

Abstract
Pharmacologic blockade of STAT3 activation in tyrosine kinase inhibitor (TKI)-resistant chronic myeloid leukemia (CML) cell lines characterized by kinase-independent resistance re-sensitized CML cells to TKI therapy, suggesting that STAT3 inhibitors in combination with TKIs are an effective combinatorial therapeutic for the treatment of CML. Benzoic acid-based STAT3 inhibitors, SH-4-54 and SH-5-07, developed in our lab, demonstrated promising activity against these resistant CML cell lines. However, pharmacokinetic studies in murine models (CD-1 mice) revealed that both SH-4-54 and SH-5-07 are susceptible to glutathione conjugation at the para position of the pentafluorophenyl group via nucleophilic aromatic substitution (SNAr). To determine whether the electrophilicity of the pentafluorophenyl sulfonamide could be tempered, an in-depth structure activity relationship (SAR) study of the SH-4-54 scaffold was conducted. The studies revealed that AM-1-124, possessing a 2,3,5,6-tetrafluorophenylsulfonamide, retained STAT3 protein affinity (Ki = 15 μM), as well as selectivity over STAT1 (Ki >250 μM). Moreover, in both hepatocytes and in in vivo pharmacokinetic studies (CD-1 mice), AM-1-124 was found to be dramatically more stable than SH-4-54 (t1/2 = 1.42 h cf. 10 minutes, respectively). AM-1-124 represents a promising STAT3-targeting inhibitor with demonstrated bioavailability, suitable for evaluation in preclinical cancer models.
Keywords: STAT3 protein, protein-protein interactions, covalent modification, Imatinib-resistance, chronic myeloid leukemia
Graphical Abstract
AM-1-124 represents a promising STAT3-targeting inhibitor with demonstrated activity in tyrosine kinase inhibitor (TKI)-resistant chronic myeloid leukemia (CML) cell lines. In contrast to SH-4-54, AM-1-124 exhibits promising bioavailability in murine models (CD-1 mice), making it a candidate for evaluation in preclinical cancer models.

Introduction
Chronic myeloid leukemia (CML) is a hematopoietic stem cell disorder caused by BCR-ABL1, a constitutively active tyrosine kinase generated as a result of a reciprocal translocation between the long arms of chromosomes 9 and 22, cytogenetically visible as the Philadelphia chromosome. BCR-ABL1 fusion results in increased cellular proliferation, loss of stromal adhesion, and resistance to apoptosis.[1] Effective CML management has been achieved with BCR-ABL1-targeting tyrosine kinase inhibitors (TKIs), initially imatinib, and subsequently second-generation inhibitors, dasatinib and nilotinib. Approximately 20–30% of patients with chronic -phase CML (CP-CML) fail imatinib treatment due to primary or acquired resistance.[2] Resistance to BCR-ABL1 TKI inhibitors occurs through two main mechanisms. First, point mutations in the BCR-ABL1 kinase domain impair TKI binding.[3] Second, BCR-ABL1-independent resistance can occur due to activation of alternative signaling pathways. Aberrant signal transducer and activator of transcription 3 (STAT3) activation has been associated with BCR-ABL1 independent resistance.[4],[5] We have reported that inhibiting STAT3 in TKI-resistant CML lines exhibiting kinase-independent resistance re-sensitized CML cells to TKI therapy, suggesting that STAT3 inhibitors in combination with TKIs are an effective combinatorial therapeutic for the treatment of CML.[5]
In addition to CML, aberrant STAT3 activation has been implicated in malignant transformation and drug resistance in a variety of cancers.[6] Various intrinsic and extrinsic factors can lead to constitutive activation of STAT3, such as acquired activating mutations,[7] silencing of STAT3 negative regulators,[8] and activation by autocrine production of IL-6.[9] SRC family kinases are upstream of STAT3,[10] and have also been linked to imatinib resistance in CML cell lines and patient samples.[11] Recently, Traer and Eiring et al.[5b, 6a] found that a subset of CML patients with TKI resistance exhibited STAT3 activation. Given that diverse upstream signals can activate STAT3, in the context of drug resistance, direct targeting of STAT3 may be a universal approach to overcome resistance.[12]
In physiological conditions the JAK/STAT3 pathway is initiated by activation of extracellular receptors by cytokines, which in turn, leads to the phosphorylation of JAK, and subsequent Tyr phosphorylation of the JAK-associated receptor. Tyr phosphorylation sites serve to recruit STAT3 protein via its phospho-Tyr-binding Src-homology 2 (SH2) domain. STAT3, once phosphorylated at Tyr705 by JAK (pSTAT3), dissociates from the receptor, and forms a pSTAT3:pSTAT3 homodimer via reciprocal SH2:Tyr705 interactions. The homodimer translocates to the nucleus, binds to promoter regions on DNA to promote transcriptions of genes involved in cell growth, proliferation, and differentiation.
Inhibiting STAT3 phosphorylation and/or dimerization is expected to ablate the function of STAT3 as a transcriptional activator. Past efforts have utilized peptidomimetics,[13] metal-based complexes,[14] oligonucleotides,[15] and small molecule inhibitors.[16] We have previously developed SH2 domain-targeting small molecule STAT3 inhibitors, BP-5-087.[5] and SH-4-54 (Table 1).[6] In the context of CML, a salicylic acid-based inhibitor, BP-5-087 was used successfully in combination with imatinib in the kinase-independent TKI-resistant CML lines AR230R. Experiments demonstrated that pSTAT3 inhibition with BP-5-087 re-sensitized AR230R cells to imatinib. While promising data with BP-5-087 was generated, BP-5-087’s liabilities included poor lipophilic efficiency (LipE) (BP-5-087, LipE = 4.79 cf. −2.25 for SH-4-54), poor solubility (logP = 4.11 (StarDrop)),[17] and low ligand efficiency (LE) (BP-5-087, LE = 0.150 cf. 0.165 for SH-4-54 (StarDrop)).
Table 1.
Chemical structures, calculated LogP values, and inhibitor IC50 values of SH-4-54, SH-5-07, and BP-5-087 against AR230 and AR230R cells (MTS assay)[5]

| |||
|---|---|---|---|
| SH-4-54 | SH-5-07 | BP-5-87 | |
| IC50 AR230 (μm) | 2.9 | 8.1 | 19.1 |
| IC50 AR230R (μm) | 2.6 | 7.0 | 17.4 |
| LogP[a] | 4.11 | 3.57 | 5.75 |
Values calculated with StarDrop.
Thus, two alternative inhibitors developed in our lab, SH-4-54 and SH-5-07.[4] SH-4-54 and SH-5-07 were previously shown to bind STAT3 with a Kd of 300 and 612 nM (surface plasmon resonance (SPR)), respectively. Both compounds exhibited nM cytotoxic potency in glioblastoma brain tumour stem cells (GBM BTSC line 30M derived from a GBM patient) with concomitant effects against pSTAT3.[4] Preclinical data from our lab in orthotopically engrafted models of glioblastoma multiforme showed reduction in tumour growth with SH-4-54 at 10 mg/kg by IV. In AR230 and AR230R CML cell lines, SH-4-54 exhibited excellent potency, with IC50 values of 2.9 and 2.6 μM, respectively. Herein, we describe identification of more drug-like STAT3 inhibitors for combination with imatinib to treat CML.
Results and Discussion
Pharmacokinetic Analyses of SH-4-54 and SH-5-07
For PK/ADME characterization, SH-4-54 was subjected to metabolic stability studies in cryopreserved hepatocytes (human, mouse, rat, and dog) and bioavailability measured in vivo in CD-1 mice (AdmeScope, Oulu Finland). In all species, SH-4-54 was rapidly metabolized with a t1/2 of 5 minutes (Figure 1A). Bioavailability studies in CD-1 mice treated with 20 mg/kg SH-4-54 (10% DMA, 65% PEG 400, 25% saline vehicle) via intraperitoneal (IP) injection indicated rapid plasma clearance of the parent compound. SH-4-54 showed a t1/2 of ~10–15 mins (IP) with a Cmax of 1657 +/− 829 ng/mL (Figure 1B). Similarly, with IV dosing, SH-4-54 decreased rapidly from 124 ng/mL at 5 min. to 4.43 ng/mL at 15 min. post treatment (5 mg/kg, 10% PEG 400) with a Cmax of only 124 ng/mL (data in SI). In the case of SH-5-07, plasma levels of compound were barely within the detectable range following IP administration, with a Cmax of 299 +/− 74 ng/mL (Figure 1C). To further evaluate the pharmacokinetic profile, SH-5-07 was dosed via both IV and PO routes of administration at 5 and 20 mg/kg, respectively. In both cases, there was negligible accumulation of compound in the plasma (~700 ng/mL after 30 mins (IV), data included in the SI). From these results, we hypothesize that the anti-tumor activity of SH-4-54 observed in vivo is likely due to metabolites.
Figure 1.

A) Hepatocyte stability data measured as the disappearance of SH-4-54 using UPLC/Q-TOF-MS; B) Mean plasma concentration of SH-4-54 vs. time profiles after IP injection in CD-1 mice; C) Mean plasma concentration of SH-5-07 vs. time profiles after IP injection in CD-1 mice. Studies performed by contract research organizations (ADMEScope Ltd. and Pharmaron).
