

Abstract
While biomaterials provide a platform to control the delivery of vaccines, the recently discovered intrinsic inflammatory characteristics of many polymeric carriers can also complicate rational design because the carrier itself can alter the response to other vaccine components. To address this challenge, we recently developed immune-polyelectrolyte multilayer (iPEMs) capsules electrostatically assembled entirely from peptide antigen and molecular adjuvants. Here, we use iPEMs built from SIINFEKL model antigen and polyIC, a stimulatory toll-like receptor agonist, to investigate the impact of pH on iPEM assembly, the processing and interactions of each iPEM component with primary immune cells, and the role of these interactions during antigen-specific T cell responses in coculture and mice. We discovered that iPEM assembly is pH dependent with respect to both the antigen and adjuvant component. Controlling the pH also allows tuning of the relative loading of SIINFEKL and polyIC in iPEM capsules. During in vitro studies with primary dendritic cells (DCs), iPEM capsules ensure that greater than 95% of cells containing at least one signal (i.e., antigen, adjuvant) also contained the other signal. This codelivery leads to DC maturation and SIINFEKL presentation via the MHC-I antigen presentation pathway, resulting in antigen-specific T cell proliferation and pro-inflammatory cytokine secretion. In mice, iPEM capsules potently expand antigen-specific T cells compared with equivalent admixed formulations. Of note, these enhancements become more pronounced with successive booster injections, suggesting that iPEMs functionally improve memory recall response. Together our results reveal some of the features that can be tuned to modulate the properties of iPEM capsules, and how these modular vaccine structures can be used to enhance interactions with immune cells in vitro and in mice.
Keywords: rational design, immunology, self-assembly, vaccine, polyelectrolyte multilayer
Introduction
Self-assembled biomaterials offer many features that can be exploited to enhance vaccination. Some of these capabilities include codelivery, controlled release, and encapsulation or condensation of antigens or molecular adjuvants into particulate forms that more efficiently trigger pathogen-sensing pathways that promote adaptive immunity.1−3 One important class of such pathways are toll-like receptors (TLRs). These signaling cascades have evolved to detect pathogen-associated molecular patterns (PAMPs) that are common in viruses and bacteria, but not in humans.4 Since ligands for these pathways –TLR agonists–drive strong pro-immune responses, TLR agonists have recently been the subject of enormous investment as new, molecular vaccines adjuvants that offer better definition compared with more classical adjuvants, such as alum.5,6 For example, CpG sequences (TLR9 ligands) are oligonucleotide sequences that are unmethylated and exhibit a high concentration of cytosine and guanine residues.7 Since these features are absent in most mammalian DNA, TLR9 activation drives strong inflammatory responses that support adaptive responses; this modality has motivated intense clinical exploration of CpG as a vaccine adjuvant.7 Similarly, polyIC, a TLR3 agonist, is based on double-stranded RNA often associated with viruses.4 Because CpG, polyIC, and other TLR agonists are recognized in a pattern-specific manner, these ligands offer more targeted strategies for directing activation of specific immune pathways during vaccination.
Polymers and other biomaterials offer significant potential to enhance vaccination by codelivering antigens and combinations of one or more TLR agonists. However, from a translational perspective, the increasing complexity of vaccines is also creating challenges in vaccine characterization and elucidation of mechanism of action.8 Amplifying these hurdles, biomaterial studies over the past decade have revealed many common polymers, including poly(lactide-co-glycolide), exhibit inflammatory characteristics that cause inflammation or modulate the signaling of TLRs and other stimulatory pathways.9−12 Underscoring the role of polymer properties in driving intrinsic immunogenicity, several studies have investigated the link between polymer-mediated immune stimulation and physicochemical properties, such as molecular weight,12,13 hydrophobicity,14 or shape.15,16 One outcome of these findings is certainly the potential to harness the intrinsic immunogenicity of biomaterials to enhance immunity. From another perspective, however, these intrinsic effects complicate vaccine design and characterization because the carrier itself can alter the response to the antigens, adjuvants, or other vaccine components in a given formulation. Thus, design of self-assembled materials that mimic attractive features of biomaterials (e.g., codelivery, tunable loading) while eliminating traditional carriers or excipients could enhance vaccination by supporting simpler, more defined compositions.
Polyelectrolyte multilayers (PEMs) provide unique features to support well-defined vaccines. These materials are prepared through electrostatic, layer-by-layer assembly of oppositely charged components to create ordered, multilayer structures. PEMs have been widely employed for catalysis, sensing, and drug delivery.17−20 Recently, PEMs have been studied as vaccine carriers administered by injection or transcutaneous delivery.20−23 In these studies, PEMs have been used to adsorb or encapsulate vaccine components on or within multilayers composed of polymers such as poly(sodium styrenesulfonate) (PSS), poly(allylamine hydrochloride) (PAH), dextran sulfate, and poly-l-arginine.24−26 These polymeric microcapsules have generated exciting preclinical results in the context of cancer and viral pathogens.21,25−28
We recently reported the design of immune-polyelectrolyte multilayers (iPEMs) composed entirely of model peptide antigen (SIINFEKL) and polyIC (TLR3 agonist) as an adjuvant.1,29 In these studies, SIINFEKL was appended with arginine residues to serve as a cationic anchor for assembly with the natural-anionic polyIC. By depositing iPEMs on a sacrificial core and subsequently exposing these particles to a chelator, hollow iPEM capsules composed entirely of SIINFEKL and polyIC can be prepared.1 These iPEMs eliminate all supports and carrier components, while providing tunable control over the relative composition of immune signals (i.e., antigen, adjuvant) used for assembly. iPEMs also confer functional benefits for immune response in mice, expanding circulating antigen-specific CD8+ T cells and promoting antitumor immunity in response to tumor challenge.1 A unique consequence of the iPEM strategy is an effective cargo loading level of 100%, since all structural components of the iPEMs also serve as the vaccine components. Common polymer or lipid particles encapsulating or adsorbing vaccine cargo typically allow entrapment efficiencies of 1–10%. This observation highlights cargo loading in iPEMs as a feature that is not readily achievable with other approaches, as well as a limitation encountered by many traditional carriers: the need for a large quantities of carrier to deliver an effective dose of the active components.
