

Abstract
Genome-wide association studies (GWASs) have mined many common genetic variants associated with human complex traits like diseases. After that, the functional annotation and enrichment analysis of significant SNPs are important tasks. Classic methods are always based on physical positions of SNPs and genes. Expression quantitative trait loci (eQTLs) are genomic loci that contribute to variation in gene expression levels and have been proven efficient to connect SNPs and genes. In this work, we integrated the eQTL data and Gene Ontology (GO), constructed associations between SNPs and GO terms, then performed functional enrichment analysis. Finally, we constructed an eQTL-based SNP Ontology and SNP functional enrichment analysis platform. Taking Parkinson Disease (PD) as an example, the proposed platform and method are efficient. We believe eSNPO will be a useful resource for SNP functional annotation and enrichment analysis after we have got significant disease related SNPs.
Genome-wide association study (GWAS) is an examination of many common genetic variants in different individuals to see if any variant is associated with a trait. GWAS studies typically focus on associations between single nucleotide polymorphisms (SNPs) and traits like major complex diseases1. Since two SNPs with significantly altered allele frequency between the Age-related Macular Degeneration (ARMD) and healthy controls was firstly found in 20052, more than 100,000 risk SNPs associated to hundreds of diseases in human have been mined via GWAS3. There are several GWAS databases for human diseases and traits, such as GWAS Catalog3, GWAS Central4 and GWASdb5,6.
After getting the significant SNPs, functional analysis is an important task. Generally, SNPs are considered to be functional through related genes, and the most popular method is SNP functional enrichment analysis. Gene ontology (GO) is a major bioinformatics initiative to unify the representation of gene and gene product attributes7,8. There are several SNP functional database, such as SNP Function Portal9 and F-SNP database10; and SNP functional enrichment analysis methods, such as I-GSEA4GWAS11, SNP-based pathway enrichment analysis12, SNPsnap13 and SNP2GO14. Similar to gene functional enrichment analysis, these methods can be divided into two categories, significant SNPs based methods and SNP sets based methods. A common ground in these methods is that the SNP functions are explained by the related genes according to physical positions on chromosome.
Expression quantitative trait loci (eQTLs) are genomic loci that contribute to variation in expression levels of mRNAs15. The first genome-wide gene expression QTL study was carried out in yeast and published in 200216. Plenty of eQTL studies followed in plants and animals, including humans. Studies have shown that SNPs reproducibly associated with complex disorders are significantly enriched for eQTLs relative to frequency-matched SNPs17. Systematic integrations of eQTLs and GWAS have been used to identify risk genes in Schizophrenia18, Psoriasis19, and Muscle traits20. Therefore, eQTL data is an important and useful source for SNP functional annotation.
In this study, taking eQTL as medium between SNPs and their functions, we integrated eQTL and GO information and constructed a human SNP Ontology database and SNP functional enrichment analysis platform. It will be an efficient tool after GWAS analysis for a complex trait.
Material and Methods
eQTL data
The eQTL data were collected from several open databases and literatures. The gene expression patterns are specific among tissue types, and so do the eQTL patterns. Therefore, a classification by tissue types is necessary. We classified them into 12 tissues (Table 1). We combined the data from different studies of same tissue type. For each data, we set a significant threshold of FDR < 0.05. We retained only the SNPs with reference names and genes with gene symbols. In each tissue type, the numbers of samples, SNPs and genes are all after the screening.
