

abstract
Due to the excellent safety profile of poly (D,L-lactide-co-glycolide) (PLGA) particles in human, and their biodegradability, many studies have focused on the application of PLGA particles as a controlled-release vaccine delivery system. Antigenic proteins/peptides can be encapsulated into or adsorbed to the surface of PLGA particles. The gradual release of loaded antigens from PLGA particles is necessary for the induction of efficient immunity. Various factors can influence protein release rates from PLGA particles, which can be defined intrinsic features of the polymer, particle characteristics as well as protein and environmental related factors. The use of PLGA particles encapsulating antigens of different diseases such as hepatitis B, tuberculosis, chlamydia, malaria, leishmania, toxoplasma and allergy antigens will be described herein. The co-delivery of antigens and immunostimulants (IS) with PLGA particles can prevent the systemic adverse effects of immunopotentiators and activate both dendritic cells (DCs) and natural killer (NKs) cells, consequently enhancing the therapeutic efficacy of antigen-loaded PLGA particles. We will review co-delivery of different TLR ligands with antigens in various models, highlighting the specific strengths and weaknesses of the system. Strategies to enhance the immunotherapeutic effect of DC-based vaccine using PLGA particles can be designed to target DCs by functionalized PLGA particle encapsulating siRNAs of suppressive gene, and disease specific antigens. Finally, specific examples of cellular targeting where decorating the surface of PLGA particles target orally administrated vaccine to M-cells will be highlighted.
Keywords: delivery system, micro/nano particles, PLGA particle, protein/peptide vaccine, TLR ligands
Introduction
Poly (DL-lactide-co-glycolide) (PLGA) micro/nano particles (MPs/NPs) present a promising vaccine delivery system. PLGA, a polymer ester of two α hydroxyacids (lactic and glycolic acids) (Fig. 1) is well-characterized, and because of its excellent safety profile in human,1 has been FDA approved for vaccine and drug delivery as well as tissue engineering.2 Though aluminum salts have been approved over the last 80 years as a vaccine adjuvant for improving humoral responses; they are poor to induce cellular responses, which are required for vaccines against intracellular pathogens. Therefore, many researchers have focused on developing other adjuvants and delivery platforms, and among them PLGA particles have been attracted much attention regarding their great advantages.1 PLGA particles are biodegradable and metabolized to their constituent monomers in aqueous media.3 However, according to the results of in vivo biodistribution studies in BALB/c mice following oral administration of PLGA NPs, it is necessary to minimize the amount of particles that reach the liver. Semete et al. have been suggested surface modification of PLGA particles with hydrophilic molecules as a strategy to enhance circulation time and reduce particle localization in the liver.4
Figure 1.
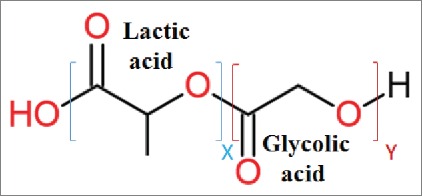
PLGA polymer structure, PLGA a copolymer of poly lactic acid (PLA) and poly glycolic acid (PGA). X and Y indicate the number of each unit repeats.
PLGA particles protect loaded peptides/proteins from proteolytic degradation, and confer them proper plasma half-life.5It has been established that PLGA particles have no adverse effect on DC function like maturation, migration, cytotokine secretion, and costimulatory properties.6 Furthermore, they can demonstrate nonspecific DC targeting by controlling their size, hydrophobicity, and charge. Nano/micro-sized particulate vaccine delivery systems have the ability to simultaneously deliver both the antigen and the immunopotentiator to the same dendritic cells (DCs) or macrophages.7 Co-delivery of both the antigen and immunostimulant (IS) may lead to enhanced potency, adjuvant dose reduction, and consequently minimizing toxicity of the IS.8,9 However, the application of PLGA particles faces some challenge such as acidic feature of PLGA microenvironment, protein exposure to water-oil interface, harsh parameters during particle preparation, and protein instability during encapsulation that may affect protein integrity and immunogenicity.10
Many strategies, including the addition of stabilizing excipients, the optimization of process conditions, chemical modifications such as glycosylation and pegylation have been developed to overcome antigen instability during encapsulation and release process.11 Moreover, application of protein adsorption instead of encapsulation not only dissolves many of problems regarding protein instability during particle preparation, but also facilities PLGA particle sterilization by gamma irradiation before protein loading.1
This review will focus on the application of PLGA particles as delivery system for peptide/protein based vaccine, and will touch on different methods for the preparation of protein/peptide PLGA particles. The mechanisms of protein release from PLGA particles and variables affecting on protein release will be described. In addition, intracellular trafficking of PLGA particles and the impact of administration routes and particle size on immunity responses will be explained.
Herein, the studies conducted so far using antigens targeting different infectious disease formulated into PLGA particles are discussed. Furthermore, we will cover examples of antigen and immunostimulating agent (e.g., TLR ligands) co-delivery using PLGA based particulate delivery system. Surface modification of PLGA particles particularly for induction of mucosal immunity, enhancing induced immunity, and affording the ability of oral or nasal administration will be highlighted. Accordingly, this review will underscore the functionalization of PLGA particle surfaces with DCs and M-cells targeting agent.
MPs or NPs definitions
It has been well established that particle size has an undeniable impact on particle immunogenicity. Particle size also influences the type of immune response achieved.12
Before addressing the advantages of MPs or NPs as vaccine delivery platform, it is worthy to clarify MP and NP size ranges. In spite of the fact that the border between polymeric NPs and MPs is defined by their size, there is no harmony among different references in this regard, and those represent various classifications according to particle size. Some of the MPs or NPs classifications according to their size are mentioned in Table 1. In general, in order to define size range of MPs and NPs, we suppose particles more than 1000 nm in diameter as MPs and particles with the size less than 1000 nm as NPs.
Table 1.
Various classifications of micro/nanoparticles based on their size in literatures.
| Particle size | Known as | Year | Ref. |
|---|---|---|---|
| >1000 nm | NPs | 1998 | 13 |
| 2-8 μm | MPs | 2007 | 14 |
| 200-600 nm | NPs | ||
| 1-250 μm | MPs | 2008 | 15 |
| 10-1000 nm | NPs | ||
| 800-900 nm | MPs | 2008 | 16 |
| 100 nm | NPs | ||
| 1–1000 μm | MPs | 2010 | 17 |
| > 1 μm | NPs | ||
| 0.1-100 μm | MPs | 2012 | 18 |
| 1-100 nm | NPs |
MPs; Microparticles, NPs; Nanoparticles
Established methods for protein loaded MPs/NPs preparation
Various methods have been applied to prepare MPs/NPs according to inherent nature of used polymer, physicochemical features of loaded drugs and also purposes of their use. Here, the applicable methods for the preparation of PLGA-based MPs/NPs encapsulating proteins are described. All following methods excluding nanoprecipitation can be applied for PLGA MPs and NPs fabrication by tuning up process parameters such as volume of organic phase to aqueous phase ratio and rate of stirring.3
Emulsification-solvent evaporation method
Emulsion-solvent evaporation technique has been known as the most common method to prepare protein and peptide loaded MPs/NPs. This technique is categorized into single and double emulsion solvent evaporation methods. Sequential steps of these two methods are shown in Figure 2 (a,b). Poly vinyl alcohol (PVA) is frequently used as a surfactant and can be applied for both single and double emulsion solvent evaporation methods, which is typically removed from particles during formulation of the vaccine.15,19 In order to harden the oil droplets, the organic solvent is evaporated under reduced or atmospheric pressure according to required particle size.3 The prepared MPs/NPs are then separated by ultracentrifugation, washed and dried through freeze-drying.20 Schematic illustration of single emulsion solvent evaporation method is shown in Figure 3. Single emulsion-solvent evaporation is not an efficient method for entrapment of hydrophilic drugs such as proteins and peptides into PLGA particles as they diffuse into aqueous phase before PLGA polymer solidification, resulting in higher degradation/inactivation of the protein antigen.20Thus, to preserve biological activity of loaded proteins, aqueous phase double emulsion solvent evaporation technique has been successfully developed for protein and peptide encapsulation.3,20,21
Figure 2.
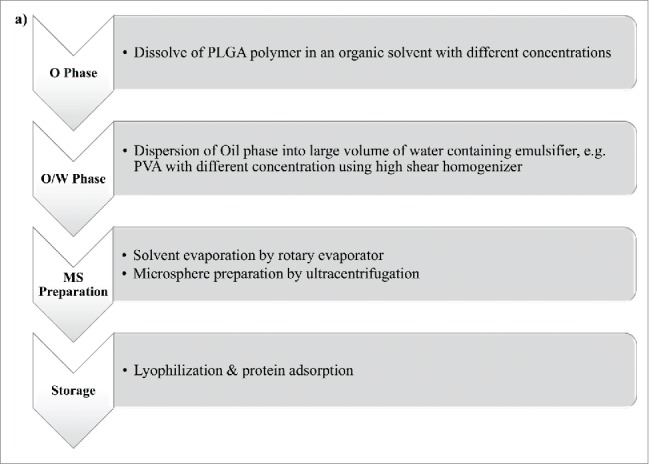
Flow diagrams of single (a) and double (b) emulsion solvent evaporation methods. a) Single emulsion solvent evaporation (Oil/Water emulsion) flow diagram. Oil phase (O phase), Oil in water phase (O/W phase), microsphere (MS), b) Double emulsion solvent evaporation (Water/Oil/Water emulsion) flow diagram. Water in oil phase (W/O phase), water in oil in water (W/O/W), microsphere (MS).
Figure 3.

Schematic illustration of the single emulsion evaporation method.
Spary-drying
Spary drying is a suitable method for peptide or protein loading on PLGA MPs/NPs with a few process parameters that should be considered prior to scaling-up step. The salient drawback of spray drying comes from the particle attachment to interior surfaces of the cyclone which decreases the process yield.3,22Besides, water in oil emulsion containing protein is atomized in a stream of hot air and leading to the formation of MPs/NPs subsequent to the solvent evaporation.15,22
Phase separation (Coacervation)
Phase separation methods can be utilized to fabricate protein loaded PLGA MPs using liquid-liquid phase separation. Hydrophilic drugs such as protein or peptides are dispersed into PLGA polymers dissolved in organic solvent 15 and then a non-solvent agent (e.g., silicon) that decreases the solubility of PLGA in its solvent, and also induces coacervation, is added to emulsion. Protein is encapsulated in PLGA- rich liquid phase (coacervate) and newly formed microspheres (MSs) are immersed into heptane to quench and solidify micro droplets.3,22
Nanoprecipitation
Nanopercipitation is one of the most common methods which can be also used for the encapsulation of peptides and proteins into PLGA NPs. PLGA polymer is dissolved in acetone and then protein is added to an aqueous solution containing emulsifier such as Pluronic F683. PLGA polymer and protein are diffused into the aqueous phase, acetone is evaporated under reduced pressures, and then protein-encapsulated NPs are produced with size ranges of ∼200 nm which are smaller than NPs formed via other approaches.20
Microfluidic approaches for MPs/NPs preparation
Microfluidics has been introduced as a promising alternative to conventional laboratory techniques. Generally, microfluidics drives a small volumes of fluids in the range of microliters (10−6) to picoliters (10−12) through microchannel network with lowest dimensions from tens to hundreds micrometers. In microfluidics devices, continuous and disperse phases flow into two separate inlets, and the disperse phase is confined into isolated droplets or narrow stream. T-junction, flow-focusing and concentric capillaries are well known designs of microfluidics which can be made from different materials like glass, silicon, polydimethylsiloxane (PDMS), stainless steel, etc.20,23
PRINT technology
PRINT (Particle Replication In Non-wetting Templates) is a continuous, roll-to-roll, high-resolution molding technology providing monodisperse particles ranging from NPs to MPs. In PRINT technology, an elastomeric mold containing wells or cavities of predefined shape and size is utilized to fabricate particles with precise structures.23,24 The biopharmaceutical cargo such as proteins can be incorporated in PRINT particles by encapsulation or adsorption. For example, Hemagglutinin (HA) antigens in three commercial trivalent influenza vaccine (TIV), were electrostatically bound to the surface of cylindrical cationic PLGA-based NPs prepared by PRINT technology.25
Overall, double emulsion solvent evaporation and spary drying are the most widely used methods for antigen loading in PLGA particles in laboratory scale. However, it is worth mentioning that for movement from laboratory to market it is necessary to consider GMP requirements and also consistency in particle preparation that can be assured by novel methods such as microfluidic approaches and PRINT technology. The size of PLGA particles obtained by these preparation methods mostly can be adjusted via process parameters. The advantages and disadvantages of all above methods are mentioned in Table 2.
Table 2.
Advantages and disadvantages of conventional methods, microfluidic approaches and PRINT technology in PLGA particles preparation.
| Method | Pros | Cons | Considerable bottleneck | Ref. | |
|---|---|---|---|---|---|
| Conventional methods | Emulsification-solvent evaporation |
|
✓ Negative effect of sonication and organic solvent on protein integrity | ✓ Heterogeneity ✓ Polydispersity ✓ Batch-to-batch variations | 3,20,21 |
| Spary-drying | ✓ Low harsh effect on protein biological activity ✓ Fast and reproducible | ✓ Low process yield ✓ Negative effect of organic solvent on protein integrity | 3,15,22 | ||
| Phase separation (Coacervation) | ✓ Negative effect of organic solvent on protein integrity | 3,22 | |||
| Nanoprecipitation | ✓ High simplicity ✓ Fast and reproducible | ✓ Negative effect of organicsolvent on protein integrity | 20 | ||
| Microfluidic approaches | ✓ Precise control of process parameters | ✓ Instrument dependent | — | 20,23 | |
| ✓ Automation ✓ Providing homogenous environment | |||||
| ✓ High efficiency | — | ||||
| PRINT Technology | ✓ Capable to modify particle surface properties ✓ Precise control over size and shape | ✓ Technology dependent | 23-25 | ||
| ✓ Compatible to GMP requirements | |||||
| ✓ large scale | |||||
| ✓ High uniform particles production |
Protein loading into/on MPs/NPs
MPs/NPs are classified according to their structure into following groups: nanocapsule, nanosphere (NS), microcapsule, and microsphere (MS).19 Encapsulation and adsorption are the most common approaches applied for protein loading into or on MPs/NPs.19The protein can be either adsorbed on the particle surfaces or encapsulated inside. The features of MPs/NPs and their protein loading efficiencies are influenced by a wide range of parameters, such as: type of solvents and surfactants, surfactant concentration, PLGA concentration in solvent, stirring rate and fabrication methods.3,20
Protein/peptide encapsulation
In all methods mentioned above, the encapsulation of desired protein into MPs/NPs and particle preparation are carried out simultaneously. Proteins are encapsulated into MSs/NSs by their dispersion either into organic solvents dissolving PLGA or in an aqueous solution using high shear homogenization or ultrasonic device.15 Some examples of peptide or protein encapsulated into MPs/NPs are pointed out in Table 3.
Table 3.