To develop insight into the physicochemical and immunological function of iPEMs, here we studied the impact of assembly conditions on iPEM growth and cargo levels, then used these materials to assess codelivery of the antigens and adjuvants comprising the iPEMs into primary dendritic cells (DCs). These effects were correlated to primary DC activation, antigen presentation, and T cell function during coculture of DCs with transgenic T cells. Finally, we investigated the durability and magnitude of recall responses during successive immunization of mice with iPEMs. Development of iPEMs or other well-defined structures that provide direct control over the relative loading of vaccine components without additional carriers or supports could enable more rationally designed vaccines. This knowledge could lead to multifunctional vaccines in which immune signals serve both as specific signaling components and as carriers.
Materials and Methods
iPEM Assembly on Planar Substrates
SIINFEKL (SIIN) and SIIINFEKL-R9 (SIIN*) were synthesized by Genscript with >98% purity, with or without a FITC label on the N-terminus. Poly(ethylenimine) (PEI, MW 25 000) was purchased from Polysciences, Inc. and poly(sodium 4-styrenesulfonate) (SPS, MW 70 000) was purchased from Sigma-Aldrich. Low molecular weight polyinosinic-polycytidylic acid (polyIC) was purchased from Invivogen. Label-IT nucleic acid labeling kits (Cy5) were purchased from Mirus Bio LLC. PolyIC was labeled according to the Mirus Bio instructions. For experiments with planar substrates, silicon (Silicon Inc.) and quartz (VWR) substrates were cut into 25 mm × 5 mm substrates using a dicing saw (Model 1006, Micro Automation). The substrates were cleaned by sequential washing with acetone, ethanol, methanol, and deionized (DI) water. Substrates were then cleaned and charged using an oxygen plasma Jupiter III (March).
The prepared planar substrates were coated with a precursor layer of 10 bilayers of PEI and SPS as previously reported.18,30 Briefly, 20 mM solutions of SPS and PEI containing 25 mM NaCl were adjusted to pH 5 with NaOH or HCl. Substrates were then sequentially dipped in PEI solution for 5 min, two DI water rinse solutions for 1 min each, SPS solution for 5 min, and two 1 min DI water rinse solutions. This process was carried out for a total of 10 cycles using a DR3 dipping robot (Riegler & Kirstein GmbH, Germany). Substrates were dried under filtered air and stored at room temperature. iPEMs were assembled on planar substrates using solutions of polyIC and either SIIN, or SIIN*; the latter contained nona-arginine (R9) to serve as a cationic anchor. Peptide and polyIC solutions were prepared at 0.2 mg/mL in PBS with 0.5 M NaCl. The pH was adjusted as indicated in the text using 0.1 M NaOH.31 The wash buffer was 0.05 M NaCl in PBS with the pH adjusted as indicated using 1 M NaOH. Films were assembled by dipping quartz or silicon substrates coated with the precursor layer into peptide solution for 5 min, followed by two 1 min rinses in wash buffer, 5 min in polyIC solution, and two additional 1 min rinses in fresh wash buffer. This sequence was repeated for up to 16 cycles to form (SIIN/polyIC)8 or (SIIN*/polyIC)8. To prepare iPEMs capsules from SIIN* and polyIC, first 1 mg/mL solutions of SIIN* and polyIC were prepared in 0.5 M NaCl in PBS with pH adjusted as indicated in the text using 0.1 M NaOH. iPEMs were then synthesized by coating sacrificial colloidal CaCO3 supports with iPEMs composed of SIIN* and polyIC.1 Each deposition step was carried out by incubating the cores in the appropriate signal for 1 min, collecting particles by centrifugation, then performing two 1 min wash steps in PBS. After the final deposition step, the core was removed using two washes with pH 4 EDTA, then the final capsules were washed and resuspended in PBS.1
iPEM Characterization
iPEM build up on silicon and quartz chips was measured by LSE stokes ellipsometry (Gaertner Scientific Corporation) and Evolution 60 UV–visible spectrophotometer (Thermo Scientific) to assess PEM thickness and relative cargo loading, respectively. Quartz chips were scanned every 1 nm, measuring absorbance from 200 to 700 nm. Five different areas throughout each chip were measured every 2 bilayers. iPEM-coated particles or capsules were measured using ImageJ to calculate diameters of at least 50 particles for each sample or condition.
