

Abstract
Cystic fibrosis (CF) results from mutations in the CF transmembrane conductance regulator (CFTR) gene, which codes for the CFTR channel protein. The most common mutation in CF is F508del, which produces a misfolded protein with diminished channel activity. The development of small-molecule CFTR-modulator compounds offers an exciting and novel approach for pharmacological treatment of CF. The corrector lumacaftor helps rescue F508del-CFTR to the cell surface, and potentiator ivacaftor increases F508del-CFTR channel activity. The combination of lumacaftor-ivacaftor (Vertex Pharmaceuticals Incorporated) represents the first FDA-approved therapy for CF patients with two copies of the F508del mutation. Although this combination therapy is the first treatment to directly target the F508del-CFTR mutation, patients taking this drug displayed only modest improvements in lung function. This article summarizes recent data from clinical trials and research discoveries relating to the lumacaftor-ivacaftor treatment, and considers options for identifying future therapies that will be most efficacious for all CF patients.
Keywords: CFTR, cystic fibrosis, lumacaftor, ivacaftor, F508del, modulator
1. Introduction
Cystic fibrosis (CF) is the most common life-limiting genetic disease in Caucasians. The underlying defect in CF is abnormal epithelial ion transport resulting from mutations in the CFTR protein, which mediates Cl− and HCO3− transport of secretory and absorptive epithelial cells in multiple organs including lungs, pancreas, liver, intestine, and sweat glands. Although CF affects many organ systems, the primary cause of morbidity and mortality is airway disease related to disturbances of airway surface liquid (ASL) homeostasis. The absence of CFTR activity results in decreased Cl− transport and enhanced Na+ uptake via the epithelial sodium channel (ENaC) in airway epithelial cells, leading to excessive water absorption and the characteristic thick secretions [1–3]. Recent findings highlight the importance of CFTR-dependent HCO3− transport in mucus formation and clearance [4–6]. The thick and viscous mucus leads to mucus stasis, airway obstruction, persistent infection, inflammation, and a progressive decline in lung function [7–9]. Major clinical advances in treating the symptoms and delaying disease progression have significantly improved survival of CF patients [10]. Much of the progress in extending life expectancy has been due to comprehensive treatments including antibiotics to eradicate and/or manage bacterial lung infections as well as improved strategies to increase mucociliary clearance and nutritional status [11, 12].
Although substantial clinical gains have been made using therapies that targeted the consequences of CFTR dysfunction, the ultimate therapeutic goal is to restore normal (or near normal) CFTR function via drugs that modulate the activity of CFTR (CFTR modulators). However, this strategy is complicated by the fact that there are nearly 2000 different mutations that can cause CF and interfere with normal CFTR function in different ways. The most common CFTR mutation, F508del, produces a protein with a trafficking defect that prevents significant amounts of the protein from reaching the apical cell surface, and those proteins that do reach the surface are defective in channel gating [13]. This mutation is found in approximately 90% of CF patients in the US, with nearly 50% of patients homozygous for this mutation. Because the majority of CF patients carry the F508del mutation, the identification of therapeutics that correct this defect represents an attractive initial approach. Importantly, cross-sectional studies of patients with various residual levels of CFTR activity suggest that even 5% of normal CFTR activity could convey clinical benefit [14].
2. Overview of the market
According to the FDA website (www.fda.gov), an orphan disease is a disease that affects fewer than 200,000 people in the U.S. CF is therefore considered an orphan disease because it affects approximately 30,000 people in the U.S., as reported by the Cystic Fibrosis Foundation (www.cff.org). Of these individuals with CF, only 5% have a CFTR gating mutation such as G551D that may benefit from ivacaftor (Kalydeco; Vertex) treatment [15] but 50% are F508del-CFTR homozygotes that may benefit from the lumacaftor-ivacaftor combination treatment (Orkambi; Vertex) [16]. Nevertheless, costs of care for individual CF patients are high [17], and the CF drug market was predicted to reach $3.9 billion by 2019 based on annual revenues of individual therapeutic agents in the hundreds of millions of dollars [18]. However, this prediction was made prior to the introduction of CFTR modulators and may have underestimated the market, since these modulators have been priced aggressively with annual costs over $250,000–$300,000 [19, 20]. Ivacaftor has estimated annual revenues over $600 million, and the more recently released lumacaftor-ivacaftor combination treatment has quarterly revenues that would support annual revenues of $1 billion dollars or more. These figures suggest that the market for CFTR modulators is potentially lucrative.
Both ivacaftor and the lumacaftor-ivacaftor combination drug have the market advantage of being the only drugs for their indication, as there are currently no other CFTR modulators approved by regulatory agencies. However, there are a large number of potential competitor compounds in various stages of clinical development, including some that are in late phase clinical trials. As described in more detail below, the clinical benefit seen in patients treated with the lumacaftor-ivacaftor combination therapy was modest, leaving opportunities for other drugs that have greater clinical impact [21]. Furthermore, the lumacaftor-ivacaftor drug was not effective in patients heterozygous for the F508del mutation, suggesting that a more potent modulator may be applicable to a larger group of patients.
There are several CFTR modulators currently in development (Table 1). Examples being tested in clinical trials include the following: N91115 (Nivalis), an S-nitrosoglutathione reductase (GSNOR) inhibitor predicted to function as a CFTR stabilizer, that is in phase 2 trials for F508del homozygous patients who are taking the lumacaftor-ivacaftor combination drug. QR-010 (ProQR Therapeutics), which repairs the genetic defect in RNA, is in phase 1b for F508del homozygous patients. QBW251 (Novartis), a CFTR modifier, is in phase 2 clinical trials for F508del homozygous and heterozygous patients. Riociguat (Bayer), stimulator of soluble guanylate cyclase, is in phase 2 for F508del homozygous patients, and is predicted to improve sweat chloride content and increase FEV1. AbbVie/Galapagos has developed a triple-combination therapy with two corrector compounds (GLPG2222 and GLPG2665) and one potentiator (GLPG1837). VX-661 (Vertex) is a corrector compound expected to have a lower likelihood of drug interactions with ivacaftor, and is generally well tolerated by patients. VX-661 combined with ivacaftor is in phase 3 trials in patients homozygous for F508del allele, or heterozygous for F508del in which the copy of CFTR on their other chromosome is grouped according to the following responses to therapies: 1) is not predicted to respond to therapy, 2) has residual function, or 3) responds to ivacaftor. The results of a phase 2 study using VX-661 with and without ivacaftor in F508del homozygous patients showed reductions in sweat chloride with VX-661 alone and in the presence of ivacaftor, and showed significant but modest improvements in lung function (FEV1) [22], similar to what was observed with the lumacaftor-ivacaftor combination therapy. If successful, these drugs being tested in the clinic may enter the market soon.