To identify the in vivo metabolites, cryopreserved hepatocytes from mouse, rat, dog, and human were treated with SH-4-54 and analysed using LC/TOF-MS analysis (Admescope, Finland). In each species, SH-4-54 metabolism was found to proceed predominantly via glutathione conjugation at the para-position of the pentafluorobenzene via nucleophilic aromatic substitution (SNAr), M6 (Scheme 1) with further metabolism of the gluthathione (GSH)-adduct to the S-cysteinyl glycine (M4, Scheme 1) and S-cysteine (M2, Scheme 1). In addition, N-Me demethylation was observed, as well as phase II acylation at the resultant amine (M1 and M9, Scheme 1). The cyclohexyl ring was also found to be hydroxylated in several metabolites (M10 and M7, Scheme 1). The major metabolite was found to be M6, having ~47% of the total combined peak area of SH-4-54 (in human) and a 71–90% share in mouse, rat, and dog (Data provided in the SI, Table 3). These results were recapitulated in vivo with SH-4-54; employing NMRI mice dosed intravenously (1 and 5 mg/kg) and orally (5 and 25 mg/kg) (Admescope Ltd, Finland). As in the hepatocyte study, M6 was found to be the major metabolite (Data provided in the SI, Table 4). Combined, these results supported the hypothesis that the pentafluorobenzene sulfonamide (PFBS) in both SH-5-07 and SH-4-54 might behave like an electrophilic ‘warhead’ toward thiol-based nucleophiles in vivo, specifically glutathione. These data led to site-directed structural modifications to eliminate the observed PK issues and facilitate the development of a more chemically stable drug.
Scheme 1.

Suggested structures for the observed SH-4-54 metabolites from mouse plasma and mouse, rat, dog and human hepatocytes.
First, to prevent oxidation at the R position, we evaluated removal or replacement of the N-Me substituent. This focused library included a demethylated analog of SH-4-54, DR-3-111, as well as molecules containing a bulky isopropyl substituent (PSL-3-40) and a non-native, N-cyclopropyl ring (DR-3-036) for which metabolic stability precedent exists.[18] The cytotoxicity results against AR230 and AR230R CML cells are outlined in Table 2. The demethylated analog, DR-3-111, showed an approximate 5-fold reduction in activity (AR230R cells: SH-4-54, IC50 = 2.6 μM cf. DR-3-111, IC50 = 15.6 μM). While PSL-3-40, possessing a bulky isopropyl substituent conferred similar losses in activity, DR-3-036, equipped with a cyclopropyl group, exhibited similar IC50 values to SH-4-54 (AR230R: SH-4-54 IC50 = 2.6 μM cf. DR-3-036 IC50 = 2.6 μM) (Table 2).
Table 2.
Phenotypic screening of R analogs of SH-4-54 in CML cell lines.

| |||||
|---|---|---|---|---|---|
| Compound ID | R | R1 | IC50 AR230 (μm) | IC50 AR230R (μm) | LogP[a] |
| DR-3-111 | H | c-Pent | 11.4 | 15.6 | 4.8 |
| SH-4-54 | Me | c-Hex | 2.9 | 2.6 | 4.53 |
| PSL-3-40 | i-Pr | c-Hex | 8.7 | 14.2 | 4.68 |
| DR-3-036 | c-Pr | c-Hex | 4.3 | 2.6 | 4.52 |
Values calculated by StarDrop
Next, while retaining the N-cyclopropyl group, the size and composition of the cyclohexyl ring on the para-aminobenzyl substituent was optimized with the overarching goal of improving metabolic stability whilst maintaining potency in the phenotypic screens. A range of cyclic and acyclic functional groups with varying size, ring strain, hydrophobicity/hydrophilicity, hetero-atom composition, and flexibility were prepared and screened against AR230 and AR230R cells (Table 3). Several interesting phenotypic trends were observed.
Table 3.
Phenotypic screening of R1 analogs in CML cell lines.

| |||||
|---|---|---|---|---|---|
| Compound ID | R1 | IC50 AR230 (μm) | IC50 AR230R (μm) | Fold Difference | LogP[a] |
| PSL-3-115 | H | 5.2 | 2.3 | 2.26 | 3.39 |
| PSL-3-122 | F | 6.1 | 3 | 2.03 | 3.36 |
| DR-4-003 | Cl | 4.1 | 1.7 | 2.41 | 3.68 |
| DR-3-186 | CF3 | 1.4 | 0.72 | 1.94 | 3.95 |
| PSL-3-132 | c-Pr | 4.2 | 2.4 | 1.75 | 3.86 |
| DR-4-020 | c-Bu | 8.6 | 4.2 | 2.05 | 4.05 |
| DR-3-162 | t-Bu | 3.2 | 0.8 | 4 | 4.24 |
| DR-3-093 | c-Pent | 3.3 | 1.2 | 2.75 | 4.23 |
| DR-3-165 | c-Hept | 2.7 | 1.1 | 2.45 | 4.8 |
| DR-3-119 |
|
5.8 | 3.2 | 1.81 | 4.84 |
| PSL-3-089 |
|
7.4 | 5.2 | 1.42 | 3.29 |
| DR-4-042 |
|
4.7 | 4.2 | 1.12 | 3.29 |
Values calculated by StarDrop.
Varying the ring size from 6 to both 5 and 7 did not have a marked effect on activity or selectivity in the pSTAT3-dependent cell line, AR230R (~2-fold selectivity over AR230) (Table 3). However, DR-3-093, containing a 5-membered ring, with a lower logP value (logP = 4.23), was considered more drug-like as compared to the cycloheptyl derivative, DR-3-165 (logP = 4.8). Of particular note, DR-3-162, possessing a para-t-Bu group, was 3-fold more potent (AR230R, IC50 = 0.8 μM), and 4-fold more selective for AR230R over parental AR230 cells. The analogous, less bulky, trifluoromethyl analog, DR-3-186, was equipotent (AR230R, IC50 = 0.72 μM), but marginally less selective (<2-fold). Introduction of heteroatoms, N or O, or a combination thereof, to the 6-membered ring, while favourably reducing logP, negligibly impacted potency and selectivity. Further studies are required to determine whether this trend is a result of the respective analogs’ cell penetrating properties.
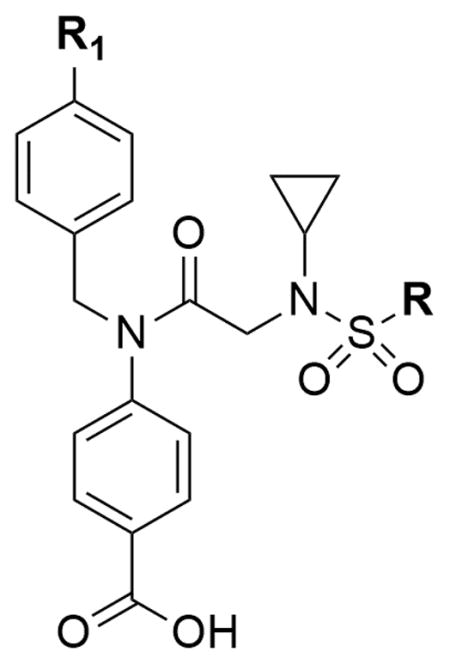
With several promising candidates in hand, most notably DR-3-093 and DR-3-162, focus was directed toward reducing the electrophilicity of the pentafluorobenzene ring. For SAR purposes, we elected to retain the cyclopentyl ring of DR-3-093 and the tert-butyl group of DR-3-162. To prevent glutathionation of prospective analogs, we screened a number of different aryl and heterocyclic substituents (Table 4). Towards this end, the electron withdrawing para-fluoro atom of the PFBS was substituted with other electron withdrawing substituents with varying electronegativities. As can be seen, replacing the PFBS with either the 2- or 3-pyridyl substituents resulted in a complete loss of activity. Moreover, retaining the problematic para-fluoro atom (AES-1-094), while at the same time reducing the overall fluorine content of the sulfonyl group, failed to elicit a significant effect on cell viability. Substituting the para-fluorine with less electron withdrawing groups proved to be a more productive route to recovering activity. While introducing either a bromo- or chloro heteroatom yielded modest cytotoxic activity, ultimately, the introduction of hydrogen at the para-aryl position afforded the most potent compound of the series, AM-1-020 (AR230R, IC50 = 13.1 μM).
Table 4.
Phenotypic screening of R1 analogs in CML cell lines.
| |||||
|---|---|---|---|---|---|
| Compound ID | R | R1 | IC50 AR230 (μm) | IC50 AR230R (μm) | LogP[a] |
| RG1-066 | 3-pyridyl | c-Pent | >32 | >32 | 3.61 |
| RG1-085 | 2-pyridyl | c-Pent | >32 | >32 | 3.61 |
| AM1-116 | 4-fluoro-3-pyridyl | c-Pent | >32 | >32 | 3.74 |
| AM1-117 | 4-fluoro-2-pyridyl | c-Pent | >32 | >32 | 3.74 |
| AM1-020 | 2,3,5,6-tetrafluoro-phenyl | c-Pent | >32 | 13.1 | 4.28 |
| AM1-112 | 4-chloro-2,3,5,6-tetrafluorophenyl | c-Pent | >32 | 30.7 | 4.6 |
| AM1-077 | 4-bromo-2,3,5,6-tetrafluorophenyl | c-Pent | 25.07 | 20.3 | 4.74 |
| AM1-066 | 4-trifluoromethyl-2,3,5,6-tetrafluorophenyl | c-Pent | 20.4 | 21.6 | 4.84 |
| AM1-030 | 4-methyl-2,3,5,6-tetrafluorophenyl | c-Pent | >32 | 18.6 | 4.54 |
| AES1-094 | 4-fluorophenyl | t-Bu | >32 | 10.6 | 1.96 |
Values calculated by StarDrop.
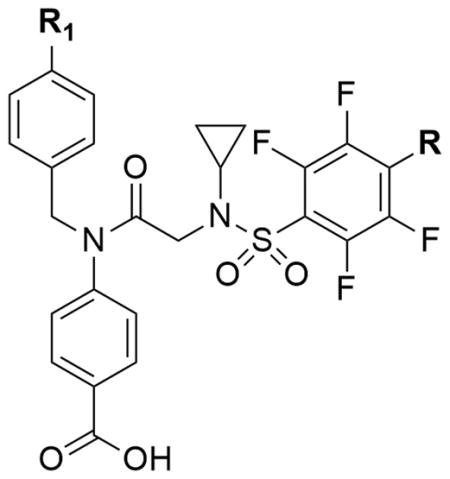
To further improve cell-based activity, the 2,3,5,6-tetrafluorobenzene group was substituted into the most potent of the R1 series of compounds (Table 5) in the hope that a more potent compound, lacking the electrophilic warhead, might be discovered. The t-Bu (DR-3-162) R1 group, with IC50 value of 0.8, was incorporated into the AM-1-020 scaffold, yielding AM-1-124 (Table 5).