In Vitro DC Studies
All animal and cell experiments were approved by the institutional animal care and use committee (IACUC) at University of Maryland, College Park. Splenic DCs were isolated from C57BL/6 mice purchased from The Jackson Laboratory (Bar Harbor, ME) with CD11c positive magnetic isolation kit (Miltenyi Biotec) following the manufactuer’s protocol. After isolation, the cell pellet was resuspended in 1 mL of DC medium (RPMI1640, 10% FBS, 0.5% pen/strep, 50 μM 2-mercaptoethanol (2-ME)) prior to use. Capsules were serially diluted and added to DCs (1 × 105 cells per well in 96 well plates) to reach final capsule concentrations of 42, 21, 10, 5, 3, 1 μg/mL. Untreated DCs, soluble peptide SIIN (5 μg/mL), LPS (1 μg/mL), polyIC (10 μg/mL), LPS (1 μg/mL) + SIIN (5 μg/mL), and polyIC (10 μg/mL) + SIIN (5 μg/mL) were used as controls in in vitro studies. After 24 h, flow cytometry was used to assess DC maturation. Briefly, cells were washed with 200 μL of FACS buffer (1% BSA in PBS), collected by centrifugation, then blocked with anti-CD16/32 (BD Biosciences) prior to surface marker staining. To assess DC maturation, DCs were stained for CD11c (APC-Cy7, BD Biosciences), CD40 (PE, BD Biosciences), CD80 (PerCP-Cy5.5, BD Biosciences), CD86 (PE-Cy7, BD Biosciences), IE/IA (Alexa Fluor 647, BD Biosciences), and viability (DAPI, Invitrogen). In antigen presentation studies, cells were stained with Anti-Mouse OVA257–264 (SIINFEKL) peptide bound to H-2Kb (PE-Cy7, Bio Legend) and CD11c (APC-Cy7, BD Biosciences). Antibody stained samples were analyzed by flow cytometry (CantoII, BD) and Flowjo v. Ten (Treestar). FlowJo software was used for all data analysis.
For iPEM internalization studies, DCs were isolated and treated as above with iPEM capsules synthesized from fluorescently labeled SIIN* and polyIC. After 2 h incubation, cells were washed with PBS and fixed with 4% paraformaldehyde, then stained with wheat germ agglutinin Texas Red conjugate (5 μg/mL in PBS) (Invitrogen) and Hoescht (Invitrogen). Confocal microscopy (Leica SP5X) was used to visualize colocalization of both fluorescently tagged SIIN* (FITC) and polyIC (Cy5) in DCs. To access DC uptake of SIIN* and polyIC quantitatively, DCs were incubated with iPEMs for 24 h, then cells were washed with PBS and stained for CD11c (APC-Cy7, BD, Bio) and a viability dye (DAPI). Samples were analyzed by flow cytometry (CantoII, BD) and Flowjo v. Ten (Treestar).
In Vitro CD8+ T Cell Expansion
OT-I mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J) were purchased from The Jackson Laboratory (Bar Harbor, ME). CD11c-enriched DCs were isolated and treated with capsules as described above for 24 h. Positive and negative control wells received free SIINFEKL peptide (5 μg/mL) or an irrelevant myelin-derived peptide (MEVGWYRSPFSRVVHLYRNGK, 5 μg/mL; Genscript), respectively. Myelin peptides have been used as specificity controls in other recent studies investigating myelin or ovalbumin-specific immune responses.32 After an additional 24 h, CD8+ T cells were isolated from OT-I mice using a CD8 negative selection isolation kit from Stemcell. The purified cells were washed twice to remove any serum and labeled with 5 μM of a proliferation dye (eFluor 670; eBioscience) for 10 min at 37 °C in the dark. After 10 min, the dye was neutralized with 5 times the volume of T cell medium (RPMI11640, 10% FBS, 1× nonessential amino acids, 10 mM HEPES, 2 mM l-glutamine, 0.5% penicillin streptomycin, 50 μM 2-ME) followed by washing three times with T cell medium. The resulting cells (3 × 105cells/50 μL) were added into the wells containing DCs/capsules and DCs/controls, then incubated for 48 h. After incubation, cells were split into two portions for analysis by (i) proliferation and (ii) intracellular cytokine staining (ICCS).
To assess T cell proliferation, cells were first blocked with anti-CD16/32 as described above and stained with anti-CD3 (PE-Cy7, BD Biosciences) and anti-CD8a (APC, BD Biosciences) for CD8+ T cells. Flow cytometry was used to analyze the signal of eFluor 670 dye as an indication of fluorescence dilution resulting from T cell proliferation. For ICCS, medium was exchanged with fresh T cell medium containing 1/1000 dilution of brefeldin A (BFA) (eBioscience), then incubated for 4 h at 37 °C. Cells were washed with ice cold FACS buffer, collected by centrifugation at 800 g for 5 min, and blocked with anti-CD16/32 for 10 min followed by staining for CD3 (PE-Cy7, BD Biosciences) and CD8a (APC, BD Biosciences) for 20 min at 4 °C. After surface staining, cells were washed twice with ice cold FACS buffer, then fixed and permeabilized using a fixation and permeabilization kit (BD Biosciences). Briefly, 100 uL of fixation solution was added to each well and incubated at 4 °C for 20 min, followed by washing twice with 200 μL of permeabilization wash buffer, and collection by centrifugation at 800 g for 5 min. Anti-IFN-γ (PE, BD Biosciences) antibody was diluted into permeabilization wash buffer and cells were stained for 30 min at 4 °C. After staining, cells were washed twice with 200 μL of permeabilization washing buffer and resuspended in 100 μL FACS buffer prior for flow cytometry analysis.
Immunizations Studies
Six to eight week old C57BL/6 female mice purchased from the Jackson Laboratory were used in all immunization studies. To assess the ability of iPEMs to expand antigen-specific CD8+ T cells during successive immunizations, mice were immunized by intradermal (i.d.) injection with iPEM capsules (n = 6), a soluble mixture of SIINFEKL/polyIC (n = 6), or left untreated (n = 6). Both capsule groups and vaccines formulated as soluble mixtures were prepared using the same dose of antigen (60 μg) and polyIC (240 μg). Mice were injected on day 0 and boosted with identical treatments at day 15 and day 28. For quantification of SIINFEKL-specific T cells, MHC-I SIINFEKL tetramer (MBL International Corporation) staining was conducted on peripheral blood every 7 days. Briefly, peripheral blood was collected into ETDA-coated tubes and treated twice with ACK Lysing buffer (Thermo Scientific) followed by a 1 mL PBS wash.1 Cells were blocked with CD16/32 (BD Biosciences) for 10 min followed by MHC-I tetramer (PE, H2-Kb, SIINFEKL) staining for 30 min. After tetramer staining, cell were stained with CD8a (APC, BD Biosciences) for 20 min and washed twice with FACS buffer. DAPI was added for viability assessment prior to analysis by flow cytometry (CantoII, BD).
