

Abstract
G protein-coupled receptors constitute the largest family of membrane receptors and modulate almost every physiological process in humans. Binding of agonists to G protein-coupled receptors induces a shift from inactive to active receptor conformations. Biophysical studies of the dynamic equilibrium of receptors suggest that a portion of receptors can remain in inactive states even in the presence of saturating concentrations of agonist and G protein mimetic. However, the molecular details of agonist-bound inactive receptors are poorly understood. Here we use the model of bitopic orthosteric/allosteric (i.e. dualsteric) agonists for muscarinic M2 receptors to demonstrate the existence and function of such inactive agonist·receptor complexes on a molecular level. Using all-atom molecular dynamics simulations, dynophores (i.e. a combination of static three-dimensional pharmacophores and molecular dynamics-based conformational sampling), ligand design, and receptor mutagenesis, we show that inactive agonist·receptor complexes can result from agonist binding to the allosteric vestibule alone, whereas the dualsteric binding mode produces active receptors. Each agonist forms a distinct ligand binding ensemble, and different agonist efficacies depend on the fraction of purely allosteric (i.e. inactive) versus dualsteric (i.e. active) binding modes. We propose that this concept may explain why agonist·receptor complexes can be inactive and that adopting multiple binding modes may be generalized also to small agonists where binding modes will be only subtly different and confined to only one binding site.
Keywords: chemical biology, drug design, G protein-coupled receptor (GPCR), molecular dynamics, molecular pharmacology, dualsteric ligands, dynamic ligand binding, dynophores, ligand binding ensembles, partial agonist
Introduction
Specific and coordinated cell-to-cell communication regulates the flow of information between cells, and proper information processing ensures physiological functions of biological systems. G protein-coupled receptors (GPCRs),8 constituting the largest class of membrane proteins in mammals, are essential mediators of chemically and light-encoded information (1–4). GPCRs sense a great variety of extracellular stimuli, e.g. neurotransmitters and hormones, and subsequently translate this information into an intracellular response via G proteins, β-arrestins, and possibly GPCR-interacting proteins (2–5). Because of their abundance and relevance in regulating the majority of (patho-)physiological processes in humans, GPCRs have for a long time represented the most important drug targets being addressed by at least a third of all currently marketed drugs (6, 7).
Agonist binding leads to receptor activation, which is followed by intracellular G protein recruitment and subsequent cell signaling. Breakthroughs in GPCR crystallography have led to inactive and active crystal structures of the same receptor protein. Among these are rhodopsin (8–10) and more recently the β2-adrenergic (11–14), M2 muscarinic (15, 16), and μ-opioid receptors (17, 18). These structures most likely represent energetically favored, relatively stable inactive and active receptor·ligand complexes. Despite the diversity of crystallized receptors, a common mechanism of receptor activation can be inferred from these structures in conjunction with a wealth of older biochemical data. Agonist-mediated receptor activation leads to an outward tilt of the intracellular parts of transmembrane helices V and VI, facilitating intracellular G protein binding, which in many cases is accompanied by an inward movement of the extracellular parts of transmembrane helices V and VI. This results in a contraction of the ligand binding site, closing it off toward the extracellular space.
Despite the wealth of GPCR structures, however, little is known about the structural dynamics of their inactive-to-active transitions and different agonist efficacies. Recent biophysical studies on various receptors (Class A and Class C) have shown that GPCRs reside in a dynamic equilibrium of distinct receptor conformations comprising inactive and active receptor states (19–22). These studies suggest a common mechanism for agonist efficacy: agonists shift the preexisting equilibrium of different receptor conformations toward more active states (21, 22).
Interestingly, the dynamic equilibrium of receptors remains heterogeneous even in the presence of saturating concentrations of full agonists and always contains a fraction of receptors in inactive states (19, 21–23). In fact, agonists alone are not sufficient to stabilize the fully active receptor state as seen in the crystal structures (19–24). The fully active state is only reached upon addition of both saturating concentrations of agonist and a G protein or nanobody (19, 21, 23). However, even under these conditions, e.g. in the presence of the full agonist isoproterenol and the nanobody Nb80, a significant percentage of β2-adrenergic receptors still remains in inactive states (21).
In line with this concept, partial agonists would stabilize fewer receptors in active states, and concomitantly a greater fraction of partial agonist-bound receptors would remain in inactive states (22, 24). However, it is puzzling how agonist-bound receptors can adopt active and inactive states. Based on the above mentioned evidence that receptors are “floppy,” it is reasonable to hypothesize that agonists may have multiple binding modes; some of these agonist binding modes might then stabilize active agonist·receptor complexes, whereas other agonist binding modes might stabilize inactive agonist·receptor complexes.
However, because purely orthosteric partial agonists are likely to show only slightly different binding modes, it is technically challenging to investigate this phenomenon with current methods. In this study, we make use of a special case of agonism to identify multiple binding topographies of agonists and to provide a proof of principle that ligand binding ensembles can form the molecular basis of different agonist efficacies. This special case is dualsteric partial agonists for the muscarinic M2 receptor (M2AChR) that comprise two pharmacophores targeting the orthosteric and allosteric binding sites, respectively (25). These dualsteric agonists have been suggested to possess two pharmacologically distinct binding modes (26–28), a feature defined as dynamic ligand binding (28). The “extreme” molecular nature of the dualsteric agonists, i.e. spanning two binding sites, is anticipated to allow the identification of distinct ligand binding topographies.
By combining pharmacological methods, molecular docking, and all-atom molecular dynamics simulations based on M2AChR crystal structures, we here identify two distinct binding topographies of a set of dualsteric partial agonists. One binding mode resembles that of the co-crystallized orthosteric agonist iperoxo (16) and forms an active agonist·receptor complex. The other binding mode is purely allosteric and forms an inactive agonist·receptor complex by adopting binding topographies similar to those of common allosteric modulators (29). We show that both types of agonist·receptor complexes are present in a receptor population at the same time. Manipulation of the dynamic equilibrium of active and inactive agonist·receptor complexes by ligand design and by mutations reveals that the fraction of inactive agonist·receptor complexes decreases overall agonist efficacy.
Our findings suggest that an ensemble of active and inactive agonist·receptor complexes in which agonists adopt multiple binding topographies is a molecular basis for different agonist efficacies. Albeit having studied a highly specific case of GPCR agonism, our findings provide general evidence for the concept that agonists can principally stabilize inactive agonist·receptor complexes; this may also apply to agonists that target only one binding site of the receptor.
Results
Direct Pharmacological Evidence for Multiple Binding Modes of a Dualsteric Agonist
Dualsteric (also termed bitopic orthosteric/allosteric) ligands (30) for muscarinic receptors are composed of orthosteric and allosteric moieties covalently connected via linkers of different chemical nature (31–34). Recent studies suggested that at least some of these bipharmacophoric ligands may have more than one binding mode (28, 34). The experimental results are compatible with a theoretical scenario, i.e. dynamic ligand binding, in which such a ligand would adopt a dualsteric pose (Fig. 1a) and a purely allosteric binding mode (Fig. 1a). In this study, we provide direct evidence that iper-6-naph indeed displays two distinct binding modes at M2AChRs. Iper-6-naph is a prototypical dualsteric ligand (Fig. 1a) that is composed of the orthosteric agonist iperoxo linked via a hexamethylene chain to an allosteric modulator derived from naphmethonium. Equilibrium binding of iper-6-naph using N-[3H]methylscopolamine ([3H]NMS) as the orthosteric radiotracer shows partial displacement of [3H]NMS in a HEPES buffer (10 mm HEPES, 10 mm MgCl2, 100 mm NaCl) (Fig. 1b). [3H]NMS displacement indicates ligand competition between iper-6-naph and the radioligand at the orthosteric site. However, the incomplete [3H]NMS displacement at high concentrations of iper-6-naph is indicative of the formation of ternary complexes consisting of [3H]NMS and iper-6-naph bound to the receptor's orthosteric and allosteric sites, respectively. The binding of iper-6-naph to the allosteric site hampers [3H]NMS dissociation, which is detected as incomplete displacement (Fig. 1b). To directly prove that iper-6-naph can indeed adopt a purely allosteric binding pose, we conducted [3H]NMS displacement experiments in a 5 mm Na+, K+, Pi buffer (for exact composition see “Experimental Procedures”). This buffer has been shown to increase the affinity of various allosteric modulators, most likely due to the absence of Mg2+ cations, which have been suggested to compete with allosteric modulators for the allosteric binding site at muscarinic M2 receptors (35). Under these conditions, iper-6-naph leads to a concentration-dependent increase of [3H]NMS binding, which is direct evidence for an allosteric binding topography of iper-6-naph (Fig. 1b). On the molecular level, enhancement of [3H]NMS binding is due to positive cooperativity of the allosteric moiety 6-naph with [3H]NMS (Fig. 1c). The 5 mm Na+, K+, Pi buffer enhances both allosteric affinity and efficacy of 6-naph (Fig. 1c). However, the 5 mm Na+, K+, Pi buffer has little to no effect on the binding of the orthosteric agonist iperoxo (Fig. 1d). These data demonstrate that iper-6-naph adopts two distinct binding modes at M2AChRs that reside in dynamic equilibrium.