Table 5.
Analogs in CML cell lines.
| ||||||
|---|---|---|---|---|---|---|
| Compound ID | R1 | R | IC50 AR230 (μm) | IC50 AR230R (μm) | Fold Difference | LogP[a] |
| SH-4-54 | c-Hex | F | 2.9 | 2.6 | - | 4.11 |
| AM-1-124 | t-Bu | H | 26.9 | 4.8 | 5.6 | 4.30 |
Values calculated by StarDrop.
Most promisingly, AM-1-124 (R1 = t-Bu) exhibited IC50 values of 26.9 and 4.8 μM in AR230 and AR230R cell lines, respectively. AM-1-124, not possessing a PFBS group, with only a modest reduction in potency, and 5-fold improvement in selectivity, represented an interesting lead analog of SH-4-54.
To experimentally assess whether GSH conjugation remained a major metabolic impediment, a time-course GSH conjugation assay was employed. AM-1-124 (100 μM) and SH-4-54 (100 μM) were incubated with a 100-fold excess of GSH (10 mM) and the reaction sampled every 15 min to determine the rate of conversion to the GSH adduct. As expected, AM-1-124 negligibly reacted with glutathione, exhibiting a t1/2 > 100 h (SI Figure 1). These data indicated that the ortho-fluoro position of the 2,3,5,6-tetrafluorobenzenesulfonamide was not reactive to thiol-based nucleophiles, likely due to steric hindrance or a general reduction in stabilization of the anionic SNAr intermediate. Control compound, SH-4-54 was rapidly converted to its corresponding GSH adduct, with a t1/2 of 79 minutes (95% confidence interval = 74.1 – 88.8 minutes, SI Figure 1). The relatively slow rate of GSH uptake when compared to the preceding mouse hepatocyte data, likely points to the involvement of the GSH-conjugating enzyme, glutathione S-transferase.[19]
19F NMR comparative examination of SH-4-54 and AM-1-124 binding to STAT3
Since both SH-4-54 and AM-1-124 possess fluorine substituents, 19F NMR studies can be readily used to track defluorination, as would be observed in the case of a covalent mode-of-action. First, as a control, SH-4-54 was incubated with glutathione at 37 °C and the reaction monitored in real time using a 600 MHz spectrometer (Varian) equipped with an H(F)CN room temperature probe. As can be seen in Figure 2C/D, the peak corresponding to the para-arylfluoride disappeared, and was accompanied by the simultaneous production of fluoride ions. In contrast, under similar reaction conditions, AM-1-124 was inert to glutathione, with negligible changes to both the ortho- and meta-arylfluoride of the tetrafluorophenyl and no observed fluoride ion produced (Figure 2F,G). Next, upon treatment of STAT3 with excess of either SH-4-54 or AM-1-124, binding of inhibitor was observed as indicated by broadening of the peaks corresponding to the ortho- and meta-arylfluorides of the fluorophenyl rings (Figure 2E,H). The observed broadening is a result of the faster dipole-dipole (T2) relaxation due to protein-drug interactions.[20] Interestingly, the binding of SH-4-54 to STAT3 resulted in fluoride ion production. This demonstrates that SH-4-54 covalently modifies the STAT3 protein, an interaction that was undetectable in previously employed binding assays. Most promisingly, this is distinct from the binding interaction with AM-1-124, where no fluoride ion production upon STAT3 binding was observed. This provided unequivocal evidence that the interactions between STAT3 and AM-1-124 are non-covalent (Figure 2E,H). The mode of STAT3 inhibition by SH-4-54 is being further explored and will be reported elsewhere.
Figure 2.

19F NMR studies were recorded at 37°C on a 600 MHz spectrometer equipped with an H(F)CN room temperature probe. All samples were prepared in 100 mM HEPES pH 7.2, 150 mM NaCl with a final concentration of 10% H2O and 1% DMSO. All spectra were normalized according to the fluorine peak of an added reference, 5-fluoro-tryptophan. A) Shows the 1D 19F NMR spectra of the reference. B) Shows the spectra of a sample containing 100 μM NaF as an F- reference. C) The 19F 1D NMR spectra of 100 μM SH-4-54. D) 100μM of SH-4-54 with 10 equivalents of glutathione incubated for 15 hours at 37°C. E) 100μM of SH-4-54 with 0.2 equivalents of STAT3 incubated for 1 hour at 37°C. F) 100μM of AM-1-124. G) 100μM of AM-1-124 with 10 equivalents of glutathione incubated for 15 hours at 37°C. H) 100μM of AM-1-124 with 0.4 equivalents of STAT3 incubated for 1 hours at 37°C.
STAT3 vs. STAT1 SH2 domain fluorescence polarization
To confirm the 19F NMR studies, previously reported competitive fluorescence polarization (FP) binding experiments with STAT3[21] were performed with AM-1-124. In addition, to examine selectivity, AM-1-124 was also assessed in a STAT1[22] FP assay. FP experiments revealed that AM-1-124 was able to selectively disrupt a STAT3:IL-6R/gp130 peptide complex (Ki = 15.3 μM, Figure 3A) over that of the homologous STAT1:IFN-γ receptor complex (Ki > 250 μM, Figure 3B).
Figure 3.

Competitive FP data for AM-1-124 vs. STAT3 or STAT1 in the presence of fluoresceinated peptide (conditions in si). A) STAT3 Ki = 15.3 μM (IC50 = 30 μM, 95% Confidence Interval = 27.8–32.3 μM); B) STAT1 Ki > 250 μM (IC50 > 1000 μM, 95% Confidence Interval = 292–1167 μM).
Immunofluorescent staining of AM-1-124 treated AR230R cells
To confirm that the cytotoxicity of AM-1-124 in the AR230R cell line is associated with STAT3 inhibition we performed immunofluorescent staining with antibodies against STAT3 and pSTAT3. AR230R cells were serum starved overnight to reduce STAT3 activation, treated for 2 hours with AM-1-124 (10 μM), then cytokine stimulated with IL-6 and IFN-μ. The cells were then fixed 1 hour after cytokine treatment and cytospun onto glass slides for fluorescent staining. As seen in Figure 4 (and fluorescent microscopy images shown in SI, Figure 9), AM-1-124 pre-treatment partially inhibited cytokine-induced pSTAT3 activation. Furthermore, AM-1-124 reduced the nuclear STAT3 protein localization, which is correlated with transcriptional activity (Figure 4).[23] These experiments demonstrate that AM-1-124 inhibits STAT3 in the AR230R cell line and provides an explanation why this compound has much better activity in the TKI-resistant/STAT3 dependent AR230R cell line compared to the STAT3-independent AR230 parental line.
Figure 4.

AR230R cells were serum starved overnight, pre-treated with AM-1-124 (10 μM) or vehicle and stimulated with the cytokines IL-6 (20 ng/mL) and IFN-μ (100 ng/ml). Fluorescence intensity of pSTAT3 (A) and total STAT3 (B) was measured in both cytoplasmic and nuclear compartments.
Metabolic Stability of AM-1-124 in Mouse Hepatocytes
As described above, SH-4-54 was rapidly metabolized in CD-1 mouse hepatocytes, with degradation of the parent molecule occurring within 10 mins. The majority of metabolites were glutathione conjugates (SI, Table 3). To evaluate liver stability relative to SH-4-54, AM-1-124 was incubated with similar mouse hepatocytes for 120 mins and analyzed at regular intervals by LC-MS to assess metabolic degradation. As can be seen from Figure 5, AM-1-124 was shown to have a t1/2 of 38.6 mins cf. 10.1 mins for SH-4-54 in CD-1 mouse hepatocytes (Pharmaron, information included in the SI). Encouraged, we next evaluated the bioavailability of AM-1-124 in CD-1 mice.
Figure 5.

Time-course metabolism study comparing SH-4-054 to AM-1-124 in CD-1 male mouse hepatocytes. Time-point measurements of remaining parent compound were taken using LC-MS/MS. Studies performed by a contract research organization (Pharmaron).
AM-1-124 Plasma Protein Binding Study
AM-1-124 was also assessed for plasma protein binding (PPB) using plasma protein isolated from CD-1 mice (Pharmaron, China). In the assay, plasma protein treated with AM-1-124 was dialyzed with PBS buffer for a period of 6 hours under typical cell growing conditions (i.e. apparatus incubated at 37°C under a 5% CO2 atmosphere). Both the plasma protein and PBS buffer compartments were analysed to find the percentage of free compound. Ketoconazole was used as a positive control. It was found that AM-1-124 was 99.7% bound to mouse plasma proteins (CD-1 mice, information included in the SI).
In vivo bioavailability of AM-1-124 (IP Route of Administration)
AM-1-124 and SH-4-54 were administered to a cohort of 3 mice via IP injection (20 mg/kg, 10% DMA, 65% PEG 400, 25% saline). Plasma samples were collected at 15, 30, 60, 90, 180, and 360 mins post-treatment and analyzed for parent compound by LC-MS (Pharmaron). As expected, SH-4-54 was rapidly cleared (Figure 6), thus, recapitulating the findings of ADMEScope. However, AM-1-124 was detectable at concentrations above 2000 ng/mL for the first 120 mins of the study (Figure 6), exhibiting an in sero half-life of 1.42 h. This data supports our hypothesis that removal of the para-fluoro substituent affords in vivo stability and significantly reduces the metabolic liabilities of SH-4-54.
Figure 6.
