er but only 19% product (entry 2).14 Other chiral ferrocene-based ligands such as Walphos-8 and Josiphos-6 provided lower er (entries 3 and 4), as did (S)-BINAP (entry 5).
Table 1. Optimization of Silver-Catalyzed Propargylation Reaction.

| entry | deviation from standard conditions | yielda (%) | erb |
|---|---|---|---|
| 1 | none | 51 | 99:1 |
| 2c | catalyst preformed in MeOH and reaction run in THF | 19 | 96:4 |
| 3 | Walphos-8 instead of Walphos-1 | 35 | 76:24 |
| 4 | Josiphos-6 instead of Walphos-1 | 70 | 74:26 |
| 5 | (S)-BINAP instead of Walphos-1 | 55 | 40:60 |
| 6 | reaction performed at 4 °C | 54 | 99:1 |
| 7 | reaction performed at rt | 57 | 98:2 |
| 8 | reaction performed at rt with 4 equiv 2 | 76 | 99:1 |

Determined using 1H NMR by comparison to PhTMS as internal standard.
Determined using chiral SFC.
Preparation of Ag/Walphos catalyst performed according to ref (10b).
We further modified the reaction conditions to improve the yield. We found that at ambient temperature 3a was still formed in a modest yield; fortunately, the er remained high (Table 1, entry 7). We hypothesized that protodeborylation of allenylboronic acid pinacol ester 2 to allene (C3H4) was competitive with the desired addition reaction and thus resulted in modest yields.15 Use of an additional 2 equiv of allenylboronic acid pinacol ester 2 via slow addition increased the yield, providing 3a in 76% yield and 99:1 er (entry 8).
Having determined optimized conditions for this reaction, we proceeded to evaluate the substrate scope. A wide range of arylketimines underwent enantioselective propargylation in high yield and >95:5 er (Scheme 1). Ketimines containing electron-withdrawing groups formed products in excellent er (3b–d). We were gratified to find that substrates with electron-donating groups participated in this reaction, as the starting ketimines are generally less reactive. 4-Methoxyphenyl-substituted sultam 3e was generated in good yield and excellent er upon using 6 equiv of allenylboronic acid pinacol ester 2. In addition, several heterocycles were tolerated in the reaction. Ketimines containing furan, thiophene, and benzothiophene functional groups reacted smoothly to provide the corresponding homopropargylic sulfonamides (3f–h) in high er. The absolute configurations of 3a, 3f, and 3h were determined by X-ray crystallographic analysis.16
Scheme 1. Scope of Diaryl Sultams.

6 equiv allenylboronic acid pinacol ester 2 were employed.
Absolute configuration assigned by X-ray crystallographic analysis.16
We were pleased to find that alkylketimines were also well tolerated in the reaction (Scheme 2), since we were concerned that these substrates would tautomerize to enamines in the presence of potassium tert-butoxide. Several alkylketimines reacted to give homopropargylic products in excellent er (5a–d). Furthermore, we found that other functional groups are compatible with this method: sultam 5c, containing an acetal protecting group, was formed in high yield. The absolute configurations of 5b and 5c were determined by X-ray crystallographic analysis.17
Scheme 2. Scope of Alkyl Sultams.

Absolute configuration assigned by X-ray crystallographic analysis.17
To emphasize the utility of the pendant terminal alkyne, we synthesized derivatives of several alkyl and aryl homopropargylic sulfonamides (Scheme 3). We prepared compound 5d for an enyne ring-closing metathesis (Scheme 3a).18 In the presence of 5 mol % of Grubbs I catalyst and under an atmosphere of ethylene, the desired spirocycle 6 was obtained in 76% yield. The Sonogashira cross-coupling reaction of compound 5b with ethyl 4-iodobenzoate proceeded in high yield (Scheme 3b). Lindlar reduction of 3a provided the corresponding enantioenriched diaryl allyl sultam 8, a moiety that has not been previously reported (Scheme 3c).7e,19 We further highlighted the versatility of the alkyne moiety by fully reducing 3a to alkane 9 in 89% yield using palladium on carbon, without reduction of the cyclic benzylic sulfonamide (Scheme 3d).