Table 1.
CFTR-targeting drugs currently in the drug pipeline or in clinical trials.
| Company | Compound |
|---|---|
| AbbVie/Galapagos | Triple-combination therapy with two corrector compounds (GLPG2222 and GLPG2665) and one potentiator (GLPG1837) |
| Bayer | Riociguat to correct F508del function |
| Calista Therapeutics | CAL inhibitor CT-007 to stabilize CFTR at the cell membrane |
| Concert Pharmaceuticals | Potentiator compound CTP-656, a deuterium-modified version of ivacaftor |
| DiscoveryBioMed | Dual acting correcting and activating CFTR ligand |
| Flatley Discovery Lab | Corrector compounds FDL160, FDL169 FDL042(FDL282), FDL392, and FDL304 |
| Genzyme/Sanofi | Non-viral gene transfer agent (PGM169/GL67A), corrector compounds for F508del-CFTR |
| Nivalis Therapeutics | N91115 to stabilize rescued F508del-CFTR |
| Novartis | Potentiator compound (QBW251) and other potentiator and corrector compounds (picolinamide) |
| Parion | Corrector compounds |
| PTC Therapeutics | Ataluren for treatment of CFTR nonsense mutations* |
| Pfizer | Corrector compounds (PYR-41 decreases the CFTR ubiquitination) and potentiator compounds (CP-628006) |
| ProQR Therapeutics | QR-010, a 33mer antisense oligonucleotide designed to repair F508del-CFTR encoded mRNA |
| Proteostasis Therapeutics | CFTR amplifier (PTI-428) that increases the levels of CFTR; combination of PTI-428 with corrector and potentiator compound (PTI‐NC-733) |
| Reata Pharmaceuticals | Corrector compounds |
| Shire Rare Disease Unit | Technology to deliver normal CFTR to the lungs |
| Southern Research | Repurposing of known drugs for translational readthrough of rare CF nonsense mutations* |
| Traffick Therapeutics/AmorChem/NuChem Therapeutics | Correctors (NU001 and NU002) that are synergistic with VX-809 or VX‐661 and VX-770 |
| Vertex Pharmaceuticals | Next-generation correctors VX-152 or VX-440 that will be combined with corrector VX-661 and potentiator VX-770 |
These compounds do not target F508del and therefore do not overlap with the therapeutic action of the lumacaftor-ivacaftor drug.
3. Introduction to the drug
CFTR modulators aimed at improving function of F508del mutation are generally divided into 2 main categories: correctors that rescue misfolded protein to the cell surface, and potentiators that improve channel activity [23]. Using high-throughput screening strategies, a series of small-molecule corrector compounds were identified that have corrector or potentiator activity that were then optimized through medicinal chemistry [24–28]. The initial corrector compound for clinical development was lumacaftor (structure shown in Figure 1, bottom), which has the chemical name 3-[6-({[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropyl]carbonyl}amino)-3-methylpyridin-2-yl]benzoic acid. The molecular formula of lumacaftor is C24H18F2N2O5 and the molecular weight is 452.41. In CF patients, the half-life of lumacaftor alone is approximately 26 hours [29]. Nearly all of lumacaftor (approximately 99%) is bound to plasma proteins, mainly albumin. In humans, the majority of lumacaftor is not extensively metabolized and is therefore excreted unchanged. However, in vitro and in vivo data have shown that the small amount of lumacaftor that is metabolized is done so via oxidation and glucuronidation [29]. Lumacaftor is thought to increase the conformational stability of CFTR by improving interactions of the intramolecular domains of CFTR protein during folding [30–33]. Data suggest that lumacaftor binds directly to CFTR [34] and restores F508del maturation and function up to 15% of WT CFTR in vitro [30–33, 35–37]. However, studies in cultured cells demonstrated that the channel activity of F508del-CFTR rescued to the cell surface remained low due to the residual gating defect [38]. This problem could be addressed through addition of the potentiator ivacaftor, which has the chemical name N-(2,4-di-tertbutyl-5-hydroxyphenyl)-1,4-dihydro-4-oxoquinoline-3-carboxamide. The molecular formula of ivacaftor is C24H28N2O3 and the molecular weight is 392.49. In healthy subjects, the half-life of ivacaftor when combined with lumacaftor is approximately 9 hours. Ivacaftor is a hydrophobic molecule and most of it (approximately 99%) is bound to plasma proteins, mainly alpha 1-acid glycoprotein and albumin [39]. In humans, ivacaftor undergoes extensive metabolism. In vitro and in vivo data indicate that metabolism of ivacaftor occurs primarily through CYP3A [40]. In cell cultures treated with lumacaftor and ivacaftor, it was found that ivacaftor accumulated in cells [41]. Ivacaftor was initially developed to improve activity of CFTR mutants with gating defects and was first approved to treat CF patients with the G551D mutation [42–45]. Ivacaftor appears to directly affect CFTR channel gating and was later approved for other gating mutations (G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, and G1349D) as well as R117H. Ivacaftor also activates rescued F508del-CFTR that is present at the cell surface [45, 46], and the combination of lumacaftor with acute addition of ivacaftor yielded significantly improved F508del-CFTR channel activity in vitro [37]. Based on these studies and early phase clinical trials described below, the lumacaftor-ivacaftor combination therapy was predicted to result in an improvement in ASL hydration and produce more hydrated mucus that will enhance microbe clearance from the lungs, as reviewed in [16, 47, 48] and illustrated in Figure 2. The lumacaftor-ivacaftor drug is a tablet for oral administration containing 200 mg of lumacaftor and 125 mg of ivacaftor. For pharmacodynamics of ivacaftor-lumacaftor, changes in sweat chloride activity were measured in patients in the phase 2 clinical trial [49]. The difference between lumacaftor-ivacaftor treatment (lumacaftor 400 mg/ivacaftor 250 mg every 12 hours) and placebo was −11 mmol/L (95% CI −18, −4). However, this decrease in sweat chloride levels did not directly correlate with an improvement in lung function, as measured by FEV1 [29].