Some examples of peptide or protein-loaded PLGA MPs/NPs by encapsulation
| Protein/ Peptide | MP/NP/MS Mean Size | TLR ligand Combination | Disease | Preparation Technique | PLGA type, Co plolymer ratio | In vivo/ in vitro | Immunity Response | Ref. |
|---|---|---|---|---|---|---|---|---|
| tetanus toxoid (TT) | NP: 0.5 µm MP: 3.9 µm | — | Tetanus | Double emulsion (W/O/W) | PLGA 50:50 (14, 45 kDa) and PLA (45 kDa) | In vivo | Humoral immunity | 26 |
| recombinant tuberculosis (TB) antigen, Ag85B, TB10.4 and TB10.4Ag85B | MP: 3.3 µm | — | Tuberculosis | Emulsion/ Spary drying | PLGA 75:25 (84.7 kDa) Intrinsic viscosity: 0.68 dL/g | In vitro | Cellular immunity | 27 |
| OVA/ PA/ HA | NP: 300 nm | MPL, R837 | Influenza | Double emulsion (W/O/W) | PLGA | In vivo | Cellular and humoral immunity | 28 |
| SBm7462 peptide | MS: 1.63 mm | — | Double emulsion (W/O/W) | Resomer®RG 506 PLGA 50:50 Intrinsic viscosity: 0.8 dl/g | In vivo | 29 | ||
| Synthetic SPf66 peptide | MS: 1.2–1.3 µm | — | Malaria | Double emulsion (W/O/W) | Resomer®RG 506 PLGA50:50 Intrinsic viscosity: 0.8 dl/g | In vivo | Anti-SPf66 antibody detection | 30 |
| BSA | MS: 1.3 µm | MPL, poly I:C | – | Double emulsion (W/O/W) | Resomer®RG 503 PLGA 50:50 (40.6 kDa)Intrinsic viscosity: 0.41 dL/g | In vivo | Cellular and homoral immunity | 31 |
| Tetanus toxoid (TTxd)/ diphtheria toxoid (DTxd) | MS: 10 µm | — | Tetani and Diphteria | Double emulsion (W/O/W) | PLGA 50:50 (40–75 kDa) | In vivo | Homoral immunity | 32 |
| HLA-A*0201-restricted peptide FluM58–66 (Influenza A M1 protein 58–66, | Spray-drying | Resomer®RG 502 H PLGA 50:50 (14 kDa) | In vitro | – | 33 | |||
| Yersinia pestis F1 antigen | MS: 3.8 µm | — | Pestis | Double emulsion (W/O/W) | PLGA/polyethylene glycol(PEG) (PLGA/PEG) | In vivo | Humoral immunity | 34 |
| OVA | NP: 0.56–1.4 µm | — | — | double emulsion (W/O/W) | PLGA | In vitro | DC uptake and cross presentation | 35 |
| NP | PLGA 50:50 (45 kD) PLGA 50:50 (15 kD) | In vivo | Humoral immunity | 26 | ||||
| HBsAg | MS | — | Hepatitis B | Double emulsion (W/O/W) | PLGA50:50, PLGA75:25 | In vivo | Humoral immunity | 36 |
| HBsAg | MS: 5 µm, 12 µm | — | Hepatitis B | Double emulsion (W/O/W) | PLGA 85:15 (85.2 kDa) PLGA 50:50 (43.5 kDa) | In vivo | Humoral immunity | 37 |
| SAG1 | MP: 4.25–6.58 µm | — | Toxoplasmosis | Double emulsion (W/O/W) | PLGA 50:50 | In vivo | Cellular and humoral immunity | 38 |
| SAG1/SAG2 | MP: 1.27–1.65 µm | — | Toxoplasmosis | Double emulsion (W/O/W) | PLGA 50:50 | In vivo | Cellular and humoral immunity | 39 |
Protein/peptide adsorption to MPs/NPs
Protein adsorption on the surface of blank MPs/NPs is accomplished by incubation of PLGA MPs/NPs with protein.40,41 To achieve protein adsorption, the blank PLGA MSs/NSs and desired protein are incubated overnight under agitation at 4°C and defined conditions (e.g., protein to PLGA W/W%, buffer composition, and pH value).42 These conditions are generally applied to proteins 40,41 and should be determined through preliminary experimental evaluation.43 Due to thecolloidal nature of MPs/NPs suspension, the protein-adsorbed MPs/NPs are separated from buffer containing free proteins by ultracentrifugation or gel filtration. Then, the particle pellets are washed with distilled water and lyophilized.19 Some examples of peptide or protein loaded MPs/NPs by adsorption are summarized in Table 4.
Table 4.
Some examples of peptide or protein-loaded PLGA MPs/NPs by adsorption.
| Protein/ Peptide | MP/NP/MS Mean Size | TLR ligand Combination | Disease | Preparation Technique | PLGA type, Co plolymer ratio | In vivo/ In vitro | Immunity Response | Ref. |
|---|---|---|---|---|---|---|---|---|
| Men B | 0.5–1.9 µm | CpG | meningitides B | Double emulsion (W/O/W) | Resomer®RG 503 PLGA 50:50 Intrinsic viscosity: 0.4 dL/g | In vivo | Humoral Immunity | 43 |
| Men B, HIV-1 gp120 protein | 1 µm | MPL/RC529 | meningitides B, HIV | Double emulsion (W/O/W) | Resomer®RG 503 PLGA 50:50 Intrinsic viscosity: 0.4dL/g | In vivo | Humoral Immunity | 40 |
| lysozyme | 8 –12 µm | — | — | Single and Double emulsion (O/W) and (W/O/W) | Resomer®RG 502 H PLGA 50:50 | In vivo | — | 44 |
| sCT | 14.8 ± 1.3 µm | — | — | Single emulsion (O/W) | Resomer®RG 503 PLGA 50:50 (34 kDa) | — | — | 45 |
| OVA, CAN, LYZ, LDH, BSA, MB1, MB2, gp120 | 1 µm | — | — | Double emulsion (W/O/W) | Resomer®RG 503, RG 502 H PLGA 50:50 | In vitro | — | 41 |
| p55 gag protein | 1 µm | — | HIV-1 | Double emulsion (W/O/W) | Resomer®RG 503 PLGA 50:50 | In vivo | Cellular and humoral | 42 |
Assessment of encapsulation and adsorption efficiency
For successful protein loading into/on PLGA particles, both high loading capacity and suitable ratio between the desired protein and PLGA particles are crucial parameters. Protein encapsulation and adsorption efficiency in protein- loaded MPs/NPs can be calculated by direct or indirect method. In direct method, the amount of desired protein is frequently determined by dissolving defined quantity of protein–loaded MPs/NPs in a solution containing 5% SDS and 0.1–0.2 M sodium hydroxide at room temperature, known as digestion method. Afterwards, the amount of protein is calculated through protein assay methods, such as BCA.36
The estimation of encapsulation 19 and adsorption efficiencies are made according to the equations below:
The indirect quantification of proteins adsorbed on or encapsulated into MSs/NSs is calculated by subtracting the amount of protein which is not incorporated into MPs/NPs (free protein) from total amount of applied protein.
Process yield determination
An important parameter that should be calculated for each process is production yield. The process yield is estimated through the following equation46:
PLGA degradation and protein release
One of the most remarkable features of PLGA polymers, as a unique vaccine delivery system, is the possibility of tuning the physico-chemical properties of the PLGA polymer in order to achieve the desired release profile dictated by the target product profile.15,22 Most researchers believe that PLGA degradation is solely driven by a hydrolytic mechanism that exclusively happens by hydrolysis of ester linkages in polymer backbone subsequent to water uptake.3,47,48 A few investigators have indicated a possible enzymatic role in PLGA particle degradation, but the results have not been convincing so far.49 PLGA particles degrade into lactic and glycolic acids monomers, both of which enter the tricarboxylic acid cycle and are subsequently eliminated from the body as carbon dioxide and water.3,19
The mechanism of protein release from PLGA particles
PLGA particulate vaccine can provide sustained antigen release which is critical to elicit potent immune responses. It is considered that the impact of PLGA particulate vaccine on immune responses relies on the kinetics of antigen release.50 To manipulate protein release rates from the particles and to obtain the desired release profile, it is necessary to know the possible mechanisms of protein release from PLGA particles.19,51 However, underlying mechanisms of release have not been clearly understood.3 Overall, protein release happens through a combination of bulk and surface diffusion as well as bulk and surface erosion of the PLGA polymer, which can be modulated through many PLGA attributes.51,52 It is hypothesized that the release rate is diffusion-controlled initially giving way to degradation/erosion at the end of release period. Diffusion can occur through water filled pores within the PLGA particle.51 Moreover, some researchers mentioned desorption of bound protein from particle surface as another release mechanism.19,22 PLGA particles commonly exhibited classic tri-phasic release patterns 51 including initial burst (phase I), slow release phase (lag time) associated with a little diffusion-driven release (phase II), followed by a faster release (phase III) attributed to erosion- accelerated release. In the phase III, a sustained protein release in a zero-order manner occurs.47,53 Phase III is sometimes called a second burst release. In contrast, mono-phasic or bi-phasic release profiles can also exist.51 Makadia and Siegel 3 described drug release through PLGA particle degradation as a biphasic curve with an initial burst release (first phase) followed by progressively increasing drug release (second phase). It is assumed that the burst release happens due to weakly bound or adsorbed proteins to particle surface.54 It has been mentioned that the lag time relies on PLGA molecular weight (MW) and end- group caps.52,53 Water absorption by PLGA polymer starts immediately upon exposure of PLGA particles to water, or in vivo administration. The hydrolytic cleavage of ester bonds to alcohol and carboxylic groups results in further PLGA degradation and acidic oligomers accumulation. Carboxylic acid build-up causes localized pH drop that enhances PLGA autocatalysis and also presents a key role in creating channels through which protein release can happen.51,55
In addition, it has been well demonstrated that molecular weight (MW) of PLGA MSs influences the release mechanism. As release from low MW (e.g., 5 kDa) PLGA MSs is mostly driven by diffusion, release from high MW (e.g., 25 kDa) PLGA, MSs occurs via a combination of diffusion and erosion.47 Furthermore, it has been revealed that larger particles have a smaller initial burst release than smaller particles.19
Factors affecting protein release rate
In the following sections, parameters influencing protein release behavior and polymer degradation from PLGA particles will be discussed. Here, the factors influencing protein release are categorized into polymer, particle, protein, and environmental related ones, all of which can be modulated to tune the hydrolytic degradation behavior of MPs/NPs and subsequently protein release rates (Table 5).
Table 5.
Factors adjusting the hydrolytic degradation rate of PLGA MPs/NPs.
| Factors |
| Polymer related factors |
| Lactide/glycolide ratio |
| MW, Polymer end group, Glass transition temperature (Tg), Glass transition temperature (glassy, rubbery), and cristalinity |
| Particle related factors |
| Particle size, surface structure and morphology, Shape, Porosity, Loading percent |
| Protein related factors |
| MW, Solubility, Distribution Coefficient, Hydrophobicity |
| Number of free thiol groups and/or disulfide bonds |
| Environmental related factors |
| pH, Temperature, Ionic strength, Sink condition, Salts, Additives, Plasticizing agents and Surfactants |
Polymer related factors
Lactide/glicolide ratio
PLGA polymer composition is the primary factor contributing to particle degradation and antigen release rate. PLGA polymer composition impacts the hydrophilicity, glass transition temperature (Tg), and hydration rate of the PLGA polymer.15,56,57 The most widely used PLGA with monomer composition of 50:50 has the fastest biodegradation rate which completely occurs in ∼50–60 days. The poly-glycolide acid is more hydrophilic due to the absence of methyl side group than ploy-lactide.55,58 The study conducted by Park 59 has shown that more glycolic acid percentage causes more water uptake, and consequently the overall weight loss of PLGA particles and faster degradation rate happen. Many in vivo studies have been evaluated the antigen release and the amount of induced immunity by PLGA polymers with various molecular weights and L/G ratios.26,36For example, humoral response against HBsAg encapsulated in PLGA MSs with different L/G ratios (PLGA 50:50, 75:25, a mixture of PLGA 50:50 and 75:25, as well as PLGA 50:50-COOH) was investigated as a single dose vaccination.36 It was clearly demonstrated that the HBsAg release from HBsAg-PLGA MSs was related to the surface morphology of the MSs, polymer composition (L/G ratio), and MW of the polymer (viscosity). The PLGA 50:50-COOH MSs released HBsAg faster than the PLGA 50:50 and PLGA 75:25 MSs, respectively.26 The glass transition temperature also relies on the composition and MW of the PLGA polymer. It has been shown that a decline in lactide quantity and MW is associated with a decrease in the degree of crystalinity of PLGA polymer.22,60
Molecular weight and polymer end group
Protein release rate in PLGA particles directly is proportional to PLGA MW, so that particles with higher MW generally tend slower degradation rates.57,59 Tracy et al.56 showed that higher MW PLGA polymers need more time to hydrolyze into soluble oligomers and have lower release rate.
End groups in PLGA polymer also impact the water uptake, and subsequently degradation rate of particles.61 End groups in PLGA polymer impact the water up-take, and subsequently degradation rate of particles.3 The PLGA end group has a greater influence than MW on particle degradation. In general, low L/G ratio, low MW and uncapped PLGA polymer make a less hydrophobic polymer with increased rate of water absorption, hydrolysis, and erosion.56 The relationship among polymer related factors affecting particle degradation rate is shown in Figure 4.
Figure 4.
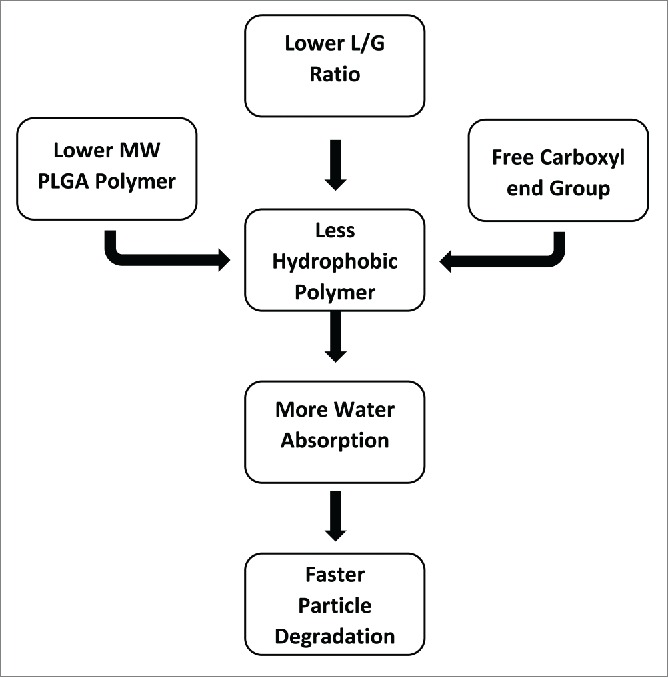
Flow diagram of PLGA polymer related factor affecting on PLGA particle degradation.
Particle related factors (Particle size and morphology)
Many studies have been carried out to demonstrate the correlation of particle size and surface morphology with release profile of antigen- loaded PLGA particles.3,19,48 For example, it was demonstrated that when particle hydrophobicity, MW of PLGA and L/G ratio (PLGA 75:25) increase, the surface porosity of a particle decreases.62 It was also found that larger particles have a smaller initial burst release than smaller particles.19 Saini et al.58 showed that PLGA (50:50) MSs are spherical with a smooth surface, whereas PLGA (75:25) MSs were spherical with rough surface. In another research,63 it was observed that in vitro polymer degradation rates are not substantially various for different particle sizes. Furthermore, protein release did not correlate with the polymer degradation rate or particle sizes.63 Siepmann et al. showed that particle size affects the release profile of encapsulated antigens through diffusion path length and autocatalysis. As particle size becomes greater, diffusion path length increases, which negatively influences the release rate. On the other hand, larger particles experience more autocatalysis.64 This implies that there are multiple factors contributing to antigen release and particle degradation, and they can vary in vitro and in vivo. In another study, the impact of formulation on particle size and subsequently release profile of PLGA particles was confirmed.37 It was demonstrated that HBsAg-encapsulated PLGA particles prepared by polyethylenimine (PEI) as external aqueous phase resultsin larger size particles and slightly higher release rates compared to those prepared by stearylamine (SA). Moreover, it was exhibited that all positively charged PLGA MSs except one formulation, showed slower release compared to unmodified PLGA MSs. Yang et al. attributed the difference of release rates from PLGA 50:50, 15:85, and L-PLA particle to their difference in surface morphology, particle porosity, and surface roughness, as PLGA 50:50 exhibited the slowest release pattern. PLA particles demonstrated rougher surface and higher porosity than those of PLGA particles.65 However, the results of this study are in contrast with the majority of the studies mentioned in this review, in which the role of L/G ratio was emphasized as an influencing factor in release rate. Furthermore, it has been demonstrated that the morphology of polymer 19,22and also route of administration 22 have a considerable impact on the protein release rate.