FIGURE 1.

Iper-6-naph adopts two distinct binding modes. a, theory of dynamic ligand binding. The partial agonist iper-6-naph consists of two pharmacophores, one active (green triangle) and one inactive (blue square) moiety connected by a flexible linker (black zigzag line). Dynamic ligands may bind to a receptor population in two distinct orientations, the dualsteric binding pose and the purely allosteric binding pose (see the receptor on the left and right sides, respectively). In the dualsteric pose, the active moiety binds to the orthosteric binding site (green ellipsoid), and the inactive moiety binds to the allosteric binding site (cyan ellipsoid). Receptors bound to iper-6-naph in the dualsteric pose induce receptor activation. In the purely allosteric binding pose, iper-6-naph resides entirely in the allosteric binding site, precluding receptor activation. b–d, equilibrium binding of iper-6-naph (b), the allosteric ligand 6-naph (c), and the orthosteric agonist iperoxo (d) competing against the orthosteric probe [3H]NMS to M2AChRs from CHO membranes in HEPES buffer (filled squares) and in Na+/K+/Pi buffer (open squares). Total binding in the absence of test compounds was set to 100%. Data are means ± S.E. from at least four independent experiments conducted in triplicate. Error bars represent S.E.
Structural Characteristics of a Ligand Binding Ensemble
The concept that ligands can adopt multiple distinct binding modes at the same GPCR has been defined as dynamic ligand binding (28) (Fig. 1a). However, the structural basis for this phenomenon remains unclear. To demonstrate that a GPCR is indeed capable of accommodating a ligand in multiple binding modes, i.e. forming a ligand binding ensemble, we here apply an interdisciplinary approach combining molecular modeling techniques guided by pharmacological experiments and vice versa. Our modeling strategy consists of a combination of all-atom molecular dynamics (MD) simulations and three-dimensional pharmacophores (36, 37).
The M2AChR displays strong allosteric coupling between the ligand binding site and the intracellular G protein-binding interface (16). Structurally, this is represented by a pronounced contraction of the orthosteric binding site (16). In fact, iperoxo binding leads to a complete closure of the orthosteric binding site toward the extracellular space (16). The so-called tyrosine lid formed by Tyr-1043.33, Tyr-4036.51, and Tyr-4267.39 separates the orthosteric binding site from the allosteric binding site in the active M2AChR crystal structures (Fig. 2a). However, the side chain conformations of these tyrosine residues appear very flexible in MD simulations, and the receptor tolerates agonist binding within an orthosteric binding site that is not completely closed. Based on the available crystal structures of the M2AChR in active (Protein Data Bank codes 4MQS and 4MQT) (16) and inactive conformations (Protein Data Bank code 3UON) (15), we first generated receptor·ligand complexes by docking experiments. To dock iper-6-naph into the active M2AChR structure (Protein Data Bank code 4MQT), the tyrosine lid was remodeled by side chain sampling based on MD simulations of the iperoxo-bound crystal structure (Fig. 2, b and c). MD-based side chain sampling detects an open tyrosine lid suitable for docking bitopic agonists. The only differences between the crystal structure and the obtained active-like receptor model are the side chain conformations of Tyr-1043.33, Tyr-4036.51, and Tyr-4267.39.
FIGURE 2.

Remodeling of the tyrosine lid. a, transmembrane view of the active M2AChR crystal structure with the orthosteric agonist iperoxo (green surface) and the positive allosteric modulator LY2119620 (cyan surface). The tyrosine lid that separates the orthosteric and allosteric binding sites is shown in red. Allosteric key residues are shown in orange. b, top view snapshots from an MD simulation of the iperoxo-bound crystal structure unveil the conformational flexibility of the tyrosine lid, especially tyrosine Tyr-4267.39. c, based on the sampling of side chain conformations, the tyrosine lid was remodeled for binding mode investigations on dualsteric agonists.
Docking and subsequent all-atom MD simulations initiated from an active receptor·ligand complex reveal a dualsteric binding topography of iper-6-naph (Fig. 3a). The iperoxo moiety of iper-6-naph binds to the orthosteric binding site, whereas the 6-naph moiety protrudes toward extracellular domains, thereby engaging residues of the common allosteric binding site (Fig. 3a). The dualsteric binding pose of iper-6-naph stabilizes an active ligand·receptor complex because the agonistic properties of iper-6-naph are mediated by the orthosteric site (25) and hence are only compatible with a binding pose in which the iperoxo moiety targets the orthosteric binding site. Based on the MD simulations, additional molecular descriptors can be inferred that argue for an active ligand·receptor complex. First, the orientation of the iperoxo moiety of iper-6-naph is almost identical to that observed in the iperoxo-bound M2AChR crystal structure (Fig. 3, a and b). In both iperoxo and iper-6-naph MD simulations of the active M2AChR complexes, the positively charged nitrogen of iperoxo interacts with Asp-1033.32 and displays π-cation interactions with Tyr-1043.33, Tyr-4036.51, and Tyr-4267.39 (Fig. 3, a and b). The triple bond exhibits hydrophobic contacts with Tyr-1043.33, Trp-1554.57, and Trp-4006.48. Additionally, Asn-4046.52 forms a hydrogen bond with the oxygen of the 4,5-dihydroisoxazole moiety of iperoxo (Fig. 3a). Second, the volume of the orthosteric binding site of iper-6-naph·receptor complexes (249.6 Å3) is similar to the orthosteric volume of the active iperoxo·receptor structure (222.3 Å3) and that obtained by MD simulation of active iperoxo·receptor complexes (242.7 Å3). In line with this, the distance between the Cα atoms of Tyr-1043.33 and Asn-4046.52 as an indicator for the extent of contraction of the orthosteric binding site upon activation is similar in the iper-6-naph (13.1 Å) and iperoxo MD simulations (12.4 Å). In contrast, the crystal structure of the inactive M2AChR shows a larger volume of the orthosteric binding site (383.6 Å3) and a larger distance between Tyr-1043.33 and Asn-4046.52 (13.6 Å).
FIGURE 3.

Structural characteristics of a ligand binding ensemble. Transmembrane view of representative conformations of M2AChR complexes with the dynamic ligand iper-6-naph (a and c) in two distinct binding orientations, dualsteric (a) and purely allosteric (c). The boxed parts of a and c illustrate in more detail ligand-receptor interactions by three-dimensional pharmacophores. Yellow spheres indicate lipophilic contacts, red arrows indicate hydrogen bond acceptors, the purple disk represents a π-stacking interaction, and positively charged centers are shown as blue spheres. b, comparison of iperoxo in its co-crystallized conformation (dark gray) with the orthosteric part of iper-6-naph from an MD simulation (light gray). d, comparison of the binding orientation in the allosteric vestibule of naphmethonium (dark gray) with iper-6-naph (light gray) with key residues for allosteric ligand binding. e, effects of iper-6-naph on [3H]NMS equilibrium binding in HEPES buffer (orange curve) and iper-6-naph-induced M2AChR-mediated G protein activation measured as incorporation of [35S]GTPγS into membranes of CHO-M2AChR cells (green curve). ACh-stimulated [35S]GTPγS binding (gray curve) defined the maximum effect of the system (set to 100%). Basal [35S]GTPγS binding in the absence of ligands was set to 0%. [3H]NMS and [35S]GTPγS binding were plotted on the left and right ordinates, respectively. Data represent mean ± S.E. from four to eight independent experiments conducted in triplicate. f, the fractional population of active agonist·receptor complexes (factive; orange bar) and the overall efficacy of iper-6-naph (Emax; green bar) were plotted on the left and right ordinates, respectively. factive was retrieved by fitting all data points in e globally to an operational model of agonism for dynamic ligands (see “Experimental Procedures”). Error bars represent S.E.
Docking of iper-6-naph into the inactive M2AChR crystal structure (Protein Data Bank code 3UON) and subsequent MD simulations reveal a second binding mode of iper-6-naph that is purely allosteric (Fig. 3c). Overall, the purely allosteric binding pose of iper-6-naph is similar to that of the allosteric modulator naphmethonium (Fig. 3d). Iper-6-naph in its purely allosteric binding mode interacts with two centers formed by aromatic residues in the allosteric vestibule (Fig. 3c). The first center is lined by Tyr-177EL2 and Trp-4227.35, and the second is lined by Tyr-802.60 and Tyr-832.63 as reported previously for prototypical allosteric modulators (29). In line with this, the ammonium group of the iperoxo moiety forms cation-π interactions with Tyr-802.60 and Tyr-832.63, and the other ammonium group displays cation-π interactions with Trp-4227.35 (Fig. 3, c and d). In addition, the triple bond is located opposite to Tyr-177EL2, and the two methyl groups of the small allosteric linker chain show lipophilic contacts to Ala-1915.43, Tyr-4036.51, and Tyr-4267.39 (Fig. 3c). The naphthalene ring is embedded in a pocket built by Thr-1875.39, Phe-1885.40, and Val-4076.55. Moreover, the ammonium group of iperoxo forms a charge interaction with Glu-175EL2, and the other ammonium group features cation-π interactions with Trp-993.28 and Tyr-4267.39 (Fig. 3, c and d).
