

Abstract
Objective
Calcific aortic valve disease is a significant clinical problem for which the regulatory mechanisms are poorly understood. Enhanced cell-cell adhesion is a common mechanism of cellular aggregation, but its role in calcific lesion formation is not known. Cadherin-11 (Cad-11) has been associated with lesion formation in vitro, but its function during adult valve homeostasis and pathogenesis is not known. This study aims to elucidate the specific functions of Cad-11 and its downstream targets RhoA and Sox9 in extracellular matrix remodeling and aortic valve calcification.
Approach and Results
We conditionally overexpressed Cad-11 in murine heart valves using a novel double transgenic Nfatc1Cre;R26-Cad11Tg/Tg mouse model. These mice developed hemodynamically significant aortic stenosis with prominent calcific lesions in the aortic valve leaflets. Cad-11 overexpression upregulated downstream targets RhoA and Sox9 in the valve interstitial cells, causing calcification and extensive pathogenic extracellular matrix remodeling. Aortic valve interstitial cells overexpressing Cad-11 in an osteogenic environment in vitro rapidly form calcific nodules analogous to in vivo lesions. Molecular analyses revealed upregulation of osteoblastic and myofibroblastic markers. Treatment with a ROCK inhibitor attenuated nodule formation, further supporting that Cad-11 driven calcification acts through the small GTPase RhoA/ROCK signaling pathway.
Conclusions
This study identifies one of the underlying molecular mechanisms of heart valve calcification and demonstrates that overexpression of Cad-11 upregulates RhoA and Sox9 to induce calcification and extracellular matrix remodeling in adult aortic valve pathogenesis. The findings provide a potential molecular target for clinical treatment.
Introduction
Calcific aortic valve disease (CAVD) is an increasingly prevalent and extremely debilitating disease that currently affects over 25% of Americans over the age of 65.1, 2 Individuals suffering from even early onset of CAVD have increased risk for heart attacks, atherosclerosis, stroke, and heart failure.3 The development of calcific lesions on the aortic valve in CAVD causes thickening and stiffening of the thin, fibrous leaflets due to disruptions in the extracellular matrix (ECM) and leads to stenosis and regurgitation. CAVD can only be diagnosed in later stages through echocardiography as no clinically useful biomarkers have been identified.4, 5 Statins targeted at lowering serum cholesterol have been disappointing in clinical trials,6 further highlighting the lack of available biological diagnostic and therapeutic agents available for treatment. The only existing therapy for valve disease is biological or mechanical prosthetic replacement, as there are no medications to slow the progression of the disease.7
Recent investigations into the mechanisms responsible for aortic valve disease have suggested that a complex interplay of molecular signaling and cell phenotypes influenced by the microenvironment and hemodynamics contribute to valve degeneration.8–10 Heart valves consist of an organized, trilaminar ECM to ensure proper biomechanical function, and disruptions in ECM organization can cause serious valve impairment.8 Inflammatory cytokines, anisotropic strain, reduced nitric oxide signaling, and oxidative stress have all been shown to contribute to ECM remodeling and calcification.11–15 Calcific lesions exhibit characteristics of both dystrophic (apoptotic cell aggregates) and osteogenic (osteoblast, bone-like) calcification.16 Additionally, reactivation of certain developmental pathways may promote valve remodeling preceding osteoblastic differentiation in valve interstitial cells (VICs).10
Cell-cell and cell-matrix interactions at cadherin and integrin junctions are critical for maintaining valvular integrity. Cadherin-11 (also Cad-11, Cdh-11, OB-cadherin) is a cell-cell adhesion protein that mediates cell migration and promotes differentiation of mesenchymal cells to osteo- and chondro-lingeages.17 Dysregulated Cad-11 expression contributes to inflammation, cartilage degradation, and metastasis in diseases such as pulmonary fibrosis, rheumatoid arthritis, and multiple cancer types.18–23 Prior reports have demonstrated increased Cad-11 expression in aortic VICs in adult ApoE−/− mice and calcified human valves.24, 25 Furthermore, Cad-11 depletion disrupts embryonic valve formation and remodeling through inactivation of GTP-RhoA and Sox9, but prevents calcification in adult mice.26 These results suggest a potential link between cell-cell and cell-matrix interactions that may influence matrix production and calcification.
Cadherin-binding mediated signaling functions upstream of Rho family GTPases (CDC42, RhoA, and Rac1), which regulate cell protrusion, contractility, and stress fibers leading to myofibroblast activation.27, 28 RhoA/Rho-associated protein kinase (ROCK) expression is associated with calcification in VICs and vascular smooth muscle cells, and inhibition of ROCK attenuates calcification.29–32 ROCK additionally activates the transcription factor and ECM-regulator Sox9 via serine phosphorylation to promote cartilage matrix production,33 and recent reports highlight an additional role for Sox9 in valve calcification.34–36 These results suggest a multifaceted mechanism that links inter and intracellular signaling with the extracellular microenvironment to promote valve pathogenesis since increased cell-cell contacts, myofibroblast contractility, and collagen/proteoglycan deposition are all common features of CAVD.8 We therefore hypothesized that Cad-11 mediates the mechanosensitivity of VICs via RhoA/ROCK to ensure proper homeostatic ECM maintenance through Sox9.
In this study, we identified a mechanism by which Cad-11 contributes to adult valve calcification and ECM remodeling in vitro and in vivo via RhoA/Sox9. We report that mice overexpressing Cad-11 in the valves display extensive ECM remodeling and develop calcific lesions on the aortic valves with increases in myofibroblastic and osteoblastic markers at 10 months of age. These valves are functionally deficient when evaluated with echocardiography. This calcific phenotype was recapitulated with Cad-11 overexpression in porcine aortic valve interstitial cells (PAVICs) but is attenuated with ROCK inhibition in vitro. These expression patterns were similarly found to be present in calcified human valves. Collectively, these results demonstrate that dysregulation of Cad-11 contributes to valve pathogenesis and identify Cad-11/RhoA/ROCK/Sox9 signaling as a potential pathway for therapeutic targets in treating and slowing the progression of aortic valve disease.
Materials and Methods
Materials and Methods are available in the online-only data supplement.
Results
Cad-11 Overexpression Induces ECM Remodeling but is Not Embryonic Lethal
To study the effects of Cad-11 in aortic valve calcification, we created a novel double transgenic mouse model that conditionally overexpresses Cad-11 in the heart valves. Cadherin-11 transgenic floxed mice (Rosa26-LSL-Cad11Tg/+) were created using targeted embryonic stem cell microinjection into C57BL/6 mice. Full-length Cad-11 cDNA preceded by a floxed stop region was inserted into the ROSA26 locus via BAC transgenesis. These mice were crossed with Nfatc1Cre mice in order to achieve valve-specific conditional overexpression of Cad-11 (Fig. 1A) and have been previously described in which the Cre reporter gene is expressed in endocardial cells and in the cushion mesenchyme derived from the endocardium but is not expressed in the epicardium or myocardium.37, 38 Presence of transgenes were confirmed using PCR (Fig. 1B), and homozygous floxed mice express approximately a five-fold increase of Cad-11 compared to wildtype mice (Fig. SIA). We crossed Nfatc1Cre;R26-Cad11Tg/+ mice, genotyped litters at birth, and found that homozygous (Nfatc1Cre;R26-Cad11Tg/Tg), heterozygous (Nfatc1Cre;R26-Cad11Tg/+), and wildtype (Nfatc1Cre;R26-Cad11+/+) mice were present at expected Mendelian ratios (Table SI, p=0.18). These mice had normal litter sizes that averaged 10.7 mice per litter (n=6) and had no visible physical defects. Additionally, Cad11 OX mice display no significant differences in aortic valve thickness or cross-sectional area at one month (Fig. SIIA). These data show that that valve-specific Cad-11 overexpression does not affect valve development and that Cad11 OX offspring are viable.
Figure 1. Cad-11 overexpression results in AoV thickening and ECM remodeling.