Scheme 3. Synthetic Transformations of Homopropargylic Sultams.

Enantiomeric ratio could not be determined using chiral SFC instrumentation.
Cyclic sulfamate ketimines (e.g., 10) are a related class of N-sulfonylketimines that react with nucleophiles to provide sulfamidates that can be easily transformed to 2-hydroxyphenylmethylamines.7 However, these ketimines are typically less reactive in addition reactions. We wanted to challenge our method and found that homopropargylic sulfamidates 11a and 11b were formed in good yield and >99:1 er (Scheme 4).
Scheme 4. Scope of Alkyl Sulfamidates.

We sought to establish a reasonable mechanism for this propargylation reaction; two of the most likely possibilities are presented in Figure 1.20 Our approach to distinguish between these mechanisms was to compare product distributions from reactions employing isomeric borolane reagents, allenyl borolane 2 and propargyl borolane 12. Importantly, both mechanisms take into account our experimental observation that allenyl borolane 2 and propargyl borolane 12 are not in equilibrium under the reaction conditions during the time frame of the reaction (vide infra).21
Figure 1.

Possible mechanisms for silver-catalyzed propargylation reaction. (a) Proposed catalytic cycle involving transmetalation of silver catalyst with borolane reagent. (b) Lewis acid catalysis.
Mechanism A involves transmetalation and isomerization of the allenylmetal intermediates.22 Transmetalation of the silver catalyst with the borolane reagent forms the key nucleophilic allenylsilver complex (13) in situ. Allenylsilver complex 13 is in equilibrium with propargylsilver complex 14.23,24 Addition of allenylsilver complex 13 to the ketimine via SE2′ mechanism is favored to form alkyne 3a. Therefore, if mechanism A is operative, using either allenyl borolane 2 or propargyl borolane 12 would result in formation of alkyne 3a via equilibration of 13 and 14 (Figure 1a).
An alternative pathway is mechanism B, involving direct addition of the borolane reagent to the ketimine.20 In this scenario, the silver catalyst acts as a chiral Lewis acid in the reaction (Figure 1b (1)). Coordination of the silver catalyst to form intermediate 16 followed by SE2′ addition of allenyl borolane 2 results in formation of alkyne 3a. In this possible mechanism, isomerization of the allenyl- and propargylboron species is slower than attack on the activated electrophile (16).21 Therefore, using propargyl borolane 12 would provide a different product, allene 18 (Figure 1b (2)).
To rule out one of these two possible mechanisms, we set out to examine reactions employing propargyl borolane 12.25 We synthesized 12 using a procedure recently published by Fandrick and co-workers.22c Using propargyl borolane 12 in the reaction yielded alkyne 3a in 64% (Table 2).26 We found that the er of the product remained high, with a slight decrease when using propargyl borolane 12. To determine whether or not propargyl borolane 12 is in equilibrium with allenyl borolane 2 under the reaction conditions, the reaction was performed in deuterated DMF and the ratio of propargyl to allenyl borolane was analyzed by 1H NMR before workup. The ratio of propargyl borolane 12 to allenyl borolane 2 remained similar before and after the reaction, most consistent with negligible equilibration of 12 and 2 over the time course of the propargylation reactions. While other mechanistic possibilities exist, these results are most consistent with mechanism A, where the silver catalyst undergoes transmetalation with the borolane reagent.
Table 2. Silver-Catalyzed Propargylation Reaction Using Allenylboronic Acid Pinacol Ester 2 or Propargylboronic Acid Pinacol Ester 12.


Determined by 1H NMR.
Isolated yield.
Determined using chiral SFC.
Determined using 1H NMR by comparison to PhTMS as internal standard.
In summary, we have developed an enantioselective silver-catalyzed propargylation reaction of cyclic N-sulfonylketimines. Using a catalyst prepared from AgPF6 and Walphos-1, we found that many aryl and alkyl homopropargylic amines were formed in high yield and excellent er. Derivatization of the terminal alkyne yielded spirocyclic, alkenyl, or alkyl products. Mechanistic experiments employing propargyl borolane reagent are most consistent with a mechanism in which the silver catalyst undergoes transmetalation with the borolane reagent to generate a nucleophilic allenylboron reagent.
