

Abstract
“Stapled” peptides are typically designed to replace two non-interacting residues with a constraining, olefinic staple. To mimic interacting leucine and isoleucine residues, we have created new amino acids that incorporate a methyl in the γ-position of the stapling amino acid S5. We have incorporated them into a sequence derived from steroid receptor coactivator 2, which interacts with estrogen receptor α. The best peptide (IC50 = 89 nM) replaces isoleucine 689 with an S-γ-methyl stapled amino acid, and has significantly higher affinity than unsubstituted peptides (390 and 760 nM). Through x-ray crystallography and molecular dynamics studies, we show that the conformation taken up by the S-γ-methyl peptide minimizes syn-pentane interactions between α- and γ-methyl groups.
Keywords: peptides, receptors, peptidomimetics, amino acids, conformational analysis
Graphical Abstract
γ-Branched stapling amino acids were synthesized and incorporated into peptides to produce high-affinity inhibitors of the estrogen receptor/steroid receptor coactivator interaction. Some branched stapled peptides were more effective than the unfunctionalized. The influence of 1,5-interactions on peptide conformation was characterized by circular dichroism, x-ray crystallography and molecular dynamics.
Several strategies exist for stabilizing or mimicking[1] biologically active peptide sequences and secondary structures with small molecules or constrained peptides.[2] Among the most widely used mimics of α-helices are “stapled” peptides.[3] They feature a side-chain to side-chain olefin cross-link that may imbue peptides with enhanced conformational stability,[4] metabolic stability, and, somewhat controversially, cell permeability.[5] To synthesize stapled peptides, two or more strategically chosen residues of a native peptide sequence are replaced with non-natural α-methyl-α-alkenyl amino acids. Ring-closing metathesis forms a macrocycle between the i and i+3, i+4, or i+7 positions.[6] Because the constraint may interfere with the ability of the peptide to bind to its receptor, stapled peptides are typically designed so the constraint is placed on a non-interacting face of an α-helix.[3a] Recently, others have reported successfully replacing interacting helical residues with a staple.[7] Although it lacks the branching functionality of valine, leucine, and isoleucine, the staple has the ability to fill protein surface grooves. As we show in this work, incorporating hydrophobic functionality at the constraint may more accurately mimic native sequences to increase affinity, and, importantly, it may also further stabilize bioactive conformations.
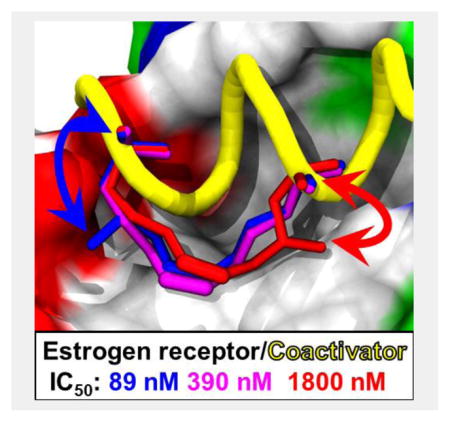
Phillips et al. reported an early example of replacing interacting residues with a hydrocarbon staple.[7a] The crystal structure of stapled peptide PFE-SP2 bound to estrogen receptor α (ERα) showed that an i, i+4 hydrocarbon staple can replace isoleucine and leucine residues on the binding face of a steroid receptor coactivator 2 (SRC2) peptide. This replacement yields an increase in α-helicity and affinity. SRC2 interacts with the surface of ERα over two turns of an α-helix using an ILXXLL motif (X is any amino acid).[8] This protein-protein interaction has been well-investigated,[9] if recalcitrant, to treat endocrine therapy-resistant breast cancers. Recently identified ERα mutants that are constitutively active and implicated in metastases have brought renewed focus to this therapeutically important interaction.[10]
We designed stapled peptides that incorporate branched stapling residues as functionalized constraints. Specifically, we designed amino acids based on stapling amino acid S5 that incorporate a methyl group in the γ-position to mimic branched hydrophobic amino acids Ile689 and Leu693 of the I689LXXLL694 motif of SRC2. Because S5 contains an α-methyl group for helical stability, incorporation of a γ-methyl group establishes 1,5-interactions, which, when appropriately positioned, could bolster helical conformations imposed by the constraint. We synthesized requisite amino acids λR and λS by joining one of Schöllkopf’s bis-lactim ethers with enantio-enriched branched alkenyl sidechains, which were synthesized using Evans’ N-acyloxazolidinone chemistry (see supporting information).[11] These amino acids, in combination with S5, were incorporated into residues 687–697 of SRC2. Solid phase peptide synthesis and ring closing metathesis were carried out as previously reported, and four stapled peptides containing λR/S and/or S5 were successfully synthesized (see Figure 1A). The Z-alkene configuration was consistent with 1H NMR H-C=C-H coupling constants of 10–11 Hz (see supporting information). SRC2-P6 failed to undergo ring-closing metathesis, even under forcing conditions, suggesting that substituting the i+4 stapling residue with R-γ-substitutions results in syn-pentane interactions that are non-productive for ring-closing.