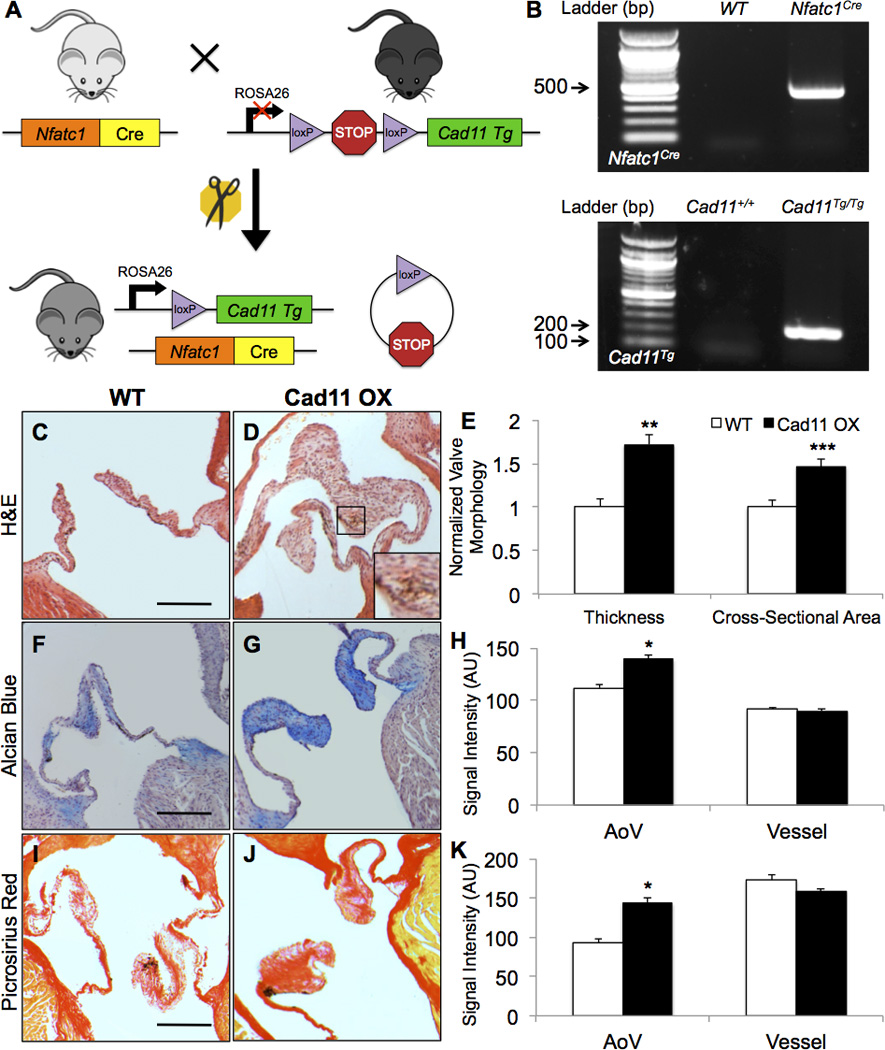
A, Diagram of the double transgenic mouse model. B, Presence of transgenes were confirmed using PCR. C–E, Nfatc1Cre;R26-Cad11Tg/Tg (Cad11 OX) mice display thickened valve morphology and greater valve cross-sectional area compared to Nfatc1Cre;R26-Cad11+/+ wildtype (WT) control mice at 10 months as shown with H&E staining. Magnified area in D shows an area of intense nuclear staining indicating cellular aggregation (n=16, **p<0.001, ***p<0.0001). Scale bar=200μm. F–K, Cad11 OX AoVs contain more glycosaminoglycans and collagen as shown by Alcian Blue and Picrosirius Red staining, respectively. Staining intensity of aortic vessels was measured to ensure consistent staining (n≥7, *p<0.005) Scale bar=200μm. Significance was determined using the Student’s t-test at p<0.05.
Histological analysis reveals that aortic valves (AoVs) of 10 month old, valve-specific Cad-11 overexpressing Nfatc1Cre;R26-Cad11Tg/Tg (Cad11 OX) mice are significantly thickened (>70%) and have greater cross-sectional area (>46%) compared to wildtype Nfatc1Cre;R26-Cad11+/+ (WT) control mice (Fig. 1C–E). Given these structural abnormalities in the Cad11 OX mice, we suspected defects in the extracellular architecture and makeup of the AoVs. We profiled the three main ECM proteins that compose the AoV: glycosaminoglycans, collagen, and elastin. Cad11 OX AoVs express significantly higher glycosaminoglycans and collagen compared to controls (Fig. 1F–H), but have no discernable changes in elastin compared to WT AoVs (Fig. SIB). Cell-matrix adhesions are also elevated as indicated by increased β1 Integrin (Fig. SIC). Analysis of the cellular makeup reveals that Cad11 OX AoVs have greater cell number but equal cell density compared to WT mice (Fig. SID,E). This is likely due to increased proliferation beginning at 1 month, as shown by increased Ki67 positive cells (Fig. SIIB). Together, these results demonstrate that Cad-11 affects the ECM and morphology of mature AoVs postnatally.
Cad-11 Overexpression Results in Calcification and Aortic Stenosis
Increased glycosaminoglycans and collagen are hallmarks of CAVD,8 which led us to stain for the calcification in the aortic valves. Cad11 OX valves exhibit 8.3% mineralization (Fig. 2A–C) and 8.5% calcification (Fig. 2D–F) while these deposits are minimal in WT valves. Myofibroblastic (αSMA) and osteoblastic (Runx2, Osteocalcin) markers are upregulated in Cad11 OX valves, indicating active dystrophic and osteogenic calcification (Fig. SIIIA–G). We further assessed AoV function with echocardiography using Doppler-ultrasound by examining outflow ejection velocity. Cad11 OX mice display elevated ejection velocity (white arrowheads) and regurgitant blood flow (white arrows) that were absent in WT mice (Fig. 2G–I). Cad11 OX mice suffer from left ventricular hypertrophic cardiomyopathy with thickening of both the interventricular septum and left ventricular wall, contributing to an enlarged heart (Fig. SIVA–F). No differences were found in mitral morphology or calcification (Fig. SVA,B), and no differences were found in aortic root diameters (Fig. SIF), supporting aortic valve leaflet-driven calcification as the cause of these CAVD-like symptoms. These results strongly suggest that Cad-11 plays a role in the progression of AoV calcification that promotes valvular and ventricular degeneration.
Figure 2. Cad-11 overexpression leads to formation of calcific lesions and aortic stenosis.
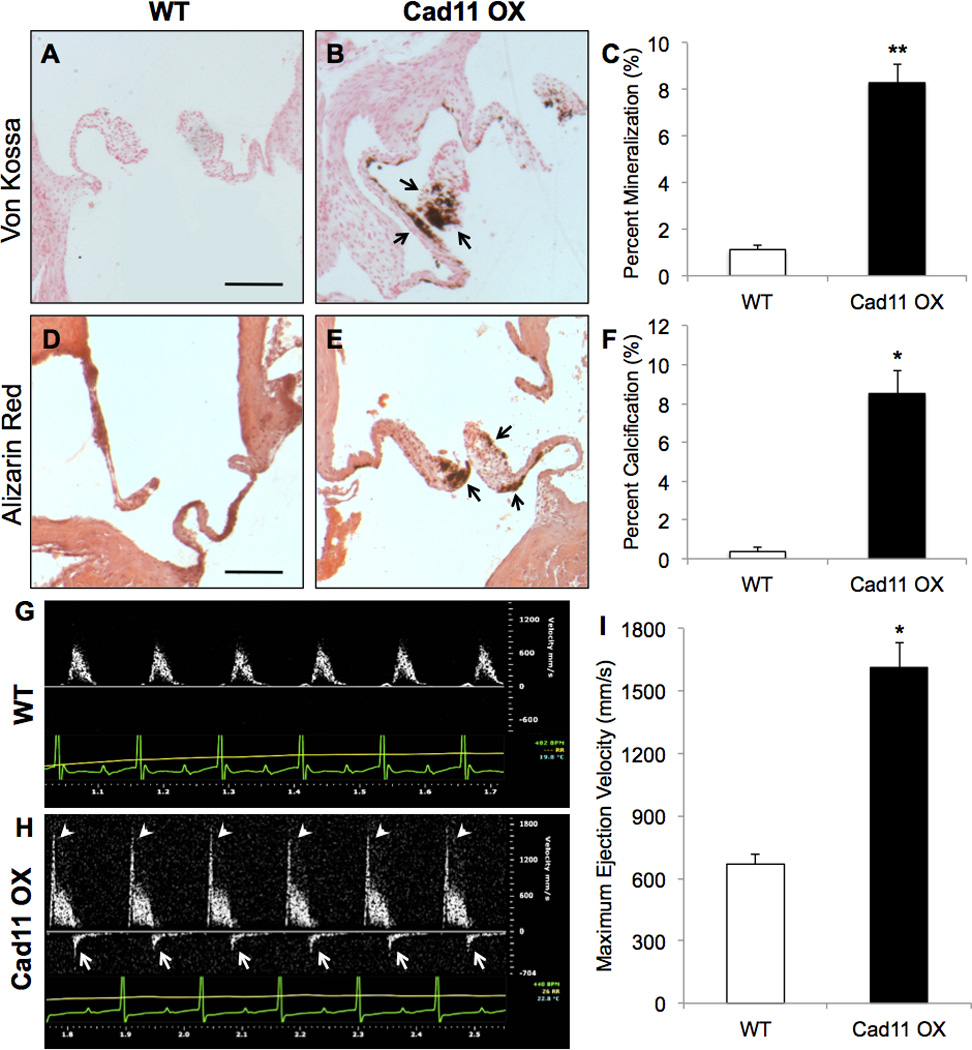
A–C, Von Kossa staining reveals greater mineralization (arrows, B) in adult Cad11 OX mice compared to WT mice at 10 months (n≥9, **p<8E-9) Scale bar=200μm. D–F, Alizarin Red staining reveals greater calcification (arrows, E) in adult Cad11 OX mice compared to WT mice at 10 months (n=4, *p<0.0005) G–I, Blood ejection velocity through the AoV was evaluated using Doppler ultrasound at 10 months. Arrows in H indicate regurgitation while arrowheads indicate elevated outflow velocity compared to WT mice (n=10, *p<0.0005). Significance was determined using the Student’s t-test at p<0.05.