Acknowledgments
This work was supported by NIH NCI (F31CA177212 to C.A.O) and DoEd GAANN (PA200A120070 to T.B.D.E). We thank Dr. Daniel Fandrick (Boehringer Ingelheim) for helpful discussions regarding the synthesis of propargylboronic acid pinacol ester 12. Dr. Joseph Ziller and Jordan Corbey (University of California, Irvine) are acknowledged for X-ray crystallography data. Dr. John Greaves and Dr. Beniam Berhane (University of California, Irvine) are acknowledged for mass spectrometry data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.5b02692.
The authors declare no competing financial interest.
Supplementary Material
References
- Ninety-four of the top 200 pharmaceuticals by U.S. retail sales in 2012 contained functional groups that could be prepared from α-chiral amines, while 25 of the top 200 pharmaceuticals contained α-chiral amines. See:; a Njardarson Group . Top Pharmaceuticals Poster. http://jon.oia.arizona.edu/top-pharmaceuticals-poster (accessed May 30, 2015).; b McGrath N. A.; Brichacek M.; Njardarson J. T. J. Chem. Educ. 2010, 87, 1348. 10.1021/ed1003806. [DOI] [Google Scholar]
- For reviews on catalytic enantioselective methods for generating chiral amines, see:; a Nugent T. C.; El-Shazly M. Adv. Synth. Catal. 2010, 352, 753. 10.1002/adsc.200900719. [DOI] [Google Scholar]; b Kobayashi S.; Mori Y.; Fossey J. S.; Salter M. M. Chem. Rev. 2011, 111, 2626. 10.1021/cr100204f. [DOI] [PubMed] [Google Scholar]
- For a discussion and review, see:Riant O.; Hannedouche J. Org. Biomol. Chem. 2007, 5, 873. 10.1039/b617746h. [DOI] [PubMed] [Google Scholar]
- For a lead reference, see:Yin L.; Otsuka Y.; Takada H.; Mouri S.; Yazaki R.; Kumagai N.; Shibasaki M. Org. Lett. 2013, 15, 698. 10.1021/ol3035609. [DOI] [PubMed] [Google Scholar]
- a For enantioselective hydrogenation, see:Oppolzer W.; Wills M.; Starkemann C.; Bernardinelli G. Tetrahedron Lett. 1990, 31, 4117. 10.1016/S0040-4039(00)97557-9. [DOI] [Google Scholar]; b For homoenolate additions, see:Rommel M.; Fukuzumi T.; Bode J. W. J. Am. Chem. Soc. 2008, 130, 17266. 10.1021/ja807937m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c For enantioselective arylation, see:Nishimura T.; Noishiki A.; Tsui G. C.; Hayashi T. J. Am. Chem. Soc. 2012, 134, 5056. 10.1021/ja300697c. [DOI] [PubMed] [Google Scholar]; d For formal [3 + 2] cycloadditions with TMM, see:Trost B. M.; Silverman S. M. J. Am. Chem. Soc. 2012, 134, 4941. 10.1021/ja210981a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synthesis of N-sulfonylketimines from saccharin:Davis F. A.; Towson J. C.; Vashi D. B.; ThimmaReddy R.; McCauley J. P. Jr.; Harakal M. E.; Gosciniak D. J. J. Org. Chem. 1990, 55, 1254. 10.1021/jo00291a028. [DOI] [Google Scholar]