Figure 1. Drug Structures.
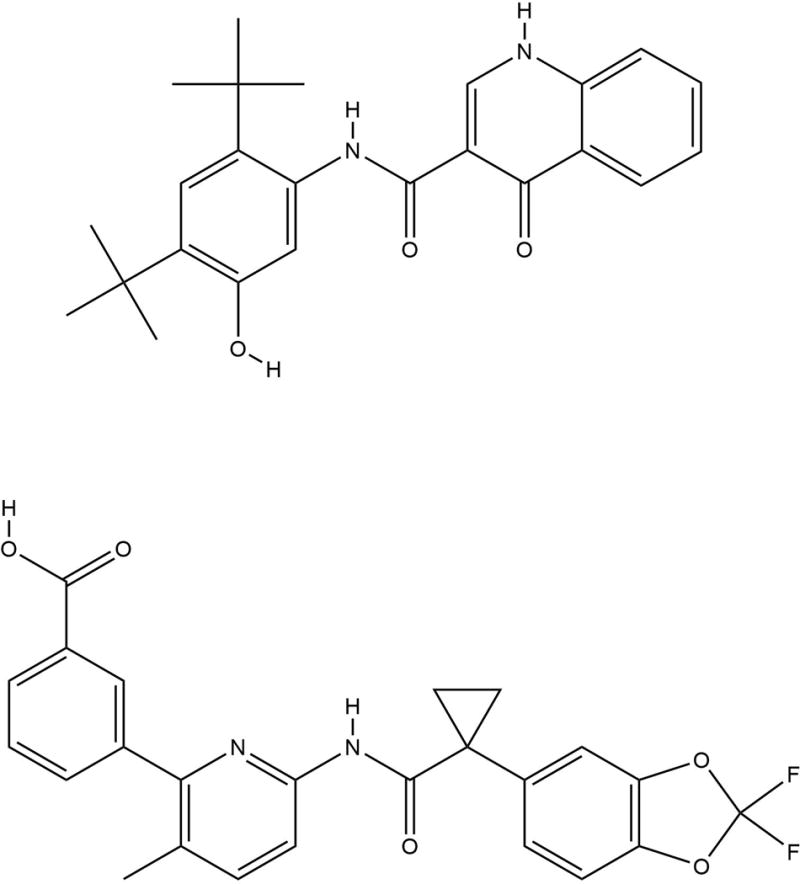
Chemical structures of ivacaftor (top) and lumacaftor (bottom) are shown.
Figure 2. Correction and Potentiation of CFTR.
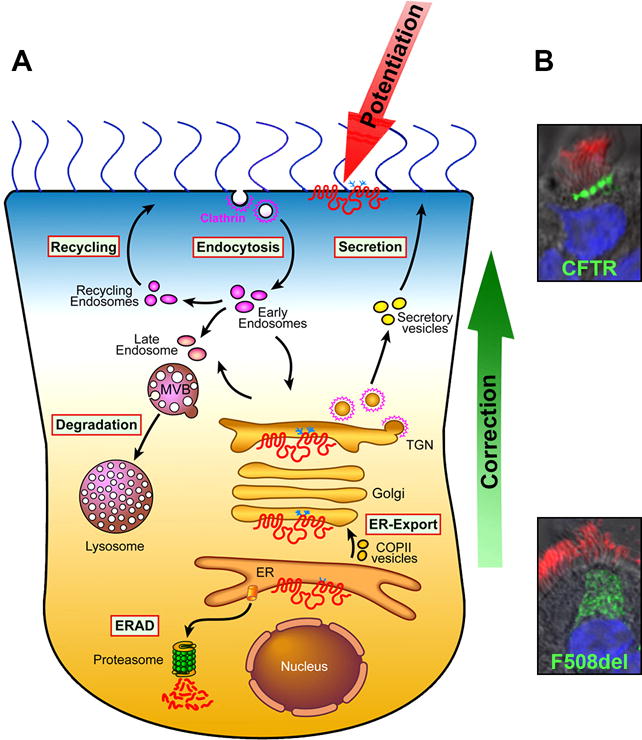
A. Correctors promote transfer of mutant CFTR from the ER to the apical membrane, whereas potentiators enhance activity of apical CFTR. B. Trafficking defect of F508del-CFTR. Confocal immunofluorescence images show apical WT and intracellular-retained F508del-CFTR (green) expressed as Extope-variant in primary HBE cells. Cilia are stained in red and nuclei in blue.
4. Clinical efficacy
Although lumacaftor alone showed a strong increase in formation of mature F508del-CFTR in vitro [37], a phase 2a clinical study of CF patients homozygous for the misfolded F508del-CFTR mutation treated with lumacaftor did not show any significant changes in lung function at any of four dose levels tested (25, 50, 100, or 200 mg once daily) [50]. This was not entirely surprising, since CFTR rescued to the cell membrane by lumacaftor was known to have reduced channel activity and rapid turnover from the cell surface [38, 51]. Therefore, a second Phase 2 clinical trial was conducted that combined lumacaftor at various doses with ivacaftor to address the gating defect [49], summarized in Table 2. In CF patients homozygous for the misfolded F508del-CFTR mutation, treatment with a combination of lumacaftor and ivacaftor resulted in modest but meaningful improvements in lung function, with the most significant effects at the higher lumacaftor doses (600 mg daily or 400 mg twice daily). However, in the same phase 2 trial, combination therapy using ivacaftor with lumacaftor in F508del heterozygous patients did not result in a significant change in FEV1 [49].
Table 2.