Cad-11 Overexpression Upregulates GTP-RhoA and Sox9
We examined the expression of RhoA (GTP-bound) and Sox9 due to their previously implicated roles in cardiovascular calcification and glycosaminoglycan/collagen production, respectively. Immunofluorescent staining reveals upregulation of both RhoA and Sox9 in Cad11 OX AoVs beginning at 1 month (Fig. SIIC,D). At 10 months, GTP-RhoA expression is 4.3 times higher in Cad11 OX leaflets compared to WT controls (Fig. 3C). Interestingly, RhoA expression is more highly expressed in the valve endothelium (Fig. 3A, white arrows) compared to the interstitium, while RhoA is robustly expressed throughout the entire valve in Cad11 OX mice (Fig. 3B, white arrowheads). Cad11 OX mice have a 2.25 fold increase in overall and nuclear Sox9 expression (Fig. 3D–G). These results collectively suggest that GTP-RhoA and Sox9 are downstream targets of Cad-11 and promote calcification and ECM remodeling.
Figure 3. GTP-RhoA and Sox9 are upregulated in Cad11 OX AoVs.
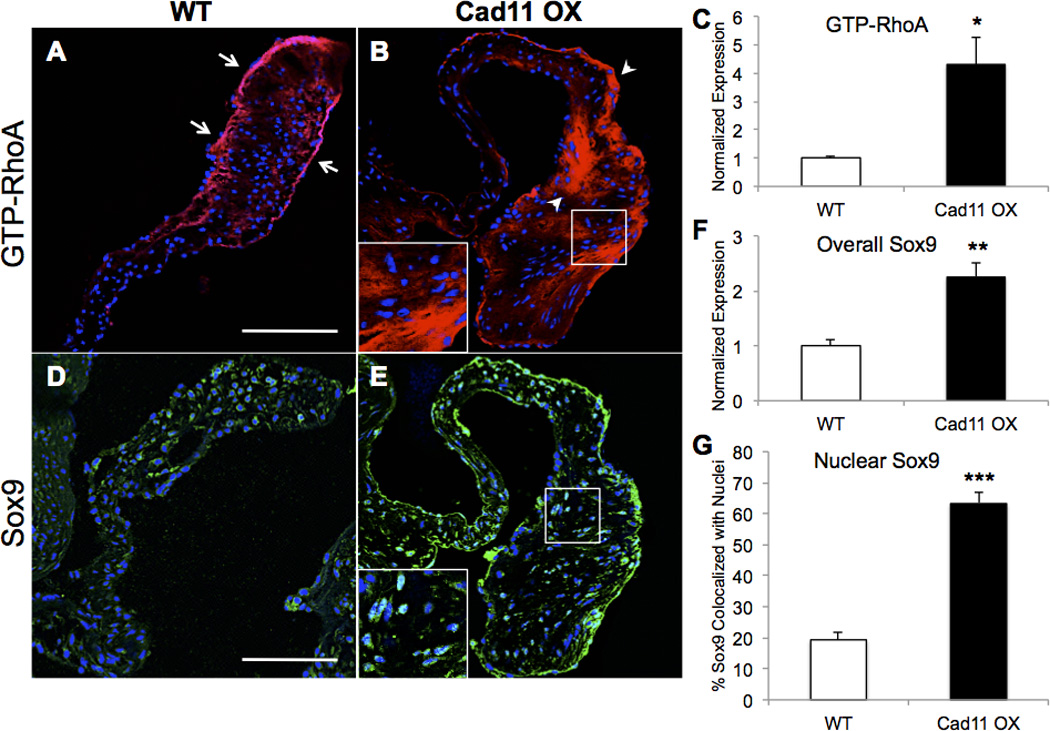
A–C, Active GTP-bound RhoA (GTP-RhoA) expression is higher in Cad11 OX mice at 10 months throughout the aortic valve. Normal GTP-RhoA expression in WT mice is mostly restricted to the valve endothelium (white arrows) whereas expression in Cad11 OX is high in both the endothelium and interstitium (white arrowheads) (n≥5, *p<0.01). D–G, Nuclear and overall Sox9 expression are higher in Cad11 OX mice at 10 months as measured by whole valve fluorescence and colocalization with DRAQ5-stained nuclei. Significance was determined using the Student’s t-test (n≥5, **p<0.005, ***p<1E-8) Scale bar=100μm.
Cad-11 Overexpression Increases Cell Adhesion, Migration, Compaction, and Stress Fiber Bundles
We first investigated the effects of Cad-11 on GTP-RhoA and Sox9 activity in vitro by transfecting primary isolated porcine valve interstitial cells (PAVICs) with a Cad-11 human ORF plasmid, consistently achieving ~100 fold change and ~5 fold increase in gene and protein expression, respectively (Fig. SVIA–D). We found that overexpression of Cad-11 increases cell migration and compaction in wound closure and collagen compaction assays (Fig. SVIIA,C). Interestingly, control PAVICs have individual migrating cells while Cad-11 transfected cells migrate mostly as a collective front and display increased cell-cell adhesion as indicated by smaller cellular aggregates (Fig. SVIIB). These behaviors implicate a greater role for Cad-11 in calcification through cell-matrix compaction and cell-cell adhesivity.
We examined the effects of Cad-11 overexpression on GTP-RhoA/Sox9 expression and cell morphology to determine mechanisms that affect compaction and migration. We found that overexpression of Cad-11 increases GTP-RhoA and Sox9 expression in PAVICs after two days (Fig. 4A–D). Sox9 is highly colocalized to the nuclei of Cad-11 transfected PAVICs but is attenuated with the ROCK inhibitor (Y27632), showing that Cad-11 induces Sox9 nuclear localization in a ROCK-dependent manner (white arrowheads, Fig. 4E,F). Control cells seeded at a low density and stained for F-actin exhibit a normal fibroblast shape with multiple lamellipodia, few filopodia, and some stress fiber bundles (Fig. 5A,D). In contrast, Cad-11 overexpressing PAVICs display multiple filopodia and parallel stress fibers (Fig. 5B,E). Addition of Y27632 to Cad-11 transfected cells did not affect filopodia protrusions (white arrowheads), as shown by the ratio of filopodia to lamellipodia (Fig. 5C,F,G). High density seeding reveals Cad-11 transfected cells to have an almost 3-fold increase in stress fiber bundle-positive cells compared to control cells (white arrows), which is mitigated with Y27632 (Fig. 5H–K).
Figure 4.
