

Abstract
The epithelial sodium channel (ENaC) mediates passive sodium transport across the apical membranes of sodium absorbing epithelia, like the distal nephron, the intestine, and the lung airways. Additionally, the channel has been involved in the transduction of mechanical stimuli, such as hydrostatic pressure, membrane stretch, and shear stress from fluid flow. Thus, in vascular endothelium, it participates in the control of the vascular tone via its activity both as a sodium channel and as a shear stress transducer. Rather recently, ENaC has been shown to participate in the processes of wound healing, a role that may also involve its activities as sodium transporter and as mechanotransducer. Its presence as the sole channel mediating sodium transport in many tissues and the diversity of its functions probably underlie the complexity of its regulation. This brief review describes some aspects of ENaC regulation, comments on evidence about ENaC participation in wound healing, and suggests possible regulatory mechanisms involved in this participation.
1. Introduction
The capacity to respond to different types of injuries by healing processes is a major achievement of biological evolution and represents a crucial property for the homeostasis of an organism. Tissue restitution is a complex physiological response triggered by an injury. In the whole organism it is accomplished by the coordinated action of many cell types and involves distinctive sequential phases, that is, hemostasis, inflammation, proliferation, and remodeling [1, 2]. However, tissue culture studies have shown that isolated tissues already have the ability to restitute their integrity and have thus been proven to be a valuable tool in the study of wound healing. The healing responses are also activated during diverse pathological situations in the absence of an external injuring aggression, such as inflammatory bowel disease [3], gastric ulcers [4], asthma [5], tissue fibrosis [6, 7], and cancer [7, 8].
The epithelial sodium channel (ENaC) has been primarily considered to be a major participant in sodium and water homeostasis [9, 10]. This view was supported by its predominant presence in the distal nephron and other salt transporting epithelia. However, the channel can be found in a wide variety of cellular types and tissues [11] and has been suggested to participate in diverse other physiological processes, such as chemo- and mechanosensation [12], control of vascular tone [13–17], and sodium sensing and membrane potential regulation in the nervous system [18]. It has also been found that ENaC has a role in the processes of wound healing. The main objective of this work is to review evidence about the participation of ENaC in these processes. Prior to this, we briefly describe the general functional, structural, and regulatory characteristics of this channel with emphasis on aspects of interest for this review.
2. The Epithelial Sodium Channel
2.1. Physiological Roles
ENaC is a constitutively active, non-voltage-gated sodium channel typically located in the apical membranes of epithelial cells and is highly sensitive to inhibition by amiloride [11, 19, 20]. It is predominantly selective for sodium ions and exhibits a low single-channel conductance [21]. In salt-absorbing epithelia, ENaC mediates sodium entry in favor of its electrochemical gradient. In this type of cells, the sodium pump is fundamentally localized at the basolateral membrane, where it performs active extrusion of sodium. The sequential localization of the two transport systems along the apical to basolateral axis of the cell determines transepithelial flow of sodium ions, which is coupled to an equivalent flow of an accompanying monovalent anion, most commonly chloride [22]. The overall process is therefore pumped by the sodium-potassium ATPase. This basic scheme accounts for sodium absorption in many epithelia, especially the distal nephron, the colon, and the lungs [19], where the channel participates in fundamental physiological functions. In this way, aldosterone-regulated sodium reabsorption in the kidney plays a major role in the control of extracellular fluid volume, blood pressure, and ionic renal secretion [23, 24]. Several inherited diseases involving alterations in sodium homeostasis, such as Liddle's syndrome and pseudohypoaldosteronism type I, are a consequence of ENaC mutations [11, 25]. The above-described scheme also accounts for sodium absorption in the colon [22]. In the lungs, the absorption of sodium contributes to the maintenance of the normal composition of the surface liquid in the airways [26, 27]. ENaC has been identified in other epithelial tissues, such as vascular endothelia, where it contributes to the maintenance of the vascular tone [13–16]. It is present in corneal endothelium [28], where it plays a fundamental role in salt and fluid transport necessary for the maintenance of the organ transparency [29]. Besides its role in sodium metabolism, ENaC has also been proposed to participate in the transduction of varied mechanical stimuli, such as shear stress, hydrostatic pressure, and membrane stretch [30]. It is interesting to note that ENaC seems to be the only channel mediating apical sodium transport in some tissues, like colon [31], cortical collecting duct of the kidney [24], and airway epithelia [32]. Together with its physiological importance, this characteristic may explain the very complex regulatory mechanisms controlling the channel activity, as commented in the following sections.
2.2. Basic Structural Aspects and Interactions
ENaC is a member of the ENaC/degenerin superfamily, which also includes the acid-sensing ion channels (ASICs), the pickpocket proteins found in Drosophila and other Diptera, and the nematode degenerins. It is an heterotrimer composed of three homologous subunits, α (or δ), β, and γ. For details concerning distribution, gene coding, and protein structure of ENaC, the reader can consult several recent comprehensive reviews [11, 25, 33]. Of interest to this work is the fact that each ENaC subunit possesses a very large extracellular domain, which is the site of many regulatory effects (see below). The intracellular domains correspond to the carboxyl and amino terminals of each subunit. At these levels, it has been suggested that ENaC interacts with a multiprotein regulatory complex that includes, among others, the ubiquitin protein ligase Nedd4 (neuronal precursor cell-expressed developmentally downregulated 4) and the serum and glucocorticoid-regulated kinase 1 (SGK1) [34]. Besides these regulatory sites, at the intracellular level, ENaC establishes diverse interactions with the cytoskeleton. Its localization at the apical membrane is maintained by its direct binding to spectrin [35] and ankyrin [36]. ENaC also binds to the actin cytoskeleton, either directly [37] or through intermediary proteins [38–40]. There is evidence that these bindings of ENaC to the actin cytoskeleton modify the channel conductance [38, 40–42]. Moreover, the interactions of ENaC with the cytoskeleton mediate some of its mechanosensitive responses [43].
2.3. ENaC Regulation
ENaC is subject to a complex regulation that involves a myriad of mechanisms, summarized in Figure 1. The reader is referred to more detailed reviews on this topic [30, 34, 44–46]. The regulation of ENaC activity can be schematically considered to occur at three levels: (i) by modulation of the kinetic properties of the channel (i.e., modifying the open probability or the single-channel conductance), (ii) by regulation of its amount at the plasma membrane, and (iii) by the effects of first and second messengers on the signaling networks that modulate the channel's activity. The latter two levels include regulatory effects at its synthesis, storage, trafficking to the plasma membrane domains, and membrane insertion and retrieval. In essence, the complexity of ENaC regulation emerges from the fact that the three levels are strongly interrelated (Figure 1). As an example, it has been proposed that SGK1 increases ENaC activity by direct phosphorylation [47], by phosphorylation of Nedd4-2 [48–50], and by augmenting the transcription of the α subunit [51]. In turn, SGK1 is at the crossroad of different signaling pathways, like those triggered by aldosterone, insulin, IGF-1, and mTOR [46, 52].
Figure 1.

Scheme of the basic mechanisms of ENaC regulation. The scheme indicates the effects of several modulators on the channel activity and/or expression. The three levels of regulation of ENaC are integrated in the scheme: (i) modulation of the kinetic properties of the channel in situ at the plasma membrane or at its trafficking, (ii) regulation of its synthesis and retrieval from the plasma membrane, and (iii) the effects of first and second messengers on the signaling networks that modulate the channel's activity, with emphasis on aldosterone. MT: methyltransferases. See the main text for a detailed description of the regulatory effects.
The channel can be directly inhibited at the extracellular domains by sodium [53–56] and chloride [57, 58] and activated by acidic pH [59]. At this level, several proteases affect the open probability of the channel [30, 32, 44]. On the intracellular side, the channel is inhibited by calcium [60] and acidic pH [60–62] and activated by phosphatidylinositol(3,4,5)-trisphosphate (PI(3,4,5)P3) and phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) [63–67]. H2O2 determines a rise in PI(3,4,5)P3 and thus produces ENaC stimulation [68]. Intracellular sodium determines inhibition of epithelial ENaC, an effect known as feedback inhibition [69, 70] which is mediated by sodium interference with ENaC proteases [71, 72].
At the plasma membrane, the channel molecules can be both in mature (active) or immature (inactive) forms, depending on their posttranslational processing [73]. ENaC is synthesized in inactive form and is activated by limited proteolysis of the extracellular domains [74]. ENaC can be activated during its trafficking to the membrane by the trans-Golgi serine protease furin and inserted already in active form [73, 75].
The retrieval of ENaC involves, among others, the ubiquitin ligase Nedd4 [34, 76]. Phosphorylation of Nedd4 by SGK1 inhibits ENaC ubiquitination and thus determines an increase in the channel activity [45]. CK2 (casein kinase 2) and GRK2 (G protein-coupled receptor kinase 2) stimulate ENaC activity by direct phosphorylation at sites that interfere with Nedd4, while other kinases (e.g., the extracellular signal-regulated kinases 1 and 2, ERK1/2) perform phosphorylation of channel sites that favor Nedd4 interaction (ibid). In addition, intracellular sodium stimulates Nedd4 [77]. After internalization, the ENaC molecules can either be recycled or be degraded. Besides aldosterone, the main regulator of ENaC synthesis (see below), other effectors promote or inhibit the channel transcription. For instance, PKA (protein kinase A) has been suggested to activate the transcription of α-ENaC, while PKC (protein kinase C) and ERK1/2 seem to inhibit the transcription of the three subunits and of the α subunit, respectively [45].
In the whole organism, ENaC activity is subject to the control of several hormones and growth factors [25], most significantly aldosterone, vasopressin, and insulin. For the regulation of ENaC activity, aldosterone behaves both as a transcriptional and as a nontranscriptional effector (for detailed reviews, see [24, 25, 78]). Briefly, aldosterone binds to the cytoplasmic mineralocorticoid receptor (MCR) and translocates to the nucleus, where, among other target genes, it promotes transcription of ENaC subunits in a tissue-specific manner [24, 79]. It also stimulates transcription of SGK1 [45, 46]. It is to be noted that binding to the MCR also stimulates the epidermal growth factor receptor that, in turn, prevents excessive activation of ENaC via ERK signaling [80, 81]. Aldosterone possesses several nongenomic effects affecting ENaC activity, such as activation of ERK1/2 and p38, activation of members of the PKC family, and stimulation of cAMP and cytosolic calcium increases [82]. Other nongenomic effects include methylation of β ENaC [83–85] and vascular endothelial cell swelling [86]. From the above, aldosterone can be considered as a crucial element in the crosstalk between regulatory pathways of ENaC activity.