Protein related factors
In addition to the aforementioned parameters affecting protein release from PLGA particles, the physicochemical characteristics of loaded protein (i.e. MW, solubility, distribution coefficient, hydrophobicity, and the number of free thiol groups and/or disulfide bonds) as well as protein loading19 influence their release rate and degradation behavior. It was demonstrated that release from MSs is not dependent on average protein size within the MS, but the mechanism of release is somewhat related to protein MW, pore size, distribution, and location of pores. In low protein loading values, release mechanism of larger proteins was driven by diffusion through pores, while for smaller proteins, release was initially dependent on diffusion through pores and finally on degradation. However, due to the presence of more interconnecting channels at higher loaded MSs, any difference in the release mechanism related to the protein MW was not observed in high protein loading values.66
Environmental related factors
Various parameters such as pH, temperature, ionic strength, sink condition, salts, plasticizing agents and surfactants can dramatically influence the rate of protein release from PLGA MPs/NPs.51
The pH of release medium has a key effect on protein release by acting on the polymer degradation rate and protein stability due to its large influence on protein conformation.55,59 Zolnik et al.47 demonstrated that the degradation and release pattern of PLGA MSs under both neutral and acidic pH exhibits a tri-phasic pattern with a similar burst release and lag phases, following with a zero order phase being accelerated at pH 2.4. Moreover, it was exhibited that MS at pH 2.4 had smooth surface without any channels and degraded more homogenously than that at pH 7.4. In a study conducted by Silva et al.,62 it was exhibited that the pH of a first emulsion (W1) in a double emulsion solvent evaporation method had a considerable effect on encapsulation efficiency and burst release. As the pH changed from acidic to alkaline, not only the high burst release drastically decreased to 10% but also the encapsulation efficiency enhanced to about 40%. It was speculated that pH alteration to values above the peptide isoelectric point, would affect the peptide charge distribution and subsequently its hydrophobicity.
The increase in ionic strength of the release medium often induces a decrease in the release rates. Presumably, the increased ionic strength reduces the swelling of the polymer matrix by dropping the diffusion of the protein from the MSs.62 Furthermore, most of the in vitro release studies have been done in PBS pH 7.4 at 37°C due to its similarity to physiological conditions, in which there is no preliminary stability studies. However, some researchers performed protein stability studies to assess the in vitro release in an optimal release medium (e.g., acetate, citrate or Tris–HCl buffers).55
Increased temperature, which raises all chemical reactions, augments the mobility of the PLGA polymer and subsequently the rate of pore closure. Moreover, in vivo studies indicated faster release and shorter lag phase in enhanced temperature due to the presence of enzymes, lipids, non-sink condition and possible impact of immune system.51 In conclusion, due to the presence of many parameters influencing particle degradation and also the interaction between protein and PLGA polymer, accurate design of a particle vaccine delivery system with a controlled release rate is a complex challenge. Hence, in vivo studies are required to verify the real behavior of any particular protein-PLGA delivery system.48,51
Impact of administration route of PLGA particles on immunogenicity
The in vivo prediction of PLGA particles behavior is not as simple as in vitro because of the complexity of environment and the presence of various enzymes. In various routes of administration, PLGA particles are internalized by different types of cells. Therefore, this parameter plays a key role in type and magnitude of the induced immunity.7,67-69 It has been reported that oral administration is the best route of mucosal immunization; however, orally administeredPLGA MPs are more likely to be hydrolyzed due to low pH of stomach and its extensive range of enzymes. On the other hand, efficient uptake of PLGA MPs by specialized mucosal associated lymphoid tissue (MALT) has been established.70 A novel approach to make PLGA particle more stable in gastrointestinal fluids relies on using PLGA co-polymer or tri-block co-polymer with hydrophilic moieties such as polyethylene glycol (PEG).71
In an early research, it was found that oral administration of B. pertusis antigens- loaded PLGA particles requires higher dose of antigens and 2 booster immunizations in comparison with parenteral administration.72 It was also indicated that polarized Th1 response is induced following intraperitoneal (IP) or intramuscular (IM) immunization by B. pertusis antigens- loaded PLGA particles. However, subcutaneous (SC) route was the least effective parenteral routs in induction of Th1 response. Hamdy et al. 7 also emphasized the strong relationship between the route of administration and the type of cell involving in PLGA particle phagocytosis. It was shown that PLGA particles were internalized by DCs through SC or intradermal (ID) routes, whereas IP administration resulted in PLGA uptake by macrophages. In order to have better understanding of the impact of administration routes on eliciting immune responses, it is required to compare the in vivo behavior of PLGA particles with the same size and loaded protein but using different immunization routes.
Influence of particle size on immunogenicity
Undoubtedly, finding optimum particle size for eliciting potent immune response against antigens has been a challenging issue until present. Surprisingly, the outcomes of different studies evaluating the effect of particle size on eliciting immune response are incompatible. In astudy conducted by Gutierro et al., a higher total IgG response was elicited by 1,000 nm-sized BSA-loaded PLGA particles in BALB/c mice than that obtained with 200 and 500 nm particles. These findings can be attributed to enhanced access of 1000 nm-sized particle to the APC, although antigen processing and presentation are similar in all PLGA particles.73 In a research using SPf66-loaded PLGA particles_ENREF_74, it was deduced that intranasal administration of 1.5 μm-sized SPf66-loaded PLGA MPs results in higher IgG antibody level compared to the conventional alum adjuvant and to the other routes. However, SPf66 loaded NPs showed a poor immunogenicity only superior to the free peptide group.74
Wendorf et al. revealed that PLGA NPs and MPs, with 110 nm and 800–900 nm in diameter, respectively, are similar in eliciting immune response according to IgG titers in mice.16The results of a study conducted by Thomas et al. confirmed that HBsAg-loaded PLGA MSs in ∼5 μm diameter could elicit a more robust immune response when compared to those with a 12 μm diameter.75 Cruz et al. demonstrated that both DC-targeted PLGA MPs and NPs caused antigen protection against rapid degradation after particle ingestion by DCs. However, DC-SIGN-targeted and PEG-coated MPs and NPs demonstrated different targeting behavior toward human DCs. PLGA NPs was effectively directed to DCs through DC-specific antibodies. In contrast, uptake of PLGA MPs by DCs was mainly accomplished through nonspecific phagocytosis.76 In another research, it was clearly demonstrated that staphylococcal enterotoxin B toxoid loaded-PLGA particles smaller than 10 µm were remarkably more immunogenic than the larger ones (10–110 µm) according to serum IgG response.77,78 Furthermore, it was shown that PLGA NPs less than 500 nm could elicit more potent CTL responses compared to MPs with size larger than 2 µm.7
Moreover, particle size may determine whether the particles passively drain to secondary lymphatic organs or are up-taken and transported there by the peripheral APCs. It was proven while larger particles (500–2000 nm) were mostly associated with DCs at the in injection site, small particles (20–200 nm) were found in LN-resident DC and macrophages, suggesting their free drainage to the LN. However, small (20 nm) and large (1000 nm) NPs showed differential localization in the popliteal LN.79 In addition, particle drainage was size dependent, as particles with a range of 20–200 nm were drained via lymphatic system within few hours. However, DCs took up larger particles (200–500 nm) and carried them to lymph node during 24 hours after administration.68
Altogether, PLGA particle size and size distributions are important attributes to determine induced immunity responses byantigen- loaded PLGA particles. It can be concluded that PLGA particles with the size between 200 nm and 2 µm can elicit better immunity responses. Moreover, it should be kept in mind and emphasized that immunity responses are complex and governed by multiple parameters including administration routes of vaccination, physicochemical characteristics of PLGA particles (e.g., size, surface charge and morphology that influence antigen release profile), antigen features, and of course the presence of specific DC targeting moieties or other particle surface modifications.
In vivo and in vitro studies using PLGA particles
The advantages of using PLGA particles as delivery vehicles, have been investigated in diseases such as hepatitis B, malaria, leishmaniasis, tuberculosis, toxoplasmosis and etc. Many studies in different disease models, indicated that long lasting immunity can be achieved by administration of PLGA particles, which can avoid the requirement of multiple injections of the conventional vaccines.36,58,80 For example, protective humoral and cellular immunities that were achieved by a single-shot HBsAg adsorbed PLGA MPs were similar to the response induced by booster injection of alum adsorbed HBsAg vaccine.58 Furthermore, a single subcutaneous injection of HBsAg-loaded PLGA MSs in mice led to comparable antibody responses to those of three injections of HBsAg aluminum vaccine.36 In a study by Jaganathan et al., PLGA MSs bearing recombinant HBsAg was stabilized by using the combination of protein stabilizer (trehalose) and antacid (Mg(OH)2). Addition of a protein stabilizer and an antacid resulted in an enhancement in antigen stability during entrapment and release from the MSs.80 It was found that a single injection of PLGA MSs gave rise to an almost equivalent anti-HBsAg antibody levels comparing to two injections of alum-adsorbed HBsAg vaccine in guinea-pigs. Therefore, this delivery system is able to be administrated as a single-shot carrier adjuvant in development of vaccines and can prevent the need for multiple doses in next generation vaccines.80 In addition, a remarkably high and long-lasting immune response associated with strong protective capacity was achieved with a single dose administration of SPf66 encapsulated PLGA MSs in Aotusmonkeys.81 These results provide additional evidence that PLGA particles can be applied as a delivery system to induce protective immunity after a single dose administration of a vaccine.81 Furthermore, small-sized PLGA particle in the presence of CpG can be useful for developing Th1-polarized immunity in asthmatic response induced by house dust mite (HDM) allergens.82 PLGA functions more efficiently and demonstrates longer potency and fewer side effects as compared to the conventional Al(OH)3 adjuvant. Therefore, PLGA-allergen nanoparticles can act suitably as candidates for allergy vaccines.83
The breadth of research being conducted in the field emphasizes the importance of PLGA particles as delivery system for protein/peptide antigens. A subset of studies using PLGA particles as delivery system of various antigens are summarized in Table 6. This table provides a useful tool to compare and correlate in vitro and in vivo results.
Table 6.
In vitro and in vivo results of studies using PLGA particle containing different disease's antigens.
| Antigen | In vitro result | In vivo result | Ref. |
|---|---|---|---|
| PLGA-Hepatitis B antigen | |||
| PLGA NPs containing HBsAg (Pulmonary immunization) |
|
|
84 |
| PLGA-Maralia antigen | |||
| PLGA 50:50 MSs loaded with SPf66 (a 45 amino acid peptide derived from different merozoite stage-specific protein fragments of Plasmodium falciparum) |
|
|
30 |
|
|||
| PLGA MPs loaded with SPf66 (Intranasal administration) |
|
|
74 |
| PLGA-Leishmania antigen | |||
| PLGA NSs loaded with an experimental autoclaved Leishmania major (ALM) |
|
|
85 |
| Co-administration of Quillaja saponins (QS) with PLGA NSs encapsulated with ALM |
|
||
| PLGA NPs loaded with a plasmid DNA encoding kinetoplastid membrane protein (KMP-11) followed by PLGA NPs loaded with the recombinant KMP-11 protein in the presence of CpG | — |
|
86 |
| PLGA NPs loaded with KMP-11 |
|
— | 87 |
| PLGA- Tuberculosis antigen | |||
| PLGA MPs encapsulated with immunodominant antigens 6-kDa early secretory antigenic target (ESAT-6) and antigen 85B (Ag85B) |
|
|
88 |
| PLGA MPs encapsulated with TB10.4-Ag85, a recombinant fusion protein of TB antigens, in respirable sizes |
|
— | 89 |
| PLGA-Chlamydia antigen | |||
| PLGA 85:15 NPs encapsulated with rMOMP-187 (a peptide derivative of Chlamydia trachomatis major outer membrane protein) |
|
— | 90 |
| PLGA 50:50 NPs encapsulated with rMOMP |
|
|
91 |
|
|||
| PLGA- Toxoplasma antigen | |||
| PLGA MPs encapsulated with surface antigen 1 (SAG1, the major immunodominant surface antigen of T. gondii tachyzoites) |
|
|
38 |
| PLGA MPs encapsulating immunodominant epitopes of a chimeric protein consisting of SAG1 and SAG2 |
|
|
39 |
| PLGA-Allergy antigens | |||
| PLGA NPs loaded with Caryotamitis profilin (rCmP) (allergens in pollen, latex, and plant foods) |
|
|
83 |
| PLGA particles coated with Dermatophagoidespteronyssinus (Der p) |
|
|
82 |
|
|||
|
|||
|
|||
Immunomodulation with PLGA particles containing TLR ligand
The systemic administration of toll like receptor (TLR) ligands is associated with high levels of serum cytokine and toxicity, which may cause immunosuppression and prevent immune responses against subsequent infections. Particulate delivery systems can reduce the required dose of antigen or adjuvant, protect degradation and enable co-administration of vaccine antigens and immunomodulators to the same cell type (Fig. 5).70,92 Therefore, the safety profile of adjuvants can be improved by dose reduction and also by alleviating the toxicity at non-targeted tissues.8 It was found that co-delivery of antigens along with TLR ligands via PLGA MPs/NPs can result in selective stimulation of DCs to produce natural killer (NK) cell-activating cytokines. Activation of both CTL and NK cells by a single vaccine strategy can lead to targeting and killing MHC class I positive and also negative tumor cells.7Additionally, TLR ligand binding to modified PLGA MPs can be applied for DC maturation or specific amplification of the immunological response to encapsulated antigens in DC-based cell therapies or in vaccination trials.93
Figure 5.
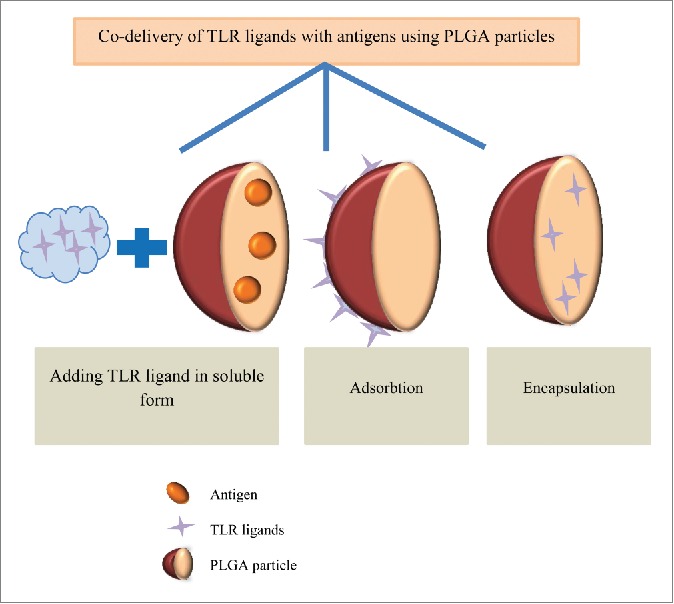
Co-delivery of immunepotentiators and antigens in PLGA particles. To enhance the therapeutic efficacy of PLGA particles, different TLR ligands (e.g., CpG, poly(I:C) and MPLA) can be adsorbed, encapsulated or added into soluble form along with antigen-loaded PLGA particles.
CpG, a bacterial DNA representing pathogen-associated molecular pattern (PAMP), activates DCs maturation and facilitates cross presentation of antigens by targeting TLR9. Unlike alum which is a Th2 adjuvant, CpG effectively induces potent Th1 responses. In a review article by Malyala et al., different formulations of CpG in PLG MPs; including addition of CpG in soluble form, adsorption to charged PLGA MPs and encapsulation within MPs have been discussed.8
Polyriboinosinic acid–polyribocytidylic acid (poly(I:C)), a double-stranded (ds) RNA virus associated danger signal, interacts with TLR3, which is located in the membrane of the endosomal compartments of most APCs.94This ligand requires internalizing for its action; therefore, unexpected activation of non-targeted cells is prevented.92 Poly(I:C) as Th1 immunomodulator has the ability to activate monocytes and NK cells to produce pro-inflammatory cytokines and chemokines which results in DC maturation and consequently stimulation of both humoral and cellular immunity.95
Monophospholipid A (MPLA), a nontoxic analog of lipid A, is a TLR4 agonist. Many researchers have applied this Th1-immunomodulator for the enhancement of Th1 and CTL responses.96 In the following section, co-delivery of antigens and various TLR ligands will be explained. Some studies using different immunomodulators are indicated in Table 7.
Table 7.