Statistical Analysis
Data was analyzed by multiple group comparison using one way analysis of variance (ANOVA) with a Tukey post-test in GraphPad Prism v.6.02. Statistical significance was defined at p values <0.05 (95% confidence interval) and indicated as * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001, and **** = p ≤ 0.0001.
Results and Discussion
We first studied the effect of pH on the assembly of iPEMs formed through layer-by-layer (LbL) deposition of polyIC (anionic) and either native SIIN (zwitterionic), or SIIN modified with R9 (SIIN*) to serve as a cationic anchor. We began these experiments using planar substrates to support initial characterization of film growth and assembly, for example, by ellipsometry and solid-state UV/Visible spectrophotometry measurements. After deposition of eight bilayers (i.e., 16 layers) on planar substrates (Figure 1a), polyIC/SIIN* films reached a thickness of up to 64.1 ± 0.85 nm, depending on the pH of the solution used to assemble PEMs (Figure 1a). In contrast, exposure of substrates to polyIC and SIIN did not result in significant increases in film thickness at any combination of pH values (Figure 1a). These results are consistent with fundamental studies demonstrating that substitution of charged amino acid residues into polypeptides can be used to tune the assembly and stability of PEMs.33−35 Thus, for future studies, SIIN* was used as a model antigen to investigate iPEM assembly and processing by immune cells and the subsequent responses.
Figure 1.

Antigens and adjuvants can be assembled into iPEMs in a pH-dependent manner. (a) Quantification of final film thickness following deposition of eight bilayers of iPEMs composed of SIIN/polyIC or SIIN*/polyIC on silicon substrates. Thicknesses were measured by ellipsometry at different pH conditions for peptide, wash, and polyIC solutions. SIIN represents SIINFEKL and SIIN* represents SIINFEKL-R9. (b) Stepwise film growth of iPEMs composed of SIIN*/polyIC as a function of the pH of deposition solutions. (c) Representative absorbance traces demonstrating increasing antigen and adjuvant loading during deposition of iPEMs on quartz substrates using SIIN* (pH 8), wash (pH 8), and polyIC (pH 10) solutions. SIIN* (dotted green line, 508 nm); polyIC (dotted red line, 260 nm). (d) Relative loading of SIIN* and polyIC based on UV/vis measurements. Values represent the mean ± s.e.m. (n = 5). Concentrations of antigen and adjuvant in deposition solutions were 0.2 mg/mL in (a) and 1.0 mg/mL in panels b–d.
To determine how pH impacts the growth and loading of immune signals in iPEMs during film build-up, film thickness was measured every 2 bilayers during deposition of polyIC and SIIN* solutions prepared at different pH values. While all pH conditions initially resulted in film growth over two bilayers, continued growth was greatest with more basic pH values (Figure 1b). Film growth was linear and most efficient (i.e., thickest) using antigen and wash solutions at pH 8, and adjuvant solutions at pH 10. These conditions generated film thicknesses of 136 ± 2.2 nm (Figure 1b) and this growth correlated to an increase in the loading of polyIC and SIIN* peptide measured directly on iPEM-coated quartz substrates (Figure 1c, 1d). Compared to the lower cargo loading concentrations (0.2 mg/mL) employed in Figure 1a, the higher concentrations (1.0 mg/mL) used in Figure 1b resulted in higher absolute cargo loading of each iPEM component at equivalent pH conditions. Thus, assembly is pH dependent and can be manipulated to control the loading efficiency of each immune signal, as well as through number of layers deposited and the deposition concentration of each component.
The importance of pH in assembling polyelectrolytes on planar substrates has been well established for biologically derived polysaccharides (e.g., heparin and chitosan),36 peptides,34 and nucleic acids assembled with synthetic polymers.18,37 For iPEMs, the impact of pH likely results from a combination of factors. First, while polyIC is polyanionic, past studies have clearly established that the pKa values and isoelectric points of RNA nucleotides shift as a function of nucleic acid confirmation and secondary or tertiary structure,38 even becoming neutral at pH 7 in some cases.39 Similarly, while SIIN* is expected to be positively charged at the pH values tested (pI ∼13), many past studies with both strong and weak PEM electrolyte pairs reveal film thickness is often still a function of pH.40,41 This occurs because higher or lower amounts of cargo are assembled as the ionization state changes, as well as due to swelling of the film that can alter film density without changing the relative amount of film components assembled. The latter effect is impacted by the balance between electrostatic and hydrogen bonding interactions at a given pH value.40,41 Interestingly, these past reports also reveal a strong dependence on the pH of wash solutions and solutions used for postassembly resuspension, effects we also observed in our experiments (Figure 1a and b).
We next studied the impact of pH on assembly of injectable iPEM capsules useful for in vivo injection studies. polyIC and SIIN* were coated layer-by-layer on sacrificial CaCO3 templates using deposition solutions with different pH values (Figure 2a). As observed with planar substrates, film thickness and cargo loading after 3 bilayers were a function of pH, with more basic solutions (SIIN*, pH 8; polyIC, pH 10; wash, pH 8) again resulting in higher cargo loading levels (Figure 2a). After iPEM coating, we observed larger particle sizes (∼12.9–14.4 μm) at lower pH conditions (SIIN*, pH 7; polyIC, pH 7; wash, pH 7), but not with the most basic set of pH values (Figure 2a, 5.5 μm). Confocal microscopy imaging of iPEM-coated particles formed from fluorescently labeled polyIC and SIIN* revealed these effects resulted from bridging and aggregation of iPEM-coated particles that was pH dependent (Figure 2b). However, the optimized pH conditions (SIIN*, pH 8; polyIC, pH 10; wash, pH 8) minimized these effects and also supported the highest level of cargo loading (Figure 2b). Together, the results above with coated templates are in agreement with the pH-dependencies we observed when assembling SIIN*/polyIC iPEMs on planar substrates (Figure 1).