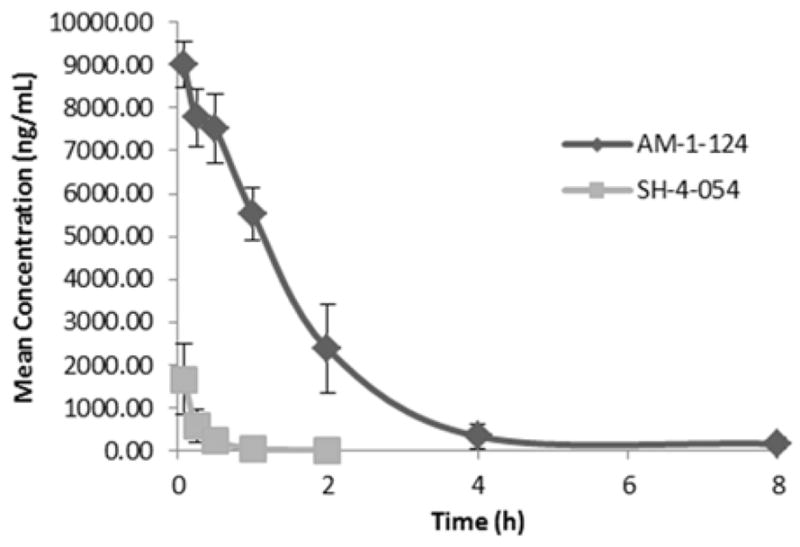
In vivo metabolism study comparing SH-4-054 to AM-1-124 in CD-1 male mouse via IP. Time-point measurements of remaining parent compound were taken using LC-MS/MS. Studies performed by a contract research organization (Pharmaron).
Conclusions
In summary, we have conducted a focused SAR on SH-4-54, a promising in vitro anti-pSTAT3 inhibitor with limited bioavailability in vivo (t1/2 = 10 mins), to afford AM-1-124. AM-1-124 has a lower molecular weight than SH-4-54, demonstrated a more selective cytotoxic effect against imatinib resistant CML cells AR230R cf. AR230 cells (5.6-fold selectivity cf. 1.1-fold for SH-4-54), was ~4-fold more resistant to metabolism in mouse hepatocytes (t1/2 = 38.9 min cf. 10.2 min for SH-4-54), and possessed a significantly improved half-life in bioavailability studies in CD-1 mice (IP (20 mg/kg, t1/2 = 1.42 h cf. 15 mins for SH-4-54).
Experimental Section
Experimental Details All NMR chemical shifts (δ) are reported in parts per million after calibration to residual isotopic solvent and coupling constants (J) are reported in Hz. Compounds AM-1-124 and DR-3-186 were fully characterized using 1D and 2D NMR, images for which are provided in supporting information. Compound purity was assessed using a HP 1100 HPLC equipped with a Phenomenex Luna 5 μm C18(2) 100 Å, 150 × 4.6 mm column. Measures of absorbance were taken at 254 nm. Gradient solvent mixtures were administered at a flow rate of 1.2 mL/min. The linear gradient consisted of a changing solvent composition of either (I) 100% H2O with 0.1% TFA (v/v) / 0% acetonitrile to 0% H2O with TFA / 100% acetonitrile over 30 minutes or (II) 100% H2O with 0.1% TFA (v/v) / 0% acetonitrile to 0% H2O with TFA / 100% acetonitrile over 60 minutes. For reporting HPLC data, percentage purity is given in parentheses after the retention time (rt) for each condition. All final products were lyophilized from water/acetonitrile prior to testing. Biologically evaluated compounds are ≥ 95 % chemical purity as measured by HPLC. Traces of the HPLC results are provided in supporting information. High Resolution Mass Spectrometry (HRMS) was performed on an AB/Scienx QStar mass spectrometer with an ESI source.
4-(N-(4-Cyclopentylbenzyl)-2-((N-cyclopropyl-2,3,5,6-tetrafluorophenyl)sulfonamido)acetamido)benzoic acid (AM-1-20)
1H NMR (400 MHz, CDCl3) δ 0.81 – 0.83 (d, J = 5.2 Hz, 4H), 1.48 – 1.62 (m, 2H), 1.62 – 1.73 (m, 2H), 1.73 – 1.86 (m, 2H), 2.04 (td, J = 10.7, 4.2 Hz, 2H), 2.88 – 3.03 (m, 2H), 3.95 (d, J = 35.5 Hz, 2H), 4.80 (s, 2H), 7.00 (d, J = 7.6 Hz, 2H), 7.10 – 7.21 (m, 4H), 7.26 – 7.35 (m, 1H), 8.12 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 23.2, 29.9, 33.0, 33.2, 52.4, 52.8, 67.0, 125.6, 126.5, 128.15, 128.22, 128.3, 128.5, 128.6, 131.4, 134.2, 135.5, 136.3, 141.7, 165.1, 166.3; 19F NMR (376 MHz, CDCl3) δ −134.51 to −134.99 (m, 2F), −131.80 to −132.27 (m, 2F); HRMS (ESI−) m/z [M-H]-Calcd for C30H27F4N2O5S: 603.1573, found: 603.1582; HPLC rt (II) = 41.5 min (96.7%).
4-(N-(4-Cyclopentylbenzyl)-2-((N-cyclopropyl-2,3,5,6-tetrafluoro-4-methylphenyl)sulfonamido)acetamido)benzoic acid (AM-1-030)
1H NMR (500 MHz, CDCl3) δ 0.79 – 0.86 (m, 4H), 1.51 – 1.60 (m, 2H), 1.63 – 1.73 (m, 2H), 1.75 – 1.85 (m, 2H), 1.99 – 2.10 (m, 2H), 2.35 (t, J = 1.9 Hz, 4H), 2.96 (m, 2H), 4.00 (s, 2H), 4.81 (s, 2H), 7.02 (d, J = 7.7 Hz, 2H), 7.16 (t, J = 9.0 Hz, 4H), 8.11 (d, J = 8.5 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 1.2, 8.2, 25.6, 30.2, 34.7, 45.7, 52.5, 53.2, 117.5 (t, 2JCF = 14.4 Hz), 121.4 (t, 2JCF = 19.2 Hz), 127.5, 128.7, 129.6, 132.2, 133.4, 144.1 (dm, 1JCF = 257.1 Hz), 144.2 (dm, 1JCF = 257.1 Hz), 145.33 (dm, 1JCF = 249 Hz), 145.46 (dm, 1JCF = 252.2 Hz), 145.57, 146.41, 166.85, 170.83; 19F NMR (564 MHz, CDCl3) δ = −136.9 (m, 2F), −141.5 (m, 2F); HRMS (ESI−) m/z [M-H]-Calcd for C31H29F4N2O5S: 617.1739, found: 617.1736; HPLC rt (II) = 43.3 min (98.3%).
4-(N-(4-Cyclopentylbenzyl)-2-((N-cyclopropyl-2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenyl)sulfonamido)acetamido)benzoic acid (AM-1-066)
1H NMR (500 MHz, CDCl3) δ = 0.86 (d, J = 6.0 Hz, 4H), 1.51 – 1.63 (m, 2H), 1.64 – 1.74 (m, 2H), 1.76 – 1.87 (m, 2H), 1.99 – 2.11 (m, 2H), 2.87 – 3.02 (m, 2H), 4.00 (s, 2H), 4.78 (s, 2H), 6.98 (d, J = 7.7 Hz, 2H), 7.11 to 7.19 (m, 4H), 8.13 (d, J = 8.4 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 7.4, 24.6, 29.3, 33.7, 44.7, 51.9, 52.3, 112 (m), 119.5 (q, 1JCF = 272 Hz), 122.8 (m), 126.5, 127.7, 128.7, 131.2, 132.1, 143.5 (dm, 1JCF = 258.7 Hz) 145.6, 165.5, 169.4; 19F NMR (564 MHz, CDCl3) δ = −56.6 (t, 25.9 Hz, 3F), −132.5 (m, 2F), −138.4 (bs, 2F); HRMS (ESI−) m/z [M-H]-Calcd for C31H26F7N2O5S: 671.1454, found: 671.1446; HPLC rt (II) = 46.0min (96.5%).
4-(N-(4-Cyclopentylbenzyl)-2-((N-cyclopropyl-2,3,5,6-tetrafluoro-4-bromophenyl)sulfonamido)acetamido)benzoic acid (AM-1-77)
1H NMR (500 MHz, CDCl3) δ = 0.78 – 0.93 (m, 4H), 1.50 – 1.62 (m, 2H), 1.65 – 1.76 (m, 2H), 1.76 – 1.88 (m, 2H), 2.01 – 2.11 (m, 2H), 2.97 (m, 2H), 4.01 (s, 2H), 4.80 (s, 2H), 7.00 (d, J = 7.6 Hz, 2H), 7.16 (m, 4H), 8.12 (d, J = 8.5 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 8.2, 25.6, 30.2, 34.6, 45.7, 52.7, 53.2, 105.3 (t, 2JCF = 22.5 Hz), 119.6 (t, 2JCF = 14.9 Hz), 127.5, 128.6, 129.6, 132.1, 133.2, 144.4 (dm, 1JCF = 261 Hz), 144.6 (dm, 1JCF = 260 Hz), 145.28, 145.3 (dm, 1JCF = 251.4 Hz), 145.5 (dm, 1JCF = 261 Hz), 146.5, 166.7, 179.7; 19F NMR (564 MHz, CDCl3) δ −133.7 (d, 3J = 15.1 Hz), −131 (d, 3J = 16.8 Hz); HRMS (ESI−) m/z [M-H]-Calcd for C30H27BrF4N2O5S: 681.0685, found: 681.0677; HPLC rt (II) = 44.9 min (98.1%).
4-(N-(4-Cyclopentylbenzyl)-2-((N-cyclopropyl-2,3,5,6-tetrafluoro-4-chlorophenyl)sulfonamido)acetamido)benzoic acid (AM-1-112)
1H NMR (500 MHz, CDCl3) δ = 0.83 (m, 4H), 1.45 (m, 2H), 1.55 (m, 2H), 1.68 (m, 2H), 1.76 – 1.84 (m, 2H), 1.99 – 2.12 (m, 2H), 2.93 – 3.00 (m, 2H), 3.99 (s, 2H), 4.77 (s, 2H), 6.99 (d, J = 7.7 Hz, 2H), 7.14 (m, 4H), 8.11 (d, J = 8.0 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 8.3, 25.6, 30.2, 34.7, 45.7, 52.7, 52.7, 53.2, 53.5, 117.2 (t, 2JCF = 19.8 Hz), 119 (t, 2JCF = 15.2 Hz), 120.4, 127.5, 128, 128.4, 128.7, 132, 133.3, 144.6 (dm, 1JCF = 259 Hz), 144.8, 146.5, 148.4, 149.1, 152.3, 166.7, 170.3; 19F NMR (564 MHz, CDCl3) δ = −138.9 (m, 2F), −134.1 (m, 2F); HRMS (ESI−) m/z [M-H]-Calcd for C30H27ClF4N2O5S: 637.1179, found: 637.1182; HPLC rt (II) = 45.2 min (95.8%).