Summary of phase 2 (Boyle et al [49]) and phase 3 (Wainwright et al [52]) studies with ivacaftor-lumacaftor combination treatment in CF patients ages 12 and older who are homozygous for F508del-CFTR.
| Study | FEV1* | BMI† | CFQ-R‡ | Pulmonary exacerbations§ | Number of participants |
|---|---|---|---|---|---|
| Phase 2 study, 8 weeks [37] | |||||
| Lumacaftor 200 mg/day | 3.8 (−0.4 to 8.1) | 23 | |||
| Ivacaftor 250 mg twice/day | P= 0.077 | ||||
| Lumacaftor 400 mg/day | 2.7 (−1.7 to 7.0) | 21 | |||
| Ivacaftor 250 mg twice/day | P= 0.225 | ||||
| Lumacaftor 600 mg/day | 5.6 (1.2 to 10.0) | 21 | |||
| Ivacaftor 250 mg twice/day | P= 0.013 | ||||
| Lumacaftor 400 mg twice/day | 4.2 (−1.3 to 9.6) | 11 | |||
| Ivacaftor 250 mg twice/day | P= 0.132 | ||||
| Phase 3 study, 24 weeks [38] | |||||
| TRAFFIC | |||||
| Lumacaftor 600 mg/day | 4.0 (2.6 to 5.4) | 0.16 (−0.04 to 0.35) | 3.9 (0.7 to 7.1) | 0.72 (0.52 to 1.00) | 183 |
| Ivacaftor 250 mg twice/day | P< 0.001 | P= 0.11 | P= 0.02 | P= 0.05 | |
| Lumacaftor 400 mg twice/day | 2.6 (1.2 to 4.0) | 0.13 (−0.07 to 0.32) | 1.5 (−1.7 to 4.7) | 0.66 (0.47 to 0.93) | 182 |
| Ivacaftor 250 mg twice/day | P< 0.001 | P= 0.19 | P= 0.36 | P= 0.02 | |
| TRANSPORT | |||||
| Lumacaftor 600 mg/day | 2.6 (1.2 to 4.1) | 0.41 (0.23 to 0.59) | 2.2 (−0.9 to 5.3) | 0.69 (0.52 to 0.92) | 185 |
| Ivacaftor 250 mg twice/day | P< 0.001 | P< 0.001 | P= 0.17 | P= 0.01 | |
| Lumacaftor 400 mg twice/day | 3.0 (1.6 to 4.4) | 0.36 (0.17 to 0.54) | 2.9 (−0.3 to 6.0) | 0.57 (0.42 to 0.76) | 187 |
| Ivacaftor 250 mg twice/day | P< 0.001 | P< 0.001 | P= 0.07 | P< 0.001 | |
| TRAFFIC + TRANSPORT Pooled | |||||
| Lumacaftor 600 mg/day | 3.3 (2.3 to 4.3) | 0.28 (0.15 to 0.41) | 3.1 (0.8 to 5.3) | 0.70 (0.56 to 0.87) | 368 |
| Ivacaftor 250 mg twice/day | P< 0.001 | P< 0.001 | P= 0.007 | P= 0.001 | |
| Lumacaftor 400 mg twice/day | 2.8 (1.8 to 3.8) | 0.24 (0.11 to 0.37) | 2.2 (0.0 to 4.5) | 0.61 (0.49 to 0.76) | 369 |
| Ivacaftor 250 mg twice/day | P< 0.001 | P< 0.001 | P= 0.05 | P< 0.001 |
Difference versus placebo for absolute change in percentage predicted FEV1 (percentage points; 95% CI).
Difference versus placebo in absolute change from baseline BMI; mean (95% CI).
Difference versus placebo in absolute change from baseline in CFQ-R respiratory domain; mean (95% CI). The minimal clinically important difference for stable patients on this scale is 4.0 points [53].
Rate ratio (95% CI)
Based on the phase 2 results, two phase 3 clinical trials, TRAFFIC and TRANSPORT, were conducted using a combination of lumacaftor (600 mg daily or 400 mg twice daily) and ivacaftor (250 mg twice daily) in patients 12 years and older homozygous for the F508del-CFTR mutation [52], summarized in Table 2. Both trials met their primary outcome measure of statistically significant improvement in FEV1, although the increase of 2.6–4% [52] were modest and significantly below those observed with ivacaftor in CF patients with gating mutations (10.6–12.5%) [15, 54]. The TRAFFIC and TRANSPORT studies did show significant reduction in pulmonary exacerbations as an important secondary outcome measure, as well as other secondary measures including the Cystic Fibrosis Questionnaire-Revised (CFQ-R) quality of life respiratory scale and body mass index. Adverse events were generally similar between placebo and treatment groups, with the exception of increased chest tightness noted more frequently in the treatment groups.
The phase 3 TRAFFIC and TRANSPORT clinical studies lasted for 24 weeks, and the ongoing PROGRESS extension study is designed to address longer term effects. Preliminary results from PROGRESS suggest that improvements in lung function (FEV1) and secondary outcomes in CF patients aged 12 and older with 2 copies of the F508del mutation taking lumacaftor-ivacaftor were maintained for an additional 24 weeks, though there were no further improvements [55].
5. Post-marketing surveillance
Since the lumacaftor-ivacaftor combination therapy was only recently approved in July of 2015, at this time there are no post-marketing surveillance data available beyond the PROGRESS extension study mentioned above. The results of safety and tolerability analyses in the PROGRESS extension study indicated that adverse events were similar to what was observed during the first 24 weeks of the study, with the most commonly observed events including shortness of breath, chest tightness, viral infection of the upper respiratory tract, and gastrointestinal symptoms, as listed in adverse events tables of the phase 2 and phase 3 trials [49, 52]. An observational study, PROSPECT, is ongoing and focuses on examining biomarkers of CFTR function and banking of specimens. This study will evaluate potential outcome markers including FEV1 and sweat chloride in CF patients that have either partial or absent CFTR function (Part A) or are F508del patients receiving lumacaftor-ivacaftor combination treatment (Part B).
6. Regulatory affairs
The lumacaftor-ivacaftor combination therapy was approved for use in the US by the FDA, in Europe by the EU, and in Australia by the TGA in CF patients ages 12 and older who are homozygous for the F508del mutation [56–58]. There are approximately 8,500 CF patients in the US, 12,000 CF patients in Europe, and 1,000 CF patients in Australia who fit these criteria.
7. Conclusion
Up until recently, treatments for CF addressed only symptoms, but it is now clear that compounds that directly modulate the activity of CFTR represent a viable therapeutic strategy. In some ways, treatment with lumacaftor-ivacaftor represents a breakthrough therapy, since it is the first CFTR modulator that provides benefit for patients carrying the most common mutation in CF. However, despite very promising in vitro studies, the results from clinical studies showed only modest improvement. As discussed further below, additional laboratory studies have revealed that potentiator ivacaftor might destabilize F508del-CFTR rescued by lumacaftor. Such studies offer opportunities to develop more potent drugs and drug combinations for F508del and other CFTR mutations, with the ultimate goal of restoring CFTR activity in all patients with CF.