- For enantioselective arylation, see:; a Jiang C.; Lu Y.; Hayashi T. Angew. Chem., Int. Ed. 2014, 53, 9936. 10.1002/anie.201406147. [DOI] [PubMed] [Google Scholar]; b Yang G.; Zhang W. Angew. Chem., Int. Ed. 2013, 52, 7540. 10.1002/anie.201302861. [DOI] [PubMed] [Google Scholar]; c Wang H.; Jiang T.; Xu M.-H. J. Am. Chem. Soc. 2013, 135, 971. 10.1021/ja3110818. [DOI] [PubMed] [Google Scholar]; d Jiang T.; Wang Z.; Xu M.-H. Org. Lett. 2015, 17, 528. 10.1021/ol503537w. [DOI] [PubMed] [Google Scholar]; e For enantioselective allylation, see:Luo Y.; Hepburn H. B.; Chotsaeng N.; Lam H. W. Angew. Chem., Int. Ed. 2012, 51, 8309. 10.1002/anie.201204004. [DOI] [PubMed] [Google Scholar]; f For enantioselective alkenylation, see:Luo Y.; Carnell A. J.; Lam H. W. Angew. Chem., Int. Ed. 2012, 51, 6762. 10.1002/anie.201202136. [DOI] [PubMed] [Google Scholar]
- For recent examples in synthesis of polyketides, see:; a Mailhol D.; Willwacher J.; Kausch-Busies N.; Rubitski E. E.; Sobol Z.; Schuler M.; Lam M.-H.; Musto S.; Loganzo F.; Maderna A.; Fürstner A. J. Am. Chem. Soc. 2014, 136, 15719. 10.1021/ja508846g. [DOI] [PubMed] [Google Scholar]; b Reznik S. K.; Marcus B. S.; Leighton J. L. Chem. Sci. 2012, 3, 3326. 10.1039/c2sc21325g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a For a comprehensive review, see:Ding C.-H.; Hou X.-L. Chem. Rev. 2011, 111, 1914. 10.1021/cr100284m. [DOI] [PubMed] [Google Scholar]; b For a synopsis of catalytic enantioselective propargylation reactions of ketones and imines, see:Wisniewska H. M.; Jarvo E. R. J. Org. Chem. 2013, 78, 11629. 10.1021/jo4019107. [DOI] [PubMed] [Google Scholar]
- For catalyst-controlled, enantioselective propargylation of imines, see:; a Kagoshima H.; Uzawa T.; Akiyama T. Chem. Lett. 2002, 31, 298. 10.1246/cl.2002.298. [DOI] [Google Scholar]; b Wisniewska H. M.; Jarvo E. R. Chem. Sci. 2011, 2, 807. 10.1039/c0sc00613k. [DOI] [Google Scholar]; c Vieira E. M.; Haeffner F.; Snapper M. L.; Hoveyda A. H. Angew. Chem., Int. Ed. 2012, 51, 6618. 10.1002/anie.201202694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of diastereoselective propargylation of imines, see:; a Gonzalez A. Z.; Soderquist J. A. Org. Lett. 2007, 9, 1081. 10.1021/ol070074g. [DOI] [PubMed] [Google Scholar]; b Fandrick D. R.; Johnson C. S.; Fandrick K. R.; Reeves J. T.; Tan Z.; Lee H.; Song J. J.; Yee N. K.; Senanayake C. H. Org. Lett. 2010, 12, 748. 10.1021/ol9028258. [DOI] [PubMed] [Google Scholar]; c García-Muñoz M. J.; Zacconi F.; Foubelo F.; Yus M. Eur. J. Org. Chem. 2013, 2013, 1287. 10.1002/ejoc.201201410. [DOI] [Google Scholar]; d Guo T.; Song R.; Yuan B.-H.; Chen X.-Y.; Sun X.-W.; Lin G.-Q. Chem. Commun. 2013, 49, 5402. 10.1039/c3cc42481b. [DOI] [PubMed] [Google Scholar]; e Chen D.; Xu M.-H. Chem. Commun. 2013, 49, 1327. 10.1039/c2cc38600c. [DOI] [PubMed] [Google Scholar]
- Kohn B. L.; Ichiishi N.; Jarvo E. R. Angew. Chem., Int. Ed. 2013, 52, 4414. 10.1002/anie.201206971. [DOI] [PubMed] [Google Scholar]
- For silver-catalyzed enantioselective allylation reactions of ketones, see:Wadamoto M.; Yamamoto H. J. Am. Chem. Soc. 2005, 127, 14556. 10.1021/ja0553351. [DOI] [PubMed] [Google Scholar]
- Formation of silver phosphine complexes in methanol followed by solvent replacement with THF is employed in related transformations. See refs (10b)(12), and (13).