Figure 2.

Effect of pH on iPEM assembly, diameter, and cargo loading. (a) Diameters and cargo loading of iPEMs assembled on sacrificial templates using different pH conditions for peptide solutions (PBS with 0.5 M NaCl), wash buffers (PBS with 0.05 M NaCl), and polyIC solutions (PBS with 0.5 M NaCl). Values represent the mean ± s.e.m., SIIN* represents SIINFEKL-R9. (b) Representative confocal microscopy images of iPEM-coated particles assembled using different pH conditions. Exposure times were fixed for all samples. (c) Diameter of iPEM-coated templates during assembly. d) Antigen and adjuvant loading in iPEM capsules after removal of the template and washing.
Using the optimal pH values above, we investigated the growth and physicochemical properties of (SIIN*/polyIC)3 during the assembly process on CaCO3 templates with initial diameters of 5.0 ± 0.10 μm. During coating with these conditions–which limited aggregation–no significant size change was observed after deposition of 1, 2, or 3 bilayers (Figure 2c), results that were in good agreement with the nanoscale thicknesses of the films (Figure 1b,c). Cargo loading in capsules after core removal with EDTA (pH 4) was ∼200 μg SIIN*/mg of iPEM capsules and 800 μg polyIC/mg of iPEM capsules, a composition of approximately 20% antigen and 80% adjuvant (Figure 2d). Compared with other biomaterial vaccines–polymer particles encapsulating antigens or adjuvant, for example–this loading modality illustrates a unique feature of iPEMs: the 100% cargo loading per mass of carrier that is achieved as a result of assembling the iPEMs entirely from immune signals.
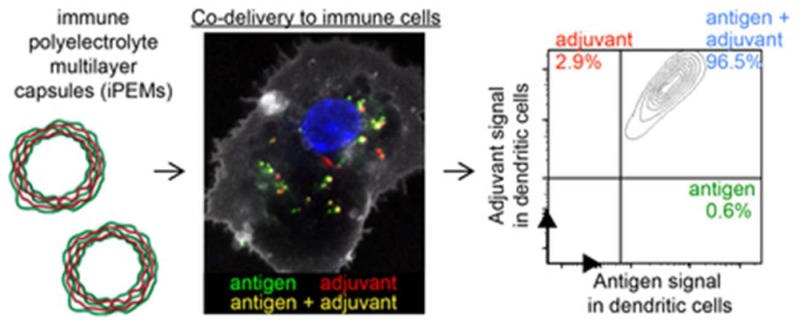
To test if iPEM capsules codeliver each iPEM component to DCs, mouse CD11c+ DCs isolated from spleens were treated with fluorescent iPEMs assembled from three bilayers of SIIN* (FITC) and polyIC (Cy5). While both bone marrow-derived DCs and splenic DCs are widely used in the literature for in vitro studies, we selected splenic DCs to minimize manipulation and culture steps, and to mimic as closely as possible the populations existing in immune tissues typically targeted by nanoparticles and other vaccines (i.e., spleen and LNs). After 24 h, up to 78% of DCs internalized iPEMs in a dose dependent manner (Figure S1). Strikingly, iPEMs supported a high degree of codelivery to these cells, particularly at moderate and high dose (Figure 3a and b). For example, at the highest dose, 96.9 ± 0.65% of cells exhibiting signal for at least one iPEM component also exhibited signals for both components (Figure 3a,3b). This feature is particularly important in vaccination, where generation of effective adaptive immune responses requires DCs or other antigen presenting cells (APCs) to encounter both an antigen that can be processed and presented to T cells, as well as an inflammatory cue that promotes expression of costimulatory molecules on APCs to serve as a second activating signal for T and B cells. Viability measurements in these studies also revealed iPEM capsules do not cause any significant toxicity to DCs compared with untreated DCs (Figure 3c). Confocal microscopy analysis of cells treated similarly to those above confirmed uptake of iPEMs capsules by DCs, with capsules dispersed through the cytosolic regions (Figure 3d). Of note, the fluorescent signals corresponding to both SIIN* and polyIC were easily observed within the cytosol, and exhibited a high degree of colocalization (indicated by yellow color) between the fluorescent signals associated with each of these components (Figure 3d).
Figure 3.

iPEM capsules are internalized by primary DCs with a high frequency of codelivery. (a) Representative histograms demonstrating SIIN* and polyIC uptake during treatment of splenic DCs with (SIIN*/polyIC)3 iPEM capsules for 24 h. SIIN* represents SIINFEKL-R9. (b) Distribution of iPEM components in cells, calculated among cells positive for at least one iPEM signal (red, polyIC; green, SIIN*; blue, polyIC+SIIN*+) to demonstrate the high degree of codelivery. (c) Viability of DCs treated with iPEM capsules relative to cells treated with LPS following a 24 h incubation. (d) Confocal microscopy images of DCs indicating internalization and colocalization of iPEM capsule components after a 2 h incubation (white, membrane; green, SIIN*; blue, nucleus; red, polyIC; scale bar = 20 μm). Values for all panels indicate the mean ± s.e.m.