8. Expert commentary
The lumacaftor-ivacaftor drug received the designation of breakthrough therapy [59, 60]; however, the clinical impact on lung function and pulmonary exacerbations was less than that of ivacaftor in G551D patients and more similar to previous CF therapies targeted at downstream pathophysiology including dornase alfa (to cleave DNA, which breaks down thick secretions), hypertonic saline (to hydrate viscous mucus), or azithromycin (to fight bacteria) [20, 61–63]. In vitro studies on the action of lumacaftor and ivacaftor indicated that corrected F508del-CFTR is destabilized in the presence of these 2 compounds [41, 64], which may explain why the lumacaftor-ivacaftor combination treatment was less successful than anticipated and exemplifies the value of bench research to guide clinical drug development [65, 66]. There was hope that the newer corrector VX-661 might avoid the destabilizing effect of ivacaftor, but in vitro studies suggest that this may not be the case [41, 64] as ivacaftor interfered with restoring F508del-CFTR function whether lumacaftor or VX-661 was used as the corrector.
For future translation of promising in vitro data to clinical efficacy, the bioavailability of the agents’ exact drug concentrations in lung tissue should be considered. Matthes et al. [67] suggested that the inhibitory effects of the lumacaftor-ivacaftor combination therapy is due to high concentrations of free ivacaftor present in plasma, and that improving CFTR function would require lowering potentiator concentration and using more efficacious correctors. It was recently discovered that some other potentiators did not appear to markedly inhibit the correction of F508del [64, 68]. While it is important to identify potentiators that do not interfere with corrector action or CFTR stability, the goal should be to develop drug combinations that exhibit additive or synergistic effects to obtain a marked improvement in CFTR function in vitro, and ultimately, patient lung function.
The clinical improvements seen in trials with lumacaftor-ivacaftor may be due to activity of this drug that is not directly related to the CFTR molecule. There is evidence that ivacaftor displays antimicrobial activity [69], which may enhance lung function by clearing infective microbes from the lung. An increase in CFTR activity is predicted to improve viscoelastic properties of mucus in CF patients, which in turn will lead to enhanced lung function [70]. However, whether the lumacaftor-ivacaftor drug might also directly affect mucus properties has not yet been investigated. It should also be considered that in vitro studies using ivacaftor have resulted in decreased activity of ENaC [41], which leads to an increase in airway surface hydration that may contribute to the observed improvement in lung function [71]. Although ENaC is regulated by CFTR, the drug activity of ivacaftor may be directly affecting ENaC. Indeed, modulation of ENaC activity is a strategy that is of interest to companies seeking to identify pharmaceuticals that repair the CF defect [72].
In October 2015, Vertex described next-generation correctors, VX-152 and VX-440, which are predicted to further improve rescue and activity of F508del-CFTR [73]. In HBE cells from patients homozygous for the F508del mutation, combining VX-152 or VX-440 together with VX-661 plus ivacaftor (triple combinations) resulted in chloride transport that was approximately three-fold greater than what was observed upon the lumacaftor-ivacaftor combination treatment of cells with the same genotype [73]. Importantly, similar findings were observed in HBE cells from patients heterozygous for F508del mutation in which the copy of CFTR on the other chromosome contains a mutation that results in minimal CFTR function. Treatment using these triple combinations also resulted in a significant increase in cilia beat frequency compared to treatment of cells with the same genotype with the lumacaftor-ivacaftor drug. These in vitro data using cell cultures suggest that a triple combination that includes a next-generation corrector with VX-661 plus ivacaftor may improve CFTR function in patients that are homozygous for the F508del mutation and in patients that are heterozygous for the F508del mutation when the other CFTR gene codes for a protein that displays minimal CFTR function. These correctors will be tested both alone and in combination with VX-661 plus ivacaftor in Phase 1 studies. Until more efficacious potentiators are identified, increasing corrector activity with multiple correctors [32] or addition of stabilizers may compensate for the detrimental effects of ivacaftor.
Considering the high cost of ivacaftor and the lumacaftor-ivacaftor combination therapy, an attractive goal is to identify drugs that target multiple CFTR defects to augment CFTR at the cell surface, channel function, and protein stability. Thus, discovery of new drug therapies should be aimed at identifying: 1) correctors that further improve processing of mutant CFTR to augment levels of CFTR at the cell surface, 2) correctors that also display substantial potentiator function, 3) correctors that also stabilize CFTR, 4) potentiators that do not destabilize CFTR, 5) stabilizers that can be used with correctors/potentiators. Because currently used potentiators are extremely expensive and their activity leads to destabilization of rescued F508del-CFTR at the plasma membrane, novel correctors that rescue the F508del-CFTR trafficking defect and also stabilize the protein at the cell surface may eliminate the need for potentiators.
9. Five-year view
The introduction of ivacaftor and the lumacaftor-ivacaftor combination therapy represent a turning point in treatment of CF, both because of their direct impact on CF care and what they represent for the future of CFTR modulators. The next five years will hopefully see approval of multiple other CFTR modulators that address different aspects of CFTR dysfunction. These will provide multiple options for individual CF patients, which we predict will be necessary to provide optimal care given the plethora of CFTR mutations that can cause disease.
The availability of new CFTR modulators will require us to rethink how we classify CFTR mutations. CFTR mutations have historically been grouped into different classes according to their defects. For example the misfolding mutation, F508del-CFTR, belongs to Class II while the gating mutation, G551D-CFTR, belongs to Class III. It could be anticipated that patients with other misfolding mutations may benefit from lumacaftor-ivacaftor combination treatment. However, while it would seem that different mutations that belong to the same class should respond similarly to drug treatments, recent observations indicate that the original mutation classifications do not accurately describe responses to CFTR modulators [74, 75]. Furthermore, patients with identical CFTR mutations may show diverse drug responses.
These observations suggest that a precision medicine approach will likely be needed to optimize patient therapies. One such approach involves the use of organoids readily prepared from rectal [35], bronchial, and nasal tissues derived from individual CF patients. These spheroid cultures display acute volume changes resulting from ion and fluid movement upon CFTR activation by drug treatments. Additionally, nasal and bronchial spheroid cultures can be used for examining mucus properties and ciliary function. This precision ex vivo approach would potentially allow testing of different combinations and doses of correctors and potentiators in individual patients, allowing development of optimal treatment strategies for each CF patient. Such studies are imperative for identifying and conducting preclinical testing of candidate compounds before they enter the clinical trials, maximizing the likelihood of achieving the dramatic enhancements in lung function and secondary outcomes necessary to make the therapy a worthwhile product for CF patients.