Table 1. eQTL data in 12 tissues.
| Tissue type | Samples | SNPs | Genes | Reference |
|---|---|---|---|---|
| Adipose Subcutaneous | 111 | 18963 | 241 | gtexportal52 |
| Artery Tibia | 124 | 28332 | 372 | gtexportal52 |
| Brain | 765 | 22740 | 7161 | eQTL Browser53, seeQTL54 |
| Heart | 87 | 14086 | 186 | gtexportal52 |
| Lung | 124 | 31905 | 434 | gtexportal52 |
| Muscle Skeletal | 143 | 25383 | 301 | gtexportal52 |
| Nerve Tibial | 102 | 23253 | 327 | gtexportal52 |
| Skin | 114 | 20506 | 296 | gtexportal52 |
| Blood | 5479 | 406341 | 6780 | Blood eQTL browser55, gtexportal52 |
| Liver | 427 | 2305 | 3463 | eQTL Browser53 |
| Lymphoblastoid | 1220 | 208039 | 9168 | eQTL Browser53, seeQTL54, Liming Liang56 |
| Thyroid | 112 | 33939 | 481 | gtexportal52 |
Brain data
As Parkinson Disease (PD) is a disorder of the central nervous system, we selected eQTL data in brain for a case study from Gibbs et al.21 and Myers et al.22. In Gibbs et al.’s study, four frozen tissue samples of the cerebellum (CRBLM), frontal cortex (FCTX), caudal pons (PONS) and temporal cortex (TCTX) were obtained from 150 neurologically normal Caucasian subjects resulting in 600 tissue samples. SNP genotyping was performed using Infinium HumanHap 550 beadchips (Illumina) for 561,466 SNPs. Profiling of 22,184 mRNA transcripts was performed using HumanRef-8 Expression BeadChips (Illumina). For each of the four brain regions, a regression analysis was performed using Plink23. After eQTL analysis in each brain regions, we integrated the results. In Myers et al.’s study, whole-genome genotyping for 366,140 SNPs and expression analysis of 14,078 genes were carried out on a series of 193 neurologically normal human brain samples using the Affymetrix GeneChip Human Mapping 500 K Array Set and Illumina HumanRefseq-8 Expression BeadChip platforms. A one-degree-of-freedom allelic test of association analysis was performed using Plink23. We integrated the results from these 2 studies. Finally, we got 51,131 significant correlations between 22,740 SNPs and 7,161 genes with the threshold of FDR < 0.05.
Gene annotation data
The gene annotation data was downloaded from the Gene Ontology (GO) database (www.geneontology.org/page/download-annotations)7,8.
ESNPO construction
We defined associations between SNPs and GO terms via combining the associations between SNPs and genes from eQTL and the associations between genes and GO terms from GO annotation database. A SNP and GO term with at least one common gene will be connected for an association. It was illustrated in Fig. 1.
Figure 1. ESNPO construction.

SNP functional enrichment analysis
We performed Fisher exact test to estimate the significance of associations between SNPs and GO terms. The Fisher exact test is equal to Hypergeometric test. Suppose there are N SNPs and M disease-related SNPs in eSNPO. For a given GO term, there are n SNPs and m disease-related SNPs. The p value is estimated as follows.
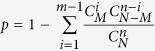 |
P value adjustment
In an analysis, multiple GO terms are tested for significance and the Type I error would increase. Therefore, a multiple test adjustment is needed after estimating p values. There are 7 p value adjustment methods adopted using p.adjust function in R. The Bonferroni correction (“bonferroni”)24 in which the p values are multiplied by the number of comparisons. Less conservative corrections are also included by Holm (“holm”)25, Hochberg (“hochberg”)26, Hommel (“hommel”)27, Benjamini & Hochberg (“BH” or its alias “fdr”)28, and Benjamini & Yekutieli (“BY”)29, respectively. There is no golden standard to compare these methods, and the most popular method is False Discovery Rate method. The False Discovery Rate (FDR) is one way of conceptualizing the rate of type I errors in null hypothesis testing when conducting multiple comparisons. In this study, we used the “fdr” method.
Database
After all, we construct a SNP Ontology and SNP functional enrichment analysis platform (http://bioinfo.hrbmu.edu.cn/esnpo/ or http://nclab.hit.edu.cn/esnpo/). It mainly includes 2 functions, eQTL-based SNP functional annotation and SNP functional enrichment analysis. After removing redundancy, we got 699,445 associations between 21,123 SNPs and 11,714 GO terms. The detailed statistics for the 12 tissues were illustrated in Table 2. The GO terms are formed by 3 components, Biological Process (BP), Cellular Component (CC) and Molecular Function(MF).