The majority of the regulatory properties of ENaC summarized in Figure 1 were described for the distal nephron and other salt-absorbing epithelia. There are significant regulatory differences, however, with other noncanonical localizations, such as vascular endothelium, where the transtissular transport of sodium does not represent the main physiological role of the channel [16].
2.4. ENaC Participation in Mechanotransduction
The above-summarized regulatory mechanisms were mostly concerned with the role of ENaC in sodium metabolism. The channel also participates in the transduction of mechanical stimuli. Evidence for ENaC activation by mechanical stimuli has been reported since the late eighties [93]. Three main types of these stimuli are transduced by ENaC: hydrostatic pressure, membrane stretch, and shear stress of fluid flow. The role of the channel in the transduction of hydrostatic pressure differences and modifications in membrane stretch has been demonstrated both in Xenopus oocyte expression experiments [94–96] and in native epithelia [97, 98]. In diverse epithelia lining tubular structures subject to fluid flow, such as renal epithelia and vascular endothelium, ENaC mediates the transduction of shear stress [17, 99–102]. As mentioned, the cellular responses to hydrostatic pressure differences and to modifications in membrane stretch, mediated by ENaC, depend upon interactions with the cytoskeleton [43]. However, activation of ENaC by shear stress does not seem to depend on the interaction of the channel with the actin cytoskeleton [103].
3. ENaC and Wound Healing
3.1. General Aspects of the Healing Processes
Confluent cultures of epithelia and other cellular types are useful experimental models to study the basic aspects of the healing responses to wounds produced by mechanical or chemical agents. Most of our present knowledge regarding tissue restitution has been obtained from this type of studies. Even under these simple in vitro conditions, the healing response is complex and involves several aspects. For the case of a mechanical scratch on an epithelial monolayer, three main initial events can be schematically described: (i) death and lysis of injured cells, with release of cellular content, (ii) development of mechanical stress, and (iii) loss of cell-cell contacts of the remaining living cells at the wound borders (Figure 2). All these events trigger different signaling pathways of the surviving cells. Since the signals are generated at the site of the wound, there will be a signaling gradient from the edge of the injury towards the undamaged monolayer, providing, among others, positional information to the healing cells. Some of the responses of the surviving cells that develop early after wounding are the fast calcium wave [104–112], the hydrogen peroxide gradient [112, 113], and the ERK1/2 waves [114]. In turn, these responses trigger signaling cascades in the healing cells.
Figure 2.
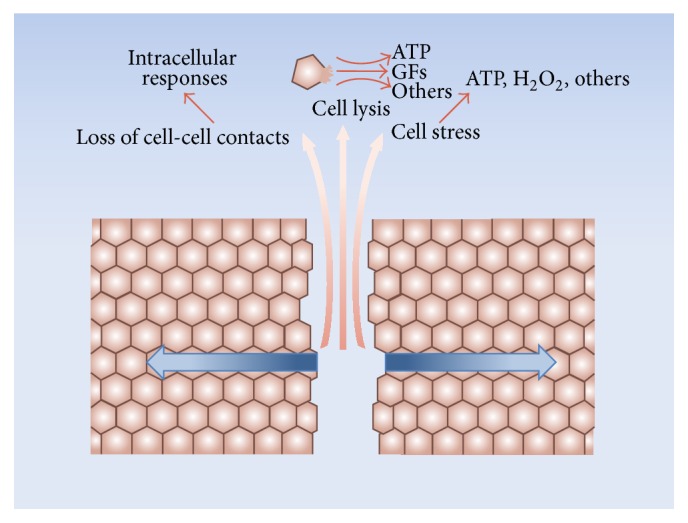
Initial events after a mechanical injury on an epithelial monolayer under culture conditions. A mechanical scratch determines three main initial events: (i) death and lysis of the injured cells, with release of cellular content, (ii) development of mechanical stress, and (iii) loss of cell-cell contacts of the remaining living cells at the wound borders. Among the intracellular molecules released, some that play a role in wound healing in in vitro conditions could be ATP [87, 88] and growth factors (GFs) [89, 90]. ATP, H2O2, and eicosanoids are released by the surviving border cells in response to stress [87, 88]. The loss of cell-cell contacts at the free wound edges stimulates migration and activates growth factor receptors [91, 92]. The three types of events trigger different responses of the surviving cells, frequently propagated to the rest of the monolayer as physical and/or chemical gradients (blue arrows).
Later in the healing process, the border cells progressively undergo dramatic morphological changes (Figure 3). The modifications are more conspicuous at the cells of the wound borders, but the neighboring cells also undergo changes up to several rows from the border. In essence, the cells modify their shapes and start to migrate into the denuded area, either individually, losing their cell-cell junctions, or as a cohesive sheet, maintaining these junctions. Besides migration, the other relevant cellular process triggered to cover the injury zone is proliferation. The extent to which proliferation contributes to wound healing depends on the particular cell type. For the case of bovine corneal endothelial (BCE) cells in culture, DNA synthesis at the leading edge starts to increase 8–10 hours after injury [115], whereas for calf pulmonary artery endothelial cells it begins at 18–24 hours [116]. The morphological changes of the healing cells depend upon the modality of wound closure. Thus, in the actin-cable mode, the cells migrate in a predominantly compact collective fashion and conserve the typical epithelial phenotype to a greater extent (Figure 3(a)), whereas in lamellipodial crawling the cells lose their apicobasal polarity and cell-cell contacts, undergoing more conspicuous morphological changes (Figure 3(b)) [117].
Figure 3.
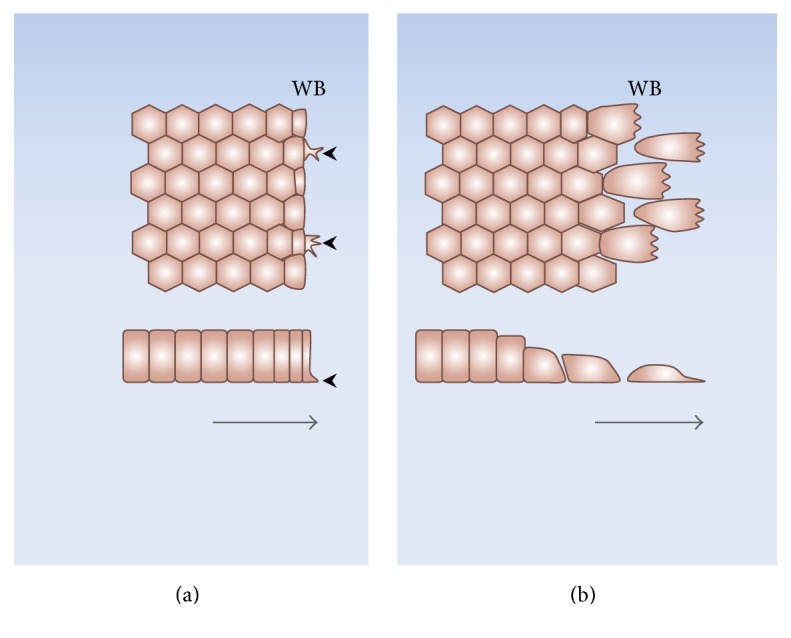
Morphological cellular changes during wound healing of cultured epithelia. In the actin-cable mode (a) the healing cells mainly migrate in compact collective fashion and conserve the epithelial phenotype to a greater extent. Some cells at the wound border develop small lamellipodia (arrowheads) that drag the tissue towards the injured area. In the lamellipodial mode (b) the border cells progressively lose their apicobasal polarity and the cell-cell contacts and acquire well-developed lamellipodia. The lateral views (lower panels) show that while in the actin-cable mode the border cells conserve their morphology, in the lamellipodial mode the cells at the wound edge flatten and detach from their neighbors. WB: wound border. The arrows indicate the direction of healing.
Another important characteristic of the processes of wound healing is the development of electric fields between the injured and uninjured zones. These electric fields represent major cues to stimulate and establish the direction of cell migration during tissue repair [118, 119]. The electrical phenomena observed during wound healing are analogous and have similar significance to those that take place during individual or collective cell migration in morphogenesis [118, 120].
In essence, wound healing is a complex process involving diverse cellular modifications, among which migration plays a leading role. The functional and morphological changes experienced by migrating cells are diverse and have been thoroughly investigated. In this respect, ionic transport has been recognized as a key player of this process [121, 122].
3.2. ENaC and Wound Healing
In recent years, evidence has emerged reporting that ENaC activation is relevant for the healing process in several cellular types. Thus, an increase in ENaC expression occurs during wound restitution in monolayers of BCE cells in culture which determines plasma membrane depolarization and increase in cytosolic sodium of the healing cells, whose inhibition significantly reduces the velocity of healing [123]. The participation of ENaC in these processes has also been proposed from studies in other cellular systems, both in culture [124–127] and in situ [128].
Work from Drummond's laboratory [124] demonstrated that vascular smooth muscle cells express ENaC and that pharmacological inhibition of the channel or silencing of its gene expression determines a decrease in the rate of wound healing. In another set of experiments, authors from this group reported that β-ENaC mediates cytotrophoblast migration and that the increase of its expression enhances individual cell migration [129]. In BeWo cells, a line of human hormone-synthesizing trophoblastic cells, Del Mónaco and coworkers [125] found that an increase in the immunofluorescence signals of the three ENaC subunits takes place during wound healing. They also observed that, in these cells, aldosterone provokes an amiloride-inhibitable rise in the velocity of healing and an augmented expression of α-ENaC. More recent work in BeWo cells from this laboratory shows that the aldosterone effect on the healing rate is at least partially due to an increase in ENaC activity by methylation [130]. From studies on a line of human keratinocytes, Yang and coworkers [126] could establish that ENaC is crucial for directional migration in an electric field by allowing stabilization of the lamella at the cathodal cellular side. Also, that electrically induced directional migration is blocked by pharmacological inhibition or genetic silencing of the channel. The importance of these results for the processes of wound healing rests on the finding that, as commented, electric fields spontaneously develop during these processes. Further work from our laboratory showed that, besides BCE, other epithelia in culture (e.g., rabbit corneal epithelia and Madin-Darby canine kidney (MDCK) cells) also exhibit an increase in ENaC expression and the consequent ionic and electrical modifications during tissue repair [127]. However, this work additionally showed that such changes do not take place in bovine aortic endothelial cells (BAEC). Nevertheless, the study also provided evidence that, in BAEC, pharmacological inhibition of ENaC decreases the velocity of healing, while aldosterone and forskolin increase it.