The measurements of codelivery described in Figure 3 highlight another unique aspect of the iPEMs used in our experiments: the R9 moiety conjugated on the antigen as a cationic anchor to support electrostatic assembly. R9 is one of a number of cell penetrating peptide (CPP) moieties that promote uptake by cells, typically through nonendocytic processes.42−44 Thus, R9 or other CPPs might provide unique opportunities to improve uptake relative to energy-dependent processes, or even to tune the route or level of uptake by controlling the number of cationic amino acid residues. Our current results do not provide the insight needed to distinguish if and how significantly this concept ultimately impacts antigen processing and immune cell activation. Thus, follow-on studies will need to investigate the route by which iPEMs are internalized, and how these mechanisms are impacted by the sequence and length of anchor. An important outcome of these studies will also be isolation of the importance of juxtaposing signals in iPEMs relative to directly improving uptake of signals (e.g., SIIN, SIIN*, TLR agonists).
To determine if iPEM capsules generate functional responses in DCs, we assessed expression of DC surface activation markers using flow cytometry. DCs treated with capsules for 24 h exhibited increased expression of CD40, CD86, and CD80, compared with untreated cells or cells treated only with peptide. This increase was dependent on capsule dose and reached maximum frequencies of 56.8 ± 3.5%, 90.0 ± 0.2%, and 88.7 ± 0.4% for CD40, CD80, and CD86, respectively (Figure 4a-4c, S2a-d). The highest activation levels were generally similar to those achieved during treatment with free forms of TLR agonists (e.g., LPS, TLR4a; polyIC, TLR3a), indicating that assembly into iPEMs does not compromise the functionality of immune signals. Interestingly, capsules did reproducibly drive increased CD40 activation compared with DCs treated with free polyIC or mixtures of free polyIC and peptide (Figure 4a). To directly test if iPEM capsules promote presentation of antigens used to build iPEMs, DCs were treated with iPEMs and stained with an antibody that binds SIINFEKL when presented in major histocompatibility complex I (MHC-I). After 24 h, a significant fraction of DCs treated with capsules expressed SIIN in MHC-I compared with untreated cells or cells treated only with TLR agonists (Figure 4d). Several recent reports demonstrate that modification of antigens with cleavable linkers allows release and processing of antigens.45,46 For example, polymeric carriers conjugated to OVA antigen using a reduction-sensitive disulfide link have been used to promote antigen cross-presentation and enhance cytotoxic T cell response.45 Uniquely, all components of iPEMs are naturally inspired (i.e., peptides, nucleic acids), thus release or degradable linkers were not expected to be necessary for immunogenicity. Our studies above confirmed this idea, as both the antigen and adjuvant were efficiently processed from iPEM capsules to promote potent (i.e., increase maturation markers) and selective (i.e., antigen presentation) interactions with DCs. Further, each of these effects were dose dependent, indicating that iPEMs drive efficient and antigen-specific cross-presented through the MHC-I pathway, a mechanism important in cell-mediated immunity against viruses, tumors, and other pathogens.
Figure 4.
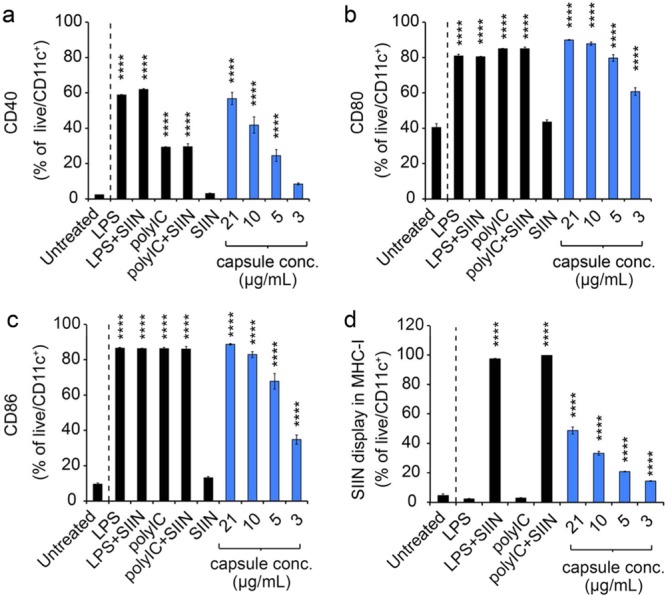
iPEMs activate DCs and promote antigen presentation. Expression of CD40 (a), CD80 (b), and CD86 (c) activation markers following treatment of primary DCs with iPEM capsules or the indicated controls for 24 h. SIIN represents SIINFEKL. d) Presentation of the SIINFEKL epitope through the MHC-I pathway assessed using anti-H-2kb-SIINFEKL following a 24 h incubation of primary DCs with iPEM capsules. Values for all panels indicate the mean ± s.e.m. Statistical comparisons indicate significance of **** = p ≤ 0.0001, and for clarity, are shown for each group compared against the control (Untreated; dashed line).
To determine if the stimulatory function of DCs driven by iPEM capsules causes functional T cell responses, CD8+ T cells from transgenic OT-I mice were isolated and cocultured with DCs. In the OT-I strain, CD8+ T cells express T cell receptors specific for SIINFEKL, providing a platform to test T cell response to antigens presented by DCs. DCs were first treated with capsules for 24 h, then cocultured with the T cells for an additional 48 h. In these studies, iPEM capsules caused strong T cell proliferation that was similar to positive control wells treated with soluble SIINFEKL peptide based on analysis of both frequency (Figure 5a) and mean fluorescent intensity (MFI; Figure S3a and S3b). These levels were starkly different than those observed in untreated cultures or cultures in which T cells were incubated with DCs treated with an irrelevant (i.e., noncognate) peptide, demonstrating a lack of proliferation. In agreement with proliferation levels, a significant fraction of T cells in cocultures treated with iPEM capsules secreted IFN-γ, while T cells in untreated samples or samples treated with irrelevant peptide did not (Figure 5b). These effects were dose-dependent, and generally lower than positive control wells treated with a 5 μg dose of SIIN peptide. Although direct comparison of dose is complicated by the presence of the arginine tag on SIIN*, one interesting question that could account for any nondose related differences is whether antigen formulated in iPEMs might require a longer time for processing and presentation by DCs. While future studies could investigate this idea by quantifying antigen presentation kinetics over time, iPEMs still led to significant levels of IFN-γ, a key effector cytokine. Thus, coupled with T cell proliferation studies (Figure 5a), these data indicate iPEMs codeliver antigens and adjuvants to DCs (Figure 3), driving DC activation and antigen processing (Figure 4), and ultimately, expansion of functional, antigen-specific CD8+ T cells (Figure 5b).