4-(N-(4-Cyclopentylbenzyl)-2-((N-cyclopropyl-6-fluoropyridine)-3-sulfonamido)acetamido)benzoic acid (AM-1-116)
1H NMR (500 MHz, CDCl3) δ = 0.72 (m, 2H), 0.79 (m, 2H), 1.49 – 1.63 (m, 2H), 1.63 – 1.75 (m, 2H), 1.75 – 1.85 (m, 2H), 2.00 – 2.10 (m, 2H), 2.62 (m, 1H), 2.96 (m, 1H), 3.94 (s, 2H), 4.82 (s, 2H), 7.01 – 7.10 (m, 3H), 7.14 – 7.21 (m, 4H), 8.15 (d, J = 8.5 Hz, 2H), 8.32 (ddd, J = 8.6, 7.2, 2.5 Hz, 1H), 8.77 (d, J = 2.6 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 8.2, 25.6, 30.3, 34.7, 45.7, 52.4, 53.3, 109.7 (d, 2JCF = 37.4 Hz), 127.5, 128.6, 129.6, 132, 133.4, 134 (d, 4JCF = 4.5Hz), 142 (d, 3JCF = 9.5Hz), 145.7, 146.5, 148.9 (d, 3JCF=16.6Hz), 165.3 (d, 1JCF = 247.3Hz), 167.4, 170.5; 19F NMR (564 MHz, CDCl3) δ = −61.1 (s, 1F); HRMS (ESI−) m/z [M-H]-Calcd for C29H29FN3O5S: 550.1806, found: 550.1806; HPLC rt (II) = 38.7 min (95.3%).
4-(N-(4-Cyclopentylbenzyl)-2-((N-cyclopropyl-5-fluoropyridine)-2-sulfonamido)acetamido)benzoic acid (AM-1-117)
1H NMR (500 MHz, CDCl3) δ = 0.66 – 0.77 (m, 4H), 1.49 – 1.59 (m, 2H), 1.62 – 1.71 (m, 2H), 1.73 – 1.82 (m, 2H), 1.99 – 2.08 (m, 2H), 2.82 – 2.90 (m, 1H), 2.94 (m, 1H), 3.99 (s, 2H), 4.80 (s, 2H), 7.03 (d, J = 7.9 Hz, 2H), 7.13 (d, J = 8.1 Hz, 2H), 7.20 (d, J = 8.1 Hz, 2H), 7.55 (m, 1H), 8.03 (dd, J = 8.7, 4.2 Hz, 1H), 8.08 (d, J = 8.5 Hz, 2H), 8.40 (d, J = 2.8 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 8, 25.5, 30.9, 34.6, 45.6, 52.3, 53.3, 124.2 (d, 3JCF = 18.2 Hz), 124.9 (d, 4JCF = 5.3 Hz), 127.3, 128.5, 128.6, 131.8, 133.6, 138.4 (d, 2JCF = 25.7 Hz), 146.2, 153.2, 160.6 (d, 1JCF = 268 Hz), 167.4, 170; 19F NMR (564 MHz, CDCl3) δ = −119.8 (s, 1F); HRMS (ESI−) m/z [M-H]-Calcd for C29H29FN3O5S: 550.1817, found: 550.1810; HPLC rt (II) = 37.1 min (95.6%).
4-(N-(4-(tert-butyl)benzyl)-2-(N-cyclopropyl-2,3,5,6-tetrafluorophenylsulfonamido)acetamido)benzoic acid (AM-1-124)
1H NMR (500 MHz, CDCl3) δ 0.83 (d, J = 5.3 Hz, 4H), 1.30 (s, 9H), 2.98 (t, J = 5.3 Hz, 1H), 4.01 (bs, 2H), 4.80 (s, 2H), 7.03 (d, J = 7.9 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 7.30 (m, 3H), 8.12 (d, J = 8.5 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 8.21, 30.30, 31.44, 34.67, 52.61, 53.14, 110.08, 121.00, 125.68, 128.44, 128.69, 129.63, 132.13, 133.03, 144.41 (dm, 1JCF = 258.1 Hz), 145.38, 146.26 (dm, 1JCF = 250.9 Hz), 151.02, 166.66, 170.69. 19F NMR (658 MHz, CDCl3) δ −136.61 (m, 2F), −135.06 (m, 2F); HRMS (ESI−) m/z [M-H]-Calcd for C32H26F3N2O5S: 591.1573, found: 591.1582; HPLC rt (I) = 24.1 min (96.4%); HPLC rt (II) = 39.4 min (96.6%).
4-(N-(4-Cyclopentylbenzyl)-2-(N-cyclopropylpyridine-3-sulfonamido)acetamido)benzoic acid (RG I 066)
1H NMR (500 MHz, Methanol-d4) δ 0.62 – 0.78 (m, 4H), 1.45 – 1.59 (m, 2H), 1.59 – 1.71 (m, 2H), 1.71 – 1.83 (m, 2H), 1.96 – 2.06 (m, 2H), 2.50 – 2.62 (m, 1H), 2.93 (tt, J=9.8, 7.5, 1H), 3.90 (s, 2H), 4.67 (s, 3H), 7.02 (d, J=7.7, 2H), 7.14 (dd, J=14.0, 7.9, 4H), 7.52 – 7.58 (m, 1H), 7.99 – 8.07 (m, 2H), 8.20 (dt, J=8.1, 1.8, 1H), 8.75 (s, 1H), 8.99 (s, 1H). 13C NMR (126 MHz, cd3od) δ = 7.57, 25.23, 30.22, 34.38, 45.51, 52.04, 52.93, 123.85, 127.13, 128.40, 131.36, 133.41, 136.36, 144.49, 146.10, 148.18, 152.54, 167.48, 167.56; HRMS (ESI+) m/z [M+H]+ Calcd for C29H33N3O5S: 534.2057, found: 534.2061; HPLC rt (I) = 22.4 min (99.1%); HPLC rt (II) = 37.1 min (99.1%).
4-(N-(4-Cyclopentylbenzyl)-2-(N-cyclopropylpyridine-2-sulfonamido)acetamido)benzoic acid (RG I 085)
1H NMR (400 MHz, CDCl3) δ 0.67 (m, 4H), 1.55 (m, 2H), 1.67 (m, 2H), 1.79 (m, 2H), 2.05 (m, 2H), 2.93 (m, 2H), 4.03 (s, 2H), 4.83 (s, 2H), 7.05 (d, J = 7.7 Hz, 2H), 7.14 (d, J = 7.8 Hz, 2H), 7.20 (d, J = 7.9 Hz, 2H), 7.48 (dd, J = 6.0 Hz, 1H), 7.92 (dd, 3JHH = 7.7 Hz, 1H), 8.05 (m, 3H), 8.68 (d, 3JHH = 4.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 25.3, 30.7, 34.4, 45.4, 52.3, 53.0, 122.9, 126.5, 127.1, 128.27, 128.32, 128.35, 128.40, 128.5, 131.6, 133.5, 137.8, 146.0, 149.5, 157.2, 167.3, 169.5; HRMS (ESI+) m/z [M+H]+ Calcd for C29H33N3O5S: 534.2057, found: 534.2062; HPLC rt (I) = 23.4 min (97.2%); HPLC rt (II) = 38.7 min (96.7%).
4-(N-(4-cyclohexylbenzyl)-2-(2,3,4,5,6-pentafluoro-N-isopropylphenylsulfonamido)acetamido)benzoic acid (PSL-3-040)
1H NMR (400 MHz, CDCl3) δ 1.11 (d, J = 6.8 Hz, 6H), 1.36–1.43 (m, 5H), 1.74–1.87 (m, 5H), 2.48–2.52 (m, 1H), 3.83 (s, 2H), 4.41 (sep, J = 6.8 Hz, 1H), 4.64 (s, 2H), 4.68 (s, 1H), 6.7 (d, J = 8.0 Hz, 2H), 6.87 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 21.1, 26.0, 26.7, 29.7, 34.3, 44.1, 45.2, 51.3, 52.9, 115.2, 118.5 (m), 125.5, 127.9, 128.4, 128.9, 130.2, 137.8 (dm, 1JCF = 260.2 Hz), 137.9, 143.2 (dm, 1JCF = 236.0 Hz), 145.0 (dm, 1JCF = 256. 2 Hz), 148.9, 155.1, 167.6. 19F NMR (658 MHz, CDCl3). −160.33 (t, J = 21.0 Hz, 2F), −148.07 (t, J = 21.0 Hz, 1F), −135.63 (d, J = 21.0 Hz, 2F); HPLC rt (II) = 31.6 min (96.6%).
4-(2-((N-Cyclopropyl)-2,3,4,5,6-pentafluorophenyl)sulfonamido)-N-(4-morpholinobenzyl)acetamido)benzoic acid (PSL-3-089)
1H 1H NMR (400 MHz, CD3OD) δ 0.76–0.82 (m, 4H), 2.81–2.83 (m, 1H), 3.22 (t, J = 4.0 Hz, 4H), 3.85 (t, J = 4.0 Hz, 4 H), 4.03 (s, 2H), 4.81 (s, 2H), 7.01 (d, J = 8.0 Hz, 2H), 7.09 (d, J = 7.8 Hz, 2H), 7.20 (d, J = 8.0 Hz, 2H), 8.03 (d, J = 7.8 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 8.1, 29.7, 30.0, 50.2, 52.5, 52.7, 66.3, 116.6, 128.4, 128.5, 129.4, 129.9, 130.0, 130.0, 132.0, 169.2, 169.3; 19F NMR (658 MHz, CDCl3) δ −159.85 (t, J = 20.3 Hz, 2F), −146.43 (t, J = 20.3 Hz, 1F), −134.11 (d, J = 20.3 Hz, 2F). HRMS (ESI+) m/z [M+H]+ Calcd for C29H28F5N3O6S: 640.1535, found: 640.1537; HPLC rt (II) = 29.1 min (96.7%).