Functionally, the allosteric binding pose of iper-6-naph stabilizes an inactive ligand·receptor complex as its topography is comparable with the allosteric ligand naphmethonium (Fig. 3d). Naphmethonium and, in particular, its building block 6-naph are allosteric inverse agonists (28). Our MD simulations indicate that iper-6-naph is able to adopt two completely distinct binding modes at the same receptor: the dualsteric iper-6-naph·M2AChR complex resembles an active receptor, whereas the purely allosteric iper-6-naph·M2AChR complex is inactive.
We use pharmacological experiments to quantify the fractions of receptors that are bound to iper-6-naph in either the dualsteric or the allosteric binding pose (Fig. 3e). [35S]GTPγS binding experiments are used to detect ligand agonism, which is mediated by iper-6-naph bound to the receptors in the dualsteric binding mode. [3H]NMS equilibrium binding experiments provide data on iper-6-naph's binding ensemble. By globally analyzing [3H]NMS and [35S]GTPγS binding data of iper-6-naph, we directly quantify the fractions of active and inactive receptors as well as iper-6-naph's affinities KA and KB (equilibrium dissociation constants) for the dualsteric active and the allosteric inactive binding modes, respectively. Moreover, the global analysis yields the dynamic efficacy, τdyn, of iper-6-naph (for fitting details see “Experimental Procedures”). This new global analysis is superior to previous analyses (25, 26, 28, 34) as it yields the allosteric affinity KB of a dualsteric ligand that has not been experimentally accessible before. Iper-6-naph displays partial M2AChR activation in relation to acetylcholine (ACh), which defines the maximal effect of the system (Fig. 3, e and f). Global analysis yields a significantly higher affinity of iper-6-naph for the dualsteric than for the allosteric binding mode (pKA and pKB are 7.40 ± 0.06 and 6.84 ± 0.06, respectively; p < 0.001, unpaired t test). Consequently, the fraction of active iper-6-naph·receptor complexes, factive (Fig. 3f), is significantly higher than the inactive fraction (79 versus 21%, p < 0.001).
Decrease of Agonist Efficacy Is Due to an Increase of Inactive Agonist-Receptor Complexes
We provide structural evidence that the ligand's preference for the purely allosteric binding mode determines its efficacy. We apply isox-6-naph, which differs from iper-6-naph only in its orthosteric moiety (for the molecular structures of all compounds see Fig. 4). Isox, the orthosteric building block of isox-6-naph, is a derivative of iperoxo displaying equal efficacy at muscarinic receptors albeit having a 100-fold lower affinity. Docking and MD simulations indicate that the topographies of iperoxo and isox in the orthosteric binding site of active M2AChR complexes are highly similar (Fig. 5, a and b). However, different hydrogen bond strengths were reported for furan and isoxazole compared with non-aromatic ring systems (38, 39). Therefore, we expected a higher occurrence of geometries related to hydrogen bonds in the simulation of the iperoxo-bound M2AChR complex. To unveil these differences, we generated three-dimensional interaction point densities (dynophores) by MD-based sampling (Fig. 5c). Following this approach, we find that iperoxo forms a hydrogen bond to the orthosteric epitope Asn-4046.52 in nearly all frames of the trajectory (98%), whereas isox shows this interaction in only 87% of the frames. Further analyses of hydrogen bond distances and angle frequencies confirm this observation, showing nearly ideal geometry for iperoxo, whereas for isox more deviations are observed (Fig. 5c). All other interactions show no difference between iperoxo and isox (Fig. 5b). Docking and MD simulations of M2AChR complexes bound to isox-6-naph in the dualsteric mode reveal overall homology to iper-6-naph (Fig. 5d). However, the dynophore analysis of iper-6-naph and isox-6-naph in their dualsteric binding modes reveals a similar occurrence of hydrogen bonds (98 versus 90%) as for the orthosteric building blocks iperoxo and isox (98 versus 87%). Hence, the topographical differences between iperoxo and isox (Fig. 5c) are preserved in the topographies of their dualsteric ligands iper-6-naph and isox-6-naph, respectively. In line with these structural data, global analysis of [3H]NMS equilibrium binding and [35S]GTPγS binding data reveals a 100-fold lower affinity of isox-6-naph than iper-6-naph in their dualsteric binding modes as reflected by pKA(isox-6-naph) = 5.12 ± 0.15 and pKA(iper-6-naph) = 7.40 ± 0.06, respectively (Fig. 5f).
FIGURE 4.

Molecular structures of all chemical probes.
FIGURE 5.

Decrease of agonist efficacy is due to the preference for the purely allosteric binding mode. a, comparison of iperoxo in its co-crystallized conformation (dark gray) with isox from an MD simulation (light gray). The yellow spheres indicate lipophilic contacts, the red sphere indicates a hydrogen bond acceptor, and the positively charged center is shown as a blue star. b, similar pharmacophoric features of iperoxo (above) and isox (below) with key residues for ligand binding. The yellow spheres indicate lipophilic contacts, the red sphere indicates a hydrogen bond acceptor, and the positively charged center is shown as a blue star. c, the combination of three-dimensional pharmacophores and MD simulations led to the new concept of dynophores (dynamic pharmacophores) that are able to reflect time-dependent changes in the interaction pattern of ligands. The yellow cloud indicates lipophilic contacts, the red cloud indicates a hydrogen bond acceptor, and the positively charged center is shown as a blue cloud. Whereas the positively charged center and the lipophilic contacts could be observed in all frames of the MD simulation, the hydrogen bond acceptor feature was present in almost all frames (98%) for iperoxo and iper-6-naph but only in 87% of all frames for isox and 90% for isox-6-naph. Below, relative frequencies of distance and angle of the hydrogen bond acceptor. Curves for iperoxo and iper-6-naph are shown in black; curves for isox and isox-6-naph are shown in gray. d, the dualsteric binding modes of iper-6-naph (dark gray) and isox-6-naph (light gray) are highly similar. e, three-dimensional pharmacophore analyses of isox-6-naph in the purely allosteric binding mode. Note that isox-6-naph forms an intramolecular π-π stacking interaction between the isoxazole moiety and the allosteric ring system. f, effects of isox-6-naph on [3H]NMS equilibrium binding in HEPES buffer (orange curve) and G protein activation (green curve). [3H]NMS and [35S]GTPγS binding data were plotted on the left and right ordinates, respectively. Data represent mean ± S.E. from four to seven independent experiments conducted in triplicate. Error bars represent S.E.
Isox-6-naph has recently been shown to be a less efficacious partial agonist than iper-6-naph (28). MD simulations of isox-6-naph in its allosteric binding mode reveal an unexpected intramolecular interaction. The isox moiety forms a π-π interaction with the allosteric moiety 6-naph that is not possible in the purely allosteric binding mode of iper-6-naph. This stabilizes an intramolecular conformation of isox-6-naph that is likely to bind with high affinity to the allosteric vestibule (Fig. 5e). In fact, the ammonium group of the isox moiety forms a charge interaction with Glu-175EL2. The other ammonium group displays cation-π interactions with Tyr-802.60 and Tyr-4267.39. The triple bond lies opposite Tyr-177EL2, and the two methyl groups of the small allosteric linker chain show lipophilic contacts to Ile-178EL2 and Tyr-4267.39. The naphthalene ring is embedded in a pocket lined by Phe-181EL2, Tyr-177EL2, and Thr-1875.39. Additionally, Ile-178EL2 is able to form a hydrogen bond to one of the carbonyl groups of the allosteric moiety (Fig. 5e). This allosteric network suggests an optimal fit of isox-6-naph to the allosteric binding site. Indeed, global pharmacological analysis (Fig. 5f) yields a significantly higher affinity of isox-6-naph for the inactive than for the active pose (pKB and pKA are 7.11 ± 0.15 and 5.12 ± 0.15, respectively; p < 0.001, unpaired t test). This results in an almost exclusive allosteric binding topography of isox-6-naph (99 versus 1% for the inactive and active fraction, respectively; p < 0.001, unpaired t test).
Overall, isox-6-naph binds to the allosteric binding site with higher affinity than to the orthosteric binding site. Isox-6-naph's preference for the purely allosteric binding mode explains its lower efficacy compared with iper-6-naph (Fig. 5f).