Figure 5.
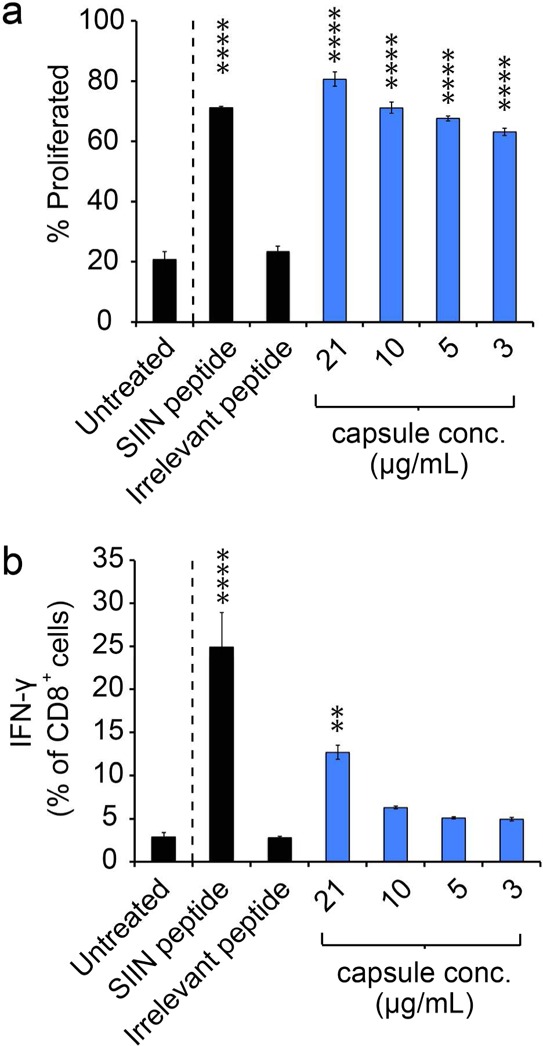
iPEM capsules induce T cell proliferation and effector cytokine secretion in primary cell coculture. (a) Expansion of SIINFEKL-specific T cells following coculture with DCs treated with iPEM capsules for 24 h. Fluorescently labeled OT-I CD8+ T cells were isolated from spleens and cultured with DCs for 48 h, then proliferation was quantified by fluorescence dilution using flow cytometry. SIIN represents SIINFEKL. (b) Secretion of IFN-γ by the cells in panel a measured by ICCS. Values for all panels indicate the mean ± s.e.m. Statistical comparisons indicate significance of ** = p ≤ 0.01; **** = p ≤ 0.0001, and for clarity, are shown for each group compared against the control (Untreated; dashed line).
We previously discovered that immunization of mice with iPEM capsules generates strong CD8+ T cells responses.1 Thus, with new understanding of the ability of these materials to drive cross-presentation, we sought to test if repeated immunizations would lead to successively higher levels of T cell response, one indicator of immunological memory. We thus immunized mice at days 0, 15, and 28 using iPEM capsules or equivalent doses of free components admixed just prior to injection. Priming injections with either formulation led to detectable antigen-specific responses at day 7, while iPEMs drove increasingly strong recall responses after each of two booster injections (red arrows) (Figure 6a). Mice receiving the first booster injection of iPEM capsules exhibited strong responses, with 4.4 ± 0.96% of circulating CD8+ T cells specific for SIINFEKL, compared with an average of 2.51 ± 0.96% in mice receiving the free form of the vaccine, and 0.35 ± 0.02% in untreated mice (Figure 6a, 5b). By day 35, 1 week after the second boost, a dramatic further increase was observed, with 10.2 ± 1.40% of CD8+ T cells specific for SIINFEKL in iPEM treated mice, compared with 4.4 ± 0.58% in mice receiving the equivalent admixed vaccines–a 3.9-fold increase (Figure 6a-c). Analysis of the rate of increase of these antigen specific T cells 7 days after the first boost was much more rapid in mice treated with iPEMs relative to the admixed vaccines (Figure 6d), rate differences that were further amplified 7 days after the second boost. This larger relative increase in antigen-specific response between boosts suggests that iPEMs contribute to more effective memory recall responses compared with the free vaccine. However, more detailed studies–for example, ex vivo peptide restimulation and phenotypic analysis of central memory and effector memory T cells–are needed to directly confirm if the improved recall responses result from iPEM-driven expansion of memory T cells.
Figure 6.

Immunization with iPEM capsules promotes enhanced recall response of antigen-specific CD8+ T cells with successive immunization. C57BL/6 mice were immunized with equivalent doses of SIINFEKL and PolyIC formulated in iPEM capsules or using a free mixture at day 0, followed by booster injections at day 15 and 28 (red arrows). SIIN represents SIINFEKL. (a) Development of SIIN-specific tetramer kinetics over 35 days. (b) Representative flow cytometry plot from each treatment group in panel (a) shown on day 21 and day 35. (c) Tetramer frequencies of individual animals at day 35 among CD8+ T cells. (d) Rate of increase (slope) in antigen-specific CD8+ T cells occurring 1 week after each booster injection are greater following immunization with iPEMs relative to equivalent soluble vaccines. Slopes were calculated by measuring tetramer frequencies just before boosting, and again after 7 days. Values for panels (a-c) indicate the mean ± s.e.m. Statistical comparisons the end of the study (day 35) are indicated by * = p ≤ 0.05, *** = p ≤ 0.001, and **** = p < 0.0001.