Figure 1.
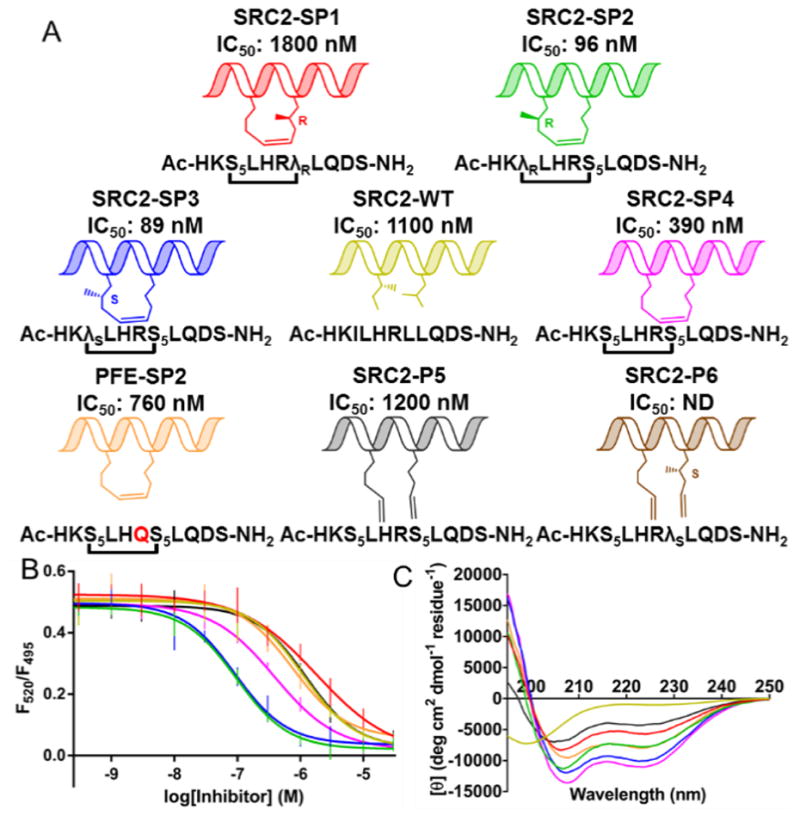
(A) Peptides (B) Time-resolved fluorescence resonance energy transfer dose-response curves for inhibition of ERα/SRC3. (C) Circular dichroism measurements were taken in 45 mM phosphate buffer pH 7.4 with 10% MeOH. SRC2-WT (yellow), SRC2-SP1 (red), SRC2-SP2 (green), SRC2-SP3 (blue), SRC2-SP4 (magenta), PFE-SP2 (orange), SRC2-P5 (gray), SRC2-P6 (brown).
A time-resolved fluorescence resonance energy transfer (TR-FRET) assay (Figure 1B) was used to measure interaction of a steroid receptor coactivator 3 (SRC3) fragment with ERα ligand-binding domain.[12] In this assay, the wild-type peptide has an IC50 of 1100 nM. The unfunctionalized stapled peptide SRC2-SP4 has an IC50 of 390 nM. This peptide is analogous to PFE-SP2, described by Phillips et al., but has a wild-type Q→R substitution, which increases the affinity two-fold. Epimers SRC2-SP2 and -SP3 were the most active, showing a 12-fold increase in potency compared to wild-type. SRC2-SP1, designed to incorporate a branched stapling residue to replace conserved Leu693, displayed minimal activity. In addition to the TR-FRET assay, surface plasmon resonance (see supporting information) was used to obtain dissociation constants for SRC2-SP1 (530 nM), SRC2-SP2 (42 nM), and SRC2-SP3 (39 nM).