Increase of Agonist Efficacy Is Due to a Decrease of Inactive Agonist-Receptor Complexes
Iper-8-naph is a dualsteric ligand in which the orthosteric and allosteric building blocks are connected via an octamethylene linker. MD simulations of iper-6-naph (Fig. 3c) and isox-6-naph (Fig. 5e) in their purely allosteric binding modes imply that a longer linker would hamper a purely allosteric binding mode of iper-8-naph. Indeed, MD simulations of allosterically bound iper-8-naph·M2AChR complexes show a suboptimal topography of iper-8-naph in the allosteric binding site (Fig. 6a). Although iper-8-naph shares some key interactions with iper-6-naph (Fig. 3c) and isox-6-naph (Fig. 5e), its overall, purely allosteric topography is distinct (Fig. 6a). Moreover, its orientation in the allosteric binding site appears to be variable. Heavy atoms of iper-8-naph show a pronounced deviation of its docking pose during the simulation time. In contrast, iper-6-naph and even more so isox-6-naph remain in their orientation (Fig. 6b). However, MD simulations of dualsterically bound iper-8-naph·M2AChR complexes show homology to the dualsteric poses of iper-6-naph and isox-6-naph. Global analysis of pharmacological data reveals that first a higher affinity of iper-8-naph for the dualsteric, active than for the allosteric, inactive pose (pKA and pKB are 8.50 ± 0.06 and 6.87 ± 0.06, respectively; p < 0.001, unpaired t test), second the fraction of active receptors is significantly greater than the fraction of inactive receptors (98 versus 2%; p < 0.001, unpaired t test), and third the efficacy of iper-8-naph resembles that of a full agonist (Fig. 6, c and d). Overall, a dualsteric ligand's preference for the dualsteric binding mode increases its efficacy.
FIGURE 6.

Increase of agonist efficacy is due to the preference for the dualsteric binding mode. a, three-dimensional pharmacophore analysis of iper-8-naph in the purely allosteric binding mode. Yellow spheres indicate lipophilic contacts, red arrows indicate hydrogen bond acceptors, and positively charged centers are shown as blue spheres. b, deviation of heavy atoms during 50 ns of MD simulation. Whereas isox-6-naph (blue curve) and iper-6-naph (green curve) remain in their orientation, iper-8-naph (gray curve) poorly fits into the allosteric vestibule. c, effect of iper-8-naph on [3H]NMS equilibrium binding in HEPES buffer (orange curves) and G protein activation (green curves). [3H]NMS and [35S]GTPγS binding were plotted on the left and right ordinates, respectively. Data represent mean ± S.E. from four to seven independent experiments conducted in triplicate. d, the fractional population of active agonist receptor·complexes (factive) stabilized by the indicated dynamic ligands (orange bars) and their overall efficacy (Emax; green bars) relative to ACh were plotted on the left and right ordinates, respectively. factive was retrieved by fitting all data points in Figs. 5f and 6c globally to an operational model of agonism for dynamic ligands. *** and ###, significantly different from iper-6-naph with regard to factive and Emax, respectively (p < 0.001, one-way analysis of variance and Bonferroni's multiple comparison test). Error bars represent S.E.
An Allosteric Triple Mutant Rescues Agonist Efficacy
For decades, the M2AChR has served as the paradigm for the study of allosteric modulation at GPCRs. Based on mutagenesis and recent computational studies, key epitopes of the allosteric binding site are well known (29, 40–42). To modulate the ensemble of dynamic ligands bound to M2AChRs, we mutated an allosteric center that is lined by Tyr-177EL2, Trp-4227.35, and Thr-4237.36 (Fig. 2a). This mutation does not affect the affinity of [3H]NMS (pKD values are 9.28 ± 0.04 and 9.21 ± 0.02 for wild type and mutant receptors, respectively). However, the affinity of the agonists ACh and iperoxo is significantly reduced (Fig. 7a). This is most likely due to the absence of Trp-4227.35, which reorientates upon receptor activation and is required to stabilize the fully active state of the M2 receptor (16). [3H]NMS equilibrium binding experiments with dualsteric ligands show a complete displacement of [3H]NMS by both ligands iper-6-naph (Fig. 7c) and isox-6-naph (Fig. 7d). This indicates that the dualsteric binding mode is preferred at allosterically mutated receptors (Fig. 7e). In line with this, [35S]GTPγS binding experiments show higher efficacy of iper-6-naph (Fig. 7, c and f) and isox-6-naph (Fig. 7, d and f) at allosteric mutant than at wild type receptors (receptor expression levels (in pmol/mg of protein) are not significantly different (p = 0.92, unpaired t test): 3.9 ± 0.75 and 3.8 ± 0.65 for wild type and mutant receptors, respectively). The efficacy of ACh and iperoxo is equal at both receptors (Fig. 7b). Global analyses of [3H]NMS and [35S]GTPγS binding data of iper-6-naph (Fig. 7c) and isox-6-naph (Fig. 7d) further reveal increased operational efficacies (log τdyn) of iper-6-naph and isox-6-naph at mutant M2AChRs (iper-6-naph, 0.34 ± 0.04 versus 0.77 ± 0.08; isox-6-naph, −2.11 ± 0.36 versus −0.14 ± 0.11 for wild type and mutant receptors, respectively; p < 0.001, unpaired t test). Taken together, mutational disruption of an allosteric center increases the ligand's preference for the dualsteric binding mode and hence rescues agonist efficacy.
FIGURE 7.

Mutational disruption of an allosteric center increases the fraction of active ligand·receptor complexes. a, equilibrium binding of ACh and iperoxo to hM2 wild type (filled squares) and triple mutant receptors (open squares) competing against the orthosteric probe [3H]NMS. Total binding in the absence of test compounds was set to 100%. Data are means ± S.E. from at least three independent experiments conducted in triplicate. b, [35S]GTPγS binding mediated by hM2 wild type (filled squares) and triple mutant receptors (open squares) reflects receptor activation induced by ACh and iperoxo. [35S]GTPγS binding in the absence of ligand was set to 0%, and maximal ligand-induced [35S]GTPγS binding was set to 100%. Data are means ± S.E. from at least four independent experiments conducted in triplicate. c and d, displacement of [3H]NMS equilibrium binding (orange curves) by iper-6-naph (c) and isox-6-naph (d) in HEPES buffer and G protein activation (green curves). For comparison, [3H]NMS equilibrium and [35S]GTPγS binding data from wild type CHO-M2AChR cells are shown in orange and green dashed lines, respectively. [3H]NMS and [35S]GTPγS binding were plotted on the left and right ordinates, respectively. Data in c and d represent mean ± S.E. from four to eight independent experiments conducted in triplicate. e, the fractional population of active M2AChRs (factive) stabilized by the indicated dynamic ligands at hM2 wild type (empty bars) and hM2 triple mutant (filled bars) receptors. f, maximal agonist efficacies (Emax) relative to Ach of the indicated ligands at wild type (empty bars) and allosteric triple mutant (filled bars) receptors. Error bars represent S.E.
Rational Design of a Full Agonist with Exclusive Dualsteric Binding Topography
Dynamic ligand binding to M2AChRs is characterized by a binding mode ensemble comprising active and inactive ligand·receptor complexes bound to the same ligand in a dualsteric and an allosteric binding mode, respectively (Fig. 3). Both ligand·receptor complexes are in equilibrium, the position of which is sensitive to the affinity to both the orthosteric and allosteric binding sites (Figs. 1, 5, 6, and 7). This implies that it should be possible to design a putatively dualsteric ligand that does not bind to the receptor in the purely allosteric mode anymore and hence should display full agonism. Based on our structural model, we estimated the very extreme distance within the allosteric vestibule to be 15.7 Å, measured between Thr-842.65 and Thr-1875.39 (Fig. 8b). Hereupon, we designed and synthesized the ligand iper-rigid-naph (for chemical synthesis, see supplemental Fig. 1). Iper-rigid-naph is a derivative of iper-6-naph comprising a linker that is longer (16.0 Å) and more rigid than the hexamethylene and octamethylene spacers (Fig. 8b). Attempts to dock iper-rigid-naph only into the allosteric vestibule of inactive M2AChRs failed due to multiple clashes with allosteric residues (Fig. 8b). In contrast, molecular docking and MD simulations of active M2AChR complexes indicate that iper-rigid-naph keeps the dualsteric binding mode, resulting in full receptor activation (Fig. 8a). The suggested loss of an allosteric binding pose of iper-rigid-naph is corroborated by pharmacological experiments (Fig. 8c). Global analysis of [3H]NMS and [35S]GTPγS binding data indicates that iper-rigid-naph exclusively stabilizes active M2AChRs (Fig. 8d). In line with this, iper-rigid-naph displays full agonism (Fig. 8d).
FIGURE 8.