As a precedent to these studies, Rajnicek and coworkers [131] found that an amiloride-sensitive endogenous sodium current was critical for Xenopus neurula wound healing. More recently, the effect of amiloride on the healing processes has been observed in other systems. Thus, amiloride inhibits reepithelization of migrating epidermal tongue in situ and in culture [132]. In both mouse and human skin, application of the drug produced significant decreases in the endogenous electrical fields generated by wounds [133]. Amiloride is a widely employed diuretic agent with negligible side effects [134]. This is possibly the reason for the lack of studies on the effects of the drug on wound healing in human patients. More direct evidence on the role of ENaC in wound healing in whole organisms is scarce, mostly due to the fact that ENaC knockout mice develop respiratory stress and die shortly after birth [126, 135, 136].
In BCE cells in culture, the ENaC-mediated effects on cellular sodium concentration and membrane potential start to become evident 1-2 hours after wounding and progressively extend to the unwounded monolayer [123]. Concomitant with these phenomena, we could observe the development of a sustained cytosolic calcium rise that temporarily and spatially overlaps with the sodium increase [111]. In this study we also showed that this slow calcium wave (SCW) results from the functional coupling between ENaC and the sodium calcium exchanger (NCX) working in the reverse mode. Also, that blockade of the calcium increase by pharmacological inhibition of NCX reduces the velocity of healing. As described (Figure 1), an elevation in cytosolic calcium constitutes an inhibitory signal of ENaC activity. However, in epithelia that develop a SCW during wound healing, this sustained calcium rise does not prevent the increase in ENaC activity [111].
Besides its role in wound healing, ENaC has been demonstrated to participate in individual cell migration of normal [137, 138] and malignant [139, 140] cells.
3.3. Possible Signals Modifying the Expression and/or Activity of ENaC during Wound Healing
As described above, there is evidence that, in some epithelia, ENaC increases both its expression and activity during wound healing. This process is noticeable a few hours after injury and progressively evolves from the wound border cells towards the rest of the monolayer. The specific signals responsible for these increases are not yet known. In the whole organism, ENaC is subject to regulation by diverse hormones and growth factors, as summarized in previous sections. However, under tissue culture conditions, the stimuli to modify ENaC expression during wound healing are restricted to those resulting from the injured tissue (Figure 2 ). As mentioned, these include release of the content of the damaged cells, loss of cell-cell contacts of the edge cells, and mechanical stress of the border and neighboring cells. These stimuli in turn trigger many signaling cascades. The calcium, hydrogen peroxide, and ERK1/2 waves are among the earliest signals detected in response to the injury (see above) and could be involved in the modification of the channel expression and/or activity (Figure 4). Besides other possible regulatory effects, the fast calcium wave has been proposed to activate immediate early genes [106] and thus constitutes a good candidate for stimulation of ENaC expression. However, for the case of BCE cells, we found that the reversible inhibition of this wave fails to block the increase in the channel expression observed during wound healing [111]. To date, there is no evidence regarding whether any of the other two waves participates in the response. Hydrogen peroxide activates ENaC in its plasma membrane location (Figure 1). However, there are no reports suggesting a possible role of H2O2 in the channel synthesis. ERK1/2 inhibits ENaC synthesis and stimulates its retrieval from the plasma membrane (Figure 1). As commented above, this could represent a mechanism of feedback inhibition to prevent an excessive increase in the channel expression [81]. Alternatively, diverse molecules released from the damaged cells could per se represent stimuli to enhance ENaC expression (Figure 2). Among these molecules, ATP was found to be responsible for the generation of the fast calcium wave [105, 108, 141–145]. Besides this role, ATP could also trigger other signaling pathways. For instance, its binding to the P2Y11 receptor activates adenylyl cyclase [146, 147], thus increasing cAMP and consequently activating PKA. In turn, PKA stimulates ENaC synthesis (Figure 1).
Figure 4.
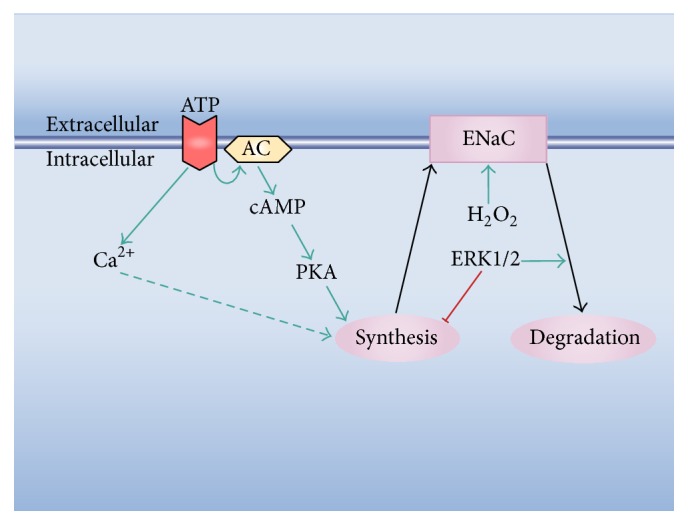
Possible signaling pathways involved in ENaC activation during wound healing. The scheme highlights possible effects of some signals triggered early in the healing process, that is, the calcium, H2O2, and ERK1/2 waves (cf. Figure 1). The dashed arrow indicates that the suggested calcium effect does not take place in some cell types. ATP binds to its plasma membrane receptor and determines an increase in cytosolic calcium. For the case of the P2Y11 receptor, ATP binding also stimulates the adenylyl cyclase (AC).
Although the increase in ENaC expression could be the sole responsible for the gradual rise in its activity during wound healing in several epithelia, other additional stimuli could be involved. A potential candidate for this role is the H2O2 wave. As mentioned, H2O2 can stimulate the channel via a phosphatidylinositide 3-kinase-dependent pathway [68] (Figure 1). ENaC activation can also be produced by actin. In this respect, it has been shown that small fragments of F-actin stimulate the channel [37, 41, 148]. Hydrogen peroxide activates cofilin, which in turn promotes actin filament severing and depolymerization [149]. In this way, H2O2 could further participate in the regulation of ENaC activity via the indirect generation of small actin fragments. In addition, modifications in the cytoskeleton produced as a consequence of the mechanical stress of the border cells (Figure 2) could also modify the channel activity. In this respect, the close interactions established between the cytoskeleton and ENaC mediate bidirectional functional effects, where the channel is capable of modifying the cytoskeletal organization via the transduction of mechanical signals and the cytoskeleton affects its activity by direct structural interactions (see above). Other mechanisms have been proposed for in situ ENaC stimulation. For instance, in injured skin, the increase in extracellular sodium activates ENaC via the sodium sensing channel Nax [150].
3.4. Possible Mechanisms Mediating the Effects of ENaC on Wound Healing
In principle, ENaC participation in the processes of wound healing can be considered to be mediated by two types of mechanisms (Figure 5): (i) those dependent on the ionic and electrical consequences of the increased channel activity, that is, increase in intracellular sodium and plasma membrane depolarization, and (ii) those dependent upon the role of the channel as a mechanotransducer and/or as an intermediate of signaling pathways regulating cytoskeletal organization.
Figure 5.

Possible effects of ENaC activation on the healing process. The scheme summarizes some effects of the increase in ENaC expression and/or activity. The direct consequences of the channel stimulation are an elevation in intracellular sodium and plasma membrane depolarization (represented by smaller plus and minus symbols). The scheme also suggests a possible effect on the cytoskeletal organization provoked by the ionic and electrical changes. In a secondary fashion, the rise in intracellular sodium and plasma membrane depolarization determine the reverse mode of operation of the sodium-calcium exchanger (NCX) with the consequent rise in cytosolic calcium. The sodium and calcium increases constitute signals that modulate diverse cellular processes.
The ENaC-mediated increase in cellular sodium and plasma membrane depolarization of the healing cells could per se represent signals to trigger cellular modifications. For instance, increases in intracellular sodium have signaling roles in smooth muscle cells [151] and astrocytes [152, 153]. Among other possible pathways, sodium has been shown to modify the interaction between α and βγ subunits of G proteins, thus modifying ionic channel activity [154, 155] and potentially affecting other G protein-dependent signaling pathways [156]. For the case of the healing processes, intracellular sodium increase and concomitant membrane depolarization mediated by the voltage-gated NaV1.2 sodium channel play a fundamental role in tail regeneration in Xenopus laevis tadpoles [157].
As commented above, during wound healing in BCE monolayers, there is an elevation in cytosolic calcium as a consequence of sodium increase and membrane depolarization, due to the fact that these conditions promote the reverse functioning of the NCX [111]. The rise in cytosolic calcium of these cells could constitute a signal to trigger diverse cellular modifications [158], in particular migration [121, 122, 159–162] and proliferation [163]. An analogous mechanism involving sodium rise and secondary calcium increase due to the coupling between ENaC and the NCX functioning in reverse mode has been suggested to promote migration in NG2 cells, a line of oligodendrocyte progenitor cells [164], microglia [165], cardiac myofibroblasts [166], tendon fibroblasts [167], and pancreatic cancer cells [168]. Besides its effect on plasma membrane NCX, cytosolic sodium increase also determines reverse functioning of the mitochondrial NCX [169] and mobilization of calcium from the endoplasmic reticulum [170].
It has been proposed that changes in the plasma membrane potential could participate in diverse cellular signaling pathways [171–173]. As a contribution to this concept, we were able to establish that, in BCE and other epithelial cells in confluence, plasma membrane depolarization determines a cytoskeletal reorganization characterized by a gradual loss of the peripheral actin ring, an increase in the amount of F-actin throughout the cytoplasm, and appearance of intercellular gaps with eventual cell detachment [174, 175]. Cell migration involves dramatic modifications in the cytoskeletal organization. It is therefore possible that the cytoskeletal changes of the healing cells are partly determined by the plasma membrane depolarization. Evidence from our studies suggests that the increase in intracellular sodium could also have a role per se in the cytoskeletal reorganization [123].