Table 2. Summary statistics of eSNPO.
| Tissue | SNPs | GO terms | BP | CC | MF |
|---|---|---|---|---|---|
| Adipose Subcutaneous | 6168 | 304 | 166 | 75 | 63 |
| Artery Tibial | 8472 | 478 | 286 | 92 | 100 |
| Brain | 21123 | 11714 | 7979 | 1168 | 2567 |
| Heart Left Ventricle | 3049 | 294 | 171 | 59 | 64 |
| Lung | 8315 | 530 | 327 | 90 | 113 |
| Muscle Skeletal | 8637 | 501 | 318 | 96 | 87 |
| Nerve Tibial | 7210 | 533 | 334 | 89 | 110 |
| Skin | 4240 | 358 | 213 | 87 | 58 |
| Blood | 353817 | 11153 | 7514 | 1136 | 2503 |
| Liver | 1976 | 7762 | 5257 | 859 | 1646 |
| Lymphoblastoid | 184971 | 12158 | 8238 | 1174 | 2746 |
| Thyroid | 8258 | 637 | 385 | 113 | 139 |
Case study
PD SNPs data
PD is a degenerative disorder of the central nervous system mainly affecting the motor system. We used 2,034 unique PD-related SNPs in Guiyou Liu et al.30. These SNPs came from these following works: 41 SNPs were from the GWAS Catalog3; 70 SNPs were from a large PD GWAS with over 3,400 cases and 29,000 controls conducted by Do et al.31; 783 SNPs were from a meta-analysis of PD GWAS with 4,238 PD cases and 4,239 controls performed by Pankratz et al.32; 1,292 SNPs were from a meta-analysis of PD GWAS using a common set of 7,893,274 variants across 13,708 cases and 95,282 controls conducted by Nalls et al.33. The threshold of p values in these studies were set to be 5.00E−08. After removing redundancy, we selected 2034 unique SNPs with P < 5.00E−08.
PD enrichment analysis
In the eQTL-based SNP enrichment analysis, of the 2,034 SNPs, there are 846 SNPs annotated in 77 terms. After Fisher exact test, there are 67 (87.0%) significant terms under the threshold of fdr < 0.01.
In the position-based SNP enrichment analysis, of the 2,034 SNPs, there are 1,318 SNPs annotated in 807 terms. After Fisher exact test, there are 396 (49.1%) significant terms under the threshold of fdr < 0.01.
Compared between the significant results from eSNPO and position-based enrichment analysis, there are 43 terms in common, including 19 Biological Process (BP) terms, 14 Cellular Component(CC) terms and 10 Molecular Function (MF) terms.
From the results, though there are fewer annotated GO terms in eSNPO than position-based method, there are higher proportion of significant results in eQTL-based method.
To evaluate the method, we performed literature verification on these significant BP GO terms. Of these 19 BP terms in common between these 2 methods, there are 5 terms about axon or neurons; 5 terms about microtubule; 4 terms about apoptotic, cell death or autophagy; 1 term about pregnancy. The axon or neurons34,35, microtubule36,37,38, apoptotic39,40,41, cell death42,43 or autophagy39,44. pregnancy45,46 were verified by other studies.
Furthermore, we further verified these significant GO terms only obtained in eQTL-based method (8 BP terms, 8 CC terms and 8 MF terms). Of these 8 BP terms, there are 2 terms about apoptotic signaling pathway47, 1 term about cell proliferation48,49, 1 term about cell adhesion50, 2 term about JUN phosphorylation51 which have been verified by other studies.