Co-administration of different immunomodulators and antigen loaded- PLGA particles.
| Antigen + immunomodulator | Results of the study | Ref. |
|---|---|---|
| CpG | ||
| Co-administration of CpG ODN adsorbed onto PLG MPs with Anthrax Vaccine Adsorbed (AVA, the licensed human anthrax vaccine) |
|
97 |
| Co-encapsulation of protamine (DNA stabilizer) with CpG and phospholipase A2 (PLA2) (the major bee venom allergen) in PLGA MP-based allergy vaccine |
|
98 |
| Co-encapsulation of ALM and CpG-ODN adjuvant in PLGA NSs |
|
99 |
| Poly(I:C) | ||
| Encapsulation of poly(I:C) or CpG-ODN with OVA antigen in PLGA NPs |
|
100 |
|
||
| Incorporation of alginate and/or poly(I:C) in PLGA MSs containing synthetic SPf66 malarial antigen |
|
95 |
|
||
| Binding of poly(I:C) to the surface of cationic, diethylaminoethyl dextran (DEAE dextran) modified PLGA MPs via electrostatic interactions |
|
93 |
| MPLA | ||
| DCs pulsing with PLGA NPs containing MPLA |
|
101 |
| DCs pulsing with PLGA NPs containing a MUC1 lipopeptide (BLP25) and MPLA |
|
|
| Loading of (HBcAg+MPLA) in PLGA MSs (single immunization) |
|
96 |
CpG
Some studies investigated the optimal way to formulate CpG in PLG particulate delivery system.102,103 For example, 3 formulations including addition of CpG in soluble form, CpG adsorption on, and CpG encapsulation into PLGA MPs with the adsorbed protein antigen from Neisseriameningitidis B were evaluated. The results indicated that CpG encapsulated formulations led to increased immunopotentiator effect which was indicated by significant higher antibody, bactericidal activity and T cell responses when compared to the traditional method of delivering CpG in the soluble form.103 In another study, the magnitude and kinetics of humoral and cellular immunity were measured in MPs containing OVA and CpG (MP/OVA/CpG), MPs comprising OVA, soluble OVA plus CpG, or OVA formulated with Alhydrogel® aluminum hydroxide adjuvant. The highest Th1- and Th2- associated antibody isotypes were achieved by MP/OVA/CpG. In addition, the highest antigen-specific IFN-γ lasted up to 42 post-vaccination was elicited by MP/OVA/CpG. The results of this study gives applicable data for designing optimal vaccination regimens in MP-based protein vaccines.102 However, it was found that polymer nature and MP characteristics are 2 affecting factors in the outcome of CpG and antigen co-encapsulating. For example, when OVA and CpG sequences were co-encapsulated into Resomer®RG PLGA 502 and 756 MPs, no improvement in cellular immune response was seen in OVA and CpG microparticulate co-delivery in comparison with CpG and OVA co-administration.104
Fischer et al. described the importance of CpG release kinetics on the immunogenicity of antigen-containing PLGA MPs. They microencapsulated the CTL-restricted OVA peptide SIINFEKL into bare, chitosan-, and protamine-coated PLGA MPs. CpG adsorption or coupling was performed onto the surface of these MPs. It was found that the duration of CpG release is related to the surface charge of the PLGA MPs. When positive zeta potential values of polycationic coating increases, the binding of CpG to them enhances and the release of both the antigen and CpG decreases. Whereas, in single immunization of mice with these different formulations, only the uncoated PLGA MP with adsorbed CpG-ODN causes significant increase in IFN-γ secreting CD8+ T cells.105
MPLA
To enhance vaccine immunogenicity, the synergistic effect of immunoadjuvants can be examined and therefore adjuvants with different mechanisms can be combined.31,106 For example, application of CpG inside PLGA MS and MPLA in injection solution (but not inside the MSs) caused significant increased immune response.106 Furthermore, IFN-γ ELISPOT responses to multiple specific CTL epitope scan be elicited by this PLGA vaccine delivery system carrying 2 specific CTL epitopes of OVA antigen. It has been also reported that combination of α-galactosylceramide (αGC), a synthetic glycolipid that can be loaded into non-classical MHC CD1d molecules and stimulate invariant NKT cells, and MPLA results in marked higher cellular immune response.31
In another approach, to prevent antigen denaturation that may occur in encapsulation process and also to mimic microbial pathogens in their structure and surface chemistry, antigens were bound to the surfaces of particles (Fig. 6). In this regard, OVA as a model antigen was conjugated to PEGylated lipids incorporated in the lipid shells of PLGA particles. Lipophilic molecular danger signals such as MPLA and αGC were incorporated into the surface bilayers of these particles. Strong antibody titers were induced by these antigen-displaying particles at antigen doses of only a few nanograms, far below the conventional doses used in mice. Conventional alum adjuvants, MPLA or αGC did not have the ability to induce immunity at such low doses of antigen. However, when MPLA was co-displayed on the membranes of particles, the highest IgG titers sustaining for over 150 days at the lowest antigen doses was elicited. Incorporating αGC into the coatings of antigen-conjugated particles resulted in rapid IgG production after a single immunization, which can be applied in emergencies such as disease pandemics.107 In another example of this strategy, a recombinant antigen derived from the circumsporozoite protein of Plasmodium vivax, VMP001, was conjugated to the lipid membrane of the PLGA core particles. Then, the lipid-enveloped PLGA NPs was incorporated with MPLA, creating a pathogen mimicking NP vaccine (VMP001-NPs). This vaccine delivery platform elicited potent and long-lasting antigen-specific humoral immune responses being characterized by enhanced avidity and capacity toward the domains within the circumsporozoite protein with the ability to neutralize live sporozoites. Totally, this vaccine platform with appropriate materials for clinical studies can be used for controlling drug release as well as enhancing protective immunity from weakly immunogenic subunit vaccines.108
Figure 6.
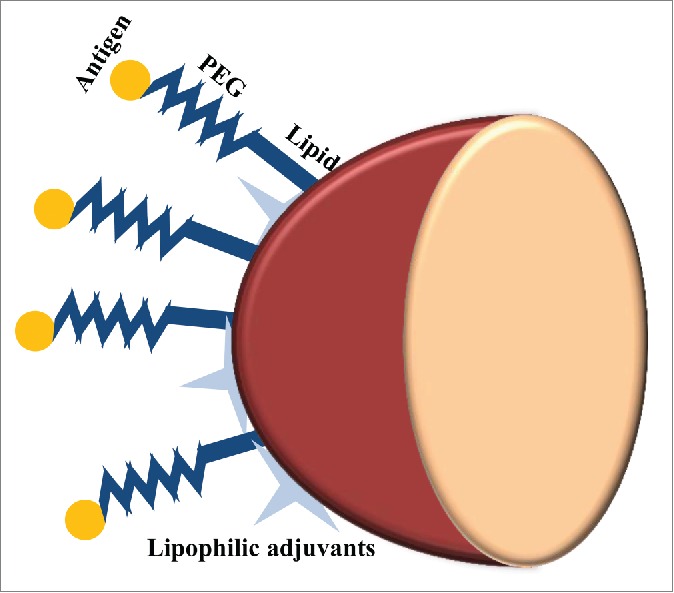
Pathogen-mimicking PLGA particles as vaccine delivery platform. In order to avoid antigen denaturation that can be caused by encapsulation process and preserve the structure and surface chemistry of antigens, they are conjugated to the PEGylated lipid shell of PLGA particles. Lipophilic molecular danger signals such as MPLA and αGC can also be incorporated into the surface of these lipid-enveloped PLGA particles.
Surface modification of PLGA particles
Surface modifications of PLGA particles with chitosan and cationic agents, which is used, respectively, to facilitate mucoadhesion and to enhance the induced immunity response are described in the following section, summarized in Table 8.
Table 8.
Examples of surface modified PLGA particles.
| Antigen | Surface polymer | Results of surface modification | Ref. |
|---|---|---|---|
| HBsAg | Chitosan | - Facilitating the mucoadhesion | 109 |
| - Production of both humoral (systemic and mucosal) and cellular immune responses | |||
| HBsAg | Chitosan | - Significant enhancement of serum specific IgG antibody responses | 110 |
| — | Chitosan and PEG | - Reporting the detail profile of induced cytokine by oral administration of modified PLGA particles | 111 |
| HBsAg | N-trimethyl chitosan (TMC) | - Induction of marked increased in anti-HBsAg titer comparing to plain PLGA MPs | 112 |
| HBsAg | TMC | - Induction of high serum antibody titers and secretory IgA levels by nasal immunization with the fast antigen releasing TMC NPs | 113 |
| BSA+imiquimod | TMC | - Induction of a high systemic humoral and mucosal immunity both at local and distal sites | 114 |
| HBsAg | Stearylamine or polyethylenimine | - Enhancement of both systemic and mucosal immunity by pulmonary administration of positive surface charged PLGA particles | 37 |
| — | Wheat germ agglutinin (WGA), RGD containing peptide, mannose-PEG3-NH2, poly-l-Lysine, (PLL) | - Increasing the uptake of PLGA MPs through macrophages phagocytosis by grafting cell-specific ligands on them | 115 |
| OVA | Protamine | - Enhancement of antibody and T-cell immune responses | 116 |
| OVA | Protamine | - Enhancing the cross-presentation of encapsulated antigen by facilitating antigen uptake and lysosomal escape | 117 |
Chitosan
The surface modification of PLGA MSs with chitosan109-111 can facilitate the mucoadhesion through the change of zeta potential from negative to positive without affecting the particle size and dispersion. Moreover, a clearance rates from the nasal cavity can be reduced by this modification.109 In addition, the surface modification of MSs with chitosan caused induction of higher potency and long lasting immune responses when compared with unmodified MSs.110
N-trimethyl chitosan (TMC) particles which carry a positive charge independent of pH, demonstrate increased solubility and are much more stable at neutral pH than chitosan particles. Nasal immunization of mice with TMC-coated PLGA particles loaded with HBsAg led to a marked enhancement in anti-HBsAg titer compared to nasal immunization with HBsAg loaded PLGA particles.112 In another study, nasal immunization with fast antigen releasing TMC based NP (prepared by ionic complexation) was superior in the elicitation of antibody responses compared to PLGA NP and TMC-coated PLGA NP (prepared by emulsification/solvent extraction). This response may be related to some features of TMC NP including mucoadhesiveness for extending the nasal residence time, the rapid release of the contained antigen for promoting antigen uptake by B-cells, and the ability to stimulate DC activation. Therefore, the particle charge and antigen release properties of NPs influence the extent and the type of immune response elicited by nasal vaccination.113
Recently, Primard et al. described a method for the co-encapsulation of a hydrophilic antigen and a hydrophobic imiquimod, an agonist of TLR-7 as immunostimulatory molecule. PLGA NPs were further coated with a mucoadhesive chitosan-derivate layer. Intranasal delivery of this vaccine delivery system resulted in a high systemic humoral and mucosal immune response, both at local and distal sites of injection. Co-delivery of imiquimod led to polarization of the immunity only at local sites, while the intensity of the immune response was preserved at distal sites. Therefore, the topical delivery of encapsulated imiquimod was safer vaccine compared to its systemic administration.114
Cationic agents
The uptake of ligand-grafted MPs results from both specific and non-specific processes and maybe dependent on the ligands coating the particles and also the particle charge. For example, in a study conducted by Brandhonneur et al. the uptake of cationic MSs was linear and characterized by non-specific uptake mechanisms. However, the uptake of negatively charged ligand-grafted MSs was enhanced 2 to 4 times depending on the ligand type, and showed non-linear specific saturatable characteristics. It was further observed that the particle-to-cell ratio influenced the rate and the efficiency of particle uptake. Therefore, suitable ligands should be evaluated for their ability to increase cellular uptake of MPs.115 It was found that HBsAg-loaded cationic surface charged-PLGA MPs led to enhanced systemic and mucosal immunity compared to unmodified particles. However, other parameters including particle diameter, drug content, and hydrophobicity may also have influence on the enhanced efficacy of positively charged PLGA MSs.37 Furthermore, coating PLGA MPs with protamine, an arginine-rich peptide with strong basic charge, resulted in increased antibodies and T-cell responses in mice.116 In addition, cellular evidence for enhanced cross-presentation of exogenous antigens encapsulated in protamine-coated PLGA NPs through facilitated antigen uptake and lysosomal escape was provided. These results suggest that protamine-modified PLGA NPs maybe used as a potent adjuvant for cellular vaccines.117
Functionalizing PLGA particles for targeting to M-cell
Oral and/or nasal vaccines have the ability to induce systemic as well as mucosal immunity and also demonstrate much higher patients' compliance comparing to parenteral vaccination. This economic route of administration overcomes the disadvantages of parenteral immunization, which include high production cost, patient discomfort and risk of disease transmission.118,119 PLGA particles can be taken up by transport system via specialized membranous epithelial cells (M-cells) located in follicle-associated epithelium (FAE) of the Peyer's patches present along the gastrointestinal tract and subsequently can elicit immunity after oral administration.118,119 The ability of M-cells to transcytose the antigen from the gut lumen to the underlying lymphoid tissues causes a mucosal immune response. Due to the presence of M-cells at a very low density in gut, efficient targeting of these cells can enhance antigen uptake in oral vaccine delivery. The distinctive glycoconjugate profile of M cells surface plays an essential role in targeting of orally administered NSs/MSs.120 Different strategies for M-cells targeting using PLGA particles (Fig. 7) will be discussed with approach summarized in Table 9.
Figure 7.
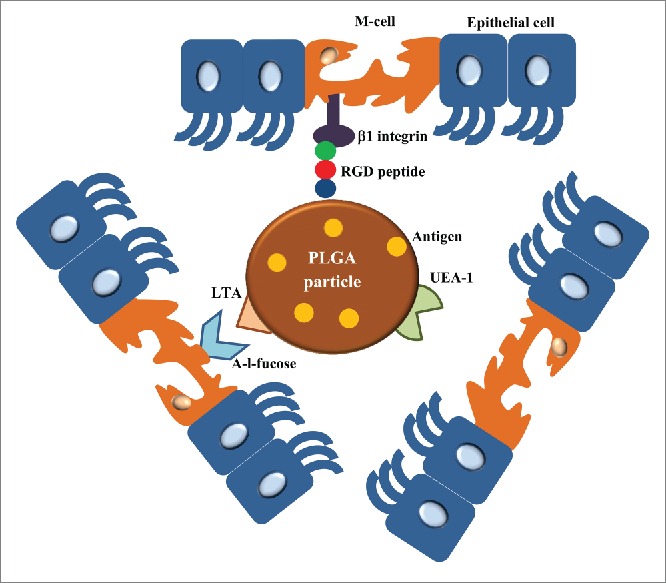
Different ways to target PLGA particles to M-cells. The surface of PLGA particles can be grafted with RGD peptide or LTA which are interacted with β1 integrin and α-l-fucose presented in the surface of M-cells, respectively. UEA-1 is another M-cell targeting agent for oral-mucosal vaccine delivery. Using these M cell targeting agent-anchored PLGA particles, the orally administrated antigens loaded in PLGA particles are targeted to M-cells to generate mucosal immunity.
Table 9.
Targeting of M-cells using modified PLGA particles.
| Antigen | Targeting strategy | Result | Ref. |
|---|---|---|---|
| HBsAg | Ulexeuropaeus 1 lectin (UEA-1) targeting mouse Peyer's patch |
|
120 |
|
|||
| OVA | RGD peptide targeting β1 integrins (expressed at the apical side of M cells) |
|
71 |
| OVA | Non-peptidic ligands (LDV peptidomimetic targeting β1 integrins, RGD peptidomimetics interacting with β1 and β3 integrins) |
|
121 |
|
|||
| HBsAg | Lotus tetragonolobus (LTA), targeting α-l-fucose receptors (presenting on the surface of M-cells) |
|
122 |
|
|||
| OVA+MPLA | UEA-1 |
|
123 |
|
|||
| a membrane protein B of Brachyspira hyodysenteriae (BmpB) | M cell homing peptide (CKS9) |
|
124 |
In vivo studies indicated that targeting of PLGA particles to Peyer's patch M-cells using different strategies including Ulexeuropaeus1 lectin (UEA-1),120 Lotus tetragonolobus (LTA) from Winged or Asparagus pea,122 and M cell homing peptide-coupled chitosan leads to the enhancement of mucosal and systemic immune responses through oral administration.124 However, M cells targeting using RGD peptide, which was grafted on PEGylated PLGA-based NPs did not result in enhancement of IgG production because of partial degradation of the RGD peptide during its trafficking in the gastrointestinal tract.71 To avoid peptide degradation, non-peptidic ligands were incorporated in PLGA NPs. Higher IgG antibodies and also cellular immunity were elicited by intraduodenal administration of non-peptic grafted PLGA NPs comparing to either non-targeted or RGD NPs.121 In another study, the TLR-agonist MPLA was further incorporated in the UEA-lipid NPs. MPLA led to up-regulating of the phagocytosis of mucosal DCs. This oral delivery system has the ability to transport vaccine antigen to M-cell which then can be taken up by underlying DCs. The in vivo oral vaccination of OVA-loaded with UEA-MPL/PLGA NPs led to the highest induction of functional mucosal IgA and serum IgG antibodies as comparing to other oral formulations. The results of this study suggest that this vaccine delivery system is a promising strategy for boosting immune responses to oral vaccines.123 Overall, M-cell targeted PLGA particles containing TLR agonist suggests a promising strategy for boosting oral immunity.