Rational design of a full agonist with exclusive dualsteric binding topography. a, transmembrane view of a representative conformation of the M2AChR in complex with iper-rigid-naph in the dualsteric binding mode taken from a 50-ns MD simulation. Agonistic moieties are shown with a green surface; antagonistic moieties are shown with a cyan surface. b, iper-rigid-naph cannot bind in a purely allosteric mode like iper-6-naph (Fig. 3c) because of the rigidified linker that is not able to enter the allosteric binding site. The maximum dilatation of the allosteric vestibule is shown as the distance between Thr-842.65 and Thr-1875.39 (lower green line in b). This conformational constraint allows iper-rigid-naph to only bind in the dualsteric mode. c, effects of iper-rigid-naph on [3H]NMS equilibrium binding in HEPES buffer (orange curve) and iper-rigid-naph-induced G protein activation (green curve). [3H]NMS and [35S]GTPγS binding are plotted on the left and right ordinates, respectively. Data are mean ± S.E. from five to nine independent experiments conducted in triplicate. d, the fractional population of active M2AChRs (factive) stabilized by iper-rigid-naph (orange bar) and its maximal efficacy (Emax; green bar) relative to ACh were plotted on the left and right ordinates, respectively. factive was retrieved by fitting all data points in c globally to an operational model of agonism for dynamic ligands (see “Experimental Procedures”). Error bars represent S.E.
Discussion
GPCRs are highly flexible membrane proteins (43, 44), adopting a multitude of distinct conformations (inactive and active) even in the absence of ligands (19–22, 24). Agonist binding leads to a shift of the receptor's conformational equilibrium toward active states. The equilibrium shift, however, is not quantitative. Even in the presence of a G protein mimetic, agonist-occupied receptors can adopt inactive states (21).
We have studied the existence, molecular details, and function of such inactive agonist·receptor complexes. Bitopic partial agonists for M2AChRs adopt a ligand binding ensemble consisting of a dualsteric and a purely allosteric binding mode (Fig. 3). Dualsterically bound agonist·receptor complexes induce G protein activation, whereas purely allosterically bound agonist·receptor complexes are inactive (Fig. 3). Multiple lines of evidence imply that both states reside in dynamic equilibrium (Figs. 1, 5, 6, and 7). First, small changes in the chemical structure of agonists (e.g. the orthosteric building block and the linker chain) strongly influence the affinities for either the active or the inactive receptor conformation (Figs. 5 and 6). Second, an allosteric triple mutant disrupts the purely allosteric binding topography of the agonists and thus fully rescues agonist efficacy (Fig. 7). Third, a putatively dualsteric agonist (iper-rigid-naph), which was designed based on our findings, adopts only the dualsteric binding pose and hence displays full agonism (Fig. 8). Our data show that the population size of inactive agonist·receptor complexes determines overall agonist efficacy.
In addition to these experimental observations, Onaran and Costa (45) and others (46, 47) have devised probabilistic models to theoretically describe a multitude of different receptor states called “receptor ensembles.” Using probability partition functions, it can be shown that ligand binding to receptors changes the frequency distribution of receptor states. This theory directly implies that a ligand would have different affinities for different receptor conformations (47). Interestingly, probabilistic models do not distinguish between the chemical natures of ligands or the binding sites that these ligands target preferentially. Hence, in principal, every ligand for GPCRs should have a multitude of microaffinities for different conformational states of the receptor. However, it is not known whether different agonist affinities go along with different agonist binding modes. To address this question experimentally, we have studied a special case of GPCR agonism because the rather extreme molecular nature of dualsteric agonists allows identifying multiple agonist binding modes with currently available techniques. Identifying multiple binding topographies of purely orthosteric agonists by crystallography appears to be technically challenging as less preferred or energetically less stable agonist·receptor complexes may not be retrieved easily in crystallization trials. Moreover, subtle differences in the agonist topography might exceed the resolution currently achieved. Although structural data of agonists bound to inactive state receptors have been reported (48, 49), it is not known whether the agonists in these structures would adopt a different binding mode in the active state receptors. Furthermore, conventional molecular docking experiments seem unapt to describe such dynamic phenomena because they aim at predicting a single, most favorable conformation of the protein·ligand complex.
However, it is tempting to hypothesize that ligand binding ensembles indeed represent a more general mechanism for partial agonism. In agreement with probabilistic models of receptor ensembles (45, 46), the conclusions drawn from this study should not be limited to bitopic agonists. In particular, our data suggest that it is of worth to study the existence of multiple binding modes of agonists that target only the orthosteric binding site. Our mechanistic model is supported by previously published data: the endogenous muscarinic AChR agonist ACh has been suggested to adopt both “productive” (i.e. signaling-competent) and “non-productive” binding modes in the orthosteric site of M1AChRs (50). Moreover, for ligand-activated nuclear receptors, ligand binding ensembles have been crystallized (51–55) and might hence appear as a general principle in protein-ligand interactions. In the case of GPCRs, biophysical techniques, single molecule studies, and the dynophore approach may be helpful to address whether purely orthosteric agonists can form a ligand binding ensemble consisting of active and inactive agonist·receptor complexes.
Taken together, the combination of pharmacological experiments and computational simulations based on the inactive and active M2AChR crystal structures have led to a molecular description of an agonist binding ensemble. This is a proof of principle that active and inactive agonist·receptor complexes can be bound by an agonist in different binding modes. The concept of ligand binding ensembles may be applicable to other GPCRs and receptor classes.
Experimental Procedures
Materials, Chemical Probes, and Buffers
All cell culture media and supplements were purchased form Sigma-Aldrich and Invitrogen. All buffer reagents were from Sigma-Aldrich, Grüssing GmbH Analytika (Filsum, Germany), and Merck Labor und Chemie Vertrieb GmbH (Bruchsal, Germany). [3H]NMS and [35S]GTPγS were purchased from PerkinElmer Life Sciences. ACh, atropine, and NMS were obtained from Sigma-Aldrich. Iperoxo (56), Isox (57), 6-naph (25), iper-6-naph (25), isox-6-naph (58), and iper-8-naph (26) have been synthesized exactly as described previously. The synthesis of iper-rigid-naph is described in the supplemental information. Binding buffer A consisted of 10 mm HEPES, 10 mm MgCl2, 100 mm NaCl, pH 7.4, and binding buffer B was composed of 5 mm Na+, K+, Pi, pH 7.4.
Site-directed Mutagenesis and Generation of Stable Cell Lines
The allosteric triple mutant hM2Y177A,W422A,T423A was generated by site-directed mutagenesis (QuikChange kit (Stratagene, La Jolla, CA)) following the manufacturer's instructions. The cDNA of the allosteric double mutant hM2Y177A,W422A in pcDNA5/FRT was used as the template. The forward and reverse primers for induction of the T423A mutation by PCR were as follows (the underlined nucleotides indicate the mutated triplet): 5′-CCCCAACACTGTGGCGGCAATTGGTTACTGGC-3′ (forward) and 5′-GCCAGTAACCAATTGCCGCCACAGTGTT GGGG-3′ (reverse). The resulting construct was verified by sequencing. The stable Chinese hamster ovary (CHO)-Flp-InTM cell line expressing the triple mutant was generated according to the manufacturer's instructions (Flp-In system, Invitrogen).
Cell Culture
Flp-In cells (Flp-In-CHO) stably expressing the hM2 receptor (CHO-hM2 wild type) or the hM2Y177A,W422A,T423A triple mutant receptor (CHO-hM2 triple mutant) were cultured in Ham's nutrient mixture F-12 supplemented with 10% (v/v) fetal bovine serum (FBS), 100 μg/ml streptomycin, 100 units/ml penicillin, and 2 mm l-glutamine. Cells were grown at 37 °C in a 5% CO2 humidified incubator.
Membrane Preparation
When stably transfected CHO cells had reached about 80–90% confluence, the medium was aspirated and replaced by fresh medium containing 5 mm sodium butyrate, and cells were incubated for another 16–18 h before membrane preparation. Medium was aspirated, and 2.4 ml of ice-cold harvesting buffer (20 mm HEPES, 10 mm Na2EDTA, pH 7.4) was added to the cells. Cells were detached from the culture dish using a cell scraper (Sarstedt, Newton, NC). The cell suspension was shredded twice for 20 s at level 6 using a Polytron homogenizer, and the resulting cell fragments were centrifuged at 40,000 × g for 10 min (4 °C). The supernatant was aspirated, and the pellet was resuspended in 15 ml of ice-cold centrifugation buffer (20 mm HEPES, 0.1 mm Na2EDTA, pH 7.4) before centrifugation in conditions mentioned above. This step was repeated once, and the remaining pellet was resuspended in ice-cold HEPES buffer (12.5 mm HEPES, 12.5 mm MgCl2, 125 mm NaCl, pH 7.4), quickly frozen, and stored in aliquots at −80 °C. The protein concentration was determined by the method of Lowry (71).