Conclusion
In this work, we constructed an eQTL-based SNP Ontology and SNP functional enrichment analysis platform (http://bioinfo.hrbmu.edu.cn/esnpo/ or http://nclab.hit.edu.cn/esnpo/). We integrated the eQTL data and GO, constructed associations between SNPs and GO terms, then performed functional enrichment analysis. Taking PD as an example, this eQTL-based method is an efficient method as the position-based method. Therefore, we believe it is a useful SNP functional enrichment analysis resource after we selected significant disease related SNPs.
However, there are still some shortages in this method. The first is there may not be enough suitable eQTL data we can use. And the second is that the scale of eSNPO is far less than the position-based method. These shortages will be solved along with more and more eQTL studies have been done.
Additional Information
How to cite this article: Li, J. et al. eSNPO: An eQTL-based SNP Ontology and SNP functional enrichment analysis platform. Sci. Rep. 6, 30595; doi: 10.1038/srep30595 (2016).
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (grant numbers: 61271346, 61300116, 61302139, 61571163, 61532014, 61402132 and 91335112) and Natural Science Foundation of Heilongjiang Province (grant number: QC2013C063).
Footnotes
Author Contributions J.L., L.W. and M.G. conceived the study. J.L., L.W. and T.J. did most of the experiments. J.W., X.L., C.W. and Z.T. constructed the web platform. X.L., C.W., R.Z. and H.L. tested the web platform. All authors reviewed the manuscript.
References
- Hirschhorn J. N. & Daly M. J. Genome-wide association studies for common diseases and complex traits. Nature reviews. Genetics 6, 95–108, doi: 10.1038/nrg1521 (2005). [DOI] [PubMed] [Google Scholar]
- Haines J. L. et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 308, 419–421, doi: 10.1126/science.1110359 (2005). [DOI] [PubMed] [Google Scholar]
- Welter D. et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research 42, D1001–1006, doi: 10.1093/nar/gkt1229 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T., Hastings R. K., Gollapudi S., Free R. C. & Brookes A. J. GWAS Central: a comprehensive resource for the comparison and interrogation of genome-wide association studies. European journal of human genetics: EJHG 22, 949–952, doi: 10.1038/ejhg.2013.274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. J. et al. GWASdb: a database for human genetic variants identified by genome-wide association studies. Nucleic acids research 40, D1047–1054, doi: 10.1093/nar/gkr1182 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. J. et al. GWASdb v2: an update database for human genetic variants identified by genome-wide association studies. Nucleic acids research 44, D869–876, doi: 10.1093/nar/gkv1317 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29, doi: 10.1038/75556 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology, C. Gene Ontology Consortium: going forward. Nucleic acids research 43, D1049–1056, doi: 10.1093/nar/gku1179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. et al. SNP Function Portal: a web database for exploring the function implication of SNP alleles. Bioinformatics 22, e523–529, doi: 10.1093/bioinformatics/btl241 (2006). [DOI] [PubMed] [Google Scholar]
- Lee P. H. & Shatkay H. F-SNP: computationally predicted functional SNPs for disease association studies. Nucleic acids research 36, D820–824, doi: 10.1093/nar/gkm904 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Chang S., Guo L. & Wang J. I-GSEA4GWAS v2: a web server for functional analysis of SNPs in trait-associated pathways identified from genome-wide association study. Protein & cell 6, 221–224, doi: 10.1007/s13238-014-0114-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng L. et al. SNP-based pathway enrichment analysis for genome-wide association studies. BMC bioinformatics 12, 99, doi: 10.1186/1471-2105-12-99 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pers T. H., Timshel P. & Hirschhorn J. N. SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics 31, 418–420, doi: 10.1093/bioinformatics/btu655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szkiba D., Kapun M., von Haeseler A. & Gallach M. SNP2GO: functional analysis of genome-wide association studies. Genetics 197, 285–289, doi: 10.1534/genetics.113.160341 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman M. V. & Kruglyak L. Genetics of global gene expression. Nature reviews. Genetics 7, 862–872, doi: 10.1038/nrg1964 (2006). [DOI] [PubMed] [Google Scholar]