Approval of other CFTR modulators may also address concerns over cost. The lumacaftor-ivacaftor combination therapy is currently priced at $259,000 per year [19, 20], with ivacaftor at $311,000 per year [19], and the two drugs are predicted to represent well in excess of $1 billion dollars in revenue for Vertex Pharmaceuticals Inc. These revenues represent an enticing motivation for investment, which is enhanced by their orphan drug status that provides the manufacturer with certain benefits such as financial incentives and market exclusivity [20, 59]. However, this orphan drug designation also results in disproportionate overpricing, which may be unsustainable by patients and health insurance companies, particularly since these therapies would ideally be utilized throughout the life of a CF patient [76]. The relatively modest benefits of the lumacaftor-ivacaftor drug coupled with its high price have led many to question whether its clinical effect can justify its cost [19]. Competition from other drugs that convey similar benefits could help reduce the cost to patients, as could more effective treatments that alter the cost-benefit ratio.
Key issues.
Cystic fibrosis results from mutations in the CFTR gene, which codes for the CFTR channel protein. The most common mutation in CF patients is F508del, which produces a misfolded protein with diminished channel activity.
Corrector and potentiator compounds directly affect CFTR function. Compounds predicted to enhance CFTR activity are currently in the clinical drug pipeline.
The lumacaftor-ivacaftor drug is an FDA-approved combination therapy administered to CF patients homozygous for F508del-CFTR; however, this therapy resulted in modest improvements in lung function.
Strategies to identify novel therapeutics must focus on drug combinations that improve CFTR maturation, function, and stability, thereby enhancing mucociliary clearance in the lungs. Preclinical laboratory testing of candidate compounds is important for understanding drug mechanisms.
The goal of identifying effective therapies is to maximize benefit to patients without unreasonable costs.
Acknowledgments
The authors were in part supported by NIH P30 DK065988 and CFF RDP BOUCHE15R, GENTZS14G0. DM Cholon has worked with CFF and Nivalis Therapeutics. M Gentzsch has worked with CFF and Nivalis Therapeutics, Parion and Galapagos N.V. CR Esther has worked with Parion.
Footnotes
Information resources
More information about CF including drugs currently in development and on-going clinical trials is available at the Cystic Fibrosis Foundation website (www.cff.org). Additional information on the lumacaftor-ivacaftor combination therapy and other Vertex drugs and Vertex press releases is available at their website (www.vrtx.com).
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of particular interest are identified as:
* = of interest
** = of considerable interest
- 1.Mall M, Bleich M, Greger R, et al. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J Clin Invest. 1998;102(1):15–21. doi: 10.1172/JCI2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mall M, Grubb BR, Harkema JR, et al. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. 2004;10(5):487–93. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 3.Stutts MJ, Canessa CM, Olsen JC, et al. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269(5225):847–50. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- 4.Chen EY, Yang N, Quinton PM, Chin WC. A new role for bicarbonate in mucus formation. Am J Physiol Lung Cell Mol Physiol. 2010;299(4):L542–9. doi: 10.1152/ajplung.00180.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shamsuddin AK, Quinton PM. Native small airways secrete bicarbonate. Am J Respir Cell Mol Biol. 2014;50(4):796–804. doi: 10.1165/rcmb.2013-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang N, Garcia MA, Quinton PM. Normal mucus formation requires cAMP-dependent HCO3- secretion and Ca2+-mediated mucin exocytosis. J Physiol. 2013;591(18):4581–93. doi: 10.1113/jphysiol.2013.257436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Accurso FJ. Early pulmonary disease in cystic fibrosis. Curr Opin Pulm Med. 1997;3(6):400–3. doi: 10.1097/00063198-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J. 2004;23(1):146–58. doi: 10.1183/09031936.03.00057003. [DOI] [PubMed] [Google Scholar]
- 9.Davis PB. Cystic fibrosis since 1938. Am J Respir Crit Care Med. 2006;173(5):475–82. doi: 10.1164/rccm.200505-840OE. [DOI] [PubMed] [Google Scholar]
- 10.Cohen-Cymberknoh M, Shoseyov D, Kerem E. Managing cystic fibrosis: strategies that increase life expectancy and improve quality of life. Am J Respir Crit Care Med. 2011;183(11):1463–71. doi: 10.1164/rccm.201009-1478CI. [DOI] [PubMed] [Google Scholar]
- 11.O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373(9678):1891–904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 12.Stern M, Bertrand DP, Bignamini E, et al. European Cystic Fibrosis Society Standards of Care: Quality Management in cystic fibrosis. J Cyst Fibros. 2014;13(Suppl 1):S43–59. doi: 10.1016/j.jcf.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 13.Dalemans W, Barbry P, Champigny G, et al. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature. 1991;354(6354):526–8. doi: 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- 14.Ramalho AS, Beck S, Meyer M, et al. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am J Respir Cell Mol Biol. 2002;27(5):619–27. doi: 10.1165/rcmb.2001-0004OC. [DOI] [PubMed] [Google Scholar]
- 15*.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–72. doi: 10.1056/NEJMoa1105185. Results of phase 3 study of ivacaftor in G551D CF patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brodsky JL, Frizzell RA. A combination therapy for cystic fibrosis. Cell. 2015;163(1):17. doi: 10.1016/j.cell.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Gool K, Norman R, Delatycki MB, et al. Understanding the costs of care for cystic fibrosis: an analysis by age and health state. Value Health. 2013;16(2):345–55. doi: 10.1016/j.jval.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Syed BA, Hamad B. The cystic fibrosis drug market. Nat Rev Drug Discov. 2014;13(10):721–2. doi: 10.1038/nrd4434. [DOI] [PubMed] [Google Scholar]
- 19.Ferkol T, Quinton P. Precision Medicine: At What Price? Am J Respir Crit Care Med. 2015;192(6):658–9. doi: 10.1164/rccm.201507-1428ED. [DOI] [PubMed] [Google Scholar]