Enhancing immunotherapeutic effect of DC-based vaccine using PLGA particles
Intracellular trafficking of PLGA MPs/NPs
Endogenous antigens are processed in the cytosol and presented by major histocompatibility complex (MHC) class I molecules, which are expressed in all nucleated cells. However, “extracellular” antigens are processed in endocytic vesicles or endosome and presented by MHC class II molecules, which are only expressed in APCs. Antigen presentation on MHC I and II results in activation of antigen specific CD8+ and CD4+ T cells, respectively. Some APCs can direct the endocytosed “exogenous” antigens into the MHC class I pathway, which is called cross-presentation.7 Targeting of vaccine antigens to DCs, the most professional APCs, leads to the enhancement of antigen-specific T cell responses.7 However, the application of DC vaccination is limited due to the rapid degradation of internalized protein by DC and also the transient nature of MHC class I epitopes on the DC surface following external loading of motif-fitting peptides. Antigen encapsulation protects antigens from degradation by lysosomal proteases through endosomal escape. The antigen encapsulation can also lead to extending Ag release and consequently more effective MHC peptide complexes presentation to CD8+ T-cells.125
PLGA MPs/NPs could efficiently promote antigen entrapment and retention in local lymph nodes providing non-specific delivery of antigens to APCs such as macrophages and DCs.126 PLGA particles possess the capability of antigen targeting toward DCs without any specific recognition because of their size similarity to pathogens which immunity system has evolved to combat them through years.7,70 The other features of PLGA MPs/NPs such as shape, surface charge, hydrophobicity, and hydrphilicity also have an influence on particle uptake by APCs.7,127 The uptake of PLGA MPs/NPs by DCs has been evaluated in several studies and it was indicated that the majority of DCs are able to internalize PLGA MSs/NSs within a 24 h incubation period. Cytochalasin B, a reagent prohibiting actin polymerization, terminates MPs/NPs uptake which clarifies the involvement of actin polymerizationin the phagocytosis of PLGA NPs.7 First, PLGA MPs/NPs associate with cell membrane; afterwards, intercellular uptake of PLGAMPs/NPs is performed through an endocytic process including phagocytosis, fluid phase, pinocytosis or by receptor-mediated endocytosis in clatherin or caveolin -coated vesicles which are concentration and time dependent.63,127 Subsequent to the internalization of PLGA MPs/NPs and primary endosome formation, PLGA particles can be processed by different pathways including phagosome to cytosol, vacuolar pathways of cross presentation, and classical endocytic pathway via MHC II antigen presentation.7 The majority of MPs/NPs are transitioned to secondary-endosomes and fuse with lysosome. MPs/NPs inside endo-lysosomes may rapidly escape from them within 10 minutes, and enter the cytoplasm where they release the encapsulated protein. A fraction of particles are transferred to recycling-endosome and convey back to cell exterior.63,128 In addition, surface charges of PLGA MPs/NPs play considerable role in their interaction with cells and subsequently their uptake and internalization.19,127 Cationic particles are particularly more effective for uptake by DCs and macrophages because of ionic interactions which happen between negatively charged cell surface and cationic particles. It should be mentioned that zeta potential of PLGA particle is changed by environmental pH.7,68 Typical intracellular pathway for PLGA MPs/NPs in DCs is depicted in Figure 8.
Figure 8.
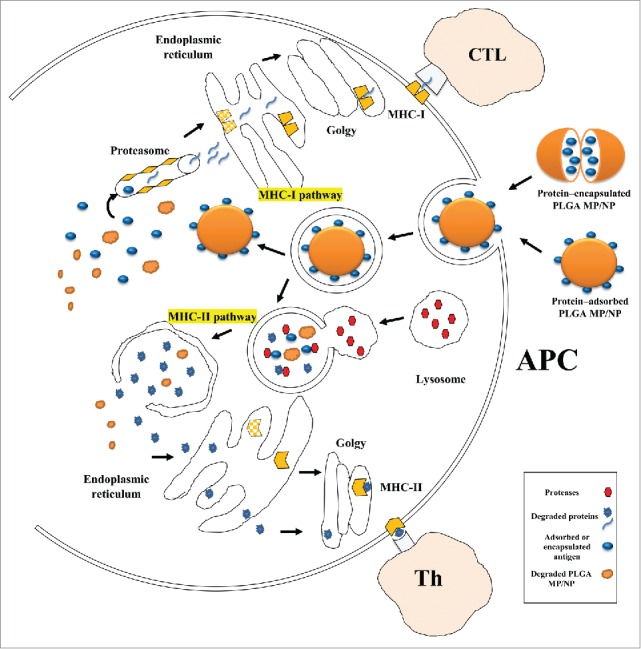
Schematic presentation of intracellular trafficking of protein/peptide - loaded PLGA particles in antigen presenting cells (mostly dendritic cell). Desired protein can be adsorbed on or encapsulated into PLGA particles, hence due to simplification only protein- adsorbed PLGA particles is shown. PLGA particles are taken up by endocytosis following binding to DC membrane. Antigens delivered by PLGA particles undergo 2 distinct intracellular pathways. In phagosome to cytosol pathway of cross-presentation, PLGA particles escape from the endosome, degrade in cytoplasm, and loaded protein is release gradually, and continue the MHC class I pathway. Secondly, degradation of protein/peptide-loaded PLGA particle happens inside endosome due to its acidic pH, thus the antigenic peptides derived from degradation can be presented via MHC class II pathway. Antigen presenting cells: APC; TCL: T cytotoxic lymphocyte; Th: T-helper lymphocyte.
Functionalizing PLGA particles for targeting to DCs
Particle-based Ag delivery systems have the ability to transfer Ag and adjuvants concomitantly to the same APC, which led to efficient induction of T cell responses.129 Furthermore, the co-administration of antigens with TLR ligands into DCs may lead to NK cells activation through NK cell-activating cytokines which are produced by activated DCs. Therefore, the requirement for systemic administration of NK cell-activating cytokines can be circumvented by this strategy.7 The review by Hamdy et al. focused on the simultaneous targeted delivery of cancer antigens and immunostimulatory adjuvants to DCs.7 In order to enhance DC targeting of PLGA NPs, particles surface can be decorated with ligands of different receptors expressed on the surface of DCs (Fig. 9a).7 DC targeting of PLGA particle by particle modification is captured in Table 10. It has been also shown that no change of MoDC in terms of expression of the surface markers including CD80, CD83, CD86, HLA-DR and macrophage mannose receptor (MMR) is found after exposure of these cells to differently coated PLGA MPs. As the phenotype and function of DCs upon phagocytosis of polyelectrolyte-coated PLGA MPs remain unaltered, these MPs can be used for ex vivo antigen loading of MoDC in cellular immunotherapy.130
Figure 9.
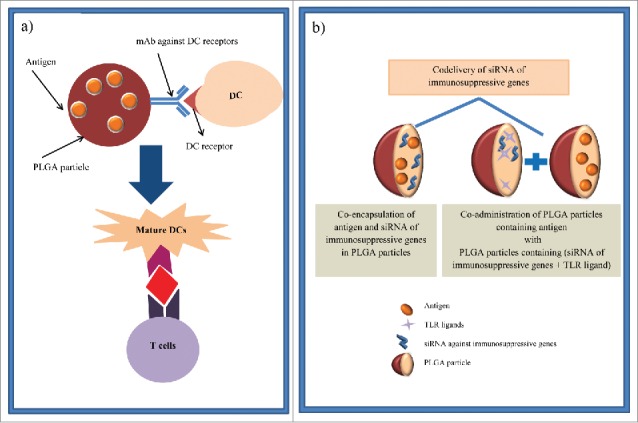
Schematic illustration of strategies to enhance the therapeutic efficacy of DC-based vaccines via PLGA particles. (a) PLGA particles are linked to specific antibodies against DC receptors (e.g., DEC-205 and CD11c) to target vaccine components which are loaded in these particles specifically to DCs. Therefore, DC maturation and consequently CD8+ activation are occurred. (b) In another strategy, siRNA of suppressive gene can either co-encapsulate in PLGA particles containing antigen or encapsulate in distinct PLGA particle with TLR ligands and co-administer with antigen loaded-PLGA particles. Then, DCs are activated by these PLGA particles and antigen specific immunity is induced.
Table 10.
Different strategies for DC targeting of PLGA particles.
| Antigen | Targeting agent | Result | Ref. |
|---|---|---|---|
| OVA peptide + TLR ligand | DC-SIGN–specific humanized Abs | ✓Enhanced DC maturation, activation and CD8+ responses | 92 |
| F(ab′)2 fragments from a rat Ab-recognizing mouse DEC-205 | ✓Enhanced CD8+ T-cell activation and CTL responses | ||
| ✓Enhanced adjuvanticity and reduced toxicity of TLR ligand | |||
| Melanoma antigen recognized by T-cells (MART-1 Ag) | Biotinylated anti-DEC-205 | ✓Enhanced antigen uptake and subsequent CD8+ T-cell stimulation by DCs | 125 |
| OVA+ Poly(I:C)+ resiquimod | mAb against receptors including CD40, DEC-205 and CD11c integrin receptor | ✓Induction of potent CD8+ immunity with higher efficacy than non-targeted NPs | 129 |
| ✓No significant difference in immunological responses of 3 different mAb-coupled-PLGA NPs | |||
| 830–844 region of tetanus toxoid (TT epitope) | Humanized antibody against DC-SIGN | ✓Induction of antigen-specific T cell responses at 10–100 fold lower concentrations than nontargeted NPs | 76 |
Different DCs receptors including C-type lectin receptors (DC-SIGN and DEC-205),76,92,125 TNF-α family receptor (CD40),129 and integrin receptor (CD11c) 129 can be targeted by PLGA particles to enhance the induced immunity. For this purpose, targeting agents such as specific humanized antibody,76,92 F(ab′)2 fragments of Ab-recognizing receptors 92 have been coated on the surface of PLGA particles. It has been found that DC targeting via humanized antibody against DC-SIGN induces similar amount of cellular response to non-targeted NPs at 10–100 fold lower concentrations.76 In addition, targeting DEC-205 receptors, which are expressed at relatively high levels on myeloid blood DCs results in efficient antigen uptake and consequently anti-tumor CD8+ T-cell stimulation that is necessary for improvement of DC vaccine potency.125
In some studies, PLGA particle containing antigen and TLR ligands such as poly (I:C) and resiquimod (TLR7/8 ligand) 92,129 have been targeted to DCs by mAb against surface DC receptors. Targeting of TLR ligands to human DC-SIGN can lead to the enhancement of DC maturation and activation as well as the induction of CD8+ T-cell responses.92 In vitro and in vivo studies using DEC-205-targeted NPs containing TLR ligands have demonstrated enhanced CTL response at 100-fold lower adjuvant dose than required in soluble form administration.92 Moreover, no significant difference has been observed in induced immunological in vivo responses with the mAb-coupled-PLGA NPs which are targeted to different DC receptors (CD40, CD11c and DEC-205). Moreover, addition of a TLR agonist has led to increased T cell responses regardless of the kind of mAb coupled to the surface of PLGA NPs.129 Overall, PLGA particles which simultaneously deliver antigens and immunomodulators (e.g., TLR agonist), and also DC targeting moieties offer a potent multivalent therapeutic strategy for inducing strong immunity in different cancers.
Immunomodulation by PLGA particles containing small interfering RNA (siRNA)
To prevent the effect of some immunosuppressive genes such as cytokine signaling 1 (SOCS1)131 and STAT3 (signal transducer and activator of transcription-3) in DC-based therapies, their mRNAs could be inhibited by small interfering RNA (siRNA) (Fig. 9b).131 It has been demonstrated that the knockdown of immunosuppressive SOCS1 genes in BMDCs using siRNA leads to enhanced level of pro-inflammatory cytokine which enhances antigen presentation of BMDCs and finally activates CD8+ OVA 1.3 T cells.131 In another study, PLGA NPs containing imiquimod (R837, a DCs activator) along with STAT3 siRNAs (a silencer of immunosuppressive genes) have been internalized in DCs and subsequently the activation of the TLR pathway and the silencing of targeted immunosuppressive genes happened. Furthermore, Dc transfection with both PLGA (R837/STAT3 siRNAs) and PLGA NPs containing OVA antigen has resulted in the activation of OVA-specific T cells and enhanced anti-tumor immunity.132 Therefore, to increase the immunotherapeutic effects of DC-based therapy, delivery of siRNA of a target immunosuppressive gene to BMDCs can be combined with an immunomodulation and antigen-specific tumor therapy via these multifunctional NPs.
Conclusion
Limitations of current vaccines particularly against intracellular parasites and cancers have encouraged the application of various vaccine delivery systems in order to improve their efficacy. Among proposed vaccine delivery systems, PLGA particles have been gaining more attention, specifically regarding their widespread advantages as an antigen/adjuvant delivery vehicle. These advantages include: PLGA inherent features that can be tuned according to the desired antigen release profile, the ease of charge or hydrophobicity modification, and the ability to target and uptake by DCs. The physicochemical properties of PLGA particulates rely on a number of factors such as: polymer composition, hydrophobicity, end group, MW, particle size, and surface charge, all of them can be adjusted in order to obtain suitable release behavior 56,57 and intracellular trafficking.7 Furthermore, PLGA particles offer novel vaccine delivery approaches that can stimulate innate immunity and direct intracellular processing of antigens inside DCs toward cross presentation pathway.63 Though, the loading of PLGA particulates by encapsulation has been applied in many studies,37,39 this strategy has some drawbacks such as harsh consequences on protein integrity, and high expense due to required aseptic manufacturing.133 Surprisingly, the adsorption of antigen onto the surface of preformed MPs/NPs as a recently developed method,7 can overcome the above mentioned limitation and offer some additional benefits.133 The size of the particle delivery system is an important factor for modulating immune responses via differential interactions with APCs. However, size alone is not the only parameter that influences cellular uptake and trafficking. Significant variances have been proved in the studies when it comes to defining the optimal particle size in order to induce maximum specific immune responses. Therefore, other particle physicochemical properties such as: ploymer composition, MW, preparation methods, antigen loading percent, as well as the route of administration must be considered when designing a PLGA vaccine delivery system.12,16,19 Moreover, it would be better if all researchers follow a standardized size definition to address a particle as MPs or NPs. Further examination is needed to determine the most appropriate size of particulate delivery system for achieving the desired goal.
Induction of both CD4+ and CD8+ T cells by PLGA particles is desirable in prophylactic and also therapeutic settings for a vaccine against intracellular pathogens. Delivering the encapsulated antigens to cytoplasm (for MHC I presentation and CD8+ T cell activation) or to the endosome (for MHC II presentation and CD4+ T cell activation) is related to the physical properties of PLGA particles. Future studies are required to determine the effect of different parameters such as hydrophobicity, L/G ratio, particle size, and surface charge on the ability of PLGA particles to activate either the MHC I or II pathway.7
In order to enhance the potency of PLGA particles, immunostimulator molecules such as TLR ligands can be formulated in PLGA particles. This formulation can prevent the toxicity of immunopotentiators in non-targeted cells. Therefore, antigens can be simultaneously delivered with immunostimulators in PLGA particles to possibly decrease the dose and toxicity of the adjuvant.8 Furthermore, NK cells can be also activated by co-delivery of antigens and TLR ligands via PLGA MPs/NPs in DCs. It can be concluded that encapsulation of immuostimulators along with antigens which results in superior response compared to soluble antigen/immunnostimulator formulations, may lead to more efficacious vaccines.8 In addition, DC targeting agents can be incorporated to PLGA particles and subsequently CD8+ cells can be stimulated via cross presentation and DC maturation. Applying the combination of DC targeting moieties and TLR ligand in PLGA particles may make these particles a promising approach for cancer vaccine formulations as CD4+, CD8+ T cells and also NK cells are activated.