The interactions between ENaC and the cytoskeleton mediate some of the responses of the channel to mechanical stimuli [43]. For the case of wound healing, physical injuries generate, among others, traction and pressure forces on the surviving cells (Figure 2) that could be transduced by cellular paths involving ENaC. The connections of the channel with the cytoskeleton can also mediate the propagation of electrical signals [176]. This property could participate in the reorganization of actin provoked by membrane depolarization (Figure 5).
3.5. Physiological Significance of ENaC Participation in Wound Healing
As described in the previous sections, there is evidence suggesting that ENaC is an important player in the processes of wound healing. Several characteristics of the channel may underlie this role:
ENaC constitutes the main, and in some epithelia the only, pathway for sodium entrance to the cells. Thus, the modulation of its activity represents the fundamental mechanism of regulation of the intracellular sodium content.
The increases in ENaC expression and/or activity determine several simultaneous cellular effects: rise in intracellular sodium concentration, plasma membrane depolarization, and modifications in cytoskeletal organization. In turn, the elevation in sodium and the plasma membrane depolarization produce secondary ionic effects, like calcium increase.
Its intricate regulation ensures a precise control of its activity.
The ionic modifications (e.g., in sodium and calcium) produced by an increment in ENaC activity constitute signals for many cellular events.
The macroscopic electric fields that could emerge as a consequence of ENaC-dependent membrane depolarization during wound healing could participate in the processes of orientation of cell migration.
In summary, the modulation of a single transport system may affect several cellular properties of importance for wound healing. In the whole organism, besides the signals that could modulate ENaC expression in vitro, other effectors could be involved, such as cytokines and growth factors. Further research on the role of ENaC in tissue restitution is certainly needed, both in vitro and, particularly, in the whole organism. This should include elucidation of the signals affecting its regulation, the mechanisms involved in its participation in the healing process, and its importance in diseases, where these mechanisms are affected.
4. Conclusions
The epithelial sodium channel (ENaC) mediates passive sodium entry across the apical membranes of transport epithelia. In kidney, intestine, and lungs this function is of critical physiological importance for the whole organism. ENaC is also involved in the transduction of mechanical stimuli, a property relevant to its participation in the control of the arterial tone. Besides these localizations, ENaC has been identified in many other epithelial and nonepithelial cell types, where its physiological roles may depend on its properties both as sodium channel and as mechanotransducer. ENaC is subject to a very complex regulatory scheme, possibly as a consequence of its physiological relevance, its presence in many cell types, and the fact that it may be the only channel mediating apical sodium transport in some epithelia.
Recent findings reveal an important contribution of ENaC to the processes of wound healing. The evidence suggests that this participation involves both its activity as a sodium channel and as a mediator of mechanotransduction. An increase in ENaC expression occurs in the edge cells of several epithelial cells in culture in the course of tissue restitution. This increase and/or regulatory effects on immature channels are the causes of the sustained rise in cellular sodium and plasma membrane depolarization observed in these cells. Pharmacological inhibition of the sodium channel or blockage of its synthesis significantly reduces the velocity of healing. The modifications in cellular sodium and in the membrane potential in turn determine secondary responses. Thus, cytosolic calcium of the healing cells, a fundamental second messenger of many processes, gradually augments as a consequence of a functional coupling between ENaC and the sodium-calcium exchanger. The cytoskeletal reorganization that the migrating cells undergo in the course of wound healing has been at least partially considered to depend on membrane depolarization. Since these findings were obtained under in vitro conditions, the basic signals involved in the increase in ENaC expression and activation during tissue restitution are restricted to those resulting from the injured cells and include release of the content of the damaged cells, loss of cell-cell contacts of the edge cells, and mechanical stress of the border and neighboring cells. At present, the nature of these signals is unknown and is the object of current investigation.
Acknowledgments
This study is supported by grants from Comisión Sectorial de Investigación Científica (CSIC), Universidad de la República, Uruguay, and Programa de Desarrollo de las Ciencias Básicas (PEDECIBA), Uruguay (to Silvia Chifflet and Julio A. Hernandez).
Competing Interests
The authors declare that there are no competing interests regarding the publication of this paper.
References
- 1.Diegelmann R. F., Evans M. C. Wound healing: an overview of acute, fibrotic and delayed healing. Frontiers in Bioscience. 2004;9:283–289. doi: 10.2741/1184. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg S. R., Diegelmann R. F. Wound healing primer. Surgical Clinics of North America. 2010;90(6):1133–1146. doi: 10.1016/j.suc.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Neurath M. F., Travis S. P. L. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut. 2012;61(11):1619–1635. doi: 10.1136/gutjnl-2012-302830. [DOI] [PubMed] [Google Scholar]
- 4.Tarnawski A. S., Ahluwalia A. Molecular mechanisms of epithelial regeneration and neovascularization during healing of gastric and esophageal ulcers. Current Medicinal Chemistry. 2012;19(1):16–27. doi: 10.2174/092986712803414088. [DOI] [PubMed] [Google Scholar]
- 5.Holgate S. T. Mechanisms of asthma and implications for its prevention and treatment: a personal journey. Allergy, Asthma and Immunology Research. 2013;5(6):343–347. doi: 10.4168/aair.2013.5.6.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White E. S., Mantovani A. R. Inflammation, wound repair, and fibrosis: reassessing the spectrum of tissue injury and resolution. The Journal of Pathology. 2013;229(2):141–144. doi: 10.1002/path.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rybinski B., Franco-Barraza J., Cukierman E. The wound healing, chronic fibrosis, and cancer progression triad. Physiological Genomics. 2014;46(7):223–244. doi: 10.1152/physiolgenomics.00158.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnold K. M., Opdenaker L. M., Flynn D., Sims-Mourtada J. Wound healing and cancer stem cells: inflammation as a driver of treatment resistance in breast cancer. Cancer Growth and Metastasis. 2015;8:1–13. doi: 10.4137/cgm.s11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garty H., Benos D. J. Characteristics and regulatory mechanisms of the amiloride-blockable Na+ channel. Physiological Reviews. 1988;68(2):309–373. doi: 10.1152/physrev.1988.68.2.309. [DOI] [PubMed] [Google Scholar]
- 10.Palmer L. G. Epithelial Na channels: function and diversity. Annual Review of Physiology. 1992;54:51–66. doi: 10.1146/annurev.ph.54.030192.000411. [DOI] [PubMed] [Google Scholar]
- 11.Hanukoglu I., Hanukoglu A. Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene. 2016;579(2):95–132. doi: 10.1016/j.gene.2015.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Shahar Y. Sensory functions for degenerin/epithelial sodium channels (DEG/ENaC) Advances in Genetics. 2011;76:1–26. doi: 10.1016/B978-0-12-386481-9.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drummond H. A., Gebremedhin D., Harder D. R. Degenerin/epithelial Na+ channel proteins: components of a vascular mechanosensor. Hypertension. 2004;44(5):643–648. doi: 10.1161/01.hyp.0000144465.56360.ad. [DOI] [PubMed] [Google Scholar]
- 14.Jernigan N. L., Drummond H. A. Vascular ENaC proteins are required for renal myogenic constriction. American Journal of Physiology—Renal Physiology. 2005;289(4):F891–F901. doi: 10.1152/ajprenal.00019.2005. [DOI] [PubMed] [Google Scholar]
- 15.Jernigan N. L., Drummond H. A. Myogenic vasoconstriction in mouse renal interlobar arteries: role of endogenous β and γENaC. American Journal of Physiology—Renal Physiology. 2006;291(6):F1184–F1191. doi: 10.1152/ajprenal.00177.2006. [DOI] [PubMed] [Google Scholar]
- 16.Kusche-Vihrog K., Jeggle P., Oberleithner H. The role of ENaC in vascular endothelium. Pflugers Archiv. 2014;466(5):851–859. doi: 10.1007/s00424-013-1356-3. [DOI] [PubMed] [Google Scholar]
- 17.Guo D., Liang S., Wang S., et al. Role of epithelial Na+ channels in endothelial function. Journal of Cell Science. 2016;129(2):290–297. doi: 10.1242/jcs.168831. [DOI] [PubMed] [Google Scholar]
- 18.Teruyama R., Sakuraba M., Wilson L. L., Wandrey N. E. J., Armstrong W. E. Epithelial Na+ sodium channels in magnocellular cells of the rat supraoptic and paraventricular nuclei. American Journal of Physiology—Endocrinology and Metabolism. 2012;302(3):E273–E285. doi: 10.1152/ajpendo.00407.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kellenberger S., Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiological Reviews. 2002;82(3):735–767. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 20.Árnadóttir J., Chalfie M. Eukaryotic mechanosensitive channels. Annual Review of Biophysics. 2010;39(1):111–137. doi: 10.1146/annurev.biophys.37.032807.125836. [DOI] [PubMed] [Google Scholar]
- 21.Weisz O. A., Johnson J. P. Noncoordinate regulation of ENaC: paradigm lost? American Journal of Physiology—Renal Physiology. 2003;285(5):F833–F842. doi: 10.1152/ajprenal.00088.2003. [DOI] [PubMed] [Google Scholar]