- 20.Mayer M. Lumacaftor-ivacaftor (Orkambi) for cystic fibrosis: behind the ‘breakthrough’. Evid Based Med. 2015 doi: 10.1136/ebmed-2015-110325. [DOI] [PubMed] [Google Scholar]
- 21.Current and Upcoming CFFT Approved Clinical Trials. 2015 Nov 16; https://www.cff.org/Our-Research/Clinical-Trials/Clinical-Trials-Happening-Now/
- 22.Treatment with VX-661 and Ivacaftor in a Phase 2 Study Resulted in Statistically Significant Improvements in Lung Function in People with Cystic Fibrosis Who Have Two Copies of the F508del Mutation. Vertex Pharmaceuticals, Inc; 2013. Apr 18, http://investors.vrtx.com/releasedetail.cfm?ReleaseID=757597. [Google Scholar]
- 23.Rowe SM, Verkman AS. Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harb Perspect Med. 2013;3(7) doi: 10.1101/cshperspect.a009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pedemonte N, Lukacs GL, Du K, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115(9):2564–71. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robert R, Carlile GW, Liao J, et al. Correction of the Delta phe508 cystic fibrosis transmembrane conductance regulator trafficking defect by the bioavailable compound glafenine. Mol Pharmacol. 2010;77(6):922–30. doi: 10.1124/mol.109.062679. [DOI] [PubMed] [Google Scholar]
- 26.Robert R, Carlile GW, Pavel C, et al. Structural analog of sildenafil identified as a novel corrector of the F508del-CFTR trafficking defect. Mol Pharmacol. 2008;73(2):478–89. doi: 10.1124/mol.107.040725. [DOI] [PubMed] [Google Scholar]
- 27.Rowe SM, Pyle LC, Jurkevante A, et al. DeltaF508 CFTR processing correction and activity in polarized airway and non-airway cell monolayers. Pulm Pharmacol Ther. 2010;23(4):268–78. doi: 10.1016/j.pupt.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang D, Ciciriello F, Anjos SM, et al. Ouabain Mimics Low Temperature Rescue of F508del-CFTR in Cystic Fibrosis Epithelial Cells. Front Pharmacol. 2012;3:176. doi: 10.3389/fphar.2012.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Highlights of Prescribing Information. Vertex Pharmaceuticals, Inc; Jul, 2015. http://pi.vrtx.com/files/uspi_lumacaftor_ivacaftor.pdf. [Google Scholar]
- 30.He L, Kota P, Aleksandrov AA, et al. Correctors of DeltaF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013;27(2):536–45. doi: 10.1096/fj.12-216119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loo TW, Bartlett MC, Clarke DM. Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem Pharmacol. 2013;86(5):612–9. doi: 10.1016/j.bcp.2013.06.028. [DOI] [PubMed] [Google Scholar]
- 32*.Okiyoneda T, Veit G, Dekkers JF, et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat Chem Biol. 2013;9(7):444–54. doi: 10.1038/nchembio.1253. Demonstrates that use of multiple correctors can enhance rescue of F508del. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren HY, Grove DE, De La Rosa O, et al. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol Biol Cell. 2013;24(19):3016–24. doi: 10.1091/mbc.E13-05-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinha C, Zhang W, Moon CS, et al. Capturing the Direct Binding of CFTR Correctors to CFTR by Using Click Chemistry. Chembiochem. 2015;16(14):2017–22. doi: 10.1002/cbic.201500123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dekkers JF, Wiegerinck CL, de Jonge HR, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–45. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- 36.Farinha CM, King-Underwood J, Sousa M, et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem Biol. 2013;20(7):943–55. doi: 10.1016/j.chembiol.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 37*.Van Goor F, Hadida S, Grootenhuis PD, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108(46):18843–8. doi: 10.1073/pnas.1105787108. First in vitro demonstration of lumacaftor-ivacaftor combination therapy for F508del. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denning GM, Anderson MP, Amara JF, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358(6389):761–4. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 39.Schneider EK, Huang JX, Carbone V, et al. Drug-drug plasma protein binding interactions of ivacaftor. J Mol Recognit. 2015;28(6):339–48. doi: 10.1002/jmr.2447. [DOI] [PubMed] [Google Scholar]
- 40.Wainwright CE. Ivacaftor for patients with cystic fibrosis. Expert Rev Respir Med. 2014;8(5):533–8. doi: 10.1586/17476348.2014.951333. [DOI] [PubMed] [Google Scholar]
- 41**.Cholon DM, Quinney NL, Fulcher ML, et al. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci Transl Med. 2014;6(246):246ra96. doi: 10.1126/scitranslmed.3008680. Demonstrates that ivacaftor interferes with lumacaftor correction of F508del in vitro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eckford PD, Li C, Ramjeesingh M, Bear CE. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem. 2012;287(44):36639–49. doi: 10.1074/jbc.M112.393637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jih KY, Hwang TC. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc Natl Acad Sci U S A. 2013;110(11):4404–9. doi: 10.1073/pnas.1215982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeh HI, Yeh JT, Hwang TC. Modulation of CFTR gating by permeant ions. J Gen Physiol. 2015;145(1):47–60. doi: 10.1085/jgp.201411272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45*.Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106(44):18825–30. doi: 10.1073/pnas.0904709106. Demonstrates that ivacaftor rescues G551D-CFTR in vitro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu H, Burton B, Huang CJ, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros. 2012;11(3):237–45. doi: 10.1016/j.jcf.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 47.Kuk K, Taylor-Cousar JL. Lumacaftor and ivacaftor in the management of patients with cystic fibrosis: current evidence and future prospects. Ther Adv Respir Dis. 2015;9(6):313–26. doi: 10.1177/1753465815601934. [DOI] [PubMed] [Google Scholar]
- 48.Brewington JJ, McPhail GL, Clancy JP. Lumacaftor alone and combined with ivacaftor: preclinical and clinical trial experience of F508del CFTR correction. Expert Rev Respir Med. 2016;10(1):5–17. doi: 10.1586/17476348.2016.1122527. [DOI] [PubMed] [Google Scholar]
- 49**.Boyle MP, Bell SC, Konstan MW, et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014;2(7):527–38. doi: 10.1016/S2213-2600(14)70132-8. Results of phase 2 clinical trial testing lumacaftor-ivacaftor on F508del homozygous CF patients. [DOI] [PubMed] [Google Scholar]