4-(N-Benzyl-2-((N-cyclopropyl)-2,3,4,5,6-pentafluorophenyl)sulfonamido)acetamido)benzoic acid (PSL-3-115)
1H NMR (400 MHz, CDCl3) δ 0.77–0.80 (m, 2H), 0.86–0.85 (m, 2H), 2.91–2.92 (m, 1H), 4.14 (s, 2H), 4.92 (s, 2H), 7.19 (d, J = 6.6 Hz, 2H), 7.33–7.26 (m, 5H), 8.06 (d, J = 8.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 8.1, 29.7, 30.0, 52.5, 53.3, 115.8 (m), 128.0, 128.3, 128.4, 128.5, 128.5, 128.5, 132.0, 135.9, 137.8 (dm, JCF = 282 Hz), 144.5 (dm, JCF = 132 Hz), 145.4 (dm, JCF = 155 Hz), 166.5, 170.0; HRMS (ESI+) m/z [M+H]+ Calcd for C25H20F5N2O5S: 554.09, found: 555.10; HPLC rt (II) = 33.2 min (98.4%).
4-(2-((N-Cyclopropyl)-2,3,4,5,6-pentafluorophenyl)sulfonamido)-N-(4-fluorobenzyl)acetamido)benzoic acid (PSL-3-122)
1H NMR (400 MHz, CDCl3) δ 0.83 (m, 4H), 2.88 – 2.92 (m, 1H), 3.97 (s, 2H), 4.78 (s, 2H), 6.96 (t, J = 8.3 Hz, 2H), 7.04 – 7.07 (m, 2H), 7.12 (d, J = 8.1 Hz, 2H), 8.13 (d, J = 8.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 8.1, 29.7, 30.0, 52.5, 115.5, 115.8, 128.5, 130.4, 130.5, 130.5, 130.7 (dm, JCF = 260.2 Hz), 131.7, 132.2, 137.6 (dm, JCF = 260.2 Hz), 143.9 (dm, JCF = 263.4 Hz), 145.0 (dm, JCF = 259.7 Hz), 161.5, 163.4, 166.6, 170.1; 19F NMR (376 MHz, CD3OD) δ −163.02 (t, J = 19.6 Hz, 2F), −150.07 (t, J = 21.9 Hz, 1F), −136.70 (d, J = 21.9 Hz, 2F), −116.66 (s, 1F). HRMS (ESI+) m/z [M+H]+ Calcd for C25H19F6N2O5S: 572.08, found: 573.09; HPLC rt (II) = 34.1 min (95.4%).
4-(2-((N-Cyclopropyl)-2,3,4,5,6-pentafluorophenyl)sulfonamido)-N-(4-cyclopropylbenzyl)acetamido)benzoic acid (PSL-3-132)
1H NMR (400 MHz, CDCl3) 0.65 – 0.68 (m, 2H), 0.82 – 0.83 (m, 4H), 0.93 – 0.98 (m, 2H), 1.89 – 1.92 (m, 1H), 2.89 – 2.95 (m, 1H), 3.97 (s, 2H), 4.75 (s, 2H), 6.96 (s, 4H), 7.12 (d, J = 7.7 Hz, 2H), 8.11 (d, J = 7.7 Hz, 2H); 13C NMR (126 MHz, CDCl3) δ 8.2, 9.6, 15.3, 29.9, 30.2, 52.7, 53.2, 116.0, 126.0, 128.6, 128.8, 129.6, 132.2, 132.9, 137.9 (dm, 1JCF = 254.0 Hz), 143.9 (dm, 1JCF = 268.0 Hz), 144.1, 145.2 (dm, 1JCF = 269.0 Hz), 145.3, 166.6, 170.4; HRMS (ESI+) m/z [M+H]+ Calcd for C28H25F5N2O5S: 595.1321, found: 595.1326; HPLC rt (II) = 36.0 (96.7%).
4-(N-(4-Cyclohexylbenzyl)-2-((N-cyclopropyl)-2,3,4,5,6-pentafluorophenyl)sulfonamido)acetamido)benzoic acid (DR-3-036)
1H NMR (700 MHz, CD3CN) δ 0.74 – 0.76 (m, 4H), 1.26 – 1.28 (m, 1H), 1.39 – 1.42 (m, 4H), 1.71 – 1.74 (m, 1H), 1.79 – 1.82 (m, 4H), 2.48 – 2.51 (m, 1H), 2.85 (p, J = 5.6 Hz, 1H), 4.00 (bs, 2H), 4.84 (s, 2H), 7.08 (d, J = 7.5 Hz, 2H), 7.15 (d, J = 7.5 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 8.00 (d, J = 8.2 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 8.1, 26.1, 26.8, 30.3, 34.4, 44.2, 52.6, 53.1, 113.9, 115.8 (m), 127.1, 128.4, 128.5, 129.4, 132.0, 133.1, 137.7 (dm, 1JCF = 255.1 Hz), 143.8 (dm, 1JCF = 262.1 Hz), 145.0 (dm, 1JCF = 257.5 Hz), 145.2, 147.9, 166.4, 170.0; 19F NMR (658 MHz, CD3CN) δ −162.0 (m, 2F), −149.4 (m, 1F), −136.7 (m, 2F); HRMS (ESI−) m/z [M-H]− Calcd for C31H28F5N2O5S: 635.1645, found: 635.1648; HPLC rt (I) = 27.5 min (95.8%); HPLC rt (II) = 45.9 min (97.1%).
4-(N-(4-Cyclopentylbenzyl)-2-((N-cyclopropyl-2,3,4,5,6-pentafluorophenyl)sulfonamido)acetamido)benzoic acid (DR-3-093)
1H NMR (700 MHz, CD3CN) δ 0.74 – 0.76 (m, 4H), 1.51 – 1.55 (m, 2H), 1.66 – 1.69 (m, 2H), 1.78 – 1.80 (m, 2H), 2.00 – 2.04 (m, 2H), 2.83 – 2.86 (m, 1H), δ 2.97 (td, J = 8.8, 7.6, 2.4 Hz, 1H), 4.01 (bs, 2H), 4.84 (s, 2H), 7.08 (d, J = 7.4 Hz, 2H), 7.18 (d, J = 7.4 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 8.00 (d, J = 8.2 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 8.1, 25.5, 30.0, 34.5, 45.6, 52.5, 53.1, 115.8 (m), 127.4, 128.5, 129.5, 132.0, 133.0, 137.7 (dm, 1JCF = 255.2 Hz), 143.8 (dm, 1JCF = 260.0 Hz), 145.0 (dm, 1JCF = 257.4 Hz), 145.3, 146.4, 166.4, 170.4; 19F NMR (658 MHz, CD3CN) δ −162.0 (m, 2F), −149.4 (m, 1F), −136.6 (m, 2F); HRMS (ESI−) m/z [M-H]− Calcd for C30H26F5N2O5S: 621.1488, found: 621.1490; HPLC rt (I) = 24.2 min (98.4%); HPLC rt (II) = 40.1 min (98.2%).
4-(N-(4-Cyclopentylbenzyl)-2-((perfluorophenyl) sulfonamido)acetamido)benzoic acid (DR-3-111)
1H NMR (700 MHz, CD3CN) δ 1.51 – 1.55 (m, 2H), 1.66 – 1.69 (m, 2H), 1.77 – 1.80 (m, 2H), 1.98 – 2.05 (m, 2H), 2.97 (tt, J = 10.0 Hz, 7.5 Hz, 1H), 3.73 (bs, 2H), 4.79 (s, 2H), 6.69 (bs, 1H), 7.08 (d, J = 7.4 Hz, 2H), 7.18 (d, J = 7.4 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 8.00 (d, J = 8.2 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 25.5, 34.5, 45.1, 45.6, 53.5, 116.4 (m), 127.4, 128.4, 128.7, 130.0, 132.2, 132.6, 137.7 (dm, 1JCF = 256.3 Hz), 143.8 (dm, 1JCF = 260.3 Hz), 144.2, 144.5 (dm, 1JCF = 256.0 Hz), 146.7, 166.4, 170.0; 19F NMR (658 MHz, CD3CN) δ −162.0 (m, 2F), −149.5 (m, 1F), −138.5 (m, 2F); HRMS (ESI−) m/z [M-H]− Calcd for C27H22F5N2O5S: 581.1173, found: 581.1169; HPLC rt (I) = 23.8 min (97.4%); HPLC rt (II) = 39.4 min (97.1%).
4-(2-((N-Cyclopropyl-2,3,4,5,6-pentafluorophenyl)sulfonamido)-N-(4-(4,4-dimethylcyclohexyl)benzyl)acetamido)benzoic acid (DR-3-119)
1H NMR (700 MHz, CD3CN) δ 0.74 – 0.76 (m, 4H), 0.95 (s, 3H), 0.98 (s, 3H), 1.33 – 1.37 (m, 2H), 1.47 – 1.49 (m, 2H), 1.59 – 1.62 (m, 4H), 2.41 (ddt, J = 15.9, 10.8, 5.5 Hz, 1H), 2.85 (p, J = 6.5 Hz, 1H), 4.00 (bs, 2H), 4.82 (s, 2H), 7.07 (d, J = 7.8 Hz, 2H), 7.17 (d, J = 7.8 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H), 8.00 (d, J = 8.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 8.1, 24.2, 29.7, 30.1, 33.1, 39.6, 44.1, 52.5, 53.1, 115.8, 127.1, 128.5, 128.6, 129.5, 132.0, 133.2, 137.7 (dm, 1JCF = 256.0 Hz), 143.8 (dm, 1JCF = 260.0 Hz), 145.1 (dm, 1JCF = 253.3 Hz), 145.3, 147.6, 166.4, 170.3; 19F NMR (658 MHz, CD3CN) δ −161.7 (m, 2F), −149.0 (m, 1F), −136.3 (m, 2F); HRMS (ESI−) m/z [M-H]− Calcd for C33H32F5N2O5S: 663.1955, found: 663.1947; HPLC rt (I) = 29.1 min (95.8%); HPLC rt (II) = 48.7 min (97.2%).