Pathogen invasion via the mucosal surface of the host requires further induction of systemic cellular, humoral and mucosal immunity to provide protection from pathogenic orgnanism.109 Different strategies such as the surface decoration of PLGA particle with RGD peptide which targets β1 integrin on M-cells,71 UEA-1,120,123 and LTA interacting with α-l-fucose receptors of M-cells 122 were applied in order to target particles to M-cells. Many researches have confirmed the induction of mucosal immunity via targeting antigen-loaded PLGA particles to M-cells,120,122-124 suggesting various infectious diseases can be managed by oral administration of M-cell targeted PLGA particles. In addition, mucosal immunity can be achieved by nasal administration of chitosan- 109and TMC- 113,114 surface modified PLGA particles.
In future studies, it is highly required to study the effect of physicochemical properties of PLGA MPs/NPs on immune response. The behavior of antigen loaded PLGA particles including bio distribution, kinetics, and cell targeting should be monitored in in vivo studies. Then, on the basis of application purpose, particle size, polymer L/G ratio and method of preparation should be selected and the decision to encapsulate or surface adsorb the antigen should be determined. When the route of administration is chosen based on the nature of disease and also patient compliance, it is necessary to decorate the surface of PLGA particles with appropriate agents to enhance the residence time of PLGA particles at target tissue. Another lesson learned from researchers' experience is surface decorating of PLGA particles with targeting agents that enable particle trafficking to the appropriate site for maximum induction of desired immune response. The use of adjuvants such as danger signals (e.g., TLR ligands) which can be adsorbed or encapsulated in PLGA particles is a relatively simple way to tailor the induced immunity. Therefore, a potent multivalent therapeutic strategy can be created by designing specifically tailored PLGA particles utilizing PLGA particles co-delivering antigens, immunomodulator, and through targeting agents; allowing vaccine researchers to produce next generation vaccines with built-in quality that provide more broadly protection against pathogenic diseases than those are used today.125
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
Authors thank Dr. R. Jalalirad (Pasteur Institute of Iran) for remarkable English revision of the manuscript.
References
- [1].Jain S, O'Hagan DT, Singh M. The long-term potential of biodegradable poly(lactide-co-glycolide) microparticles as the next-generation vaccine adjuvant. Expert Rev Vaccines 2011; 10:1731-42; PMID:22085176; http://dx.doi.org/ 10.1586/erv.11.126 [DOI] [PubMed] [Google Scholar]
- [2].Amjadi I, Rabiee M, Hosseini MS. Anticancer Activity of Nanoparticles Based on PLGA and its Co-polymer: In-vitro Evaluation. Iran J Pharm Res 2013; 12:623-34; PMID:24523742 [PMC free article] [PubMed] [Google Scholar]
- [3].Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011; 3:1377-97; PMID:22577513; http://dx.doi.org/ 10.3390/polym3031377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Semete B, Booysen L, Lemmer Y, Kalombo L, Katata L, Verschoor J, Swai HS. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine 2010; 6:662-71; http://dx.doi.org/ 10.1016/j.nano.2010.02.002 [DOI] [PubMed] [Google Scholar]
- [5].Mahboubian A, Hashemein SK, Moghadam S, Atyabi F, Dinarvand R. Preparation and in-vitro evaluation of controlled release PLGA microparticles containing triptoreline. Iran J Pharm Res 2010; 9:369; PMID:24381601 [PMC free article] [PubMed] [Google Scholar]
- [6].Waeckerle-Men Y, Groettrup M. PLGA microspheres for improved antigen delivery to dendritic cells as cellular vaccines. Adv Drug Deliv Rev 2005; 57:475-82; PMID:15560953; http://dx.doi.org/ 10.1016/j.addr.2004.09.007 [DOI] [PubMed] [Google Scholar]
- [7].Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Targeting dendritic cells with nano-particulate PLGA cancer vaccine formulations. Adv Drug Deliv Rev 2011; 63:943-55; PMID:21679733; http://dx.doi.org/ 10.1016/j.addr.2011.05.021 [DOI] [PubMed] [Google Scholar]
- [8].Malyala P, O'Hagan DT, Singh M. Enhancing the therapeutic efficacy of CpG oligonucleotides using biodegradable microparticles. Adv Drug Deliv Rev 2009; 61:218-25; PMID:19168103; http://dx.doi.org/ 10.1016/j.addr.2008.12.009 [DOI] [PubMed] [Google Scholar]
- [9].Bhatnagar S, Naqvi RA, Ali R, Rao DN. Preparation of peptide microspheres using tumor antigen-derived peptides. Methods Mol Biol 2014; 1139:443-52; PMID:24619698; http://dx.doi.org/ 10.1007/978-1-4939-0345-0_34 [DOI] [PubMed] [Google Scholar]
- [10].van de Weert M, Hennink WE, Jiskoot W. Protein instability in poly (lactic-co-glycolic acid) microparticles. Pharm Res 2000; 17:1159-67; PMID:11145219; http://dx.doi.org/ 10.1023/A:1026498209874 [DOI] [PubMed] [Google Scholar]
- [11].Brown LR. Commercial challenges of protein drug delivery. Expert Opin Drug Deliv 2005; 2:29-42; PMID:16296733; http://dx.doi.org/ 10.1517/17425247.2.1.29 [DOI] [PubMed] [Google Scholar]
- [12].Salvador A, Igartua M, Hernández RM, Pedraz JL. An overview on the field of micro-and nanotechnologies for synthetic peptide-based vaccines. J Drug Deliv 2011; 2011:181646; PMID:21773041; http://dx.doi.org/ 10.1155/2011/181646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Quintanar-Guerrero D, Allemann E, Fessi H, Doelker E. Preparation techniques and mechanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev Ind Pharm 1998; 24:1113-28; PMID:9876569; http://dx.doi.org/ 10.3109/03639049809108571 [DOI] [PubMed] [Google Scholar]
- [14].Kanchan V, Panda AK. Interactions of antigen-loaded polylactide particles with macrophages and their correlation with the immune response. Biomaterials 2007; 28:5344-57; PMID:17825905; http://dx.doi.org/ 10.1016/j.biomaterials.2007.08.015 [DOI] [PubMed] [Google Scholar]
- [15].Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly (d, l-lactide-< i>co-glycolide) and its derivatives. J Control Release 2008; 125:193-209; PMID:18083265; http://dx.doi.org/ 10.1016/j.jconrel.2007.09.013 [DOI] [PubMed] [Google Scholar]
- [16].Wendorf J, Chesko J, Kazzaz J, Ugozzoli M, Vajdy M, O'Hagan D, Singh M. A comparison of anionic nanoparticles and microparticles as vaccine delivery systems. Hum Vaccin 2008; 4:44-9; PMID:18438105; http://dx.doi.org/ 10.4161/hv.4.1.4886 [DOI] [PubMed] [Google Scholar]
- [17].Tan ML, Choong PF, Dass CR. Recent developments in liposomes, microparticles and nanoparticles for protein and peptide drug delivery. Peptides 2010; 31:184-93; PMID:19819278; http://dx.doi.org/ 10.1016/j.peptides.2009.10.002 [DOI] [PubMed] [Google Scholar]
- [18].Vert M, Hellwich K-H, Hess M, Hodge P, Kubisa P, Rinaudo M, et al.. Terminology for biorelated polymers and applications (IUPAC Recommendations 2012). Pure Applied Chem 2012; 84:377-410; http://dx.doi.org/ 10.1351/PAC-REC-10-12-04 [DOI] [Google Scholar]
- [19].Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces 2010; 75:1-18; http://dx.doi.org/ 10.1016/j.colsurfb.2009.09.001 [DOI] [PubMed] [Google Scholar]
- [20].Zhang Y, Chan HF, Leong KW. Advanced materials and processing for drug delivery: the past and the future. Adv Drug Deliv Rev 2013; 65:104-20; PMID:23088863; http://dx.doi.org/ 10.1016/j.addr.2012.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bilati U, Allémann E, Doelker E. Strategic approaches for overcoming peptide and protein instability within biodegradable nano-and microparticles. Eur J Pharm Biopharm 2005; 59:375-88; PMID:15760718; http://dx.doi.org/ 10.1016/j.ejpb.2004.10.006 [DOI] [PubMed] [Google Scholar]
- [22].Sinha V, Trehan A. Biodegradable microspheres for protein delivery. J Control Release 2003; 90:261-80; PMID:12880694; http://dx.doi.org/ 10.1016/S0168-3659(03)00194-9 [DOI] [PubMed] [Google Scholar]
- [23].Xu Q, Hashimoto M, Dang TT, Hoare T, Kohane DS, Whitesides GM, Langer R, Anderson DG. Preparation of monodisperse biodegradable polymer microparticles using a microfluidic flow‐focusing device for controlled drug delivery. Small 2009; 5:1575-81; PMID:19296563; http://dx.doi.org/ 10.1002/smll.200801855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xu J, Wong DH, Byrne JD, Chen K, Bowerman C, DeSimone JM. Future of the particle replication in nonwetting templates (PRINT) technology. Angew Chem Int Ed 2013; 52:6580-9; http://dx.doi.org/ 10.1002/anie.201209145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Galloway AL, Murphy A, DeSimone JM, Di J, Herrmann JP, Hunter ME, Kindig JP, Malinoski FJ, Rumley MA, Stoltz DM, et al.. Development of a nanoparticle-based influenza vaccine using the PRINT technology. Nanomedicine 2013; 9:523-31; PMID:23178283; http://dx.doi.org/ 10.1016/j.nano.2012.11.001 [DOI] [PubMed] [Google Scholar]
- [26].Raghuvanshi RS, Katare YK, Lalwani K, Ali MM, Singh O, Panda AK. Improved immune response from biodegradable polymer particles entrapping tetanus toxoid by use of different immunization protocol and adjuvants. Int J Pharm 2002; 245:109-21; PMID:12270248; http://dx.doi.org/ 10.1016/S0378-5173(02)00342-3 [DOI] [PubMed] [Google Scholar]
- [27].Shi S, Hickey AJ. PLGA microparticles in respirable sizes enhance an in vitro T cell response to recombinant Mycobacterium tuberculosis antigen TB10.4-Ag85B. Pharm Res 2010; 27:350-60; PMID:20024670; http://dx.doi.org/ 10.1007/s11095-009-0028-7 [DOI] [PubMed] [Google Scholar]
- [28].Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, et al.. Programming the magnitude and persistence of antibody responses with innate immunity. Nature 2011; 470:543-7; PMID:21350488; http://dx.doi.org/ 10.1038/nature09737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sales-Junior P, Guzman F, Vargas M, Sossai S, González C, Patarroyo J. Use of biodegradable PLGA microspheres as a slow release delivery system for the Boophilus microplus synthetic vaccine SBm7462. Vet Immunol Immunopathol 2005; 107:281-90; PMID:16002149; http://dx.doi.org/ 10.1016/j.vetimm.2005.05.004 [DOI] [PubMed] [Google Scholar]
- [30].Rosas JE, Hernandez RM, Gascon AR, Igartua M, Guzman F, Patarroyo ME, Pedraz JL. Biodegradable PLGA microspheres as a delivery system for malaria synthetic peptide SPf66. Vaccine 2001; 19:4445-51; PMID:11483270; http://dx.doi.org/ 10.1016/S0264-410X(01)00192-X [DOI] [PubMed] [Google Scholar]
- [31].Salvador A, Igartua M, Hernandez RM, Pedraz JL. Combination of immune stimulating adjuvants with poly(lactide-co-glycolide) microspheres enhances the immune response of vaccines. Vaccine 2012; 30:589-96; PMID:22119926; http://dx.doi.org/ 10.1016/j.vaccine.2011.11.057 [DOI] [PubMed] [Google Scholar]
- [32].Quintilio W, Takata CS, Sant'Anna OA, da Costa MHB, Raw I. Evaluation of a diphtheria and tetanus PLGA microencapsulated vaccine formulation without stabilizers. Curr Drug Deliv 2009; 6:297-304; PMID:19604144; http://dx.doi.org/ 10.2174/156720109788680886 [DOI] [PubMed] [Google Scholar]
- [33].Waeckerle-Men Y, Uetz-von Allmen E, Gander B, Scandella E, Schlosser E, Schmidtke G, Merkle HP, Groettrup M. Encapsulation of proteins and peptides into biodegradable poly (D, L-lactide-co-glycolide) microspheres prolongs and enhances antigen presentation by human dendritic cells. Vaccine 2006; 24:1847-57; PMID:16288821; http://dx.doi.org/ 10.1016/j.vaccine.2005.10.032 [DOI] [PubMed] [Google Scholar]
- [34].Huang SS, Li IH, Hong PD, Yeh MK. Development of Yersinia pestis F1 antigen-loaded microspheres vaccine against plague. Int J Nanomedicine 2014; 9:813-22; PMID:24550673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee Y-R, Lee Y-H, Im S-A, Kim K, Lee C-K. Formulation and Characterization of Antigen-loaded PLGA Nanoparticles for Efficient Cross-priming of the Antigen. Immune Netw 2011; 11:163-8; PMID:21860609; http://dx.doi.org/ 10.4110/in.2011.11.3.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Feng L, Qi XR, Zhou XJ, Maitani Y, Wang SC, Jiang Y, Nagai T. Pharmaceutical and immunological evaluation of a single-dose hepatitis B vaccine using PLGA microspheres. J Controlled Release 2006; 112:35-42; PMID:16516999; http://dx.doi.org/ 10.1016/j.jconrel.2006.01.012 [DOI] [PubMed] [Google Scholar]
- [37].Thomas C, Gupta V, Ahsan F. Influence of surface charge of PLGA particles of recombinant hepatitis B surface antigen in enhancing systemic and mucosal immune responses. Int J Pharm 2009; 379:41-50; PMID:19524654; http://dx.doi.org/ 10.1016/j.ijpharm.2009.06.006 [DOI] [PubMed] [Google Scholar]
- [38].Chuang S-C, Ko J-C, Chen C-P, Du J-T, Yang C-D. Induction of long-lasting protective immunity against Toxoplasma gondii in BALB/c mice by recombinant surface antigen 1 protein encapsulated in poly (lactide-co-glycolide) microparticles. Parasit Vectors 2013; 6:34; PMID:23398973; http://dx.doi.org/ 10.1186/1756-3305-6-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chuang S-C, Ko J-C, Chen C-P, Du J-T, Yang C-D. Encapsulation of chimeric protein rSAG1/2 into poly (lactide-co-glycolide) microparticles induces long-term protective immunity against Toxoplasma gondii in mice. Exp Parasitol 2013; 134:430-7; PMID:23624036; http://dx.doi.org/ 10.1016/j.exppara.2013.04.002 [DOI] [PubMed] [Google Scholar]
- [40].Kazzaz J, Singh M, Ugozzoli M, Chesko J, Soenawan E, O'Hagan DT. Encapsulation of the immune potentiators MPL and RC529 in PLG microparticles enhances their potency. J Control Release 2006; 110:566-73; PMID:16360956; http://dx.doi.org/ 10.1016/j.jconrel.2005.10.010 [DOI] [PubMed] [Google Scholar]
- [41].Chesko J, Kazzaz J, Ugozzoli M, O'Hagan DT, Singh M. An investigation of the factors controlling the adsorption of protein antigens to anionic PLG microparticles. J Pharm Sci 2005; 94:2510-9; PMID:16200615; http://dx.doi.org/ 10.1002/jps.20472 [DOI] [PubMed] [Google Scholar]