- 50.Clancy JP, Rowe SM, Accurso FJ, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67(1):12–8. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lukacs GL, Chang XB, Bear C, et al. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993;268(29):21592–8. [PubMed] [Google Scholar]
- 52**.Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(18):1783–4. doi: 10.1056/NEJMc1510466. Results of phase 3 clinical trials testing lumacaftor-ivacaftor on F508del homozygous CF patients. [DOI] [PubMed] [Google Scholar]
- 53.Quittner AL, Modi AC, Wainwright C, et al. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire-Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest. 2009;135(6):1610–8. doi: 10.1378/chest.08-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013;187(11):1219–25. doi: 10.1164/rccm.201301-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vertex Announces Data Presentations at European Cystic Fibrosis Society (ECFS) Conference. Vertex Pharmaceuticals, Inc; 2015. Jun 11, http://investors.vrtx.com/releasedetail.cfm?ReleaseID=917543. [Google Scholar]
- 56.FDA Approves ORKAMBI™ (lumacaftor/ivacaftor) - the First Medicine to Treat the Underlying Cause of Cystic Fibrosis for People Ages 12 and Older with Two Copies of the F508del Mutation. Vertex Pharmaceuticals, Inc; 2015. Jul 2, http://investors.vrtx.com/releasedetail.cfm?ReleaseID=920512. [Google Scholar]
- 57.Vertex Receives Australian Approval for ORKAMBI® (lumacaftor/ivacaftor), the First Medicine to Treat the Underlying Cause of Cystic Fibrosis in People Ages 12 and Older with Two Copies of the F508del Mutation. Vertex Pharmaceuticals, Inc; 2016. Mar 8, http://investors.vrtx.com/releasedetail.cfm?ReleaseID=959473. [Google Scholar]
- 58.Vertex Receives EU Approval for ORKAMBI® (lumacaftor/ivacaftor), the First Medicine to Treat the Underlying Cause of Cystic Fibrosis in People Ages 12 and Older with Two Copies of the F508del Mutation. Vertex Pharmaceuticals, Inc; 2015. Nov 20, http://investors.vrtx.com/releasedetail.cfm?ReleaseID=943778. [Google Scholar]
- 59.FDA Approves New Treatment for Cystic Fibrosis. U. S. Food and Drug Administration; 2015. Jul 2, http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm453565.htm. [Google Scholar]
- 60.Solomon GM, Marshall SG, Ramsey BW, Rowe SM. Breakthrough therapies: Cystic fibrosis (CF) potentiators and correctors. Pediatr Pulmonol. 2015;50(Suppl 40):S3–S13. doi: 10.1002/ppul.23240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elkins MR, Robinson M, Rose BR, et al. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006;354(3):229–40. doi: 10.1056/NEJMoa043900. [DOI] [PubMed] [Google Scholar]
- 62.Quan JM, Tiddens HA, Sy JP, et al. A two-year randomized, placebo-controlled trial of dornase alfa in young patients with cystic fibrosis with mild lung function abnormalities. J Pediatr. 2001;139(6):813–20. doi: 10.1067/mpd.2001.118570. [DOI] [PubMed] [Google Scholar]
- 63.Saiman L, Anstead M, Mayer-Hamblett N, et al. Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA. 2010;303(17):1707–15. doi: 10.1001/jama.2010.563. [DOI] [PubMed] [Google Scholar]
- 64**.Veit G, Avramescu RG, Perdomo D, et al. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Sci Transl Med. 2014;6(246):246ra97. doi: 10.1126/scitranslmed.3008889. Demonstrates that ivacaftor interferes with lumacaftor correction of F508del in vitro. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin WY, Yu YC. Interaction non grata between CFTR’s correctors and potentiators. Ann Transl Med. 2015;3(6):75. doi: 10.3978/j.issn.2305-5839.2015.01.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maiuri L, De Stefano D, Raia V, Kroemer G. The holy grail of cystic fibrosis research: pharmacological repair of the F508del-CFTR mutation. Ann Transl Med. 2015;3(Suppl 1):S24. doi: 10.3978/j.issn.2305-5839.2015.02.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matthes E, Goepp J, Carlile GW, et al. Low free drug concentration prevents inhibition of F508del CFTR functional expression by the potentiator VX-770 (ivacaftor) Br J Pharmacol. 2015 doi: 10.1111/bph.13365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Phuan PW, Veit G, Tan JA, et al. Potentiators of Defective DeltaF508-CFTR Gating that Do Not Interfere with Corrector Action. Mol Pharmacol. 2015;88(4):791–9. doi: 10.1124/mol.115.099689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reznikov LR, Abou Alaiwa MH, Dohrn CL, et al. Antibacterial properties of the CFTR potentiator ivacaftor. J Cyst Fibros. 2014;13(5):515–9. doi: 10.1016/j.jcf.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gianotti A, Capurro V, Scudieri P, et al. Pharmacological rescue of mutant CFTR protein improves the viscoelastic properties of CF mucus. J Cyst Fibros. 2015 doi: 10.1016/j.jcf.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 71.Gentzsch M, Dang H, Dang Y, et al. The cystic fibrosis transmembrane conductance regulator impedes proteolytic stimulation of the epithelial Na+ channel. J Biol Chem. 2010;285(42):32227–32. doi: 10.1074/jbc.M110.155259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vertex Pharmaceuticals, Inc; 2015. Jun 4, Vertex and Parion Sciences Establish Collaboration to Develop Epithelial Sodium Channel (ENaC) Inhibitors in Cystic Fibrosis and Other Pulmonary Diseases. http://investors.vrtx.com/releasedetail.cfm?ReleaseID=916591. [Google Scholar]
- 73.Vertex Announces Significant Progress in Its Development Efforts to Treat the Cause of Cystic Fibrosis in the Vast Majority of People with the Disease. Vertex Pharmaceuticals, Inc; 2015. Oct 8, http://investors.vrtx.com/releasedetail.cfm?ReleaseID=935806. [Google Scholar]
- 74.Cholon DM, Quinney NL, Chaudhry IG, et al. Pharmacological Rescue of Mutant CFTR: Exploring Mutant-Specific Therapies. Pediatr Pulmonol. 2015;(Suppl 41):194. (Abstract) [Google Scholar]
- 75.Veit G, Avramescu RG, Chiang AN, et al. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell. 2016;27(3):424–33. doi: 10.1091/mbc.E14-04-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O’Sullivan BP, Orenstein DM, Milla CE. Pricing for orphan drugs: will the market bear what society cannot? JAMA. 2013;310(13):1343–4. doi: 10.1001/jama.2013.278129. [DOI] [PubMed] [Google Scholar]