- Brem R. B., Yvert G., Clinton R. & Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science 296, 752–755, doi: 10.1126/science.1069516 (2002). [DOI] [PubMed] [Google Scholar]
- Nicolae D. L. et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS genetics 6, e1000888, doi: 10.1371/journal.pgen.1000888 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X. J. et al. Systematic Integration of Brain eQTL and GWAS Identifies ZNF323 as a Novel Schizophrenia Risk Gene and Suggests Recent Positive Selection Based on Compensatory Advantage on Pulmonary Function. Schizophrenia bulletin 41, 1294–1308, doi: 10.1093/schbul/sbv017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X. et al. Five regulatory genes detected by matching signatures of eQTL and GWAS in psoriasis. Journal of dermatological science 76, 139–142, doi: 10.1016/j.jdermsci.2014.07.007 (2014). [DOI] [PubMed] [Google Scholar]
- Ponsuksili S., Murani E., Trakooljul N., Schwerin M. & Wimmers K. Discovery of candidate genes for muscle traits based on GWAS supported by eQTL-analysis. International journal of biological sciences 10, 327–337, doi: 10.7150/ijbs.8134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs J. R. et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS genetics 6, e1000952, doi: 10.1371/journal.pgen.1000952 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers A. J. et al. A survey of genetic human cortical gene expression. Nature genetics 39, 1494–1499, doi: 10.1038/ng.2007.16 (2007). [DOI] [PubMed] [Google Scholar]
- Purcell S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81, 559–575, doi: 10.1086/519795 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn O. J. Estimation of the Medians for Dependent Variables. Olive Jean Dunn 30, 192–197 (1959). [Google Scholar]
- Holm S. A Simple Sequentially Rejective Multiple Test Procedure. Scandinavian Journal of Statistics, 6, 65–70, doi: 10.2307/4615733 (1979). [DOI] [Google Scholar]
- Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika 75, 800–803 (1988). [Google Scholar]
- Hommel G. A stagewise rejective multiple test procedure based on a modified Bonferroni test. Biometrika 75, 383–386, doi: 10.1093/biomet/75.2.383 (1988). [DOI] [Google Scholar]
- Benjamini Y. & Hochberg Y. Controlling The False Discovery Rate - A Practical And Powerful Approach To Multiple Testing. Journal of the Royal Statistical Society 57, 289–300 (1995). [Google Scholar]
- Benjamini Y. & Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Annals of statistics 29, 1165–1188 (2001). [Google Scholar]
- Liu G. et al. Identifying the Association Between Alzheimer’s Disease and Parkinson’s Disease Using Genome-Wide Association Studies and Protein-Protein Interaction Network. Molecular neurobiology 52, 1629–1636, doi: 10.1007/s12035-014-8946-8 (2015). [DOI] [PubMed] [Google Scholar]
- Do C. B. et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS genetics 7, e1002141, doi: 10.1371/journal.pgen.1002141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratz N. et al. Meta-analysis of Parkinson’s disease: identification of a novel locus, RIT2. Annals of neurology 71, 370–384, doi: 10.1002/ana.22687 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nature genetics 46, 989–993, doi: 10.1038/ng.3043 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin J. E., Uryu K., Lee V. M. & Trojanowski J. Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proceedings of the National Academy of Sciences of the United States of America 96, 13450–13455 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W. et al. Role of the Akt/GSK-3beta/CRMP-2 pathway in axon degeneration of dopaminergic neurons resulting from MPP+ toxicity. Brain research 1602, 9–19, doi: 10.1016/j.brainres.2014.08.030 (2015). [DOI] [PubMed] [Google Scholar]