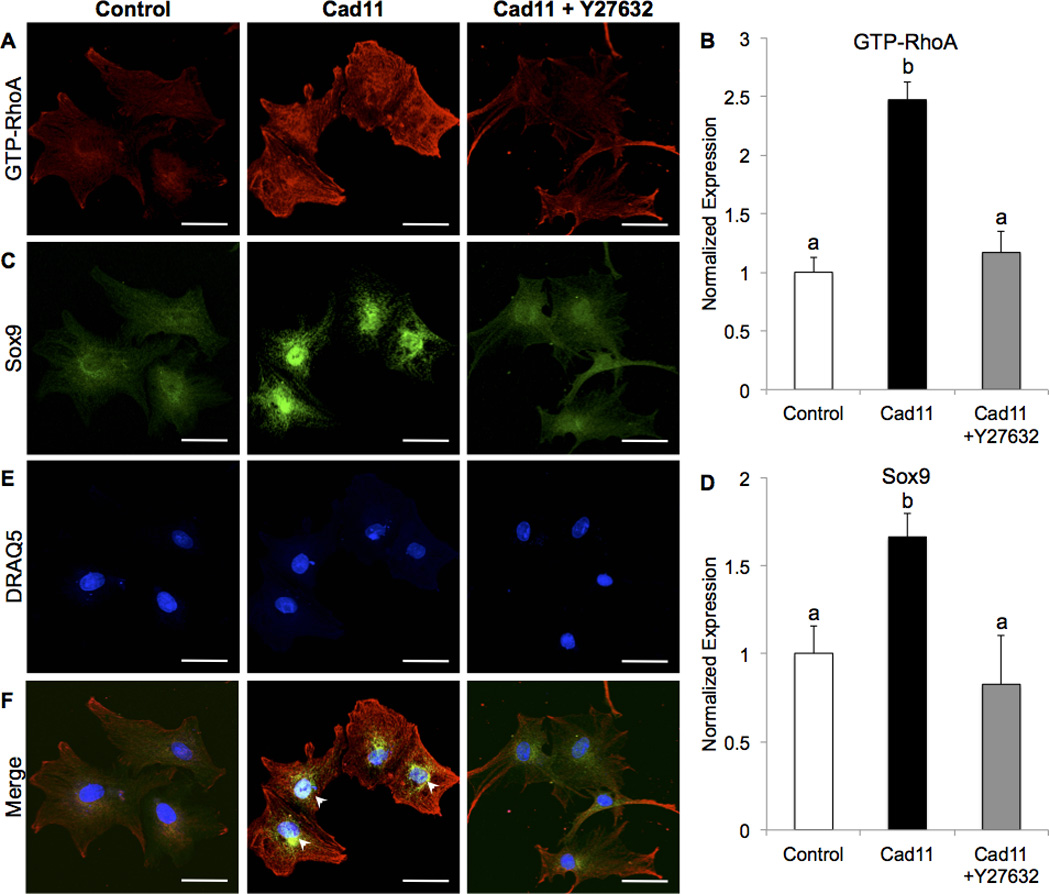
Overexpression of Cad-11 in PAVICs increases GTP-RhoA (A,B) and Sox9 (C,D) expression two days post-transfection, which is attenuated by Y27632 treatment. Sox9 is colocalized to the nucleus (white arrowheads, E,F) in Cad-11 overexpressing PAVICs and expressed mostly in the cytoplasm of control and Y27632 treated cells (n=4). Fluorescence intensity was divided by cell area and normalized to controls. Bars that do not share any letters are significantly different according to a one-way ANOVA with Tukey’s post-hoc test (p<0.005, n≥20 cells). Scale bar=50μm.
Figure 5. Cad-11 overexpression increases stress fiber bundles and protrusions.
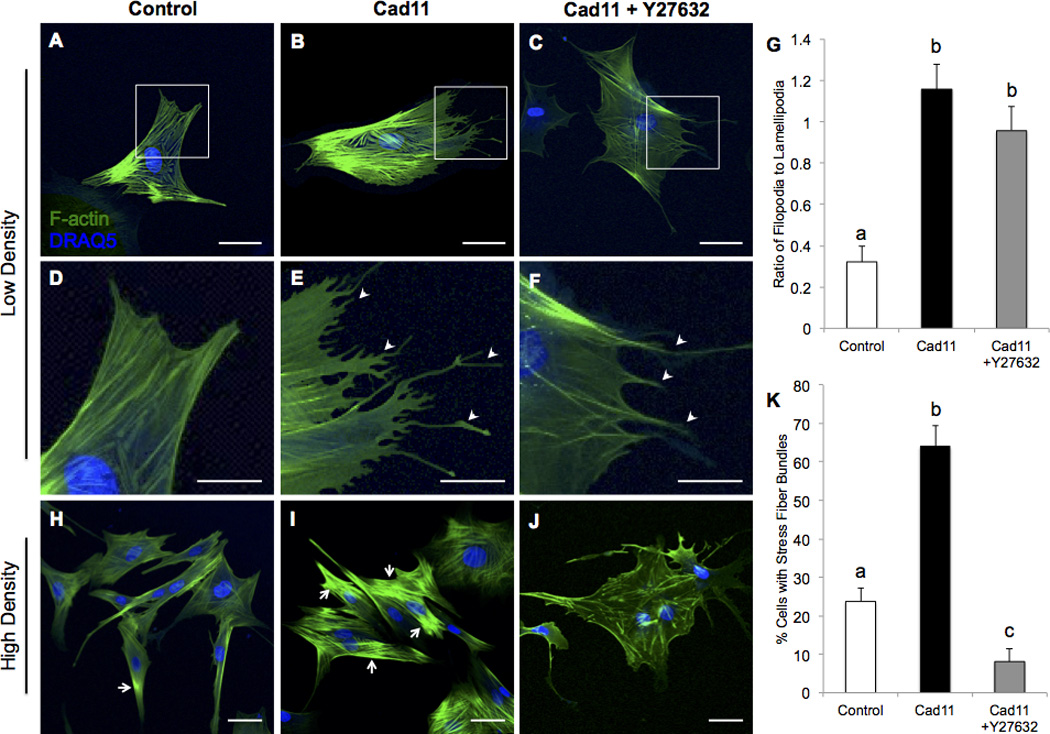
Control PAVICs seeded at a low density (A,D) contain only lamellipodia while Cad-11 overexpressing (Cad11) PAVICs (B,E) exhibit stress fibers and multiple filopodia (white arrowheads) two days post-transfection. Cad-11 overexpressing PAVICs treated with Y27632 exhibit no stress fiber bundles, but have multiple filopodia (white arrowheads, C,F) (Scale bar=50μm for A–C, Scale bar=25μm for D–F). Quantification of the ratio of filopodia to lamellipodia (G). Bars that do not share any letters are significantly different according to a one-way ANOVA with Tukey’s post-hoc test (p<0.01, n≥12 cells). Control PAVICs seeded at a high density have fewer stress fiber bundle positive cells (H, white arrows) compared to Cad-11 transfected cells (I). Cad-11 overexpressing cells treated with Y27632 have few stress fiber bundles (J). Quantification of percent stress fiber bundle positive cells (K). Bars that do not share any letters are significantly different according to a one-way ANOVA with Tukey’s post-hoc test (p<0.01, n≥13 groups of at least 4 cells, Scale bar=50μm for H–J).
Cad-11 Induced Calcification is RhoA/ROCK Dependent
We further examined the role of Cad-11 in calcification by placing PAVICs in osteogenic growth media (OGM). After 10 days, cells in regular media do not calcify, while cells in OGM show numerous calcific nodules, which are decreased with Y27632 (Fig. SVIIIA–C). Brightfield imaging shows aggregation of cells, and live/dead staining of the calcific nodules reveals that these nodules have a partially apoptotic core (Fig. SVIIID,E). Compared to cells in regular growth media, PAVICs placed in OGM display a 14.8 fold increase in Cad-11 mRNA, along with increases in αSMA, RhoA, Sox9, Runx2, and OCN (Osteocalcin). Addition of Y27632 to OGM decreases Cad-11 and RhoA expression, and restores Sox9, αSMA, Runx2, and OCN to control or near-control levels (Fig. SVIIIF).
Cad-11 transfected PAVICs were next analyzed for calcific nodule formation in PAVICs. At Day 10, Cad-11 overexpressing PAVICs in OGM average 92 nodules and greater Alizarin Red activity compared to control cells, which average 47 nodules (Fig. 6A–E), and have significantly upregulated αSMA, RhoA, Sox9, Runx2, and OCN compared to control cells as shown by qRT-PCR (Fig. 6F). Addition of Y27632 attenuates excessive calcification and gene expression even with Cad-11 overexpression (Fig. 6C–F). Cad-11 overexpression alone is sufficient to induce calcific nodule formation through dystrophic and osteogenic mechanisms, but these nodules are much smaller and reduced by Y27632 (Fig. SIXA–E). These results demonstrate that the Cad-11/RhoA/ROCK signaling pathway actively contributes to nodule formation through dystrophic and osteogenic programs and may be a potential target in therapeutic treatment to reduce calcification.