Circular dichroism (CD) analysis of the peptides (Figure 1C) indicates the wild-type sequence is disordered and that the stapled peptide SRC2-SP4 adopts an alpha helical conformation in solution. The CD spectrum for SRC2-SP1 shows that a λR substitution at Leu693 negatively impacts α-helicity; however, a λS substitution at Ile689 (SRC2-SP3) maintains helicity as does a λR substitution at Ile689 (SRC2-SP2), albeit to a lesser extent. The observation that addition of methyl groups may positively impact affinity while having a slightly negative effect on helicity may imply that constructive interactions with the surface of the receptor are more important for affinity than locking in a helical conformation.
We obtained co-crystal structures of SRC2-SP1, -SP2, -SP3, -SP4, and -P5 bound to the ligand binding domain of constitutively active Y537S ERα mutant. In all cases, the peptides bind in a similar α-helical conformation,[13] occupy the hydrophobic groove, and make contacts with the so-called “charge clamp:” flanking Lys and Glu residues that align complimentarily with the inherent dipole of the coactivator helix. The most notable difference is that the stapled peptides display a 1.2 Å shift towards the Glu end of the charge clamp, as compared to wild-type (Figure 2A–C). The γ-CH3 of SRC2-SP1 occupies the same region as Leu693 (Figure 2A), and it occupies a pseudo-equatorial conformation to alleviate unfavorable syn-pentane strain between the α- and γ-methyl groups (Figure 2D). The resulting orientation of the γ-methyl increases contact with Ile358 of ERα, which may disrupt its interaction with the Lys end of the charge clamp. Minimizing syn-pentane interactions at this position also substantially alters the χ1 torsion angle (−44°), at Leu693 relative to the more helical stapled peptides (i.e., χ1 = +61° for SRC2-SP3; see Figure 2A). Analogous to SRC2-SP1, minimization of syn-pentane interactions is seen with the γ-methyl group of SRC2-SP3, but, instead of opposing the predominant conformation, the S-γ-methyl reinforces a high-affinity conformation (Figure 2D). Additionally, the γ-methyl occupies the same region as Ile689 in the wild-type sequence (Figure 2C). The γ-methyl of SRC2-SP2 also occupies this same space (Figure 2D), even though the methyl groups are opposite in configuration. The change in the χ2 torsions between these two peptides is ~120°, which can explain how this is possible (Figure 2D).
Figure 2.

X-ray co-crystal structures of peptides bound to ERα (surface: red = acidic, blue = basic, white = nonpolar, green = polar). (A) SRC2-SP1 (red, PDB 5DXB), (B) SRC2-SP2 (green, PDB 5HYR), and (C) SRC2-SP3 (blue, PDB 5DX3) superimposed onto SRC2-WT (yellow, PDB 3ERD). Torsion angles (ωα,β) about the Cα-Cβ bond at position Leu693 are shown. (D) Hydrocarbon staples of SRC2-SP1 (red), SRC2-SP3 (blue), and SRC2-SP4 (magenta, PDB 5DXE) superimposed onto the backbone of SRC2-WT (shown as a yellow tube). SRC2-SP1, SRC2-SP2, and SRC2-SP3 adopt conformations to alleviate syn-pentane interactions between the α- and γ-methyls. The sidechains of non-cyclic SRC2-P5 also bind along the hydrophobic shelf (see supporting information).
We carried out molecular dynamics (MD) studies on SRC2-SP1, -SP2, -SP3, and -SP4 bound to ERα using the NAMD2[14] simulation package with trajectory analysis performed in VMD.[15] The structural ensembles confirmed the strong influence of syn-pentane interactions to the conformation taken up by the staple. In particular, the dihedral angles between position 693 in SRC2-SP1 and SRC2-SP2, -SP3, and -SP4 are opposite in sign, with substantially more fluctuation at this position in SRC2-SP1 (see Figure 3A). In agreement with the x-ray structure, the simulations suggest that SRC2-SP2 adopts a pseudo-eclipsed conformation of –90° at position 689. ERα accommodates the branching methyl of the λS residue at position 689, but the λR residue at position 693 introduces a steric clash with Ile358 of the protein and induces a substantial shift in peptide positioning (Figure 3C). We also carried out MD studies on SRC2-SP1–4 in solution in the absence of ERα. These data confirm that the observed χ1 torsion angles in solution correlate well with the observed angles in the crystal structure, with the caveat that SRC2-SP4 shows stable conformations at both –60° and –90° in solution (see supporting information).