4-(N-(4-(tert-butyl)benzyl)-2-((N-cyclopropyl-2,3,4,5,6-pentafluorophenyl)sulfonamido)acetamido)benzoic acid (DR-3-162)
1H NMR (700 MHz, CD3CN) δ δ 0.75 – 0.77 (m, 4H), 1.29 (s, 9H), 2.85 (p, J = 5.3 Hz, 1H), 4.02 (bs, 2H), 4.85 (s, 2H), 7.07 (d, J = 8.2 Hz, 2H), 7.17 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 8.2 Hz, 2H), 8.01 (d, J = 8.2 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 8.1, 30.0, 31.3, 34.5, 52.6, 53.1, 115.8 (m), 125.5, 128.2, 128.4, 129.6, 132.0, 132.8, 137.7 (dm, 1JCF = 253.3 Hz), 143.8 (dm, 1JCF = 256.5 Hz), 145.0 (dm, 1JCF = 253.5 Hz), 145.3, 151.0, 166.4, 170.4; 19F NMR (658 MHz, CD3CN) δ −162.0 (m, 2F), −149.4 (m, 1F), −136.7 (m, 2F); HRMS (ESI−) m/z [M-H]− Calcd for C29H26F5N2O5S: 609.1488, found: 609.1489; HPLC rt (I) = 25.5 min (98.3%); HPLC rt (II) = 42.3 min (98.3%).
4-(N-(4-Cycloheptylbenzyl)-2-((N-cyclopropyl-2,3,4,5,6-pentafluorophenyl)sulfonamido)acetamido)benzoic acid (DR-3-165)
1H NMR (700 MHz, CD3CN) δ 0.72 – 0.79 (m, 4H), 1.52 – 1.63 (m, 6H), 1.67 – 1.71 (m, 2H), 1.76 – 1.80 (m, 2H), 1.81 – 1.84 (m, 2H), 2.66 (tt, J = 10.6, 3.4 Hz, 1H), 2.85 (p, J = 6.4 Hz, 1H), 3.99 (bs, 2H), 4.82 (s, 2H), 7.07 (d, J = 7.8 Hz, 2H), 7.17 (d, J = 7.8 Hz, 2H), 7.23 (d, J = 7.8 Hz, 2H), 8.00 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 8.1, 27.2, 27.9, 30.0, 36.7, 46.7, 52.6, 53.1, 115.8 (m), 126.9, 128.4, 128.6, 129.5, 132.0, 132.8, 137.7 (dm, 1JCF = 255.0 Hz), 143.8 (dm, 1JCF = 254.4 Hz), 145.0 (dm, 1JCF = 259.2 Hz), 145.2, 149.9, 166.4, 170.4; 19F NMR (658 MHz, CD3CN) δ −161.7 (m, 2F), −149.0 (tt, J = 27.9, 5.7 Hz, 1F), −136.3 (m, 2F); HRMS (ESI−) m/z [M-H]− Calcd for C32H30F5N2O5S: 649.1799, found: 649.1790; HPLC rt (I) = 28.5 min (97.3%); HPLC rt (II) = 47.5 min (98.2%).
4-(2-(N-cyclopropyl-2,3,4,5,6-pentafluorophenylsulfonamido)-N-(4-(trifluoromethyl)benzyl)acetamido)benzoic acid (DR-3-186)
1H NMR (700 MHz, CDCl3) δ 0.82 – 0.83 (m, 4H), 2.90 – 3.05 (m, 1H), 4.02 (s, 2H), 4.88 (s, 2H), 7.18 (d, J = 8.1 Hz, 2H), 7.24 (d, J = 7.9 Hz, 2H), 7.56 (d, J = 7.9 Hz, 2H), 8.15 (d, J = 8.2 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 8.30, 30.2, 52.7, 53.1, 116.0, 124.1 (q, 1JCF3 = 272.2 Hz), 125.8 (q, 3JCF3 = 3.7 Hz), 128.4, 129.0, 130.0, 130.5 (q, 2JCF3 = 32.6 Hz), 132.5, 137.9 (dm, 1JCF = 256.1 Hz), 140.1, 144.0 (dm, 1JCF = 258.7 Hz), 145.1, 145.2 (dm, 1JCF = 257.8 Hz), 167.1, 170.21. 19F NMR (658 MHz, CDCl3) δ −159.72 (m, 2F), −146.19 (t, 1F), −134.19 (m, 2F), −62.67 (s, 3F); HRMS (ESI−) m/z [M-H]− Calcd for C26H17F8N2O5S: 621.0735, found: 621.0734; HPLC rt (I) = 23.5 min (98.3%); HPLC rt (II) = 39.2 min (98.9%).
4-(N-(4-Chlorobenzyl)-2-((N-cyclopropyl-2,3,4,5,6-pentafluorophenyl)sulfonamido)acetamido)benzoic acid (DR-4-003)
1H NMR (400 MHz, CDCl3) δ 0.82 – 0.83 (m, 4H), 2.89 – 2.91 (m, 1H), 3.98 (bs, 2H), 4.78 (s, 2H), 7.03 (d, J = 8.0 Hz, 2H), 7.14 (d, J = 8.0 Hz, 2H), 7.22 – 7.29 (m, 2H), 8.13 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 8.0, 29.9, 52.4, 52.5, 115.7, 128.2, 128.7, 129.9, 133.8, 134.3, 136.3, 137.6 (dm, 1JCF = 251.3 Hz), 144.8 (dm, 1JCF = 254.4 Hz), 144.8, 144.9 (dm, 1JCF = 274.0 Hz), 166.6, 169.6; 19F NMR (658 MHz, CDCl3) δ −159.7 (m, 2F), −146.2 (m, 1F), −134.1 (m, 2F); HRMS (ESI−) m/z [M-H]− Calcd for C25H17ClF5N2O5S: 587.047, found 587.047; HPLC rt (I) = 23.8 min (95.4%); HPLC rt (II) = 38.8 min (95.5%).
4-(N-(4-Cyclobutylbenzyl)-2-((N-cyclopropyl-2,3,4,5,6-pentafluorophenyl)sulfonamido)acetamido)benzoic acid (DR-4-020)
1H NMR (700 MHz, CD3CN) δ 0.72 – 0.79 (m, 4H), 1.79 – 1.87 (m, 1H), 1.96 – 2.14 (m, 3H), 2.26 – 2.34 (m, 2H), 2.85 (p, J = 5.3 Hz, 1H), 3.52 (p, J = 8.6 Hz, 1H), 4.00 (bs, 2H), 4.84 (s, 2H), 7.07 (d, J = 7.6 Hz, 2H), 7.16 (d, J = 7.6 Hz, 2H), 7.25 (d, J = 8.3 Hz, 2H), 8.00 (d, J = 8.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 8.1, 18.2, 29.7, 30.0, 40.0, 52.5, 53.1, 115.8 (m), 126.6, 128.5, 129.5, 132.0, 133.1, 137.7 (dm, 1JCF = 259.2 Hz), 143.8 (dm, 1JCF = 256.6 Hz), 145.1 (dm, 1JCF = 254.3 Hz), 145.2, 146.1, 166.4, 170.4; 19F NMR (658 MHz, CD3CN) δ −162.0 (m, 2F), −149.4 (tt, J = 20.4, 6.4 Hz, 1F), −136.7 (m, 2F); HRMS (ESI−) m/z [M-H]− Calcd for C29H24F5N2O5S: 607.1332, found: 607.1332. HPLC rt (I) = 25.6 min (97.6%); HPLC rt (II) = 42.4 min (98.2%).
4-(2-((N-Cyclopropyl)-2,3,4,5,6-pentafluorophenyl)sulfonamido)-N-(4-(tetrahydro-2H-pyran-4-yl)benzyl)acetamido)benzoic acid (DR-4-042)
1H NMR (700 MHz, CD3CN) δ 0.73 – 0.78 (m, 4H), 1.67 – 1.72 (m, 4H), 2.76 (tt, J = 9.1, 6.0 Hz, 1H), 2.84 (td, J = 6.4, 3.1 Hz, 1H), 3.42 – 3.57 (m, 2H), 3.97 (dt, J = 11.1, 3.1 Hz, 4H), 4.01 (bs, 2H), 4.85 (s, 2H), 7.11 (d, J = 7.5 Hz, 2H), 7.19 (d, J = 7.5 Hz, 2H), 7.26 (d, J = 8.3 Hz, 2H), 8.00 (d, J = 8.3 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 8.1, 30.0, 33.8, 41.1, 52.5, 53.1, 68.3, 115.8, 127.0, 128.4, 128.8, 129.7, 132.0, 133.8, 137.7 (dm, 1JCF = 271.8 Hz), 143.8 (dm, 1JCF = 259.0 Hz), 145.0 (dm, 1JCF = 270.0 Hz), 145.1, 145.5, 166.5, 169.9; 19F NMR (658 MHz, CD3CN) δ −162.0 (m, 2F), −149.4 (tt, J = 20.3, 6.3 Hz, 1F), −136.7 (m, 2F); HRMS (ESI−) m/z [M-H]− Calcd for C30H26F5N2O6S: 637.1435, found: 637.1431; HPLC rt (I) = 21.9 min (96.4%); HPLC rt (II) = 36.3 min (97.7%).