[3H]NMS Equilibrium Binding
CHO-hM2 wild type and triple mutant membrane homogenates (15–20 μg/ml) were incubated in binding buffer A or B supplemented with GDP (100 μm), the radioligand [3H]NMS (0.2 nm/well), and the test compounds at different concentrations for 18 h at 30 °C. The incubation time was calculated as described previously (41). Atropine (10 μm) was used to determine unspecific binding. The assay was terminated by rapid vacuum filtration. All experiments were carried out in a 96-well microtiter plate (Thermo Scientific Abgene, Germany) in a final volume of 500 μl/well. Membrane-bound radioactivity was separated from non-bound radioligand by vacuum filtration using the TomTec filtration machine. The filter mats (Printed Filtermat A for use with 1450 MicroBetaTM, glass fiber filter, PerkinElmer Life Sciences) were then washed twice with ice-cold distilled water, further dried for 2.5 min in a microwave, and covered with melt-on scintillator sheets (MeltiLexTM melt-on scintillator sheets, PerkinElmer Life Sciences) on a heating block. The filter mats were then transferred into a plastic sample bag (Sample Bag for MicroBetaTM, PerkinElmer Life Sciences) and placed into a reading cassette. Finally, a solid scintillation counter (Beckman Instruments, Palo Alto, CA) was used to quantify the radioactivity on the filter mats.
[35S]GTPγS Binding
CHO-hM2 wild type and triple mutant membrane homogenates (40 μg/ml) were incubated with [35S]GTPγS (0.07 nm) in binding buffer A supplemented with GDP (10 μm). Ligand-induced [35S]GTPγS accumulation was determined after incubation for 1 h at 30 °C.
Pharmacological Data Analysis and Calculations
Analyses of all concentration-effect curves were performed using Prism 5.01 (GraphPad Software, San Diego, CA). Data points of equilibrium binding experiments of all orthosteric ligands to the M2WT and M2 triple mutant receptors were fitted by a four-parameter logistic function. The resulting IC50 values were converted into apparent affinity constants, Ki, using the method of Cheng-Prusoff (59). Data points of equilibrium binding experiments of all allosteric ligands to the M2WT and M2 triple mutant receptors were fitted by the allosteric ternary complex model as described previously (25, 26, 28, 60). This yielded the affinity of the allosteric ligand to free receptors and its cooperativity α with the radioligand [3H]NMS.
The pharmacological parameters of dynamic ligand binding were determined by fitting [35S]GTPγS binding and [3H]NMS equilibrium binding data globally using the following system of equations.
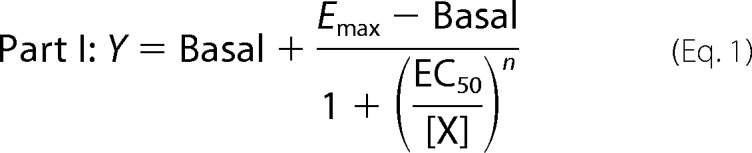 |
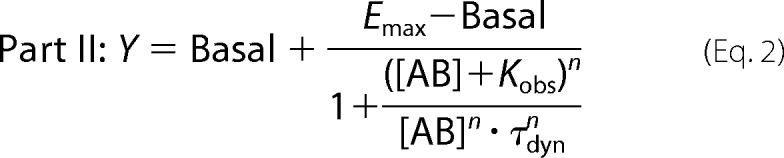 |
where
 |
and
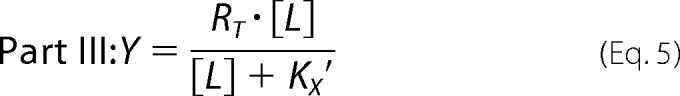 |
in which
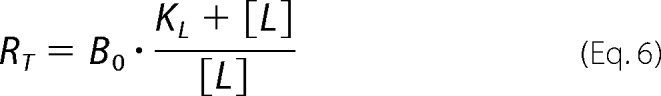 |
and
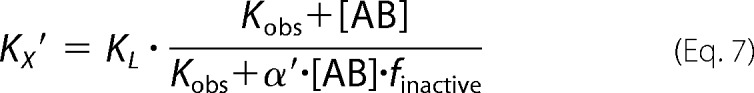 |
where “Basal” and Emax represent the unstimulated (in the absence of agonist) and maximally ACh-stimulated [35S]GTPγS accumulation (as a surrogate for the maximum response of the system), respectively. EC50 is the concentration of the reference ligand ACh that gives the half-maximal response of the system. [X] is the concentration of the reference ligand ACh. [AB] is the concentration of the dynamic ligand AB. n is the slope of the curve. Kobs represents the observed equilibrium dissociation constant of the dynamic ligand. KA and KB are the equilibrium dissociation constants of the dynamic ligand in the active and inactive poses, respectively. τdyn is the dynamic transduction coefficient of the dynamic ligand and reflects its efficacy. τmax is the maximal efficacy of the dynamic ligand that it would have if it was bound to the receptors exclusively in the active pose. factive is the fraction of dynamic ligand·receptor complexes in the active pose. RT is the total number of receptors. B0 is the fraction of binding sites labeled with [3H]NMS in the absence of competitor. KL is the equilibrium dissociation constant of the radiotracer [3H]NMS, and [L] is its concentration. α′ displays the cooperativity of the dynamic ligand in the inactive pose with the radiotracer [3H]NMS. finactive is the fraction of dynamic ligand·receptor complexes in the inactive pose.
The global fit was set up using Equations 1, 2, and 5 with its subvariables. For fitting, the following parameters were constrained to the indicated values: Basal = 0, log EC50 to that of ACh determined separately for wild type (log EC50 = −7.27) and triple mutant receptors (log EC50 = −5.94), B0 = 100; log[L] = −9.7, and log KL = −9.28. α′ was constrained to the cooperativity of the respective allosteric fragment with [3H]NMS. Emax, Kobs, and n were shared among all data sets. This global fit yields three parameters, i.e. τdyn, Kobs, and finactive. From those, the remaining parameters can be calculated consequently as follows.
 |
 |
and
 |
Error propagation was accounted for by using the following equation.
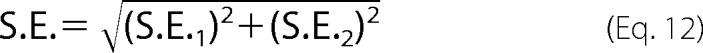 |
Docking, All-atom Molecular Dynamics Simulations, and Three-dimensional Pharmacophore Analysis
In the presented study, different modeling techniques, such as docking, all-atom MD simulations, and both static and dynamic three-dimensional pharmacophore analyses, were successfully combined (36). All protein-ligand docking experiments reported in this study were carried out with the Cambridge Crystallographic Data Centre's software GOLD version 5.1 (61). Prior to the docking experiments, ligand conformations were generated by CORINA 3.0 (62). Antagonistic ligand poses were obtained by docking into the inactive M2AChR crystal structure (Protein Data Bank code 3UON) (15), and agonistic ligand poses were obtained by docking into the active M2AChR crystal structure (Protein Data Bank code 4MQT) (16). Due to the superagonistic properties of the co-crystallized ligand iperoxo, the tyrosine lid (Tyr-1043.33, Tyr-4036.51, and Tyr-4267.39) strongly separates the orthosteric and the allosteric binding sites in the active crystal structure. Therefore, this tyrosine lid was remodeled by a side chain sampling based on MD simulations of the aporeceptor and the active crystal structure. The preparation of protein structures was performed using Molecular Operating Environment (MOE; 2014.09, Chemical Computing Group Inc.). All ligands and water molecules were removed, and correct protonation states were assigned. All residues of the inner core region and the extracellular domains were defined as potential binding sites (10 Å around the co-crystallized ligands; Protein Data Bank code 4MQT). Default settings were used for ligand docking, and GoldScore served as the scoring function. The obtained docking poses and receptor-ligand interactions were analyzed using LigandScout 3.1 (63, 64) using a three-dimensional pharmacophore approach.
All MD simulations described in this study were performed using Desmond 3.2 (65, 66). An orthorhombic box was used to build the model systems with periodic boundary conditions in an isothermal–isobaric ensemble with a constant number of particles. The system temperature was kept at 300 K, and the pressure was kept at atmospheric pressure. The definition of transmembrane regions was taken from the OPM database (67). The receptor structures were embedded in a pre-equilibrated palmitoyloleoylphosphatidylcholine membrane (bilayer) and solvated with simple point charge water and 0.15 m NaCl. All other parameters were set on default values. Each simulation consisted of an equilibration run of 4.52 ns followed by a production run of 50 ns. The simulations were carried out on the Soroban computer cluster (Freie Universität Berlin) by using 24 central processing units. The obtained trajectories were analyzed with the software VMD (68) and LigandScout 3.1 (63, 64).
Dynamic Three-dimensional Pharmacophores (Dynophores)
For the comparison of iperoxo and isox as well as their related dualsteric ligands iper-6-naph and isox-6-naph, respectively, a novel approach termed dynophore was developed that combines static three-dimensional pharmacophores with MD-based conformational sampling. Unlike previous applications of gathering pharmacophore information from molecular dynamics (33, 69), this new implementation works in a fully automated way: dynophore groups interaction points (such as hydrogen bonds, charges, and lipophilic contacts) of each trajectory frame according to their type and ligand atoms involved. All feature groups are graphically represented by three-dimensional volumetric feature density clouds, which are statistically characterized by occurrence frequency and interaction patterns with the protein. The dynophore algorithm was implemented within the LigandScout framework (63, 64, 70).
Statistical Analysis
Data are shown as mean ± S.E. for n observations. A significant difference between two distinct values was tested using an unpaired t test. Comparisons of groups were performed using one-way analysis of variance with Bonferroni's multiple comparison test.