Figure 3.

MD simulations of peptides bound to ERα. (A) The dihedral angle about the χ1 bond reveals different conformations of staple residues 689 and 693 for peptides SRC2-SP1, SRC2-SP2 and SRC2-SP3. (B) The structural conformations of the γ-methyl substituted residue are shown for the last frame of the simulation. (C) The position of the staple shifts substantially during the course of the simulation for SRC2-SP1 and is relatively stable for SRC2-SP3.
In conclusion, we have created a stapling amino acid, λS, that both mimics native branched side chains and stabilizes a helical conformation. In this study, we have used this amino acid and its epimer, λR, to prepare highly potent inhibitors of the ERα/SRC interaction. We have shown that incorporation of a γ-methyl in the R- or S-configuration at the i position of an i, i+4 stapled peptide is a tolerated modification that allows the hydrocarbon staple to effectively mimic branched hydrophobic residues, although the S-methyl results in a conformation with higher helical content than the R does. The S-methyl reinforces an α-helical conformation through minimization of syn-pentane interactions. Incorporation of a γ-methyl group at the i+4 position in either configuration appears to have a destabilizing effect on α-helicity. Although the design here is for interacting residues, incorporation of γ-methyl groups may be applicable to non-interacting stapled residues, as well. In this regard, the simulated and observed staple geometry of methyl substitutions has provided a blueprint for installing γ-methyls and other substituents in stapling amino acids for related protein-protein interactions.
Supplementary Material
Chart 1.

Branched stapling amino acids
Acknowledgments
This work was funded by American Association of Colleges of Pharmacy (to TWM), University of Illinois Cancer Center (to TWM), Searle Funds at The Chicago Community Trust (to TWM and GLG), and NIH (P41-GM104601 to ET). TES is funded by training grant T32 AT007533, sponsored by the Office of the NIH Director and the National Center for Complementary and Integrative Health. Supercomputing resources were provided by NSF XSEDE allocation MCA06N060 (to ET). We thank Kathryn Carlson and John Katzenellenbogen (University of Illinois at Urbana-Champaign) for providing reagents for TR-FRET and Hyun Lee (Research Resources Center, UIC), Gerd Prehna and Ben Ramirez (Center for Structural Biology, UIC) for help with SPR, CD and NMR studies.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Pelay-Gimeno M, Glas A, Koch O, Grossmann TN. Angew Chem Int Ed. 2015;54:8896–8927. doi: 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nevola L, Giralt E. Chem Commun. 2015;51:3302–3315. doi: 10.1039/c4cc08565e. [DOI] [PubMed] [Google Scholar]
- 3.a) Verdine GL, Hilinski GJ. Methods Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]; b) Cromm PM, Spiegel J, Grossmann TN. ACS Chem Biol. 2015;10:1362–1375. doi: 10.1021/cb501020r. [DOI] [PubMed] [Google Scholar]
- 4.Schafmeister CE, Po J, Verdine GL. J Am Chem Soc. 2000;122:5891–5892. [Google Scholar]
- 5.Chu Q, Moellering RE, Hilinski GJ, Kim Y-W, Grossmann TN, Yeh JTH, Verdine GL. Med Chem Commun. 2015;6:111–119. [Google Scholar]
- 6.Kim YW, Grossmann TN, Verdine GL. Nat Protoc. 2011;6:761–771. doi: 10.1038/nprot.2011.324. [DOI] [PubMed] [Google Scholar]