- 22.Kunzelmann K., Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiological Reviews. 2002;82(1):245–289. doi: 10.1152/physrev.00026.2001. [DOI] [PubMed] [Google Scholar]
- 23.Büsst C. J. Blood pressure regulation via the epithelial sodium channel: from gene to kidney and beyond. Clinical and Experimental Pharmacology and Physiology. 2013;40(8):495–503. doi: 10.1111/1440-1681.12124. [DOI] [PubMed] [Google Scholar]
- 24.Rossier B. C., Baker M. E., Studer R. A. Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiological Reviews. 2015;95(1):297–340. doi: 10.1152/physrev.00011.2014. [DOI] [PubMed] [Google Scholar]
- 25.Rossier B. C. Epithelial sodium channel (ENaC) and the control of blood pressure. Current Opinion in Pharmacology. 2014;15(1):33–46. doi: 10.1016/j.coph.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh A., Boucher R. C., Tarran R. Airway hydration and COPD. Cellular and Molecular Life Sciences. 2015;72(19):3637–3652. doi: 10.1007/s00018-015-1946-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haq I. J., Gray M. A., Garnett J. P., Ward C., Brodlie M. Airway surface liquid homeostasis in cystic fibrosis: pathophysiology and therapeutic targets. Thorax. 2016;71(3):284–287. doi: 10.1136/thoraxjnl-2015-207588. [DOI] [PubMed] [Google Scholar]
- 28.Mirshahi M., Nicolas C., Mirshahi S., Golestaneh N., D'Hermies F., Agarwal M. K. Immunochemical analysis of the sodium channel in rodent and human eye. Experimental Eye Research. 1999;69(1):21–32. doi: 10.1006/exer.1999.0675. [DOI] [PubMed] [Google Scholar]
- 29.Fischbarg J. Fluid transport across leaky epithelia: central role of the tight junction and supporting role of aquaporins. Physiological Reviews. 2010;90(4):1271–1290. doi: 10.1152/physrev.00025.2009. [DOI] [PubMed] [Google Scholar]
- 30.Shi S., Carattino M. D., Hughey R. P., Kleyman T. R. ENaC regulation by proteases and shear stress. Current Molecular Pharmacology. 2013;6(1):28–34. doi: 10.2174/18744672112059990027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barrett K. E. New ways of thinking about (and teaching about) intestinal epithelial function. American Journal of Physiology —Advances in Physiology Education. 2008;32(1):25–34. doi: 10.1152/advan.00092.2007. [DOI] [PubMed] [Google Scholar]
- 32.Gaillard E. A., Kota P., Gentzsch M., Dokholyan N. V., Stutts M. J., Tarran R. Regulation of the epithelial Na+ channel and airway surface liquid volume by serine proteases. Pflugers Archiv European Journal of Physiology. 2010;460(1):1–17. doi: 10.1007/s00424-010-0827-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eastwood A. L., Goodman M. B. Insight into DEG/ENaC channel gating from genetics and structure. Physiology. 2012;27(5):282–290. doi: 10.1152/physiol.00006.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soundararajan R., Lu M., Pearce D. Organization of the ENaC-regulatory machinery. Critical Reviews in Biochemistry and Molecular Biology. 2012;47(4):349–359. doi: 10.3109/10409238.2012.678285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rotin D., Bar-Sagi D., O'Brodovich H., et al. An SH3 binding region in the epithelial Na+ channel (αrENaC) mediates its localization at the apical membrane. The EMBO Journal. 1994;13(19):4440–4450. doi: 10.1002/j.1460-2075.1994.tb06766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith P. R., Saccomani G., Joe E.-H., Angelides K. J., Benos D. J. Amiloride-sensitive sodium channel is linked to the cytoskeleton in renal epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(16):6971–6975. doi: 10.1073/pnas.88.16.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazzochi C., Bubien J. K., Smith P. R., Benos D. J. The carboxyl terminus of the α-subunit of the amiloride-sensitive epithelial sodium channel binds to F-actin. Journal of Biological Chemistry. 2006;281(10):6528–6538. doi: 10.1074/jbc.M509386200. [DOI] [PubMed] [Google Scholar]
- 38.Ilatovskaya D. V., Pavlov T. S., Levchenko V., Negulyaev Y. A., Staruschenko A. Cortical actin binding protein cortactin mediates ENaC activity via Arp2/3 complex. The FASEB Journal. 2011;25(8):2688–2699. doi: 10.1096/fj.10-167262. [DOI] [PubMed] [Google Scholar]
- 39.Ilatovskaya D. V., Pavlov T. S., Negulyaev Y. A., Staruschenko A. Mechanisms of epithelial sodium channel (ENaC) regulation by cortactin: involvement of dynamin. Cell and Tissue Biology. 2012;6(1):52–59. doi: 10.1134/s1990519x1201004x. [DOI] [PubMed] [Google Scholar]
- 40.Alli A. A., Bao H.-F., Liu B.-C., et al. Calmodulin and CaMKII modulate ENaC activity by regulating the association of MARCKS and the cytoskeleton with the apical membrane. American Journal of Physiology—Renal Physiology. 2015;309(5):F456–F463. doi: 10.1152/ajprenal.00631.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cantiello H. F., Stow J. L., Prat A. G., Ausiello D. A. Actin filaments regulate epithelial Na+ channel activity. American Journal of Physiology—Cell Physiology. 1991;261(5):C882–C888. doi: 10.1152/ajpcell.1991.261.5.C882. [DOI] [PubMed] [Google Scholar]
- 42.Karpushev A. V., Ilatovskaya D. V., Pavlov T. S., Negulyaev Y. A., Staruschenko A. Intact cytoskeleton is required for small G protein dependent activation of the epithelial Na+ channel. PLoS ONE. 2010;5(1) doi: 10.1371/journal.pone.0008827.e8827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinac B. The ion channels to cytoskeleton connection as potential mechanism of mechanosensitivity. Biochimica et Biophysica Acta (BBA)—Biomembranes. 2014;1838(2):682–691. doi: 10.1016/j.bbamem.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 44.Kashlan O. B., Kleyman T. R. Epithelial Na+ channel regulation by cytoplasmic and extracellular factors. Experimental Cell Research. 2012;318(9):1011–1019. doi: 10.1016/j.yexcr.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baines D. Kinases as targets for ENaC regulation. Current Molecular Pharmacology. 2013;6(1):50–64. doi: 10.2174/18744672112059990028. [DOI] [PubMed] [Google Scholar]
- 46.Lang F., Pearce D. Regulation of the epithelial Na+ channel by the mTORC2/SGK1 pathway. Nephrology Dialysis Transplantation. 2016;31(2):200–205. doi: 10.1093/ndt/gfv270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diakov A., Korbmacher C. A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel's α-subunit. The Journal of Biological Chemistry. 2004;279(37):38134–38142. doi: 10.1074/jbc.m403260200. [DOI] [PubMed] [Google Scholar]
- 48.Flores S. Y., Loffing-Cueni D., Kamynina E., et al. Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd4-2 phosphorylation and increased Na+ transport in cortical collecting duct cells. Journal of the American Society of Nephrology. 2005;16(8):2279–2287. doi: 10.1681/asn.2004100828. [DOI] [PubMed] [Google Scholar]
- 49.Debonneville C., Flores S. Y., Kamynina E., et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. The EMBO Journal. 2001;20(24):7052–7059. doi: 10.1093/emboj/20.24.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snyder P. M., Olson D. R., Kabra R., Zhou R., Steines J. C. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na+ channel through convergent phosphorylation of Nedd4-2. Journal of Biological Chemistry. 2004;279(44):45753–45758. doi: 10.1074/jbc.m407858200. [DOI] [PubMed] [Google Scholar]
- 51.Zhang W., Xia X., Reisenauer M. R., et al. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel α . The Journal of Clinical Investigation. 2007;117(3):773–783. doi: 10.1172/jci29850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ilatovskaya D. V., Levchenko V., Brands M. W., Pavlov T. S., Staruschenko A. Cross-talk between insulin and IGF-1 receptors in the cortical collecting duct principal cells: implication for ENaC-mediated NA+ reabsorption. American Journal of Physiology—Renal Physiology. 2015;308(7):F713–F719. doi: 10.1152/ajprenal.00081.2014.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuchs W., Larsen E. H., Lindemann B. Current voltage curve of sodium channels and concentration dependence of sodium permeability in frog skin. Journal of Physiology. 1977;267(1):137–166. doi: 10.1113/jphysiol.1977.sp011805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheng S., Carattino M. D., Bruns J. B., Hughey R. P., Kleyman T. R. Furin cleavage activates the epithelial Na+ channel by relieving Na+ self-inhibition. American Journal of Physiology—Renal Physiology. 2006;290(6):F1488–F1496. doi: 10.1152/ajprenal.00439.2005. [DOI] [PubMed] [Google Scholar]
- 55.Maarouf A. B., Sheng N., Chen J., et al. Novel determinants of epithelial sodium channel gating within extracellular thumb domains. The Journal of Biological Chemistry. 2009;284(12):7756–7765. doi: 10.1074/jbc.m807060200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winarski K. L., Sheng N., Chen J., Kleyman T. R., Sheng S. Extracellular allosteric regulatory subdomain within the γ subunit of the epithelial Na+ channel. The Journal of Biological Chemistry. 2010;285(34):26088–26096. doi: 10.1074/jbc.m110.149963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Collier D. M., Snyder P. M. Extracellular chloride regulates the epithelial sodium channel. Journal of Biological Chemistry. 2009;284(43):29320–29325. doi: 10.1074/jbc.M109.046771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collier D. M., Snyder P. M. Identification of epithelial Na+ channel (ENaC) intersubunit Cl− inhibitory residues suggests a trimeric αγβ channel architecture. The Journal of Biological Chemistry. 2011;286(8):6027–6032. doi: 10.1074/jbc.m110.198127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collier D. M., Snyder P. M. Extracellular protons regulate human ENaC by modulating Na+ self-inhibition. Journal of Biological Chemistry. 2009;284(2):792–798. doi: 10.1074/jbc.m806954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gu Y. Effects of [Ca2+]i and pH on epithelial Na+ channel activity of cultured mouse cortical collecting ducts. Journal of Experimental Biology. 2008;211(19):3167–3173. doi: 10.1242/jeb.019646. [DOI] [PubMed] [Google Scholar]