Figure 6. Cad-11 overexpression increases calcification but is attenuated with ROCK inhibition.
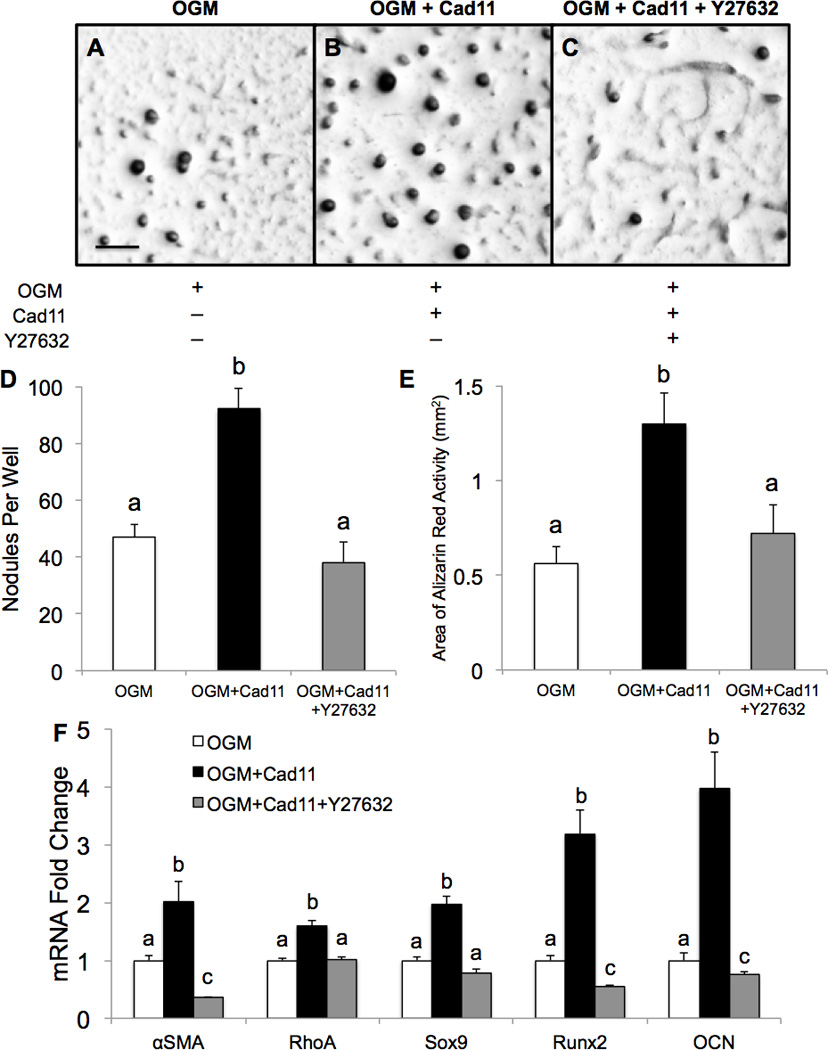
A–C, Alizarin Red staining shows calcific nodules of PAVICs placed in osteogenic growth media (OGM) after 10 days. D,E, Quantification of calcific nodules per well and Alizarin Red activity. Scale bar=0.5mm. F, qRT-PCR shows differences in gene expression among PAVICs in the three conditions compared to OGM. Bars that do not share any letters are significantly different according to a one-way ANOVA with Tukey’s post-hoc test (p<0.01, n=4).
Cad-11, RhoA, and Sox9 are Elevated in Calcified Human Aortic Valves
Histopathological examination of calcified human aortic valves (CHAV) demonstrates significant ECM remodeling and large, calcific nodules (Fig. SXA–D). Similar to Cad11 OX AoVs, increased Cad-11, GTP-RhoA, and Sox9 are apparent in CHAV (Fig. 7E–L). Greater expression of Cad-11, GTP-RhoA, and Sox9 are found in calcified regions (Fig. 7I–K) relative to non-calcified regions (Fig. 7F–H), but both regions have higher expression compared to control aortic valves (Fig. 7A–D). β1 Integrin, αSMA, and Runx2 are also elevated in CHAV compared to controls with calcified regions having higher expression than non-calcified regions (Fig. SXIA–J). Together, this suggests that the Cad-11/RhoA/Sox9 pathway is a clinically relevant mechanism that is associated with increased cell-matrix adhesions and promotes both dystrophic and osteogenic calcification.
Figure 7. Calcified human aortic valves display differences in expression between calcified and non-calcified regions and compared to controls.
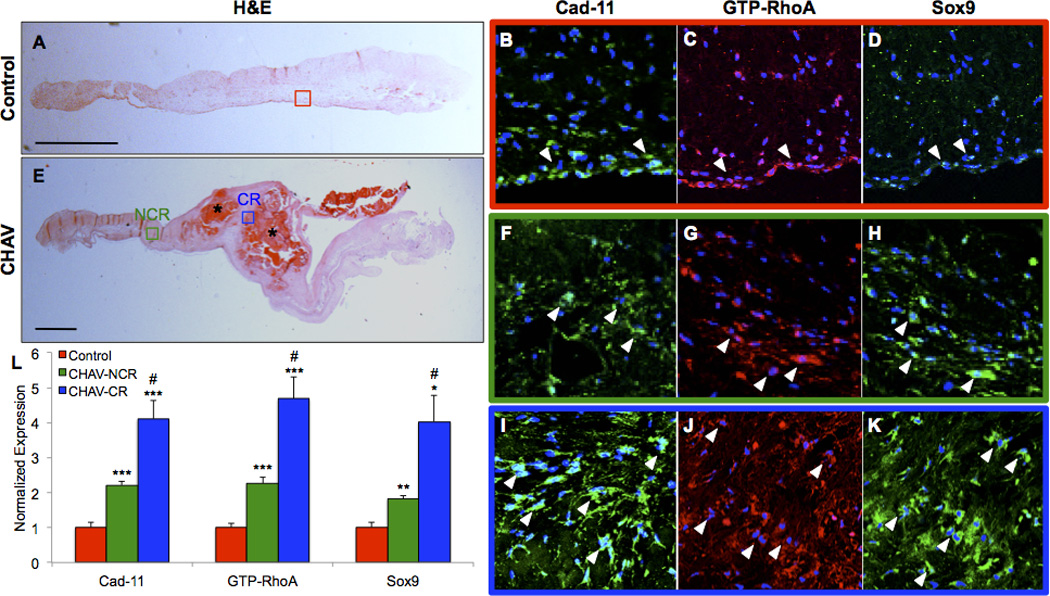
Calcification in control human aortic valves and calcified human aortic valves (CHAV) was visualized with H&E staining (A,E). Cad-11, GTP-RhoA, and Sox9 immunofluorescent staining in control aortic valves (B–D), non-calcified regions (NCR) of CHAV (F–H), and calcified regions (CR) of CHAV (I–K). Quantification of fluorescent intensity (L). White arrowheads point to areas of positive staining. Colored boxes correspond to magnified regions. CR=Calcified Region, NCR=Non-Calcified Region. Significance was determined using the Student’s t-test (n=3 Control, n=5 CHAV, *p<0.05 **p<0.01 ***p<0.005 vs. Control, #p<0.05 vs. CHAV-NCR) Scale bars=1mm
Discussion
Homeostatic maintenance of the cellular architecture in the heart valve must be preserved in order for long-term, efficient cardiac performance. Valve thickening and stiffening caused by calcification compromise this finely tuned, unidirectional diode-like function, ultimately leading to regurgitation and stenosis. CAVD has no clinically useful biomarkers for diagnosis or medications to slow the progression of the disease, so identification of signaling pathways that mediate valve homeostasis and initiate progression of disease is critical to the development of potential diagnostic and therapeutic targets.5, 7, 10 Excessive cadherin-binding mediated signaling increases the small GTPase RhoA, having profound effects on cell behavior, phenotype, and gene expression. In this study, we present evidence that Cad-11 actively participates in calcification and matrix remodeling in the aortic valve upstream of RhoA and Sox9, directing VICs towards myofibroblast and osteoblast phenotypes.