4-(N-(4-(tert-butyl)benzyl)-2-((N-cyclopropyl-4-fluorophenyl)sulfonamido)acetamido)benzoic acid (AES-1-094A)
White solid (12.9 mg, 16%); 1H NMR (400 MHz, CD3CN) δ 0.64 – 0.67 (m, 4H), 1.28 (s, 9H), 2.49 – 2.53 (m, 1H), 3.88 (bs, 2H), 4.84 (s, 2H), 7.11 (d, J = 7.9 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 7.9 Hz, 2H), 7.83 (dd, J = 7.6, 5.3 Hz, 2H), 7.99 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CD3CN) δ 9.6, 33.0, 36.5, 54.2, 54.9, 118.1, 118.4, 127.8, 127.9, 130.2, 130.7, 133.0, 133.1, 133.4, 133.48, 133.54, 136.6, 138.2, 166.0, 170.1; 19F NMR (54 MHz, CD3CN) δ −109.0; HRMS (ESI–) m/z [M-H]− Calcd for C29H30FN2O5S: 537.1869 found: 537.1865; HPLC rt (II) = 38.4 min (98.3%)
Supplementary Material
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Prof. Michael W. Deininger, Email: michael.deininger@hci.utah.edu.
Prof. Patrick T. Gunning, Email: patrick.gunning@utoronto.ca.
References
- 1.Goldman JM, Melo JV. New England Journal of Medicine. 2001;344:1084–1086. doi: 10.1056/NEJM200104053441409. [DOI] [PubMed] [Google Scholar]
- 2.de Lavallade H, Apperley JF, Khorashad JS, Milojkovic D, Reid AG, Bua M, Szydlo R, Olavarria E, Kaeda J, Goldman JM, Marin D. Journal of Clinical Oncology. 2008;26:3358–3363. doi: 10.1200/JCO.2007.15.8154. [DOI] [PubMed] [Google Scholar]
- 3.a Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]; b Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 4.Haftchenary S, Luchman HA, Jouk AO, Veloso AJ, Page BDG, Cheng XR, Dawson SS, Grinshtein N, Shahani VM, Kerman K, Kaplan DR, Griffin C, Aman AM, Al-awar R, Weiss S, Gunning PT. ACS Medicinal Chemistry Letters. 2013;4:1102–1107. doi: 10.1021/ml4003138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Bewry NN, Nair RR, Emmons MF, Boulware D, Pinilla-Ibarz J, Hazlehurst LA. Molecular Cancer Therapeutics. 2008;7:3169–3175. doi: 10.1158/1535-7163.MCT-08-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Traer E, MacKenzie R, Snead J, Agarwal A, Eiring AM, O’Hare T, Druker BJ, Deininger MW. Leukemia. 2012;26:1140–1143. doi: 10.1038/leu.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eiring AM, Page BDG, Kraft IL, Mason CC, Vellore NA, Resetca D, Zabriskie MS, Zhang TY, Khorashad JS, Engar AJ, Reynolds KR, Anderson DJ, Senina A, Pomicter AD, Arpin CC, Ahmad S, Heaton WL, Tantravahi SK, Todic A, Colaguori R, Moriggl R, Wilson DJ, Baron R, O’Hare T, Gunning PT, Deininger MW. Leukemia. 2014 doi: 10.1038/leu.2017.14. [DOI] [PubMed] [Google Scholar]; b Lai P-S, Rosa DA, Magdy Ali A, Gómez-Biagi RF, Ball DP, Shouksmith AE, Gunning PT. Expert Opinion on Therapeutic Patents. 2015;25:1397–1421. doi: 10.1517/13543776.2015.1086749. [DOI] [PubMed] [Google Scholar]
- 7.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 8.a Couronné L, Scourzic L, Pilati C, Valle VD, Duffourd Y, Solary E, Vainchenker W, Merlio J-P, Beylot-Barry M, Damm F, Stern M-H, Gaulard P, Lamant L, Delabesse E, Merle-Beral H, Nguyen-Khac F, Fontenay M, Tilly H, Bastard C, Zucman-Rossi J, Bernard OA, Mercher T. STAT3 mutations identified in human hematologic neoplasms induce myeloid malignancies in a mouse bone marrow transplantation model. 2013;98 doi: 10.3324/haematol.2013.085068. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Koskela HLM, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, Lagström S, Clemente MJ, Olson T, Jalkanen SE, Majumder MM, Almusa H, Edgren H, Lepistö M, Mattila P, Guinta K, Koistinen P, Kuittinen T, Penttinen K, Parsons A, Knowles J, Saarela J, Wennerberg K, Kallioniemi O, Porkka K, Loughran TP, Heckman CA, Maciejewski JP, Mustjoki S. New England Journal of Medicine. 2012;366:1905–1913. doi: 10.1056/NEJMoa1114885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a Nallar SC, Kalakonda S, Lindner DJ, Lorenz RR, Lamarre E, Weihua X, Kalvakolanu DV. Journal of Biological Chemistry. 2013;288:7930–7941. doi: 10.1074/jbc.M112.440610. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rossa C, Jr, Sommer G, Spolidorio LC, Rosenzweig SA, Watson DK, Kirkwood KL. PLoS ONE. 2012;7:e45197. doi: 10.1371/journal.pone.0045197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartman ZC, Yang XY, Glass O, Lei G, Osada T, Dave SS, Morse MA, Clay TM, Lyerly HK. Cancer Research. 2011;71:4380–4391. doi: 10.1158/0008-5472.CAN-11-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao X, Tay A, Guy GR, Tan YH. Molecular and Cellular Biology. 1996;16:1595–1603. doi: 10.1128/mcb.16.4.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, Talpaz M. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. 2003;101 doi: 10.1182/blood.V101.2.690. [DOI] [PubMed] [Google Scholar]; b Wu J, Meng F, Kong L-Y, Peng Z, Ying Y, Bornmann WG, Darnay BG, Lamothe B, Sun H, Talpaz M, Donato NJ. Journal of the National Cancer Institute. 2008;100:926–939. doi: 10.1093/jnci/djn188. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pene-Dumitrescu T, Smithgall TE. The Journal of Biological Chemistry. 2010;285:21446–21457. doi: 10.1074/jbc.M109.090043. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Proceedings of the National Academy of Sciences. 2006;103:16870–16875. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Samanta AK, Chakraborty SN, Wang Y, Kantarjian H, Sun X, Hood J, Perrotti D, Arlinghaus RB. Oncogene. 2009;28:1669–1681. doi: 10.1038/onc.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a Ren Z, Cabell L, Schaefera T, McMurry J. Bioorg Med Chem Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]; b Turkson J, Gunning P. University of Central Florida Research Foundation, University of Toronto Mississauga; 2011. [Google Scholar]; c Mandal PK, Liao WSL, McMurray JS. Organic Letters. 2009;11:3394–3397. doi: 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mandal PK, Limbrick D, Coleman DR, Dyer GA, Ren Z, Birtwistle JS, Xiong C, Chen X, Briggs JM, McMurray JS. Journal of Medicinal Chemistry. 2009;52:2429–2442. doi: 10.1021/jm801491w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sebti SM, Lawrence NJ, Turkson J. I. H. Lee Moffitt Cancer Center & Research Institute, I. University Of Central Florida Research Foundation, editor. 2013;WO2013063504 A1 [Google Scholar]
- 15.Grandis JR, Johnson DE, Leong P. U. O. P.-O. T. C. S. O. H. Education, editor. 2006;US8722640 B2 [Google Scholar]
- 16.a Fletcher S, Singh J, Zhang X, Yue P, Page BDG, Sharmeen S, Shahani VM, Zhao W, Schimmer AD, Turkson J, Gunning PT. ChemBioChem. 2009;10:1959–1964. doi: 10.1002/cbic.200900172. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Song H, Wang R, Wang S, Lin J. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Farjo R. 20090082470 A1. US. 2009; d Suganami E, Takagi H, Ohashi H, Suzuma K, Suzuma I, Oh H, Watanabe D, Ojima T, Suganami T, Fujio Y, Nakao K, Ogawa Y, Yoshimura N. Diabetes. 2004;53:2443–2448. doi: 10.2337/diabetes.53.9.2443. [DOI] [PubMed] [Google Scholar]; e Gunning P, Turkson J. I. University Of Central Florida Research Foundation, U. O. T. Mississauga, editor. 2012;WO2012018868 A1 [Google Scholar]; f Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip MLR, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Proceedings of the National Academy of Sciences. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Asai A, Matsuno K, Ogo N, Takahashi O, Masuda Y. G. I. A. P. V. P. S. Organization, I. Pharma Design, K. K. Y. Honsha, S. P. Kumamoto Health Science University, editor. 2011;US20120302524 A1 [Google Scholar]
- 17.Earnshaw C. Chemistry World. 2010;7:55–550. [Google Scholar]
- 18.Dorwald FZ. Lead Optimizations for Medicinal Chemists: Pharmacokinetic Properties of Functional Groups and Organic Compounds. Wiley; Germany: 2012. [Google Scholar]
- 19.Booth J, Boyland E, Sims P. Biochemical Journal. 1961;79:516–524. doi: 10.1042/bj0790516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a Fernández C, Jahnke W. Drug Discovery Today: Technologies. 2004;1:277–283. doi: 10.1016/j.ddtec.2004.10.003. [DOI] [PubMed] [Google Scholar]; b Dalvit C, Fagerness PE, Hadden DTA, Sarver RW, Stockman BJ. Journal of the American Chemical Society. 2003;125:7696–7703. doi: 10.1021/ja034646d. [DOI] [PubMed] [Google Scholar]
- 21.Schust J, Berg T. Analytical Biochemistry. 2004;330:114–118. doi: 10.1016/j.ab.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 22.a Yue P, Lopez-Tapia F, Paladino D, Li Y, Chen C-H, Hilliard T, Chen Y, Tius MA, Turkson J. Cancer Research. 2015 doi: 10.1158/0008-5472.CAN-14-3558. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wu P, Brasseur M, Schindler U. Analytical Biochemistry. 1997;249:29–36. doi: 10.1006/abio.1997.2158. [DOI] [PubMed] [Google Scholar]
- 23.Darnell JE, Jr, Kerr IM, Stark GR. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.