- Kohn B. L.; Jarvo E. R. Org. Lett. 2011, 13, 4858. 10.1021/ol2019423. [DOI] [PubMed] [Google Scholar]
- For supplementary crystallographic data, see the Supporting Information and CCDC 1405841, 1405894, and 1405895.
- For supplementary crystallographic data, see the Supporting Information and CCDC 1410049 and 1405843.
- Diver S. T.; Giessert A. J. Chem. Rev. 2004, 104, 1317. 10.1021/cr020009e. [DOI] [PubMed] [Google Scholar]
- For enantioselective allylation of alkyl cyclic N-sulfonylketimines, see:Hepburn H. B.; Chotsaeng N.; Luo Y.; Lam H. W. Synthesis 2013, 45, 2649. 10.1055/s-0033-1339499. [DOI] [Google Scholar]
- For a discussion of these mechanistic possibilities, including representative examples, see ref (9a,9b).
- Propargyl borolane 12 and allenyl borolane 2 can undergo base-catalyzed equilibration, favoring allenyl borolane 2. However, in control experiments, we have determined that under the propargylation reaction conditions (8% KOt-Bu, DMF, 6 h) in the absence of silver catalyst there is no equilibration between 12 and 2. See the Supporting Information for full details.
- For selected examples of reactions that likely proceed through a similar mechanism, see:; a Tamaru Y.; Goto S.; Tanaka A.; Shimizu N.; Kimura M. Angew. Chem., Int. Ed. Engl. 1996, 35, 878. 10.1002/anie.199608781. [DOI] [Google Scholar]; b Hameury T.; Guillemont J.; Van Hijfte L.; Bellosta V.; Cossy J. Org. Lett. 2009, 11, 2397. 10.1021/ol900494g. [DOI] [PubMed] [Google Scholar]; c Fandrick D. R.; Saha J.; Fandrick K. R.; Sanyal S.; Ogikubo J.; Lee H.; Roschangar F.; Song J. J.; Senanayake C. H. Org. Lett. 2011, 13, 5616. 10.1021/ol202343c. [DOI] [PubMed] [Google Scholar]; d Fandrick K. R.; Ogikubo J.; Fandrick D. R.; Patel N. D.; Saha J.; Lee H.; Ma S.; Grinberg N.; Busacca C. A.; Senanayake C. H. Org. Lett. 2013, 15, 1214. 10.1021/ol400124f. [DOI] [PubMed] [Google Scholar]; e Mszar N. W.; Haeffner F.; Hoveyda A. H. J. Am. Chem. Soc. 2014, 136, 3362. 10.1021/ja500373s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples of isomerization of allenyl- and propargylmetal complexes, see:; a Elsevier C. J.; Kleijn H.; Boersma J.; Vermeer P. Organometallics 1986, 5, 716. 10.1021/om00135a015. [DOI] [Google Scholar]; b Ogoshi S.; Fukunishi Y.; Tsutsumi K.; Kurosawa H. J. Chem. Soc., Chem. Commun. 1995, 2485. 10.1039/c39950002485. [DOI] [Google Scholar]; c Ogoshi S.; Nishida T.; Fukunishi Y.; Tsutsumi K.; Kurosawa H. J. Organomet. Chem. 2001, 620, 190. 10.1016/S0022-328X(00)00787-7. [DOI] [Google Scholar]
- For examples in the context of palladium-catalyzed cross-coupling reactions, see:; a Moriya T.; Miyaura N.; Suzuki A. Synlett 1994, 1994, 149. 10.1055/s-1994-22773. [DOI] [Google Scholar]; b Ma S.; Zhang A. J. Org. Chem. 2002, 67, 2287. 10.1021/jo0111098. [DOI] [PubMed] [Google Scholar]; c Marshall J. A. Chem. Rev. 2000, 100, 3163. 10.1021/cr000003u. [DOI] [PubMed] [Google Scholar]
- This strategy has been employed by Fandrick and co-workers. See ref (22c). A related strategy has been employed to elucidate the mechanistic details of transition-metal-catalyzed allylation reactions. See:; a Reference (22c); b Shaghafi M. B.; Kohn B. L.; Jarvo E. R. Org. Lett. 2008, 10, 4743. 10.1021/ol801830h. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for full experimental details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.