- 7.a) Phillips C, Roberts LR, Schade M, Bazin R, Bent A, Davies NL, Moore R, Pannifer AD, Pickford AR, Prior SH, Read CM, Scott A, Brown DG, Xu B, Irving SL. J Am Chem Soc. 2011;133:9696–9699. doi: 10.1021/ja202946k. [DOI] [PubMed] [Google Scholar]; b) Douse CH, Maas SJ, Thomas JC, Garnett JA, Sun Y, Cota E, Tate EW. ACS Chem Biol. 2014;9:2204–2209. doi: 10.1021/cb500271c. [DOI] [PubMed] [Google Scholar]; c) Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, Filipovic Z, Qureshi FZ, Cai H, Berry P, Feyfant E, Shi XE, Horstick J, Annis DA, Manning AM, Fotouhi N, Nash H, Vassilev LT, Sawyer TK. Proc Natl Acad Sci USA. 2013;110:E3445–E3454. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Glas A, Bier D, Hahne G, Rademacher C, Ottmann C, Grossmann TN. Angew Chem Int Ed. 2014;53:2489–2493. doi: 10.1002/anie.201310082. [DOI] [PubMed] [Google Scholar]; e) Baek S, Kutchukian PS, Verdine GL, Huber R, Holak TA, Lee KW, Popowicz GM. J Am Chem Soc. 2012;134:103–106. doi: 10.1021/ja2090367. [DOI] [PubMed] [Google Scholar]; f) Stewart ML, Fire E, Keating AE, Walensky LD. Nat Chem Biol. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heery DM, Kalkhoven E, Hoare S, Parker MG. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 9.a) Chang CY, Norris JD, Grøn H, Paige LA, Hamilton PT, Kenan DJ, Fowlkes D, McDonnell DP. Mol Cell Biol. 1999;19:8226–8239. doi: 10.1128/mcb.19.12.8226. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fuchs S, Nguyen HD, Phan TT, Burton MF, Nieto L, de Vries-van Leeuwen IJ, Schmidt A, Goodarzifard M, Agten SM, Rose R, Ottmann C, Milroy LG, Brunsveld L. J Am Chem Soc. 2013;135:4364–4371. doi: 10.1021/ja311748r. [DOI] [PubMed] [Google Scholar]; c) Caboni L, Lloyd DG. Med Res Rev. 2013;33:1081–1118. doi: 10.1002/med.21275. [DOI] [PubMed] [Google Scholar]; d) Gallastegui N, Estébanez-Perpiñá E. In: Nuclear Receptors: From Structure to the Clinic. McEwan IJ, Kumar R, editors. Springer International Publishing; 2015. pp. 179–203. [Google Scholar]; e) Moore TW, Mayne CG, Katzenellenbogen JA. Mol Endocrinol. 2010;24:683–695. doi: 10.1210/me.2009-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, He X, Liu S, Hoog J, Lu C, Ding L, Griffith OL, Miller C, Larson D, Fulton RS, Harrison M, Mooney T, McMichael JF, Luo J, Tao Y, Goncalves R, Schlosberg C, Hiken JF, Saied L, Sanchez C, Giuntoli T, Bumb C, Cooper C, Kitchens RT, Lin A, Phommaly C, Davies SR, Zhang J, Kavuri MS, McEachern D, Dong YY, Ma C, Pluard T, Naughton M, Bose R, Suresh R, McDowell R, Michel L, Aft R, Gillanders W, DeSchryver K, Wilson RK, Wang S, Mills GB, Gonzalez-Angulo A, Edwards JR, Maher C, Perou CM, Mardis ER, Ellis MJ. Cell Rep. 2013;4:1116–1130. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, Yelensky R, Brown M, Miller VA, Sarid D, Rizel S, Klein B, Rubinek T, Wolf I. Cancer Res. 2013;73:6856–6864. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]; c) Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, Gursky A, Siddiqui J, Tomlins SA, Roychowdhury S, Pienta KJ, Kim SY, Roberts JS, Rae JM, Van Poznak CH, Hayes DF, Chugh R, Kunju LP, Talpaz M, Schott AF, Chinnaiyan AM. Nat Genet. 2013;45:1446–1451. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, Hortobagyi G, Greene G, Berger M, Baselga J, Chandarlapaty S. Nat Genet. 2013;45:1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Schöllkopf U. Tetrahedron. 1983;39:2085–2091. [Google Scholar]; b) Evans DA, Ennis MD, Mathre DJ. J Am Chem Soc. 1982;104:1737–1739. [Google Scholar]
- 12.Gunther JR, Du Y, Rhoden E, Lewis I, Revennaugh B, Moore TW, Kim SH, Dingledine R, Fu H, Katzenellenbogen JA. J Biomol Screen. 2009;14:181–193. doi: 10.1177/1087057108329349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nettles KW, Bruning JB, Gil G, Nowak J, Sharma SK, Hahm JB, Kulp K, Hochberg RB, Zhou H, Katzenellenbogen JA, Katzenellenbogen BS, Kim Y, Joachmiak A, Greene GL. Nat Chem Biol. 2008;4:241–247. doi: 10.1038/nchembio.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Humphrey W, Dalke A, Schulten K. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.