- 61.Palmer L. G., Frindt G. Effects of cell Ca and pH on Na channels from rat cortical collecting tubule. American Journal of Physiology. 1987;253:F333–F339. doi: 10.1152/ajprenal.1987.253.2.F333. [DOI] [PubMed] [Google Scholar]
- 62.Chalfant M. L., Denton J. S., Berdiev B. K., Ismailov I. I., Benos D. J., Stanton B. A. Intracellular H+ regulates the α-subunit of ENaC, the epithelial Na+ channel. American Journal of Physiology—Cell Physiology. 1999;276(2):C477–C486. doi: 10.1152/ajpcell.1999.276.2.C477. [DOI] [PubMed] [Google Scholar]
- 63.He-Ping M., Saxena S., Warnock D. G. Anionic phospholipids regulate native and expressed epithelial sodium channel (ENaC) The Journal of Biological Chemistry. 2002;277(10):7641–7644. doi: 10.1074/jbc.c100737200. [DOI] [PubMed] [Google Scholar]
- 64.Yue G., Malik B., Yue G., Eaton D. C. Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. The Journal of Biological Chemistry. 2002;277(14):11965–11969. doi: 10.1074/jbc.m108951200. [DOI] [PubMed] [Google Scholar]
- 65.Pochynyuk O., Staruschenko A., Tong Q., Medina J., Stockand J. D. Identification of a functional phosphatidylinositol 3,4,5-trisphosphate binding site in the epithelial Na+ channel. Journal of Biological Chemistry. 2005;280(45):37565–37571. doi: 10.1074/jbc.m509071200. [DOI] [PubMed] [Google Scholar]
- 66.Kunzelmann K., Bachhuber T., Regeer R., Markovich D., Sun J., Schreiber R. Purinergic inhibition of the epithelial Na+ transport via hydrolysis of PIP2. The FASEB Journal. 2005;19(1):142–143. doi: 10.1096/fj.04-2314fje. [DOI] [PubMed] [Google Scholar]
- 67.Pochynyuk O., Tong Q., Medina J., et al. Molecular determinants of PI(4,5)P2 and PI(3,4,5)P3 regulation of the epithelial Na+ channel. Journal of General Physiology. 2007;130(4):399–413. doi: 10.1085/jgp.200709800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma H.-P. Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3-kinase-dependent pathway. The Journal of Biological Chemistry. 2011;286(37):32444–32453. doi: 10.1074/jbc.m111.254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frindt G., Silver R. B., Windhager E. E., Palmer L. G. Feedback regulation of Na channels in rat CCT. II. Effects of inhibition of Na entry. American Journal of Physiology—Renal Fluid and Electrolyte Physiology. 1993;264(3):F565–F574. doi: 10.1152/ajprenal.1993.264.3.F565. [DOI] [PubMed] [Google Scholar]
- 70.Abriel H., Horisberger J.-D. Feedback inhibition of rat amiloride-sensitive epithelial sodium channels expressed in Xenopus laevis oocytes. The Journal of Physiology. 1999;516(1):31–43. doi: 10.1111/j.1469-7793.1999.031aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Knight K. K., Wentzlaff D. M., Snyder P. M. Intracellular sodium regulates proteolytic activation of the epithelial sodium channel. The Journal of Biological Chemistry. 2008;283(41):27477–27482. doi: 10.1074/jbc.m804176200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heidrich E., Carattino M. D., Hughey R. P., Pilewski J. M., Kleyman T. R., Myerburg M. M. Intracellular Na+ regulates epithelial Na+ channel maturation. The Journal of Biological Chemistry. 2015;290(18):11569–11577. doi: 10.1074/jbc.m115.640763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hughey R. P., Bruns J. B., Kinlough C. L., Kleyman T. R. Distinct pools of epithelial sodium channels are expressed at the plasma membrane. Journal of Biological Chemistry. 2004;279(47):48491–48494. doi: 10.1074/jbc.C400460200. [DOI] [PubMed] [Google Scholar]
- 74.Hughey R. P., Mueller G. M., Bruns J. B., et al. Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. The Journal of Biological Chemistry. 2003;278(39):37073–37082. doi: 10.1074/jbc.m307003200. [DOI] [PubMed] [Google Scholar]
- 75.Hughey R. P., Bruns J. B., Kinlough C. L., et al. Epithelial sodium channels are activated by furin-dependent proteolysis. The Journal of Biological Chemistry. 2004;279(18):18111–18114. doi: 10.1074/jbc.c400080200. [DOI] [PubMed] [Google Scholar]
- 76.Staub O., Gautschi I., Ishikawa T., et al. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO Journal. 1997;16(21):6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dinudom A., Harvey K. F., Komwatana P., Young J. A., Kumar S., Cook D. I. Nedd4 mediates control of an epithelial Na+ channel in salivary duct cells by cytosolic Na+ . Proceedings of the National Academy of Sciences of the United States of America. 1998;95(12):7169–7173. doi: 10.1073/pnas.95.12.7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Terker A. S., Yarbrough B., Ferdaus M. Z., et al. Direct and indirect mineralocorticoid effects determine distal salt transport. Journal of the American Society of Nephrology. 2015 doi: 10.1681/ASN.2015070815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Snyder P. M. Minireview: regulation of epithelial Na+ channel trafficking. Endocrinology. 2005;146(12):5079–5085. doi: 10.1210/en.2005-0894. [DOI] [PubMed] [Google Scholar]
- 80.Booth R. E., Stockand J. D. Targeted degradation of ENaC in response to PKC activation of the ERK1/2 cascade. American Journal of Physiology—Renal Physiology. 2003;284(5):F938–F947. doi: 10.1152/ajprenal.00373.2002. [DOI] [PubMed] [Google Scholar]
- 81.Grossmann C., Freudinger R., Mildenberger S., Krug A. W., Gekle M. Evidence for epidermal growth factor receptor as negative-feedback control in aldosterone-induced Na+ reabsorption. American Journal of Physiology—Renal Physiology. 2004;286(6):F1226–F1231. doi: 10.1152/ajprenal.00378.2003. [DOI] [PubMed] [Google Scholar]
- 82.Dooley R., Harvey B. J., Thomas W. Non-genomic actions of aldosterone: from receptors and signals to membrane targets. Molecular and Cellular Endocrinology. 2012;350(2):223–234. doi: 10.1016/j.mce.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 83.Rokaw M. D., Wang J.-M., Edinger R. S., et al. Carboxylmethylation of the β subunit of xENaC regulates channel activity. The Journal of Biological Chemistry. 1998;273(44):28746–28751. doi: 10.1074/jbc.273.44.28746. [DOI] [PubMed] [Google Scholar]
- 84.Becchetti A., Kemendy A. E., Stockand J. D., Sariban-Sohraby S., Eaton D. C. Methylation increases the open probability of the epithelial sodium channel in A6 epithelia. The Journal of Biological Chemistry. 2000;275(22):16550–16559. doi: 10.1074/jbc.m000954200. [DOI] [PubMed] [Google Scholar]
- 85.Edinger R. S., Yospin J., Perry C., Kleyman T. R., Johnson J. P. Regulation of epithelial Na+ channels (ENaC) by methylation: a novel methyltransferase stimulates ENaC activity. The Journal of Biological Chemistry. 2006;281(14):9110–9117. doi: 10.1074/jbc.m509232200. [DOI] [PubMed] [Google Scholar]
- 86.Oberleithner H., Schneider S. W., Albermann L., et al. Endothelial cell swelling by aldosterone. Journal of Membrane Biology. 2003;196(3):163–172. doi: 10.1007/s00239-003-0635-6. [DOI] [PubMed] [Google Scholar]
- 87.Cordeiro J. V., Jacinto A. The role of transcription-independent damage signals in the initiation of epithelial wound healing. Nature Reviews Molecular Cell Biology. 2013;14(4):249–262. doi: 10.1038/nrm3541. [DOI] [PubMed] [Google Scholar]
- 88.Enyedi B., Niethammer P. Mechanisms of epithelial wound detection. Trends in Cell Biology. 2015;25(7):398–407. doi: 10.1016/j.tcb.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McNeil P. L., Muthukrishnan L., Warder E., D'Amore P. A. Growth factors are released by mechanically wounded endothelial cells. The Journal of Cell Biology. 1989;109(2):811–822. doi: 10.1083/jcb.109.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aizman I., Vinodkumar D., McGrogan M., Bates D. Cell injury-induced release of fibroblast growth factor 2: relevance to intracerebral mesenchymal stromal cell transplantations. Stem Cells and Development. 2015;24(14):1623–1634. doi: 10.1089/scd.2015.0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Poujade M., Grasland-Mongrain E., Hertzog A., et al. Collective migration of an epithelial monolayer in response to a model wound. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(41):15988–15993. doi: 10.1073/pnas.0705062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klarlund J. K., Block E. R. Free edges in epithelia as cues for motility. Cell Adhesion and Migration. 2011;5(2):106–110. doi: 10.4161/cam.5.2.13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jorgensen F., Ohmori H. Amiloride blocks the mechano-electrical transduction channel of hair cells of the chick. Journal of Physiology. 1988;403:577–588. doi: 10.1113/jphysiol.1988.sp017265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Awayda M. S., Ismailov I. I., Berdiev B. K., Benos D. J. A cloned renal epithelial Na+ channel protein displays stretch activation in planar lipid bilayers. American Journal of Physiology—Cell Physiology. 1995;268(6):C1450–C1459. doi: 10.1152/ajpcell.1995.268.6.C1450. [DOI] [PubMed] [Google Scholar]
- 95.Ismailov I. I., Berdiev B. K., Shlyonsky V. G., Benos D. J. Mechanosensitivity of an epithelial Na+ channel in planar lipid bilayers: release from Ca2+ block. Biophysical Journal. 1997;72(3):1182–1192. doi: 10.1016/s0006-3495(97)78766-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hong-Long J. I., Fuller C. M., Benos D. J. Osmotic pressure regulates -rENaC expressed in Xenopus oocytes. American Journal of Physiology. 1998;275(5):C1182–C1190. doi: 10.1152/ajpcell.1998.275.5.C1182. [DOI] [PubMed] [Google Scholar]
- 97.Lewis S. A., Ifshin M. S., Loo D. D. F., Diamond J. M. Studies of sodium channels in rabbit urinary bladder by noise analysis. The Journal of Membrane Biology. 1984;80(2):135–151. doi: 10.1007/BF01868770. [DOI] [PubMed] [Google Scholar]
- 98.Wang E. C. Y., Lee J.-M., Johnson J. P., Kleyman T. R., Bridges R., Apodaca G. Hydrostatic pressure-regulated ion transport in bladder uroepithelium. American Journal of Physiology—Renal Physiology. 2003;285(4):F651–F663. doi: 10.1152/ajprenal.00403.2002. [DOI] [PubMed] [Google Scholar]
- 99.Satlin L. M., Sheng S., Woda C. B., Kleyman T. R. Epithelial Na+ channels are regulated by flow. American Journal of Physiology—Renal Physiology. 2001;280(6):F1010–F1018. doi: 10.1152/ajprenal.2001.280.6.F1010. [DOI] [PubMed] [Google Scholar]