Author Contributions
A. B. and M. B. conceived the project. A. B. generated the allosteric triple mutant and supervised and analyzed all pharmacological experiments. M. B. developed functional receptor models, designed and conducted all of the molecular modeling experiments, and analyzed the corresponding data. F. K. and B. C. did pharmacological experiments. C. M. and C. D. synthesized and characterized iper-rigid-naph. D. S., M. B., and G. W. developed the dynophore analysis tool. U. H. and M. D. A. planned and supervised chemical syntheses and contributed to discussion. M. J. L. provided essential ideas. A. B., M. B., and M. J. L. wrote the manuscript with contributions of all authors. A. B., M. B., G. W., and K. M. supervised the overall research.
Supplementary Material
Acknowledgment
We thank the computing center of the Freie Universität Berlin (ZEDAT) for providing the computer cluster Soroban for molecular dynamics simulations.
The authors declare that they have no conflicts of interest with the contents of this article.

- GPCR
- G protein-coupled receptor
- MD
- molecular dynamics
- M2AChR
- muscarinic M2 receptor
- NMS
- N-methylscopolamine
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- ACh
- acetylcholine
- AChR
- acetylcholine receptor
- EL2
- extracellular loop 2.
References
- 1. Fredriksson R., Lagerström M. C., Lundin L.-G., and Schiöth H. B. (2003) The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63, 1256–1272 [DOI] [PubMed] [Google Scholar]
- 2. Rosenbaum D. M., Rasmussen S. G., and Kobilka B. K. (2009) The structure and function of G-protein-coupled receptors. Nature 459, 356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Katritch V., Cherezov V., and Stevens R. C. (2013) Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., and Babu M. M. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194 [DOI] [PubMed] [Google Scholar]
- 5. Ritter S. L., and Hall R. A. (2009) Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol. 10, 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Overington J. P., Al-Lazikani B., and Hopkins A. L. (2006) How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 [DOI] [PubMed] [Google Scholar]
- 7. Rask-Andersen M., Almén M. S., and Schiöth H. B. (2011) Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 10, 579–590 [DOI] [PubMed] [Google Scholar]
- 8. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., and Miyano M. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 9. Park J. H., Scheerer P., Hofmann K. P., Choe H. W., and Ernst O. P. (2008) Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187 [DOI] [PubMed] [Google Scholar]
- 10. Scheerer P., Park J. H., Hildebrand P. W., Kim Y. J., Krauss N., Choe H. W., Hofmann K. P., and Ernst O. P. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455, 497–502 [DOI] [PubMed] [Google Scholar]
- 11. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., and Stevens R. C. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosenbaum D. M., Cherezov V., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Yao X. J., Weis W. I., Stevens R. C., and Kobilka B. K. (2007) GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 318, 1266–1273 [DOI] [PubMed] [Google Scholar]
- 13. Rasmussen S. G., Choi H. J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., et al. (2011) Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haga K., Kruse A. C., Asada H., Yurugi-Kobayashi T., Shiroishi M., Zhang C., Weis W. I., Okada T., Kobilka B. K., Haga T., and Kobayashi T. (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482, 547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kruse A. C., Ring A. M., Manglik A., Hu J., Hu K., Eitel K., Hübner H., Pardon E., Valant C., Sexton P. M., Christopoulos A., Felder C. C., Gmeiner P., Steyaert J., Weis W. I., et al. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Mathiesen J. M., Sunahara R. K., Pardo L., Weis W. I., Kobilka B. K., and Granier S. (2012) Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang W., Manglik A., Venkatakrishnan A. J., Laeremans T., Feinberg E. N., Sanborn A. L., Kato H. E., Livingston K. E., Thorsen T. S., Kling R. C., Granier S., Gmeiner P., Husbands S. M., Traynor J. R., Weis W. I., et al. (2015) Structural insights into μ-opioid receptor activation. Nature 524, 315–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., et al. (2013) The dynamic process of β2-adrenergic receptor activation. Cell 152, 532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kofuku Y., Ueda T., Okude J., Shiraishi Y., Kondo K., Maeda M., Tsujishita H., and Shimada I. (2012) Efficacy of the β2-adrenergic receptor is determined by conformational equilibrium in the transmembrane region. Nat. Commun. 3, 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manglik A., Kim T. H., Masureel M., Altenbach C., Yang Z., Hilger D., Lerch M. T., Kobilka T. S., Thian F. S., Hubbell W. L., Prosser R. S., and Kobilka B. K. (2015) Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 161, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vafabakhsh R., Levitz J., and Isacoff E. Y. (2015) Conformational dynamics of a class C G-protein-coupled receptor. Nature 524, 497–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sounier R., Mas C., Steyaert J., Laeremans T., Manglik A., Huang W., Kobilka B. K., Déméné H., and Granier S. (2015) Propagation of conformational changes during μ-opioid receptor activation. Nature 524, 375–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Olofsson L., Felekyan S., Doumazane E., Scholler P., Fabre L., Zwier J. M., Rondard P., Seidel C. A., Pin J. P., and Margeat E. (2014) Fine tuning of sub-millisecond conformational dynamics controls metabotropic glutamate receptors agonist efficacy. Nat. Commun. 5, 5206. [DOI] [PubMed] [Google Scholar]
- 25. Antony J., Kellershohn K., Mohr-Andrä M., Kebig A., Prilla S., Muth M., Heller E., Disingrini T., Dallanoce C., Bertoni S., Schrobang J., Tränkle C., Kostenis E., Christopoulos A., Höltje H. D., et al. (2009) Dualsteric GPCR targeting: a novel route to binding and signaling pathway selectivity. FASEB J. 23, 442–450 [DOI] [PubMed] [Google Scholar]
- 26. Bock A., Merten N., Schrage R., Dallanoce C., Bätz J., Klöckner J., Schmitz J., Matera C., Simon K., Kebig A., Peters L., Müller A., Schrobang-Ley J., Tränkle C., Hoffmann C., et al. (2012) The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat. Commun. 3, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bock A., Kostenis E., Tränkle C., Lohse M. J., and Mohr K. (2014) Pilot the pulse: controlling the multiplicity of receptor dynamics. Trends Pharmacol. Sci. 35, 630–638 [DOI] [PubMed] [Google Scholar]
- 28. Bock A., Chirinda B., Krebs F., Messerer R., Bätz J., Muth M., Dallanoce C., Klingenthal D., Tränkle C., Hoffmann C., De Amici M., Holzgrabe U., Kostenis E., and Mohr K. (2014) Dynamic ligand binding dictates partial agonism at a G protein-coupled receptor. Nat. Chem. Biol. 10, 18–20 [DOI] [PubMed] [Google Scholar]
- 29. Dror R. O., Green H. F., Valant C., Borhani D. W., Valcourt J. R., Pan A. C., Arlow D. H., Canals M., Lane J. R., Rahmani R., Baell J. B., Sexton P. M., Christopoulos A., and Shaw D. E. (2013) Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299 [DOI] [PubMed] [Google Scholar]
- 30. Christopoulos A., Changeux J. P., Catterall W. A., Fabbro D., Burris T. P., Cidlowski J. A., Olsen R. W., Peters J. A., Neubig R. R., Pin J. P., Sexton P. M., Kenakin T. P., Ehlert F. J., Spedding M., and Langmead C. J. (2014) International Union of Basic and Clinical Pharmacology. XC. Multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol. Rev. 66, 918–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bock A., and Mohr K. (2013) Dualsteric GPCR targeting and functional selectivity: the paradigmatic M2 muscarinic acetylcholine receptor. Drug Discov. Today Technol. 10, e245–e252 [DOI] [PubMed] [Google Scholar]
- 32. Mohr K., Schmitz J., Schrage R., Tränkle C., and Holzgrabe U. (2013) Molecular alliance-from orthosteric and allosteric ligands to dualsteric/bitopic agonists at G protein coupled receptors. Angew. Chem. Int. Ed. Engl. 52, 508–516 [DOI] [PubMed] [Google Scholar]
- 33. Schmitz J., van der Mey D., Bermudez M., Klöckner J., Schrage R., Kostenis E., Tränkle C., Wolber G., Mohr K., and Holzgrabe U. (2014) Dualsteric muscarinic antagonists—orthosteric binding pose controls allosteric subtype selectivity. J. Med. Chem. 57, 6739–6750 [DOI] [PubMed] [Google Scholar]
- 34. Chen X., Klöckner J., Holze J., Zimmermann C., Seemann W. K., Schrage R., Bock A., Mohr K., Tränkle C., Holzgrabe U., and Decker M. (2015) Rational design of partial agonists for the muscarinic m1 acetylcholine receptor. J. Med. Chem. 58, 560–576 [DOI] [PubMed] [Google Scholar]