- Feng J. Microtubule: a common target for parkin and Parkinson’s disease toxins. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry 12, 469–476, doi: 10.1177/1073858406293853 (2006). [DOI] [PubMed] [Google Scholar]
- Parisiadou L. & Cai H. LRRK2 function on actin and microtubule dynamics in Parkinson disease. Communicative & integrative biology 3, 396–400, doi: 10.4161/cib.3.5.12286 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arduino D. M., Esteves A. R. & Cardoso S. M. Mitochondria drive autophagy pathology via microtubule disassembly: a new hypothesis for Parkinson disease. Autophagy 9, 112–114, doi: 10.4161/auto.22443 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglade P. et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histology and histopathology 12, 25–31 (1997). [PubMed] [Google Scholar]
- Hartmann A. et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America 97, 2875–2880, doi: 10.1073/pnas.040556597 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton W. G., Chalmers-Redman R., Brown D. & Tatton N. Apoptosis in Parkinson’s disease: signals for neuronal degradation. Annals of neurology 53 Suppl 3, S61–70;discussion S70-62, doi: 10.1002/ana.10489 (2003). [DOI] [PubMed] [Google Scholar]
- Jenner P. & Olanow C. W. Understanding cell death in Parkinson’s disease. Annals of neurology 44, S72–84 (1998). [DOI] [PubMed] [Google Scholar]
- Youdim M. B., Grunblatt E., Levites-Royak Y. & Mandel S. Drugs to prevent cell death in Parkinson’s disease. Neuroprotection against oxidative stress and inflammatory gene expression. Advances in neurology 86, 115–124 (2001). [PubMed] [Google Scholar]
- Pan T., Kondo S., Le W. & Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain: a journal of neurology 131, 1969–1978, doi: 10.1093/brain/awm318 (2008). [DOI] [PubMed] [Google Scholar]
- Golbe L. I. Parkinson’s disease and pregnancy. Neurology 37, 1245–1249 (1987). [DOI] [PubMed] [Google Scholar]
- Shulman L. M., Minagar A. & Weiner W. J. The effect of pregnancy in Parkinson’s disease. Movement disorders: official journal of the Movement Disorder Society 15, 132–135 (2000). [DOI] [PubMed] [Google Scholar]
- Inamdar A. A., Masurekar P., Hossain M., Richardson J. R. & Bennett J. W. Signaling pathways involved in 1-octen-3-ol-mediated neurotoxicity in Drosophila melanogaster: implication in Parkinson’s disease. Neurotoxicity research 25, 183–191, doi: 10.1007/s12640-013-9418-z (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglinger G. U. et al. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nature neuroscience 7, 726–735, doi: 10.1038/nn1265 (2004). [DOI] [PubMed] [Google Scholar]
- Vedam-Mai V. et al. Increased precursor cell proliferation after deep brain stimulation for Parkinson’s disease: a human study. PloS one 9, e88770, doi: 10.1371/journal.pone.0088770 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman M. A. Interactions between cell adhesion and the synaptic vesicle cycle in Parkinson’s disease. Medical hypotheses 83, 203–207, doi: 10.1016/j.mehy.2014.04.029 (2014). [DOI] [PubMed] [Google Scholar]
- Li Y., Zhang J. & Yang C. UNC-51-like kinase 1 blocks S6k1 phosphorylation contributes to neurodegeneration in Parkinson’s disease model in vitro. Biochemical and biophysical research communications 459, 196–200, doi: 10.1016/j.bbrc.2015.02.008 (2015). [DOI] [PubMed] [Google Scholar]
- Consortium G. T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660, doi: 10.1126/science.1262110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coordinators N. R. Database resources of the National Center for Biotechnology Information. Nucleic acids research 44, D7–D19, doi: 10.1093/nar/gkv1290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia K. et al. seeQTL: a searchable database for human eQTLs. Bioinformatics 28, 451–452, doi: 10.1093/bioinformatics/btr678 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature genetics 45, 1238–1243, doi: 10.1038/ng.2756 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L. et al. A cross-platform analysis of 14,177 expression quantitative trait loci derived from lymphoblastoid cell lines. Genome research 23, 716–726, doi: 10.1101/gr.142521.112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]