- 100.Carattino M. D., Sheng S., Kleyman T. R. Epithelial Na+ channels are activated by laminar shear stress. The Journal of Biological Chemistry. 2004;279(6):4120–4126. doi: 10.1074/jbc.m311783200. [DOI] [PubMed] [Google Scholar]
- 101.Morimoto T., Liu W., Woda C., et al. Mechanism underlying flow stimulation of sodium absorption in the mammalian collecting duct. American Journal of Physiology—Renal Physiology. 2006;291(3):F663–F669. doi: 10.1152/ajprenal.00514.2005. [DOI] [PubMed] [Google Scholar]
- 102.Li Q., Kresge C., Bugde A., Lamphere M., Park J. Y., Feranchak A. P. Regulation of mechanosensitive biliary epithelial transport by the epithelial Na+ channel. Hepatology. 2016;63(2):538–549. doi: 10.1002/hep.28301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karpushev A. V., Ilatovskaya D. V., Staruschenko A. The actin cytoskeleton and small G protein RhoA are not involved in flow-dependent activation of ENaC. BMC Research Notes. 2010;3, article 210 doi: 10.1186/1756-0500-3-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Klepeis V. E., Cornell-Bell A., Trinkaus-Randall V. Growth factors but not gap junctions play a role in injury-induced Ca2+ waves in epithelial cells. Journal of Cell Science. 2001;114(23):4185–4195. doi: 10.1242/jcs.114.23.4185. [DOI] [PubMed] [Google Scholar]
- 105.Sammak P. J., Hinman L. E., Tran P. O. T., Sjaastad M. D., Machen T. E. How do injured cells communicate with the surviving cell monolayer? Journal of Cell Science. 1997;110, part 4:465–475. doi: 10.1242/jcs.110.4.465. [DOI] [PubMed] [Google Scholar]
- 106.Tran P. O. T., Hinman L. E., Unger G. M., Sammak P. J. A wound-induced [Ca2+]i increase and its transcriptional activation of immediate early genes is important in the regulation of motility. Experimental Cell Research. 1999;246(2):319–326. doi: 10.1006/excr.1998.4239. [DOI] [PubMed] [Google Scholar]
- 107.Boucher I., Yang L., Mayo C., Klepeis V., Trinkaus-Randall V. Injury and nucleotides induce phosphorylation of epidermal growth factor receptor: MMP and HB-EGF dependent pathway. Experimental Eye Research. 2007;85(1):130–141. doi: 10.1016/j.exer.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shabir S., Southgate J. Calcium signalling in wound-responsive normal human urothelial cell monolayers. Cell Calcium. 2008;44(5):453–464. doi: 10.1016/j.ceca.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 109.Milara J., Mata M., Serrano A., Peiró T., Morcillo E. J., Cortijo J. Extracellular calcium-sensing receptor mediates human bronchial epithelial wound repair. Biochemical Pharmacology. 2010;80(2):236–246. doi: 10.1016/j.bcp.2010.03.035. [DOI] [PubMed] [Google Scholar]
- 110.Xu S., Chisholm A. D. A Gα q-Ca2+ signaling pathway promotes actin-mediated epidermal wound closure in C. elegans . Current Biology. 2011;21(23):1960–1967. doi: 10.1016/j.cub.2011.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chifflet S., Justet C., Hernández J. A., Nin V., Escande C., Benech J. C. Early and late calcium waves during wound healing in corneal endothelial cells. Wound Repair and Regeneration. 2012;20(1):28–37. doi: 10.1111/j.1524-475x.2011.00749.x. [DOI] [PubMed] [Google Scholar]
- 112.Razzell W., Evans I. R., Martin P., Wood W. Calcium flashes orchestrate the wound inflammatory response through duox activation and hydrogen peroxide release. Current Biology. 2013;23(5):424–429. doi: 10.1016/j.cub.2013.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Niethammer P., Grabher C., Look A. T., Mitchison T. J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459(7249):996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Matsubayashi Y., Ebisuya M., Honjoh S., Nishida E. ERK activation propagates in epithelial cell sheets and regulates their migration during wound healing. Current Biology. 2004;14(8):731–735. doi: 10.1016/j.cub.2004.03.060. [DOI] [PubMed] [Google Scholar]
- 115.Justet C., Hernandez J. A., Torriglia A., Chifflet S. Fast calcium wave inhibits excessive apoptosis during epithelial wound healing. Cell and Tissue Research. 2016 doi: 10.1007/s00441-016-2388-8. [DOI] [PubMed] [Google Scholar]
- 116.Tran P. O. T., Tran Q.-H. P., Hinman L. E., Sammak P. J. Co-ordination between localized wound-induced Ca2+ signals and pre-wound serum signals is required for proliferation after mechanical injury. Cell Proliferation. 1998;31(3-4):155–170. doi: 10.1111/j.1365-2184.1998.tb01193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Grasso S., Hernández J. A., Chifflet S. Roles of wound geometry, wound size, and extracellular matrix in the healing response of bovine corneal endothelial cells in culture. American Journal of Physiology—Cell Physiology. 2007;293(4):C1327–C1337. doi: 10.1152/ajpcell.00001.2007. [DOI] [PubMed] [Google Scholar]
- 118.Levin M. Molecular bioelectricity: how endogenous voltage potentials control cell behavior and instruct pattern regulation in vivo. Molecular Biology of the Cell. 2014;25(24):3835–3850. doi: 10.1091/mbc.e13-12-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reid B., Zhao M. The electrical response to injury: molecular mechanisms and wound healing. Advances in Wound Care. 2014;3(2):184–201. doi: 10.1089/wound.2013.0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Levin M. Endogenous bioelectrical networks store non-genetic patterning information during development and regeneration. The Journal of Physiology. 2014;592(11):2295–2305. doi: 10.1113/jphysiol.2014.271940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Schwab A., Fabian A., Hanley P. J., Stock C. Role of ion channels and transporters in cell migration. Physiological Reviews. 2012;92(4):1865–1913. doi: 10.1152/physrev.00018.2011. [DOI] [PubMed] [Google Scholar]
- 122.Schwab A., Stock C. Ion channels and transporters in tumour cell migration and invasion. Philosophical Transactions of the Royal Society of London B: Biological Sciences. 2014;369(1638) doi: 10.1098/rstb.2013.0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chifflet S., Hernández J. A., Grasso S. A possible role for membrane depolarization in epithelial wound healing. American Journal of Physiology—Cell Physiology. 2005;288(6):C1420–C1430. doi: 10.1152/ajpcell.00259.2004. [DOI] [PubMed] [Google Scholar]
- 124.Grifoni S. C., Gannon K. P., Stec D. E., Drummond H. A. ENaC proteins contribute to VSMC migration. American Journal of Physiology—Heart and Circulatory Physiology. 2006;291(6):H3076–H3086. doi: 10.1152/ajpheart.00333.2006. [DOI] [PubMed] [Google Scholar]
- 125.Del Mónaco S. M., Marino G. I., Assef Y. A., Damiano A. E., Kotsias B. A. Cell migration in BeWo cells and the role of epithelial sodium channels. Journal of Membrane Biology. 2009;232(1–3):1–13. doi: 10.1007/s00232-009-9206-0. [DOI] [PubMed] [Google Scholar]
- 126.Yang H.-Y., Charles R.-P., Hummler E., Baines D. L., Isseroff R. R. The epithelial sodium channel mediates the directionality of galvanotaxis in human keratinocytes. Journal of Cell Science. 2013;126(9):1942–1951. doi: 10.1242/jcs.113225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Justet C., Evans F., Vasilskis E., Hernández J. A., Chifflet S. ENaC contribution to epithelial wound healing is independent of the healing mode and of any increased expression in the channel. Cell and Tissue Research. 2013;353(1):53–64. doi: 10.1007/s00441-013-1635-5. [DOI] [PubMed] [Google Scholar]
- 128.Fuchigami T., Matsuzaki T., Ihara S. Possible roles of ENaC and Cl− channel in wound closure in Xenopus laevis embryos. Zoological Science. 2011;28(10):703–711. doi: 10.2108/zsj.28.703. [DOI] [PubMed] [Google Scholar]
- 129.Warrington J. P., Coleman K., Skaggs C., et al. Heme oxygenase-1 promotes migration and β-epithelial Na+ channel expression in cytotrophoblasts and ischemic placentas. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2014;306(9):R641–R646. doi: 10.1152/ajpregu.00566.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Marino G. I., Assef Y. A., Kotsias B. A. The migratory capacity of human trophoblastic BeWo cells: effects of aldosterone and the epithelial sodium channel. Journal of Membrane Biology. 2013;246(3):243–255. doi: 10.1007/s00232-013-9526-y. [DOI] [PubMed] [Google Scholar]
- 131.Rajnicek A. M., Stump R. F., Robinson K. R. An endogenous sodium current may mediate wound healing in Xenopus neurulae. Developmental Biology. 1988;128(2):290–299. doi: 10.1016/0012-1606(88)90292-8. [DOI] [PubMed] [Google Scholar]
- 132.Dubé J., Rochette-Drouin O., Lévesque P., et al. Restoration of the transepithelial potential within tissue-engineered human skin in vitro and during the wound healing process in vivo. Tissue Engineering Part A. 2010;16(10):3055–3063. doi: 10.1089/ten.tea.2010.0030. [DOI] [PubMed] [Google Scholar]
- 133.Nuccitelli R., Nuccitelli P., Ramlatchan S., Sanger R., Smith P. J. S. Imaging the electric field associated with mouse and human skin wounds. Wound Repair and Regeneration. 2008;16(3):432–441. doi: 10.1111/j.1524-475x.2008.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brater D. C. Update in diuretic therapy: clinical pharmacology. Seminars in Nephrology. 2011;31(6):483–494. doi: 10.1016/j.semnephrol.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 135.Olivier R., Scherrer U., Horisberger J.-D., Rossier B. C., Hummler E. Selected contribution: limiting Na+ transport rate in airway epithelia from α-ENaC transgenic mice: a model for pulmonary edema. Journal of Applied Physiology. 2002;93(5):1881–1887. doi: 10.1152/japplphysiol.00413.2002. [DOI] [PubMed] [Google Scholar]