- [42].Kazzaz J, Neidleman J, Singh M, Ott G, O'Hagan D. Novel anionic microparticles are a potent adjuvant for the induction of cytotoxic T lymphocytes against recombinant p55 gag from HIV-1. J Control Release 2000; 67:347-56; PMID:10825566; http://dx.doi.org/ 10.1016/S0168-3659(00)00226-1 [DOI] [PubMed] [Google Scholar]
- [43].Singh M, Kazzaz J, Chesko J, Soenawan E, Ugozzoli M, Giuliani M, Pizza M, Rappouli R, O'Hagan DT. Anionic microparticles are a potent delivery system for recombinant antigens from Neisseria meningitidis serotype B. J Pharm Sci 2004; 93:273-82; PMID:14705185; http://dx.doi.org/ 10.1002/jps.10538 [DOI] [PubMed] [Google Scholar]
- [44].Park TG, Lee HY, Nam YS. A new preparation method for protein loaded poly (D, L-lactic-co-glycolic acid) microspheres and protein release mechanism study. J Control Release 1998; 55:181-91; PMID:9795050; http://dx.doi.org/ 10.1016/S0168-3659(98)00050-9 [DOI] [PubMed] [Google Scholar]
- [45].Calis S, Jeyanthi R, Tsai T, Mehta RC, DeLuca PP. Adsorption of salmon calcitonin to PLGA microspheres. Pharm Res 1995; 12:1072-6; PMID:7494805; http://dx.doi.org/ 10.1023/A:1016278902839 [DOI] [PubMed] [Google Scholar]
- [46].Mao S, Xu J, Cai C, Germershaus O, Schaper A, Kissel T. Effect of WOW process parameters on morphology and burst release of FITC-dextran loaded PLGA microspheres. Int J Pharm 2007; 334:137-48; PMID:17196348; http://dx.doi.org/ 10.1016/j.ijpharm.2006.10.036 [DOI] [PubMed] [Google Scholar]
- [47].Zolnik BS, Burgess DJ. Effect of acidic pH on PLGA microsphere degradation and release. J Control Release 2007; 122:338-44; PMID:17644208; http://dx.doi.org/ 10.1016/j.jconrel.2007.05.034 [DOI] [PubMed] [Google Scholar]
- [48].Anderson JM, Shive MS. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev 2012; 64:72-82; http://dx.doi.org/ 10.1016/j.addr.2012.09.004 [DOI] [PubMed] [Google Scholar]
- [49].Schakenraad J, Hardonk M, Feijen J, Molenaar I, Nieuwenhuis P. Enzymatic activity toward poly (L‐lactic acid) implants. J Biomed Mater Res 1990; 24:529-45; PMID:2324125; http://dx.doi.org/ 10.1002/jbm.820240502 [DOI] [PubMed] [Google Scholar]
- [50].Demento SL, Cui W, Criscione JM, Stern E, Tulipan J, Kaech SM, Fahmy TM. Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype. Biomaterials 2012; 33:4957-64; PMID:22484047; http://dx.doi.org/ 10.1016/j.biomaterials.2012.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Fredenberg S, Wahlgren M, Reslow M, Axelsson A. The mechanisms of drug release in poly (lactic-co-glycolic acid)-based drug delivery systems—a review. Int J Pharm 2011; 415:34-52; PMID:21640806; http://dx.doi.org/ 10.1016/j.ijpharm.2011.05.049 [DOI] [PubMed] [Google Scholar]
- [52].Wischke C, Schwendeman SP. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int J Pharm 2008; 364:298-327; PMID:18621492; http://dx.doi.org/ 10.1016/j.ijpharm.2008.04.042 [DOI] [PubMed] [Google Scholar]
- [53].Makino K, Mogi T, Ohtake N, Yoshida M, Ando S, Nakajima T, et al.. Pulsatile drug release from poly (lactide-co-glycolide) microspheres: how does the composition of the polymer matrices affect the time interval between the initial burst and the pulsatile release of drugs? Colloids and Surfaces B: Biointerfaces 2000; 19:173-9; http://dx.doi.org/ 10.1016/S0927-7765(00)00148-X [DOI] [Google Scholar]
- [54].Chen Y, Mohanraj VJ, Wang F, Benson HA. Designing chitosan-dextran sulfate nanoparticles using charge ratios. AAPS PharmSciTech 2007; 8:E98; PMID:18181558; http://dx.doi.org/ 10.1208/pt0804098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Giteau A, Venier-Julienne M-C, Aubert-Pouëssel A, Benoit J-P. How to achieve sustained and complete protein release from PLGA-based microparticles? Int J Pharm 2008; 350:14-26; PMID:18162341; http://dx.doi.org/ 10.1016/j.ijpharm.2007.11.012 [DOI] [PubMed] [Google Scholar]
- [56].Tracy MA, Ward KL, Firouzabadian L, Wang Y, Dong N, Qian R, Zhang Y. Factors affecting the degradation rate of poly(lactide-co-glycolide) microspheres in vivo and in vitro. Biomaterials 1999; 20:1057-62; PMID:10378806; http://dx.doi.org/ 10.1016/S0142-9612(99)00002-2 [DOI] [PubMed] [Google Scholar]
- [57].Lima K, Rodrigues Júnior J. Poly-DL-lactide-co-glycolide microspheres as a controlled release antigen delivery system. Braz J Med Biol Res 1999; 32:171-80 [DOI] [PubMed] [Google Scholar]
- [58].Saini V, Jain V, Sudheesh MS, Jaganathan KS, Murthy PK, Kohli DV. Comparison of humoral and cell-mediated immune responses to cationic PLGA microspheres containing recombinant hepatitis B antigen. Int J Pharm 2011; 408:50-7; PMID:21291968; http://dx.doi.org/ 10.1016/j.ijpharm.2011.01.045 [DOI] [PubMed] [Google Scholar]
- [59].Park TG. Degradation of poly (lactic-co-glycolic acid) microspheres: effect of copolymer composition. Biomaterials 1995; 16:1123-30; PMID:8562787; http://dx.doi.org/ 10.1016/0142-9612(95)93575-X [DOI] [PubMed] [Google Scholar]
- [60].Passerini N, Craig D. An investigation into the effects of residual water on the glass transition temperature of polylactide microspheres using modulated temperature DSC. J Control Release 2001; 73:111-5; PMID:11337064; http://dx.doi.org/ 10.1016/S0168-3659(01)00245-0 [DOI] [PubMed] [Google Scholar]
- [61].Wiggins JS, Hassan MK, Mauritz KA, Storey RF. Hydrolytic degradation of poly (d, l-lactide) as a function of end group: carboxylic acid vs. hydroxyl. Polymer 2006; 47:1960-9; http://dx.doi.org/ 10.1016/j.polymer.2006.01.021 [DOI] [Google Scholar]
- [62].Silva A, Rosalia R, Sazak A, Carstens M, Ossendorp F, Oostendorp J, Jiskoot W. Optimization of encapsulation of a synthetic long peptide in PLGA nanoparticles: Low-burst release is crucial for efficient CD8< sup>+T cell activation. Eur J Pharm Biopharm 2013; 83:338-45; PMID:23201055; http://dx.doi.org/ 10.1016/j.ejpb.2012.11.006 [DOI] [PubMed] [Google Scholar]
- [63].Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev 2003; 55:329-47; PMID:12628320; http://dx.doi.org/ 10.1016/S0169-409X(02)00228-4 [DOI] [PubMed] [Google Scholar]
- [64].Siepmann J, Elkharraz K, Siepmann F, Klose D. How autocatalysis accelerates drug release from PLGA-based microparticles: a quantitative treatment. Biomacromolecules 2005; 6:2312-9; PMID:16004477; http://dx.doi.org/ 10.1021/bm050228k [DOI] [PubMed] [Google Scholar]
- [65].Yang Q, Owusu-Ababio G. Biodegradable progesterone microsphere delivery system for osteoporosis therapy. Drug Dev Ind Pharm 2000; 26:61-70; PMID:10677811; http://dx.doi.org/ 10.1081/DDC-100100328 [DOI] [PubMed] [Google Scholar]
- [66].Sandor M, Enscore D, Weston P, Mathiowitz E. Effect of protein molecular weight on release from micron-sized PLGA microspheres. J Control Release 2001; 76:297-311; PMID:11578744; http://dx.doi.org/ 10.1016/S0168-3659(01)00446-1 [DOI] [PubMed] [Google Scholar]
- [67].Newman KD, Elamanchili P, Kwon GS, Samuel J. Uptake of poly (D, L‐lactic‐co‐glycolic acid) microspheres by antigen‐presenting cells in vivo. J Biomed Mater Res 2002; 60:480-6; PMID:11920673; http://dx.doi.org/ 10.1002/jbm.10019 [DOI] [PubMed] [Google Scholar]
- [68].van Riet E, Ainai A, Suzuki T, Kersten G, Hasegawa H. Combatting infectious diseases; nanotechnology as a platform for rational vaccine design. Adv Drug Deliv Rev 2014; 74:28-34; PMID:24862579; http://dx.doi.org/ 10.1016/j.addr.2014.05.011 [DOI] [PubMed] [Google Scholar]
- [69].Mohanan D, Slütter B, Henriksen-Lacey M, Jiskoot W, Bouwstra JA, Perrie Y, Kündig TM, Gander B, Johansen P. Administration routes affect the quality of immune responses: a cross-sectional evaluation of particulate antigen-delivery systems. J Control Release 2010; 147:342-9; PMID:20727926; http://dx.doi.org/ 10.1016/j.jconrel.2010.08.012 [DOI] [PubMed] [Google Scholar]
- [70].O'Hagan DT, Singh M, Ulmer JB. Microparticle-based technologies for vaccines. Methods 2006; 40:10-9; http://dx.doi.org/ 10.1016/j.ymeth.2006.05.017 [DOI] [PubMed] [Google Scholar]
- [71].Garinot M, Fiévez V, Pourcelle V, Stoffelbach F, des Rieux A, Plapied L, Theate I, Freichels H, Jérôme C, Marchand-Brynaert J, et al.. PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination. J Control Release 2007; 120:195-204; PMID:17586081; http://dx.doi.org/ 10.1016/j.jconrel.2007.04.021 [DOI] [PubMed] [Google Scholar]
- [72].Conway MA, Madrigal-Estebas L, McClean S, Brayden DJ, Mills KH. Protection against Bordetella pertussis infection following parenteral or oral immunization with antigens entrapped in biodegradable particles: effect of formulation and route of immunization on induction of Th1 and Th2 cells. Vaccine 2001; 19:1940-50; PMID:11228364; http://dx.doi.org/ 10.1016/S0264-410X(00)00433-3 [DOI] [PubMed] [Google Scholar]
- [73].Gutierro I, Hernandez R, Igartua M, Gascon A, Pedraz J. Size dependent immune response after subcutaneous, oral and intranasal administration of BSA loaded nanospheres. Vaccine 2002; 21:67-77; PMID:12443664; http://dx.doi.org/ 10.1016/S0264-410X(02)00435-8 [DOI] [PubMed] [Google Scholar]
- [74].Carcaboso AM, Hernandez RM, Igartua M, Rosas JE, Patarroyo ME, Pedraz JL. Potent, long lasting systemic antibody levels and mixed Th1/Th2 immune response after nasal immunization with malaria antigen loaded PLGA microparticles. Vaccine 2004; 22:1423-32; PMID:15063565; http://dx.doi.org/ 10.1016/j.vaccine.2003.10.020 [DOI] [PubMed] [Google Scholar]
- [75].Thomas C, Gupta V, Ahsan F. Particle size influences the immune response produced by hepatitis B vaccine formulated in inhalable particles. Pharm Res 2010; 27:905-19; PMID:20232117; http://dx.doi.org/ 10.1007/s11095-010-0094-x [DOI] [PubMed] [Google Scholar]
- [76].Cruz LJ, Tacken PJ, Fokkink R, Joosten B, Stuart MC, Albericio F, Torensma R, Figdor CG. Targeted PLGA nano-but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J Control Release 2010; 144:118-26; PMID:20156497; http://dx.doi.org/ 10.1016/j.jconrel.2010.02.013 [DOI] [PubMed] [Google Scholar]
- [77].O'Hagan DT, Jeffery H, Davis S. Long-term antibody responses in mice following subcutaneous immunization with ovalbumin entrapped in biodegradable microparticles. Vaccine 1993; 11:965-9; PMID:8212845; http://dx.doi.org/ 10.1016/0264-410X(93)90387-D [DOI] [PubMed] [Google Scholar]
- [78].Eldridge JH, Staas J, Meulbroek J, Tice T, Gilley R. Biodegradable and biocompatible poly (DL-lactide-co-glycolide) microspheres as an adjuvant for staphylococcal enterotoxin B toxoid which enhances the level of toxin-neutralizing antibodies. Infect Immun 1991; 59:2978-86; PMID:1879922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol 2008; 38:1404-13; PMID:18389478; http://dx.doi.org/ 10.1002/eji.200737984 [DOI] [PubMed] [Google Scholar]
- [80].Jaganathan KS, Singh P, Prabakaran D, Mishra V, Vyas SP. Development of a single-dose stabilized poly(D,L-lactic-co-glycolic acid) microspheres-based vaccine against hepatitis B. J Pharm Pharmacol 2004; 56:1243-50; PMID:15482638; http://dx.doi.org/ 10.1211/0022357044418 [DOI] [PubMed] [Google Scholar]
- [81].Rosas JE, Pedraz JL, Hernandez RM, Gascon AR, Igartua M, Guzman F, Rodríguez R, Cortés J, Patarroyo ME. Remarkably high antibody levels and protection against P. falciparum malaria in Aotus monkeys after a single immunisation of SPf66 encapsulated in PLGA microspheres. Vaccine 2002; 20:1707-10; PMID:11906756; http://dx.doi.org/ 10.1016/S0264-410X(01)00508-4 [DOI] [PubMed] [Google Scholar]
- [82].Joshi VB, Adamcakova-Dodd A, Jing X, Wongrakpanich A, Gibson-Corley KN, Thorne PS, Salem AK. Development of a poly (lactic-co-glycolic acid) particle vaccine to protect against house dust mite induced allergy. AAPS J 2014; 16:975-85; PMID:24981892; http://dx.doi.org/ 10.1208/s12248-014-9624-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Xiao X, Zeng X, Zhang X, Ma L, Liu X, Yu H, Mei L, Liu Z. Effects of Caryota mitis profilin-loaded PLGA nanoparticles in a murine model of allergic asthma. Int J Nanomedicine 2013; 8:4553; PMID:24376349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Thomas C, Rawat A, Hope-Weeks L, Ahsan F. Aerosolized PLA and PLGA nanoparticles enhance humoral, mucosal and cytokine responses to hepatitis B vaccine. Mol Pharm 2011; 8:405-15; PMID:21189035; http://dx.doi.org/ 10.1021/mp100255c [DOI] [PubMed] [Google Scholar]
- [85].Tafaghodi M, Eskandari M, Kharazizadeh M, Khamesipour A, Jaafari M. Immunization against leishmaniasis by PLGA nanospheres loaded with an experimental autoclaved Leishmania major (ALM) and Quillaja saponins. Tropical biomedicine 2010; 27:639-50; PMID:21399606 [PubMed] [Google Scholar]
- [86].Santos DM, Carneiro MW, de Moura TR, Fukutani K, Clarencio J, Soto M, Espuelas S, Brodskyn C, Barral A, Barral-Netto M, et al.. Towards development of novel immunization strategies against leishmaniasis using PLGA nanoparticles loaded with kinetoplastid membrane protein-11. Int J Nanomedicine 2012; 7:2115-27; PMID:22619548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Santos DM, Carneiro MW, de Moura TR, Soto M, Luz NF, Prates DB, Irache JM, Brodskyn C, Barral A, Barral-Netto M, et al.. PLGA nanoparticles loaded with KMP-11 stimulate innate immunity and induce the killing of< i>Leishmania. Nanomedicine 2013; 9:985-95; http://dx.doi.org/ 10.1016/j.nano.2013.04.003 [DOI] [PubMed] [Google Scholar]
- [88].Kirby DJ, Rosenkrands I, Agger EM, Andersen P, Coombes AG, Perrie Y. PLGA microspheres for the delivery of a novel subunit TB vaccine. J Drug Target 2008; 16:282-93; PMID:18446607; http://dx.doi.org/ 10.1080/10611860801900462 [DOI] [PubMed] [Google Scholar]