Previous studies have highlighted the role of Cad-11 in maintenance of a fibroblastic phenotype with increased levels leading to inflammation and tissue disorganization.18, 21, 39–41 Calcified human aortic valves express a fifty-fold increase in Cad-11 mRNA, and Cad-11-positive cells are highly colocalized with myofibroblast and osteoblast markers in diseased human and mouse valves.24, 25 Interestingly, adult Cad-11−/− mice have thickened, hemodynamically stenotic valves and decreased GTP-RhoA expression, but lack calcification suggesting that Cad-11 insufficiency is protective of calcification potentially through reduced myofibroblastic activation. This persistent valve thickening is a consequence of defective cellular distribution in valve cushions and subsequent valve compaction and elongation.26, 42 Cad11 OX valves were also thickened, but this thickening occurs due to proliferation preceding increased ECM proteins from excessive remodeling and calcification via active dystrophic and osteogenic mechanisms. Tanaka et al. found ApoE−/− mice to develop calcified and stenotic aortic valves in an age-dependent manner characterized by elevated transaortic velocity and regurgitation.43 Cad11 OX mice also develop aortic stenosis and calcification at 10 months of age as indicated by an average ejection velocity >1600mm/s, regurgitation, presence of osteoblastic markers, and positive Von Kossa and Alizarin Red staining, resulting in remodeling of the left ventricle. These findings elevate the role of cell-cell adhesive signaling in maintaining the homeostatic integrity of the valve architecture necessary for proper function in adulthood and provide opportunities for modulation of adhesive signaling in disease treatment.
Cad-11 promotes inflammation, collective migration, invasion, and proliferation in many cell types including mesenchymal, vascular smooth muscle, cancer, fibroblasts, and neural crest cells.20–23, 44–48 Furthermore, Cad-11 promotes osteogenic differentiation in the context of bone growth and development and additionally plays a role in myofibroblastic activation in dermal and pulmonary fibrosis.49–51 Cad-11 provides the necessary cell-cell tension for aggregation and calcification of valvular myofibroblasts,25 likely mediated by RhoA and favoring a pro-calcific environment as we show here. Cad-11 increases stress fibers, compaction, and collective migration. Stress fiber bundles may indicate cell tension, which is conducive towards calcific nodule formation and demonstrate the role of Cad-11 in mechanosensation. Notably, small GTPases interact with pathogenic pathways such as TGF-β,52 suggesting that an interlocking network of mechanisms is collectively responsible for CAVD. ROCK inhibition mitigates Cad-11-induced mechanosensitivity and calcification as shown by decreased formation of stress fibers, calcific nodules, and osteoblastic genes. Together, these results indicate that Cad-11 increases cell sensitivity to mechanical tension through RhoA signaling and promotes interstitial differentiation into myofibroblasts and osteoblasts.
CHAV and Cad11 OX mice have increased Sox9 expression in the AoVs, and CHAV show higher expression in calcified regions compared to non-calcified regions. Cad-11 has been shown to direct mesenchymal cells to osteo- and chondral-lineages,17 while Sox9 promotes chondrogenesis and prevents osteogenesis by inhibiting Runx2.53 Calcification and chondrogenesis are both present in CAVD,16, 54 and Sox9 mRNA and protein expression are elevated in calcified human aortic valve leaflets, though differences between calcified and non-calcified regions were not examined.36, 55 Previous studies have shown that while reduced Sox9 promotes valve calcification, Sox9 overexpression induces chondrogenesis via β-catenin signaling.34, 35, 56 Together, these results suggest conflicting roles for Sox9 in CAVD. Chondrogenesis and osteogenesis may not be mutually exclusive as mesenchymal stem cells seeded on a collagen/fibronectin/Cad-11 coated surface co-express Sox9 and Runx2.57 We postulate that initial Cad-11/RhoA-induced myofibroblasts increase ECM production via Sox9 since prior studies have identified serine phosphorylation of Sox9 by ROCK as a mechanism of nuclear translocation.33 RhoA activity increases with matrix stiffness, and high RhoA activity directs mesenchymal progenitors to osteogenic lineages.58–60 Increased matrix stiffness and adhesivity, as indicated by increased β1 Integrin, may upregulate RhoA in a positive feedback manner until sufficient to induce osteogenic differentiation. Furthermore, increased ECM stiffness promotes both chondrogenesis and calcification, especially in VICs.8, 61–64 The temporal and spatial expression pattern of the Cad-11/RhoA/Sox9 pathway, mechanisms of Sox9 nuclear translocation, and the dependency of this pathway on matrix stiffness in CAVD remain elusive but certainly warrant further investigation as current evidence places an emphasis on cell-cell and cell-matrix adhesions in determining valve pathobiology by demonstrating the role of tissue stiffness and cellular microenvironment in controlling cell fate.
Cholesterol-lowering drugs as a means of treating CAVD have proven to be ineffective in prior clinical trials.6 Treatments that target pathways mediating cell-adhesion interactions may show more promise by mitigating cell-cell tension and maintaining a fibroblastic phenotype. Inhibition of RhoA/ROCK has been shown to mitigate calcific nodule formation in prior studies, and known functions of the small GTPases suggests that this process involves actin dynamics and cytoskeletal reorganization.29 Favorable cell phenotypes also prevent excess secretion of ECM proteins, contributing to the maintenance of healthy cell-matrix interactions. Our findings show that inhibition of RhoA/ROCK signaling relieves cell tension and is sufficient to prevent excessive calcification, mitigate Sox9 expression, and reduce mRNA levels of myofibroblastic and osteogenic markers, supporting this pathway as a target for prevention of cadherin-small GTPase mediated calcification along with its extracellular consequences. ROCK directly phosphorylates Sox9, contributing to cartilage matrix production in response to TGF-β signaling and supporting our findings that ROCK inhibition decreases Sox9 expression and nuclear localization downstream of RhoA.33 Guanine dissociation inhibitors (GDIs) and GTPase activating proteins (GAPs) both facilitate the conversion of GTP-RhoA to its inactive GDP-bound form and may function as better regulators of RhoA signaling in clinical treatment.65 Furthermore, certain domains of Cad-11 play specific functions in mediating cell polarity and cell-cell interactions, and targeting these specific domains may prove to be most effective.66, 67 Future work investigating the benefits of these inhibitors for prevention or treatment of CAVD may help clinicians arrive at a therapeutic solution to the debilitating disease.
Altogether, we establish the pathological nature of Cad-11 overexpression in aortic valve calcification, preceded by myofibroblastic and osteoblastic differentiation, and its relevance in human valvulopathy. We identify RhoA and Sox9 as downstream targets of Cad-11 whose upregulation causes excessive mechanical activation and ECM remodeling resulting in morphogenic consequences that seriously impair valve function. Finally, we identify ROCK inhibition as a potential means of molecular, therapeutic intervention to halt disease progression and restore healthy valve phenotypes that should be clinically pursued. These results demonstrate the importance of cell-cell interactions in maintaining valve architecture across the lifespan, as abnormal expression in development and adulthood both have pathological consequences.
Supplementary Material
Highlights.
Cadherin-11 overexpression induces aortic stenosis via GTP-RhoA mediated osteogenic and dystrophic calcification and Sox9 mediated extracellular matrix remodeling in vivo.
Treatment with ROCK inhibitor Y27632 inhibits calcific nodule formation, reduces stress fiber formation, and prevents Sox9 nuclear localization in vitro.
Calcified human aortic valves have increased Cadherin-11, GTP-RhoA, and Sox9, with calcified regions having greater expression than non-calcified regions.
Nfatc1Cre;R26-Cad11Tg/Tg display aortic valve calcification, aortic insufficiency, elevated ejection velocity, and left ventricular hypertrophic cardiomyopathy and can be used as a novel model of calcific aortic valve disease.
Initiation of calcification can occur via modulation of a single cell-cell adhesion protein. This is a surface accessible protein rather than genetic modulation of transcription factors and represents potential for locally targeted therapy.
Acknowledgments
We thank the Weill Hall animal facilities veterinary staff and Cornell Biotechnology Resource Center Imaging Facility staff (supported by NIH grants S10RR025502 and S10OD016191) for their skillful and technical assistance. We also thank John Schimenti (supported by NY State Stem Cell Program CO29155) for his critical assistance in the development of the Cad-11 transgenic mouse.
Sources of Funding
This study was supported by the National Institutes of Health (HL110328, HL128745, and HL18672 to JTB and HL078881, HL104444, and HL111770 to BZ), the National Science Foundation (CBET-0955712 to JTB), the American Heart Association Undergraduate Student Fellowship (CJB and KAV), and the Hunter R. Rawlings III Cornell Presidential Research Scholarship (DCS and CJB).