- 35. Burgmer U., Schulz U., Tränkle C., and Mohr K. (1998) Interaction of Mg2+ with the allosteric site of muscarinic M2 receptors. Naunyn Schmiedebergs Arch. Pharmacol. 357, 363–370 [DOI] [PubMed] [Google Scholar]
- 36. Bermudez M., and Wolber G. (2015) Structure versus function—the impact of computational methods on the discovery of specific GPCR-ligands. Bioorg. Med. Chem. 23, 3907–3912 [DOI] [PubMed] [Google Scholar]
- 37. Wolber G., Seidel T., Bendix F., and Langer T. (2008) Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov. Today 13, 23–29 [DOI] [PubMed] [Google Scholar]
- 38. Bissantz C., Kuhn B., and Stahl M. (2010) A medicinal chemist's guide to molecular interactions. J. Med. Chem. 53, 5061–5084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaur D., and Khanna S. (2011) Intermolecular hydrogen bonding interactions of furan, isoxazole and oxazole with water. Comput. Theor. Chem. 963, 71–75 [Google Scholar]
- 40. Voigtländer U., Jöhren K., Mohr M., Raasch A., Tränkle C., Buller S., Ellis J., Höltje H. D., and Mohr K. (2003) Allosteric site on muscarinic acetylcholine receptors: identification of two amino acids in the muscarinic M2 receptor that account entirely for the M2/M5 subtype selectivities of some structurally diverse allosteric ligands in N-methylscopolamine-occupied receptors. Mol. Pharmacol. 64, 21–31 [DOI] [PubMed] [Google Scholar]
- 41. Prilla S., Schrobang J., Ellis J., Höltje H. D., and Mohr K. (2006) Allosteric interactions with muscarinic acetylcholine receptors: complex role of the conserved tryptophan M2422Trp in a critical cluster of amino acids for baseline affinity, subtype selectivity, and cooperativity. Mol. Pharmacol. 70, 181–193 [DOI] [PubMed] [Google Scholar]
- 42. Bermudez M., Rakers C., and Wolber G. (2015) Structural characteristics of the allosteric binding site represent a key to subtype selective modulators of muscarinic acetylcholine receptors. Mol. Inform. 34, 526–530 [DOI] [PubMed] [Google Scholar]
- 43. Kenakin T., and Miller L. J. (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 62, 265–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Manglik A., and Kobilka B. (2014) The role of protein dynamics in GPCR function: insights from the β2AR and rhodopsin. Curr. Opin. Cell Biol. 27, 136–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Onaran H. O., and Costa T. (1997) Agonist efficacy and allosteric models of receptor action. Ann. N.Y. Acad. Sci. 812, 98–115 [DOI] [PubMed] [Google Scholar]
- 46. Kenakin T., and Onaran O. (2002) The ligand paradox between affinity and efficacy: can you be there and not make a difference? Trends Pharmacol. Sci. 23, 275–280 [DOI] [PubMed] [Google Scholar]
- 47. Kenakin T. (2013) New concepts in pharmacological efficacy at 7TM receptors: IUPHAR review 2. Br. J. Pharmacol. 168, 554–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Warne T., Moukhametzianov R., Baker J. G., Nehmé R., Edwards P. C., Leslie A. G., Schertler G. F., and Tate C. G. (2011) The structural basis for agonist and partial agonist action on a β1-adrenergic receptor. Nature 469, 241–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rosenbaum D. M., Zhang C., Lyons J. A., Holl R., Aragao D., Arlow D. H., Rasmussen S. G., Choi H. J., Devree B. T., Sunahara R. K., Chae P. S., Gellman S. H., Dror R. O., Shaw D. E., Weis W. I., et al. (2011) Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature 469, 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Page K. M., Curtis C. A., Jones P. G., and Hulme E. C. (1995) The functional role of the binding site aspartate in muscarinic acetylcholine receptors, probed by site-directed mutagenesis. Eur. J. Pharmacol. 289, 429–437 [DOI] [PubMed] [Google Scholar]
- 51. Pochetti G., Godio C., Mitro N., Caruso D., Galmozzi A., Scurati S., Loiodice F., Fracchiolla G., Tortorella P., Laghezza A., Lavecchia A., Novellino E., Mazza F., and Crestani M. (2007) Insights into the mechanism of partial agonism: crystal structures of the peroxisome proliferator-activated receptor γ ligand-binding domain in the complex with two enantiomeric ligands. J. Biol. Chem. 282, 17314–17324 [DOI] [PubMed] [Google Scholar]
- 52. Bruning J. B., Parent A. A., Gil G., Zhao M., Nowak J., Pace M. C., Smith C. L., Afonine P. V., Adams P. D., Katzenellenbogen J. A., and Nettles K. W. (2010) Coupling of receptor conformation and ligand orientation determine graded activity. Nat. Chem. Biol. 6, 837–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Srinivasan S., Nwachukwu J. C., Parent A. A., Cavett V., Nowak J., Hughes T. S., Kojetin D. J., Katzenellenbogen J. A., and Nettles K. W. (2013) Ligand-binding dynamics rewire cellular signaling via estrogen receptor-α. Nat. Chem. Biol. 9, 326–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nwachukwu J. C., Srinivasan S., Bruno N. E., Parent A. A., Hughes T. S., Pollock J. A., Gjyshi O., Cavett V., Nowak J., Garcia-Ordonez R. D., Houtman R., Griffin P. R., Kojetin D. J., Katzenellenbogen J. A., Conkright M. D., et al. (2014) Resveratrol modulates the inflammatory response via an estrogen receptor-signal integration network. Elife 3, e02057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anami Y., Itoh T., Egawa D., Yoshimoto N., and Yamamoto K. (2014) A mixed population of antagonist and agonist binding conformers in a single crystal explains partial agonism against vitamin D receptor: active vitamin D analogues with 22R-alkyl group. J. Med. Chem. 57, 4351–4367 [DOI] [PubMed] [Google Scholar]
- 56. Kloeckner J., Schmitz J., and Holzgrabe U. (2010) Convergent, short synthesis of the muscarinic superagonist iperoxo. Tetrahedron Lett. 51, 3470–3472 [Google Scholar]
- 57. Dallanoce C., Conti P., De Amici M., De Micheli C., Barocelli E., Chiavarini M., Ballabeni V., Bertoni S., and Impicciatore M. (1999) Synthesis and functional characterization of novel derivatives related to oxotremorine and oxotremorine-M. Bioorg. Med. Chem. 7, 1539–1547 [DOI] [PubMed] [Google Scholar]
- 58. Disingrini T., Muth M., Dallanoce C., Barocelli E., Bertoni S., Kellershohn K., Mohr K., De Amici M., and Holzgrabe U. (2006) Design, synthesis, and action of oxotremorine-related hybrid-type allosteric modulators of muscarinic acetylcholine receptors. J. Med. Chem. 49, 366–372 [DOI] [PubMed] [Google Scholar]
- 59. Cheng Y., and Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 60. Ehlert F. J. (1988) Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mol. Pharmacol. 33, 187–194 [PubMed] [Google Scholar]
- 61. Jones G., Willett P., and Glen R. C. (1995) Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 245, 43–53 [DOI] [PubMed] [Google Scholar]
- 62. Sadowski J., Gasteiger J., and Klebe G. (1994) Comparison of automatic three-dimensional model builders using 639 x-ray structures. J. Chem. Inf. Comput. Sci. 34, 1000–1008 [Google Scholar]
- 63. Wolber G., and Langer T. (2005) LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 45, 160–169 [DOI] [PubMed] [Google Scholar]
- 64. Wolber G., Dornhofer A. A., and Langer T. (2006) Efficient overlay of small organic molecules using 3D pharmacophores. J. Comput. Aided. Mol. Des. 20, 773–788 [DOI] [PubMed] [Google Scholar]
- 65. Bowers K. J., Chow E., Xu H., Dror R. O., Eastwood M. P., Gregerson B. A., Klepeis J. L., Kolossvary I., Moraes M. A., Sacerdoti F. D., Salmon J. K., Shan Y., and Shaw D. E. (2006) Scalable algorithms for molecular dynamics simulations on commodity clusters, in Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, November 11–17, 2006, IEEE, Piscataway, NJ [Google Scholar]
- 66. Mortier J., Rakers C., Bermudez M., Murgueitio M. S., Riniker S., and Wolber G. (2015) The impact of molecular dynamics on drug design: applications for the characterization of ligand-macromolecule complexes. Drug Discov. Today 20, 686–702 [DOI] [PubMed] [Google Scholar]
- 67. Lomize M. A., Lomize A. L., Pogozheva I. D., and Mosberg H. I. (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 [DOI] [PubMed] [Google Scholar]
- 68. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 69. Rakers C., Schumacher F., Meinl W., Glatt H., Kleuser B., and Wolber G. (2016) In silico prediction of human sulfotransferase 1E1 activity guided by pharmacophores from molecular dynamics simulations. J. Biol. Chem. 291, 58–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Seidel T., Ibis G., Bendix F., and Wolber G. (2010) Strategies for 3D pharmacophore-based virtual screening. Drug Discov. Today Technol. 7, e203–e270 [DOI] [PubMed] [Google Scholar]
- 71. Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.