- [89].Wang C, Muttil P, Lu D, Beltran-Torres AA, Garcia-Contreras L, Hickey AJ. Screening for potential adjuvants administered by the pulmonary route for tuberculosis vaccines. AAPS J 2009; 11:139-47; PMID:19277872; http://dx.doi.org/ 10.1208/s12248-009-9089-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Taha MA, Singh SR, Dennis VA. Biodegradable PLGA85/15 nanoparticles as a delivery vehicle for Chlamydia trachomatis recombinant MOMP-187 peptide. Nanotechnology 2012; 23:325101; PMID:22824940; http://dx.doi.org/ 10.1088/0957-4484/23/32/325101 [DOI] [PubMed] [Google Scholar]
- [91].Fairley SJ, Singh SR, Yilma AN, Waffo AB, Subbarayan P, Dixit S, Taha MA, Cambridge CD, Dennis VA. Chlamydia trachomatis recombinant MOMP encapsulated in PLGA nanoparticles triggers primarily T helper 1 cellular and antibody immune responses in mice: a desirable candidate nanovaccine. Int J Nanomedicine 2013; 8:2085-99; PMID:23785233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Tacken PJ, Zeelenberg IS, Cruz LJ, van Hout-Kuijer MA, van de Glind G, Fokkink RG, Lambeck AJ, Figdor CG. Targeted delivery of TLR ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood 2011; 118:6836-44; PMID:21967977; http://dx.doi.org/ 10.1182/blood-2011-07-367615 [DOI] [PubMed] [Google Scholar]
- [93].Wischke C, Zimmermann J, Wessinger B, Schendler A, Borchert HH, Peters JH, Nesselhut T, Lorenzen DR. Poly(I:C) coated PLGA microparticles induce dendritic cell maturation. Int J Pharm 2009; 365:61-8; PMID:18812217; http://dx.doi.org/ 10.1016/j.ijpharm.2008.08.039 [DOI] [PubMed] [Google Scholar]
- [94].Hafner AM, Corthésy B, Merkle HP. Particulate formulations for the delivery of poly (I: C) as vaccine adjuvant. Adv Drug Deliv Rev 2013; 65:1386-99; PMID:23751781; http://dx.doi.org/ 10.1016/j.addr.2013.05.013 [DOI] [PubMed] [Google Scholar]
- [95].Salvador A, Igartua M, Hernandez RM, Pedraz JL. Designing improved poly lactic-co-glycolic acid microspheres for a malarial vaccine: incorporation of alginate and polyinosinic-polycytidilic acid. J Microencapsul 2014; 31:560-6; PMID:24697189; http://dx.doi.org/ 10.3109/02652048.2014.885608 [DOI] [PubMed] [Google Scholar]
- [96].Chong CS, Cao M, Wong WW, Fischer KP, Addison WR, Kwon GS, Tyrrell DL, Samuel J. Enhancement of T helper type 1 immune responses against hepatitis B virus core antigen by PLGA nanoparticle vaccine delivery. J Control Release 2005; 102:85-99; PMID:15653136; http://dx.doi.org/ 10.1016/j.jconrel.2004.09.014 [DOI] [PubMed] [Google Scholar]
- [97].Xie H, Gursel I, Ivins BE, Singh M, O'Hagan DT, Ulmer JB, Klinman DM. CpG oligodeoxynucleotides adsorbed onto polylactide-co-glycolide microparticles improve the immunogenicity and protective activity of the licensed anthrax vaccine. Infect Immun 2005; 73:828-33; PMID:15664922; http://dx.doi.org/ 10.1128/IAI.73.2.828-833.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Martinez Gomez JM, Fischer S, Csaba N, Kundig TM, Merkle HP, Gander B, Johansen P. A protective allergy vaccine based on CpG- and protamine-containing PLGA microparticles. Pharm Res 2007; 24:1927-35; PMID:17541735; http://dx.doi.org/ 10.1007/s11095-007-9318-0 [DOI] [PubMed] [Google Scholar]
- [99].Tafaghodi M, Khamesipour A, Jaafari MR. Immunization against leishmaniasis by PLGA nanospheres encapsulated with autoclaved Leishmania major (ALM) and CpG-ODN. Parasitol Res 2011; 108:1265-73; PMID:21125294; http://dx.doi.org/ 10.1007/s00436-010-2176-4 [DOI] [PubMed] [Google Scholar]
- [100].Lee Y-R, Lee Y-H, Kim K-H, Im S-A, Lee C-K. Induction of potent antigen-specific cytotoxic T cell response by PLGA-nanoparticles containing antigen and TLR agonist. Immune Netw 2013; 13:30-3; PMID:23559898; http://dx.doi.org/ 10.4110/in.2013.13.1.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Elamanchili P, Diwan M, Cao M, Samuel J. Characterization of poly (D, L-lactic-co-glycolic acid) based nanoparticulate system for enhanced delivery of antigens to dendritic cells. Vaccine 2004; 22:2406-12; PMID:15193402; http://dx.doi.org/ 10.1016/j.vaccine.2003.12.032 [DOI] [PubMed] [Google Scholar]
- [102].Wang Q, Tan MT, Keegan BP, Barry MA, Heffernan MJ. Time course study of the antigen-specific immune response to a PLGA microparticle vaccine formulation. Biomaterials 2014; 35(29):8385-93 [DOI] [PubMed] [Google Scholar]
- [103].Malyala P, Chesko J, Ugozzoli M, Goodsell A, Zhou F, Vajdy M, O'Hagan DT, Singh M. The potency of the adjuvant, CpG oligos, is enhanced by encapsulation in PLG microparticles. J Pharm Sci 2008; 97:1155-64; PMID:17683059; http://dx.doi.org/ 10.1002/jps.21065 [DOI] [PubMed] [Google Scholar]
- [104].San Roman B, Irache JM, Gomez S, Tsapis N, Gamazo C, Espuelas MS. Co-encapsulation of an antigen and CpG oligonucleotides into PLGA microparticles by TROMS technology. Eur J Pharm Biopharm 2008; 70:98-108; http://dx.doi.org/ 10.1016/j.ejpb.2008.03.015 [DOI] [PubMed] [Google Scholar]
- [105].Fischer S, Schlosser E, Mueller M, Csaba N, Merkle HP, Groettrup M, Gander B. Concomitant delivery of a CTL-restricted peptide antigen and CpG ODN by PLGA microparticles induces cellular immune response. J Drug Target 2009; 17:652-61; PMID:19622019; http://dx.doi.org/ 10.1080/10611860903119656 [DOI] [PubMed] [Google Scholar]
- [106].Rubsamen RM, Herst CV, Lloyd PM, Heckerman DE. Eliciting cytotoxic T-lymphocyte responses from synthetic vectors containing one or two epitopes in a C57BL/6 mouse model using peptide-containing biodegradable microspheres and adjuvants. Vaccine 2014; 32:4111-6; PMID:24912025; http://dx.doi.org/ 10.1016/j.vaccine.2014.05.071 [DOI] [PubMed] [Google Scholar]
- [107].Bershteyn A, Hanson MC, Crespo MP, Moon JJ, Li AV, Suh H, Irvine DJ. Robust IgG responses to nanograms of antigen using a biomimetic lipid-coated particle vaccine. J Control Release 2012; 157:354-65; PMID:21820024; http://dx.doi.org/ 10.1016/j.jconrel.2011.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Moon JJ, Suh H, Polhemus ME, Ockenhouse CF, Yadava A, Irvine DJ. Antigen-displaying lipid-enveloped PLGA nanoparticles as delivery agents for a Plasmodium vivax malaria vaccine. PloS one 2012; 7:e31472; PMID:22328935; http://dx.doi.org/ 10.1371/journal.pone.0031472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Jaganathan K, Vyas SP. Strong systemic and mucosal immune responses to surface-modified PLGA microspheres containing recombinant hepatitis B antigen administered intranasally. Vaccine 2006; 24:4201-11; PMID:16446012; http://dx.doi.org/ 10.1016/j.vaccine.2006.01.011 [DOI] [PubMed] [Google Scholar]
- [110].Pandit S, Cevher E, Zariwala MG, Somavarapu S, Alpar HO. Enhancement of immune response of HBsAg loaded poly (L-lactic acid) microspheres against hepatitis B through incorporation of alum and chitosan. J Microencapsul 2007; 24:539-52; PMID:17654174; http://dx.doi.org/ 10.1080/02652040701443700 [DOI] [PubMed] [Google Scholar]
- [111].Semete B, Booysen L, Kalombo L, Venter JD, Katata L, Ramalapa B, Verschoor JA, Swai H. In vivo uptake and acute immune response to orally administered chitosan and PEG coated PLGA nanoparticles. Toxicol Appl Pharmacol 2010; 249:158-65; PMID:20851137; http://dx.doi.org/ 10.1016/j.taap.2010.09.002 [DOI] [PubMed] [Google Scholar]
- [112].Pawar D, Goyal AK, Mangal S, Mishra N, Vaidya B, Tiwari S, Jain AK, Vyas SP. Evaluation of mucoadhesive PLGA microparticles for nasal immunization. AAPS J 2010; 12:130-7; PMID:20077052; http://dx.doi.org/ 10.1208/s12248-009-9169-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Slutter B, Bal S, Keijzer C, Mallants R, Hagenaars N, Que I, Kaijzel E, van Eden W, Augustijns P, Löwik C, et al.. Nasal vaccination with N-trimethyl chitosan and PLGA based nanoparticles: nanoparticle characteristics determine quality and strength of the antibody response in mice against the encapsulated antigen. Vaccine 2010; 28:6282-91; PMID:20638455; http://dx.doi.org/ 10.1016/j.vaccine.2010.06.121 [DOI] [PubMed] [Google Scholar]
- [114].Primard C, Poecheim J, Heuking S, Sublet E, Esmaeili F, Borchard G. Multifunctional PLGA-based nanoparticles encapsulating simultaneously hydrophilic antigen and hydrophobic immunomodulator for mucosal immunization. Mol Pharm 2013; 10:2996-3004; PMID:23869898; http://dx.doi.org/ 10.1021/mp400092y [DOI] [PubMed] [Google Scholar]
- [115].Brandhonneur N, Chevanne F, Vié V, Frisch B, Primault R, Le Potier M-F, Le Corre P. Specific and non-specific phagocytosis of ligand-grafted PLGA microspheres by macrophages. Eur J Pharm Sci 2009; 36:474-85; PMID:19110055; http://dx.doi.org/ 10.1016/j.ejps.2008.11.013 [DOI] [PubMed] [Google Scholar]
- [116].Martínez Gómez JM, Csaba N, Fischer S, Sichelstiel A, Kündig TM, Gander B, Johansen P. Surface coating of PLGA microparticles with protamine enhances their immunological performance through facilitated phagocytosis. J Control Release 2008; 130:161-7; http://dx.doi.org/ 10.1016/j.jconrel.2008.06.003 [DOI] [PubMed] [Google Scholar]
- [117].Han R, Zhu J, Yang X, Xu H. Surface modification of poly(D,L-lactic-co-glycolic acid) nanoparticles with protamine enhanced cross-presentation of encapsulated ovalbumin by bone marrow-derived dendritic cells. J Biomed Mater Res A 2011; 96:142-9; PMID:21105162; http://dx.doi.org/ 10.1002/jbm.a.32860 [DOI] [PubMed] [Google Scholar]
- [118].Sarti F, Perera G, Hintzen F, Kotti K, Karageorgiou V, Kammona O, Kiparissides C, Bernkop-Schnürch A. In vivo evidence of oral vaccination with PLGA nanoparticles containing the immunostimulant monophosphoryl lipid A. Biomaterials 2011; 32:4052-7; PMID:21377204; http://dx.doi.org/ 10.1016/j.biomaterials.2011.02.011 [DOI] [PubMed] [Google Scholar]
- [119].Jain S, Harde H, Indulkar A, Agrawal AK. Improved stability and immunological potential of tetanus toxoid containing surface engineered bilosomes following oral administration. Nanomedicine 2014; 10:431-40; http://dx.doi.org/ 10.1016/j.nano.2013.08.012 [DOI] [PubMed] [Google Scholar]
- [120].Gupta PN, Khatri K, Goyal AK, Mishra N, Vyas SP. M-cell targeted biodegradable PLGA nanoparticles for oral immunization against hepatitis B. J Drug Target 2007; 15:701-13; PMID:18041638; http://dx.doi.org/ 10.1080/10611860701637982 [DOI] [PubMed] [Google Scholar]
- [121].Fievez V, Plapied L, des Rieux A, Pourcelle V, Freichels H, Wascotte V, Vanderhaeghen ML, Jerôme C, Vanderplasschen A, Marchand-Brynaert J, et al.. Targeting nanoparticles to M cells with non-peptidic ligands for oral vaccination. Eur J Pharm Biopharm 2009; 73:16-24; PMID:19409989; http://dx.doi.org/ 10.1016/j.ejpb.2009.04.009 [DOI] [PubMed] [Google Scholar]
- [122].Mishra N, Tiwari S, Vaidya B, Agrawal GP, Vyas SP. Lectin anchored PLGA nanoparticles for oral mucosal immunization against hepatitis B. J Drug Target 2011; 19:67-78; PMID:20334603; http://dx.doi.org/ 10.3109/10611861003733946 [DOI] [PubMed] [Google Scholar]
- [123].Ma T, Wang L, Yang T, Ma G, Wang S. M-cell targeted polymeric lipid nanoparticles containing a Toll-like receptor agonist to boost oral immunity. Int J Pharm 2014; 473:296-303; PMID:24984067; http://dx.doi.org/ 10.1016/j.ijpharm.2014.06.052 [DOI] [PubMed] [Google Scholar]
- [124].Jiang T, Singh B, Li H-S, Kim Y-K, Kang S-K, Nah J-W, Choi YJ, Cho CS. Targeted oral delivery of BmpB vaccine using porous PLGA microparticles coated with M cell homing peptide-coupled chitosan. Biomaterials 2014; 35:2365-73; PMID:24342722; http://dx.doi.org/ 10.1016/j.biomaterials.2013.11.073 [DOI] [PubMed] [Google Scholar]
- [125].Saluja SS, Hanlon DJ, Sharp FA, Hong E, Khalil D, Robinson E, Tigelaar R, Fahmy TM, Edelson RL. Targeting human dendritic cells via DEC-205 using PLGA nanoparticles leads to enhanced cross-presentation of a melanoma-associated antigen. Int J Nanomedicine 2014; 9:5231-46; PMID:25419128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].T O'Hagan D, Singh M, Gupta RK. Poly (lactide-co-glycolide) microparticles for the development of single-dose controlled-release vaccines. Adv Drug Deliv Rev 1998; 32:225-46; PMID:10837646; http://dx.doi.org/ 10.1016/S0169-409X(98)00012-X [DOI] [PubMed] [Google Scholar]
- [127].Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release 2012; 161:505-22; PMID:22353619; http://dx.doi.org/ 10.1016/j.jconrel.2012.01.043 [DOI] [PubMed] [Google Scholar]
- [128].Vasir JK, Labhasetwar V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv Drug Deliv Rev 2007; 59:718-28; PMID:17683826; http://dx.doi.org/ 10.1016/j.addr.2007.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Cruz LJ, Rosalia RA, Kleinovink JW, Rueda F, Lowik CW, Ossendorp F. Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8(+) T cell response: a comparative study. J Control Release 2014; 192:209-18; PMID:25068703; http://dx.doi.org/ 10.1016/j.jconrel.2014.07.040 [DOI] [PubMed] [Google Scholar]
- [130].Fischer S, Uetz-von Allmen E, Waeckerle-Men Y, Groettrup M, Merkle HP, Gander B. The preservation of phenotype and functionality of dendritic cells upon phagocytosis of polyelectrolyte-coated PLGA microparticles. Biomaterials 2007; 28:994-1004; PMID:17118442; http://dx.doi.org/ 10.1016/j.biomaterials.2006.10.034 [DOI] [PubMed] [Google Scholar]
- [131].Heo MB, Cho MY, Lim YT. Polymer nanoparticles for enhanced immune response: combined delivery of tumor antigen and small interference RNA for immunosuppressive gene to dendritic cells. Acta biomaterialia 2014; 10:2169-76; PMID:24394635; http://dx.doi.org/ 10.1016/j.actbio.2013.12.050 [DOI] [PubMed] [Google Scholar]
- [132].Heo MB, Lim YT. Programmed nanoparticles for combined immunomodulation, antigen presentation and tracking of immunotherapeutic cells. Biomaterials 2014; 35:590-600; PMID:24125775; http://dx.doi.org/ 10.1016/j.biomaterials.2013.10.009 [DOI] [PubMed] [Google Scholar]
- [133].Brito LA, O'Hagan DT. Designing and building the next generation of improved vaccine adjuvants. J Control Release 2014; 190:563-79; PMID:24998942; http://dx.doi.org/ 10.1016/j.jconrel.2014.06.027 [DOI] [PubMed] [Google Scholar]