Non-Standard Abbreviations and Acronyms
- αSMA
alpha smooth muscle actin
- AoV
aortic valve
- Cad11 OX
Cad-11 overexpression
- Cadherin-11
Cad-11
- CAVD
calcific aortic valve disease
- CHAV
calcified human aortic valve
- ECM
extracellular matrix
- OGM
osteogenic growth media
- PAVIC
porcine aortic valve interstitial cell
- ROCK
Rho-associated protein kinase
- VIC
valve interstitial cell
Footnotes
Disclosures
None.
References
- 1.Towler DA. Oxidation, inflammation, and aortic valve calcification peroxide paves an osteogenic path. J Am Coll Cardiol. 2008;52:851–854. doi: 10.1016/j.jacc.2008.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Messika-Zeitoun D, Bielak LF, Peyser PA, Sheedy PF, Turner ST, Nkomo VT, Breen JF, Maalouf J, Scott C, Tajik AJ, Enriquez-Sarano M. Aortic valve calcification: determinants and progression in the population. Arterioscler Thromb Vasc Biol. 2007;27:642–648. doi: 10.1161/01.ATV.0000255952.47980.c2. [DOI] [PubMed] [Google Scholar]
- 3.Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999;341:142–147. doi: 10.1056/NEJM199907153410302. [DOI] [PubMed] [Google Scholar]
- 4.Baumgartner H, Hung J, Bermejo J, Chambers JB, Evangelista A, Griffin BP, Iung B, Otto CM, Pellikka PA. Quinones M and Eae/Ase. Echocardiographic assessment of valve stenosis: EAE/ASE recommendations for clinical practice. Eur J Echocardiogr. 2009;10:1–25. doi: 10.1093/ejechocard/jen303. [DOI] [PubMed] [Google Scholar]
- 5.Beckmann E, Grau JB, Sainger R, Poggio P, Ferrari G. Insights into the use of biomarkers in calcific aortic valve disease. J Heart Valve Dis. 2010;19:441–452. [PMC free article] [PubMed] [Google Scholar]
- 6.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. Scottish Aortic S and Lipid Lowering Trial IoRI. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- 7.Freeman RV, Otto CM. Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies. Circulation. 2005;111:3316–3326. doi: 10.1161/CIRCULATIONAHA.104.486738. [DOI] [PubMed] [Google Scholar]
- 8.Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108:1510–1524. doi: 10.1161/CIRCRESAHA.110.234237. [DOI] [PubMed] [Google Scholar]
- 9.Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res. 2013;113:198–208. doi: 10.1161/CIRCRESAHA.113.300155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butcher JT, Mahler GJ, Hockaday LA. Aortic valve disease and treatment: the need for naturally engineered solutions. Adv Drug Deliv Rev. 2011;63:242–268. doi: 10.1016/j.addr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Farrar EJ, Butcher JT. Heterogeneous susceptibility of valve endothelial cells to mesenchymal transformation in response to TNFalpha. Ann Biomed Eng. 2014;42:149–161. doi: 10.1007/s10439-013-0894-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farrar EJ, Huntley GD, Butcher J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS One. 2015;10:e0123257. doi: 10.1371/journal.pone.0123257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:121–130. doi: 10.1161/ATVBAHA.112.300504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gould RA, Chin K, Santisakultarm TP, Dropkin A, Richards JM, Schaffer CB, Butcher JT. Cyclic strain anisotropy regulates valvular interstitial cell phenotype and tissue remodeling in three-dimensional culture. Acta Biomater. 2012;8:1710–1719. doi: 10.1016/j.actbio.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards J, El-Hamamsy I, Chen S, Sarang Z, Sarathchandra P, Yacoub MH, Chester AH, Butcher JT. Side-specific endothelial-dependent regulation of aortic valve calcification: interplay of hemodynamics and nitric oxide signaling. Am J Pathol. 2013;182:1922–1931. doi: 10.1016/j.ajpath.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohler ER, 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 17.Kii I, Amizuka N, Shimomura J, Saga Y, Kudo A. Cell-cell interaction mediated by cadherin-11 directly regulates the differentiation of mesenchymal cells into the cells of the osteo-lineage and the chondro-lineage. J Bone Miner Res. 2004;19:1840–1849. doi: 10.1359/JBMR.040812. [DOI] [PubMed] [Google Scholar]
- 18.Chang SK, Noss EH, Chen M, Gu Z, Townsend K, Grenha R, Leon L, Lee SY, Lee DM, Brenner MB. Cadherin-11 regulates fibroblast inflammation. Proc Natl Acad Sci U S A. 2011;108:8402–8407. doi: 10.1073/pnas.1019437108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider DJ, Wu M, Le TT, Cho SH, Brenner MB, Blackburn MR, Agarwal SK. Cadherin-11 contributes to pulmonary fibrosis: potential role in TGF-beta production and epithelial to mesenchymal transition. FASEB J. 2012;26:503–512. doi: 10.1096/fj.11-186098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Assefnia S, Dakshanamurthy S, Guidry Auvil JM, Hampel C, Anastasiadis PZ, Kallakury B, Uren A, Foley DW, Brown ML, Shapiro L, Brenner M, Haigh D, Byers SW. Cadherin-11 in poor prognosis malignancies and rheumatoid arthritis: common target, common therapies. Oncotarget. 2014;5:1458–1474. doi: 10.18632/oncotarget.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiener HP, Lee DM, Agarwal SK, Brenner MB. Cadherin-11 induces rheumatoid arthritis fibroblast-like synoviocytes to form lining layers in vitro. Am J Pathol. 2006;168:1486–1499. doi: 10.2353/ajpath.2006.050999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang CF, Lira C, Chu K, Bilen MA, Lee YC, Ye X, Kim SM, Ortiz A, Wu FL, Logothetis CJ, Yu-Lee LY, Lin SH. Cadherin-11 increases migration and invasion of prostate cancer cells and enhances their interaction with osteoblasts. Cancer Res. 2010;70:4580–4589. doi: 10.1158/0008-5472.CAN-09-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaur H, Phillips-Mason PJ, Burden-Gulley SM, Kerstetter-Fogle AE, Basilion JP, Sloan AE, Brady-Kalnay SM. Cadherin-11, a marker of the mesenchymal phenotype, regulates glioblastoma cell migration and survival in vivo. Mol Cancer Res. 2012;10:293–304. doi: 10.1158/1541-7786.MCR-11-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J, Bowen C, Lu G, Knapp Iii C, Recknagel A, Norris RA, Butcher JT. Cadherin-11 expression patterns in heart valves associate with key functions during embryonic cushion formation, valve maturation and calcification. Cells Tissues Organs. 2013;198:300–310. doi: 10.1159/000356762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutcheson JD, Chen J, Sewell-Loftin MK, Ryzhova LM, Fisher CI, Su YR, Merryman WD. Cadherin-11 regulates cell-cell tension necessary for calcific nodule formation by valvular myofibroblasts. Arterioscler Thromb Vasc Biol. 2013;33:114–120. doi: 10.1161/ATVBAHA.112.300278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowen CJ, Zhou J, Sung DC, Butcher JT. Cadherin-11 coordinates cellular migration and extracellular matrix remodeling during aortic valve maturation. Dev Biol. 2015;407:145–157. doi: 10.1016/j.ydbio.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kashef J, Kohler A, Kuriyama S, Alfandari D, Mayor R, Wedlich D. Cadherin-11 regulates protrusive activity in Xenopus cranial neural crest cells upstream of Trio and the small GTPases. Genes Dev. 2009;23:1393–1398. doi: 10.1101/gad.519409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noren NK, Niessen CM, Gumbiner BM, Burridge K. Cadherin engagement regulates Rho family GTPases. J Biol Chem. 2001;276:33305–33308. doi: 10.1074/jbc.C100306200. [DOI] [PubMed] [Google Scholar]