- 136.Hummler E., Barker P., Galzy J., et al. Early death due to defective neonatal lung liquid clearance in αENaC-deficient mice. Nature Genetics. 1996;12(3):325–328. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- 137.Wang S., He G., Yang Y., et al. Reduced expression of Enac in Placenta tissues of patients with severe preeclampsia is related to compromised trophoblastic cell migration and invasion during pregnancy. PloS one. 2013;8(8, article e72153) doi: 10.1371/journal.pone.0072153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cerecedo D., Martínez-Vieyra I., Alonso-Rangel L., Benítez-Cardoza C., Ortega A. Epithelial sodium channel modulates platelet collagen activation. European Journal of Cell Biology. 2014;93(3):127–136. doi: 10.1016/j.ejcb.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 139.Kapoor N., Bartoszewski R., Qadri Y. J., et al. Knockdown of ASIC1 and epithelial sodium channel subunits inhibits glioblastoma whole cell current and cell migration. The Journal of Biological Chemistry. 2009;284(36):24526–24541. doi: 10.1074/jbc.m109.037390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rooj A. K., McNicholas C. M., Bartoszewski R., Bebok Z., Benos D. J., Fuller C. M. Glioma-specific cation conductance regulates migration and cell cycle progression. The Journal of Biological Chemistry. 2012;287(6):4053–4065. doi: 10.1074/jbc.m111.311688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Klepeis V. E., Weinger I., Kaczmarek E., Trinkaus-Randall V. P2Y receptors play a critical role in epithelial cell communication and migration. Journal of Cellular Biochemistry. 2004;93(6):1115–1133. doi: 10.1002/jcb.20258. [DOI] [PubMed] [Google Scholar]
- 142.Gomes P., Srinivas S. P., Van Driessche W., Vereecke J., Himpens B. ATP release through connexin hemichannels in corneal endothelial cells. Investigative Ophthalmology and Visual Science. 2005;46(4):1208–1218. doi: 10.1167/iovs.04-1181. [DOI] [PubMed] [Google Scholar]
- 143.Gomes P., Srinivas S. P., Vereecke J., Himpens B. ATP-dependent paracrine intercellular communication in cultured bovine corneal endothelial cells. Investigative Ophthalmology & Visual Science. 2005;46(1):104–113. doi: 10.1167/iovs.04-0846. [DOI] [PubMed] [Google Scholar]
- 144.Berra-Romani R., Raqeeb A., Avelino-Cruz J. E., et al. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium. 2008;44(3):298–309. doi: 10.1016/j.ceca.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 145.Sherwood C. L., Lantz R. C., Burgess J. L., Boitano S. Arsenic alters ATP-dependent Ca2+ signaling in human airway epithelial cell wound response. Toxicological Sciences. 2011;121(1):191–206. doi: 10.1093/toxsci/kfr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.van der Weyden L., Adams D. J., Luttrell B. M., Conigrave A. D., Morris M. B. Pharmacological characterisation of the P2Y11 receptor in stably transfected haematological cell lines. Molecular and Cellular Biochemistry. 2000;213(1-2):75–81. doi: 10.1023/a:1007168215748. [DOI] [PubMed] [Google Scholar]
- 147.Torres B., Zambon A. C., Insel P. A. P2Y11 receptors activate adenylyl cyclase and contribute to nucleotide-promoted cAMP formation in MDCK-D1 cells. A mechanism for nucleotide-mediated autocrine-paracrine regulation. The Journal of Biological Chemistry. 2002;277(10):7761–7765. doi: 10.1074/jbc.m110352200. [DOI] [PubMed] [Google Scholar]
- 148.Prat A. G., Bertorello A. M., Ausiello D. A., Cantiello H. F. Activation of epithelial Na+ channels by protein kinase A requires actin filaments. American Journal of Physiology—Cell Physiology. 1993;265(1):C224–C233. doi: 10.1152/ajpcell.1993.265.1.C224. [DOI] [PubMed] [Google Scholar]
- 149.Kim J.-S., Huang T. Y., Bokoch G. M. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Molecular Biology of the Cell. 2009;20(11):2650–2660. doi: 10.1091/mbc.E09-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Xu W., Hong S. J., Zhong A., et al. Sodium channel Nax is a regulator in epithelial sodium homeostasis. Science Translational Medicine. 2015;7(312) doi: 10.1126/scitranslmed.aad0286.312ra177 [DOI] [PubMed] [Google Scholar]
- 151.Orlov S. N., Hamet P. Intracellular monovalent ions as second messengers. Journal of Membrane Biology. 2006;210(3):161–172. doi: 10.1007/s00232-006-0857-9. [DOI] [PubMed] [Google Scholar]
- 152.Chatton J., Magistretti P. J., Barros L. F. Sodium signaling and astrocyte energy metabolism. Glia. 2016 doi: 10.1002/glia.22971. [DOI] [PubMed] [Google Scholar]
- 153.Rose C. R., Chatton J.-Y. Astrocyte sodium signaling and neuro-metabolic coupling in the brain. Neuroscience. 2016;323:121–134. doi: 10.1016/j.neuroscience.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 154.Rishal I., Keren-Raifman T., Yakubovich D., et al. Na+ promotes the dissociation between GαGDP and Gβγ, activating G protein-gated K+ channels. The Journal of Biological Chemistry. 2003;278(6):3840–3845. doi: 10.1074/jbc.c200605200. [DOI] [PubMed] [Google Scholar]
- 155.Blumenstein Y., Maximyuk O. P., Lozovaya N., et al. Intracellular Na+ inhibits voltage-dependent N-type Ca2+ channels by a G protein βγ subunit-dependent mechanism. Journal of Physiology. 2004;556(1):121–134. doi: 10.1113/jphysiol.2003.056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ellisdon A. M., Halls M. L. Compartmentalization of GPCR signalling controls unique cellular responses. Biochemical Society Transactions. 2016;44(2):562–567. doi: 10.1042/bst20150236. [DOI] [PubMed] [Google Scholar]
- 157.Tseng A.-S., Beane W. S., Lemire J. M., Masi A., Levin M. Induction of vertebrate regeneration by a transient sodium current. The Journal of Neuroscience. 2010;30(39):13192–13200. doi: 10.1523/jneurosci.3315-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Brini M., Ottolini D., Calì T., Carafoli E. Calcium in health and disease. Metal Ions in Life Sciences. 2013;13:81–137. doi: 10.1007/978-94-007-7500-8-4. [DOI] [PubMed] [Google Scholar]
- 159.Wei C., Wang X., Zheng M., Cheng H. Calcium gradients underlying cell migration. Current Opinion in Cell Biology. 2012;24(2):254–261. doi: 10.1016/j.ceb.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 160.Plotnikov S. V., Waterman C. M. Guiding cell migration by tugging. Current Opinion in Cell Biology. 2013;25(5):619–626. doi: 10.1016/j.ceb.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Howe A. K. Cross-talk between calcium and protein kinase A in the regulation of cell migration. Current Opinion in Cell Biology. 2011;23(5):554–561. doi: 10.1016/j.ceb.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zheng J. Q., Poo M.-M. Calcium signaling in neuronal motility. Annual Review of Cell and Developmental Biology. 2007;23:375–404. doi: 10.1146/annurev.cellbio.23.090506.123221. [DOI] [PubMed] [Google Scholar]
- 163.Pinto M. C. X., Kihara A. H., Goulart V. A. M., et al. Calcium signaling and cell proliferation. Cellular Signalling. 2015;27(11):2139–2149. doi: 10.1016/j.cellsig.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 164.Tong X.-P., Li X.-Y., Zhou B., et al. Ca2+ signaling evoked by activation of Na+ channels and Na+/Ca2+ exchangers is required for GABA-induced NG2 cell migration. The Journal of Cell Biology. 2009;186(1):113–128. doi: 10.1083/jcb.200811071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Noda M., Ifuku M., Mori Y., Verkhratsky A. Calcium influx through reversed NCX controls migration of microglia. Advances in Experimental Medicine and Biology. 2013;961:289–294. doi: 10.1007/978-1-4614-4756-6-24. [DOI] [PubMed] [Google Scholar]
- 166.Raizman J. E., Komljenovic J., Chang R., et al. The participation of the Na+-Ca2+ exchanger in primary cardiac myofibroblast migration, contraction, and proliferation. Journal of Cellular Physiology. 2007;213(2):540–551. doi: 10.1002/jcp.21134. [DOI] [PubMed] [Google Scholar]
- 167.Sakamoto K., Owada Y., Shikama Y., et al. Involvement of Na+/Ca2+ exchanger in migration and contraction of rat cultured tendon fibroblasts. Journal of Physiology. 2009;587(22):5345–5359. doi: 10.1113/jphysiol.2009.172080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dong H., Shim K.-N., Li J. M. J., et al. Molecular mechanisms underlying Ca2+-mediated motility of human pancreatic duct cells. American Journal of Physiology—Cell Physiology. 2010;299(6):C1493–C1503. doi: 10.1152/ajpcell.00242.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fekete Á., Franklin L., Ikemoto T., et al. Mechanism of the persistent sodium current activator veratridine-evoked Ca2+ elevation: implication for epilepsy. Journal of Neurochemistry. 2009;111(3):745–756. doi: 10.1111/j.1471-4159.2009.06368.x. [DOI] [PubMed] [Google Scholar]
- 170.Borin M. L., Tribe R. M., Blaustein M. P. Increased intracellular Na+ augments mobilization of Ca2+ from SR in vascular smooth muscle cells. American Journal of Physiology. 1994;266(1):C311–C317. doi: 10.1152/ajpcell.1994.266.1.C311. [DOI] [PubMed] [Google Scholar]
- 171.Olivotto M., Arcangeli A., Carlà M., Wanke E. Electric fields at the plasma membrane level: a neglected element in the mechanisms of cell signalling. BioEssays. 1996;18(6):495–504. doi: 10.1002/bies.950180612. [DOI] [PubMed] [Google Scholar]
- 172.Hernández J. A., Cristina E. Modeling cell volume regulation in nonexcitable cells: the roles of the Na+ pump and of cotransport systems. American Journal of Physiology—Cell Physiology. 1998;275(4):C1067–C1080. doi: 10.1152/ajpcell.1998.275.4.C1067. [DOI] [PubMed] [Google Scholar]
- 173.Lang F., Busch G. L., Ritter M., et al. Functional significance of cell volume regulatory mechanisms. Physiological Reviews. 1998;78(1):247–306. doi: 10.1152/physrev.1998.78.1.247. [DOI] [PubMed] [Google Scholar]
- 174.Chifflet S., Hernández J. A., Grasso S., Cirillo A. Nonspecific depolarization of the plasma membrane potential induces cytoskeletal modifications of bovine corneal endothelial cells in culture. Experimental Cell Research. 2003;282(1):1–13. doi: 10.1006/excr.2002.5664. [DOI] [PubMed] [Google Scholar]
- 175.Chifflet S., Correa V., Nin V., Justet C., Hernández J. A. Effect of membrane potential depolarization on the organization of the actin cytoskeleton of eye epithelia. The role of adherens junctions. Experimental Eye Research. 2004;79(6):769–777. doi: 10.1016/j.exer.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 176.Tuszyński J. A., Portet S., Dixon J. M., Luxford C., Cantiello H. F. Ionic wave propagation along actin filaments. Biophysical Journal. 2004;86(4):1890–1903. doi: 10.1016/s0006-3495(04)74255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]