- 29.Gu X, Masters KS. Role of the Rho pathway in regulating valvular interstitial cell phenotype and nodule formation. Am J Physiol Heart Circ Physiol. 2011;300:H448–H458. doi: 10.1152/ajpheart.01178.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monzack EL, Gu X, Masters KS. Efficacy of simvastatin treatment of valvular interstitial cells varies with the extracellular environment. Arterioscler Thromb Vasc Biol. 2009;29:246–253. doi: 10.1161/ATVBAHA.108.179218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen NX, Chen X, O'Neill KD, Atkinson SJ, Moe SM. RhoA/Rho kinase (ROCK) alters fetuin-A uptake and regulates calcification in bovine vascular smooth muscle cells (BVSMC) Am J Physiol Renal Physiol. 2010;299:F674–F680. doi: 10.1152/ajprenal.00730.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terao Y, Satomi-Kobayashi S, Hirata K, Rikitake Y. Involvement of Rho-associated protein kinase (ROCK) and bone morphogenetic protein-binding endothelial cell precursor-derived regulator (BMPER) in high glucose-increased alkaline phosphatase expression and activity in human coronary artery smooth muscle cells. Cardiovasc Diabetol. 2015;14:104. doi: 10.1186/s12933-015-0271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haudenschild DR, Chen J, Pang N, Lotz MK, D'Lima DD. Rho kinase-dependent activation of SOX9 in chondrocytes. Arthritis Rheum. 2010;62:191–200. doi: 10.1002/art.25051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fang M, Alfieri CM, Hulin A, Conway SJ, Yutzey KE. Loss of beta-catenin promotes chondrogenic differentiation of aortic valve interstitial cells. Arterioscler Thromb Vasc Biol. 2014;34:2601–2608. doi: 10.1161/ATVBAHA.114.304579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res. 2010;106:712–719. doi: 10.1161/CIRCRESAHA.109.213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexopoulos A, Bravou V, Peroukides S, Kaklamanis L, Varakis J, Alexopoulos D, Papadaki H. Bone regulatory factors NFATc1 and Osterix in human calcific aortic valves. Int J Cardiol. 2010;139:142–149. doi: 10.1016/j.ijcard.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 37.Wu B, Zhang Z, Lui W, Chen X, Wang Y, Chamberlain AA, Moreno-Rodriguez RA, Markwald RR, O'Rourke BP, Sharp DJ, Zheng D, Lenz J, Baldwin HS, Chang CP, Zhou B. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell. 2012;151:1083–1096. doi: 10.1016/j.cell.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snider P, Simmons O, Wang J, Hoang CQ, Conway SJ. Ectopic Noggin in a Population of Nfatc1 Lineage Endocardial Progenitors Induces Embryonic Lethality. J Cardiovasc Dev Dis. 2014;1:214–236. doi: 10.3390/jcdd1030214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiener HP, Brenner MB. Building the synovium: cadherin-11 mediates fibroblast-like synoviocyte cell-to-cell adhesion. Arthritis Res Ther. 2005;7:49–54. doi: 10.1186/ar1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niessen CM, Leckband D, Yap AS. Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol Rev. 2011;91:691–731. doi: 10.1152/physrev.00004.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valencia X, Higgins JM, Kiener HP, Lee DM, Podrebarac TA, Dascher CC, Watts GF, Mizoguchi E, Simmons B, Patel DD, Bhan AK, Brenner MB. Cadherin-11 provides specific cellular adhesion between fibroblast-like synoviocytes. J Exp Med. 2004;200:1673–1679. doi: 10.1084/jem.20041545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowen CJ, Zhou J, Sung DC, Butcher JT. Cadherin-11 coordinates cellular migration and extracellular matrix remodeling during aortic valve maturation. Dev Biol. 2015 doi: 10.1016/j.ydbio.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka K, Sata M, Fukuda D, Suematsu Y, Motomura N, Takamoto S, Hirata Y, Nagai R. Age-associated aortic stenosis in apolipoprotein E-deficient mice. J Am Coll Cardiol. 2005;46:134–141. doi: 10.1016/j.jacc.2005.03.058. [DOI] [PubMed] [Google Scholar]
- 44.Becker SF, Mayor R, Kashef J. Cadherin-11 mediates contact inhibition of locomotion during Xenopus neural crest cell migration. PLoS One. 2013;8:e85717. doi: 10.1371/journal.pone.0085717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCusker C, Cousin H, Neuner R, Alfandari D. Extracellular cleavage of cadherin-11 by ADAM metalloproteases is essential for Xenopus cranial neural crest cell migration. Mol Biol Cell. 2009;20:78–89. doi: 10.1091/mbc.E08-05-0535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vallin J, Girault JM, Thiery JP, Broders F. Xenopus cadherin-11 is expressed in different populations of migrating neural crest cells. Mech Dev. 1998;75:171–174. doi: 10.1016/s0925-4773(98)00099-9. [DOI] [PubMed] [Google Scholar]
- 47.Monahan TS, Andersen ND, Panossian H, Kalish JA, Daniel S, Shrikhande GV, Ferran C, Logerfo FW. A novel function for cadherin 11/osteoblast-cadherin in vascular smooth muscle cells: modulation of cell migration and proliferation. J Vasc Surg. 2007;45:581–589. doi: 10.1016/j.jvs.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 48.Pishvaian MJ, Feltes CM, Thompson P, Bussemakers MJ, Schalken JA, Byers SW. Cadherin-11 is expressed in invasive breast cancer cell lines. Cancer Res. 1999;59:947–952. [PubMed] [Google Scholar]
- 49.Kawaguchi J, Kii I, Sugiyama Y, Takeshita S, Kudo A. The transition of cadherin expression in osteoblast differentiation from mesenchymal cells: consistent expression of cadherin-11 in osteoblast lineage. J Bone Miner Res. 2001;16:260–269. doi: 10.1359/jbmr.2001.16.2.260. [DOI] [PubMed] [Google Scholar]
- 50.Di Benedetto A, Watkins M, Grimston S, Salazar V, Donsante C, Mbalaviele G, Radice GL, Civitelli R. N-cadherin and cadherin 11 modulate postnatal bone growth and osteoblast differentiation by distinct mechanisms. J Cell Sci. 2010;123:2640–2648. doi: 10.1242/jcs.067777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schroer AK, Merryman WD. Mechanobiology of myofibroblast adhesion in fibrotic cardiac disease. J Cell Sci. 2015;128:1865–1875. doi: 10.1242/jcs.162891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edlund S, Landstrom M, Heldin CH, Aspenstrom P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell. 2002;13:902–914. doi: 10.1091/mbc.01-08-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, Lee B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci U S A. 2006;103:19004–19009. doi: 10.1073/pnas.0605170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O'Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation. 2011;124:1783–1791. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, Spelsberg TC, McCarthy PM, Rahimtoola SH, Rajamannan NM. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. 2006;47:1707–1712. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lincoln J, Kist R, Scherer G, Yutzey KE. Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev Biol. 2007;305:120–132. doi: 10.1016/j.ydbio.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong S, Guo H, Zhang Y, Li Z, Kang F, Yang B, Kang X, Wen C, Yan Y, Jiang B, Fan Y. rFN/Cad-11-modified collagen type II biomimetic interface promotes the adhesion and chondrogenic differentiation of mesenchymal stem cells. Tissue Eng Part A. 2013;19:2464–2477. doi: 10.1089/ten.tea.2012.0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khatiwala CB, Kim PD, Peyton SR, Putnam AJ. ECM compliance regulates osteogenesis by influencing MAPK signaling downstream of RhoA and ROCK. J Bone Miner Res. 2009;24:886–898. doi: 10.1359/JBMR.081240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim TJ, Seong J, Ouyang M, Sun J, Lu S, Hong JP, Wang N, Wang Y. Substrate rigidity regulates Ca2+ oscillation via RhoA pathway in stem cells. J Cell Physiol. 2009;218:285–293. doi: 10.1002/jcp.21598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163:583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Allen JL, Cooke ME, Alliston T. ECM stiffness primes the TGFbeta pathway to promote chondrocyte differentiation. Mol Biol Cell. 2012;23:3731–3742. doi: 10.1091/mbc.E12-03-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duan B, Hockaday LA, Kapetanovic E, Kang KH, Butcher JT. Stiffness and adhesivity control aortic valve interstitial cell behavior within hyaluronic acid based hydrogels. Acta Biomater. 2013;9:7640–7650. doi: 10.1016/j.actbio.2013.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yip CY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936–942. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- 64.Benton JA, Kern HB, Anseth KS. Substrate properties influence calcification in valvular interstitial cell culture. J Heart Valve Dis. 2008;17:689–699. [PMC free article] [PubMed] [Google Scholar]
- 65.DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15:356–363. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 66.Kiener HP, Niederreiter B, Lee DM, Jimenez-Boj E, Smolen JS, Brenner MB. Cadherin 11 promotes invasive behavior of fibroblast-like synoviocytes. Arthritis Rheum. 2009;60:1305–1310. doi: 10.1002/art.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kiener HP, Stipp CS, Allen PG, Higgins JM, Brenner MB. The cadherin-11 cytoplasmic juxtamembrane domain promotes alpha-catenin turnover at adherens junctions and intercellular motility. Mol Biol Cell. 2006;17:2366–2376. doi: 10.1091/mbc.E05-08-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.