Conclusion
In this study, we investigated the pH-controlled assembly of iPEMs and the subsequent interaction with, and processing by, immune cells. Of note, pH conditions were identified that support improved cargo loading with reduced aggregation. These iPEMs capsules ensure near-complete codelivery of antigen and TLR agonists to DCs, leading to DC activation and efficient antigen cross-presentation ex vivo. In mice, iPEMs raise potent antigen-specific CD8+ T cells responses and promote strong memory recall responses. These features, along with tunable control over cargo loading and the elimination of all carriers or excipients, could support vaccines with simpler compositions and improved definition compared to existing approved adjuvant formulations.
Acknowledgments
We acknowledge A. Beaven at the University of Maryland Imaging Core Facility for assistance in confocal microscopy. This work was supported in part by NSF CAREER Award 1351688 and the University of Maryland Division of Research (Tier 1). Y.C.C. is a trainee on NIH Grant T32 CA154274. J.M.G. is a grantee of the Pediatric Oncology Student Training award from Alex’s Lemonade Stand Foundation. J.I.A. is a trainee on NIH Grant T32 AI089621 and a Graduate Fellow supported by the American Association of Pharmaceutical Scientists Foundation. L.H.T. is a fellow supported by the NSF Graduate Research Fellowship Program Grant DGE1322106. C.M.J. is a Damon Runyon-Rachleff Innovator supported by the Damon Runyon Foundation (DRR3415), and a Young Investigator of the Alliance for Cancer Gene Therapy (15051543) and the Melanoma Research Alliance (348963).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.6b06275.
MFI measurements for uptake and activations studies conducted in DCs and for T cell expansion studies (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Chiu Y. C.; Gammon J. M.; Andorko J. I.; Tostanoski L. H.; Jewell C. M. Modular Vaccine Design Using Carrier-Free Capsules Assembled from Polyionic Immune Signals. ACS Biomater. Sci. Eng. 2015, 1, 1200–1205. 10.1021/acsbiomaterials.5b00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo M.; Wei C. J.; Yassine H. M.; McTamney P. M.; Boyington J. C.; Whittle J. R.; Rao S. S.; Kong W. P.; Wang L.; Nabel G. J. Self-Assembling Influenza Nanoparticle Vaccines Elicit Broadly Neutralizing H1N1 Antibodies. Nature 2013, 499, 102–106. 10.1038/nature12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudra J. S.; Mishra S.; Chong A. S.; Mitchell R. A.; Nardin E. H.; Nussenzweig V.; Collier J. H. Self-Assembled Peptide Nanofibers Raising Durable Antibody Responses against a Malaria Epitope. Biomaterials 2012, 33, 6476–6484. 10.1016/j.biomaterials.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhagen F.; Kinjo T.; Bode C.; Klinman D. M. Tlr-Based Immune Adjuvants. Vaccine 2011, 29, 3341–3355. 10.1016/j.vaccine.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duthie M. S.; Windish H. P.; Fox C. B.; Reed S. G. Use of Defined TLR Ligands as Adjuvants within Human Vaccines. Immunol. Rev. 2011, 239, 178–196. 10.1111/j.1600-065X.2010.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed S. G.; Orr M. T.; Fox C. B. Key Roles of Adjuvants in Modern Vaccines. Nat. Med. 2013, 19, 1597–1608. 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- Shirota H.; Klinman D. M. Recent Progress Concerning CpG DNA and Its Use as a Vaccine Adjuvant. Expert Rev. Vaccines 2014, 13, 299–312. 10.1586/14760584.2014.863715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T. Y.; Singh M.; Miller A. T.; De Gregorio E.; Doro F.; D’Oro U.; Skibinski D. A.; Mbow M. L.; Bufali S.; Herman A. E.; Cortez A.; Li Y.; Nayak B. P.; Tritto E.; Filippi C. M.; Otten G. R.; Brito L. A.; Monaci E.; Li C.; Aprea S.; Valentini S.; Calabro S.; Laera D.; Brunelli B.; Caproni E.; Malyala P.; Panchal R. G.; Warren T. K.; Bavari S.; O’Hagan D. T.; Cooke M. P.; Valiante N. M. Rational Design of Small Molecules as Vaccine Adjuvants. Sci. Transl. Med. 2014, 6, 263ra160. 10.1126/scitranslmed.3009980. [DOI] [PubMed] [Google Scholar]
- Sharp F. A.; Ruane D.; Claass B.; Creagh E.; Harris J.; Malyala P.; Singh M.; O’Hagan D. T.; Petrilli V.; Tschopp J.; O’Neill L. A.; Lavelle E. C. Uptake of Particulate Vaccine Adjuvants by Dendritic Cells Activates the Nalp3 Inflammasome. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 870–875. 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demento S. L.; Eisenbarth S. C.; Foellmer H. G.; Platt C.; Caplan M. J.; Mark Saltzman W.; Mellman I.; Ledizet M.; Fikrig E.; Flavell R. A.; Fahmy T. M. Inflammasome-Activating Nanoparticles as Modular Systems for Optimizing Vaccine Efficacy. Vaccine 2009, 27, 3013–3021. 10.1016/j.vaccine.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva C. A.; Chalouni C.; Williams A.; Hartl D.; Lee C. G.; Elias J. A. Chitin is a Size-Dependent Regulator of Macrophage TNF and IL-10 Production. J. Immunol. 2009, 182, 3573–3582. 10.4049/jimmunol.0802113. [DOI] [PubMed] [Google Scholar]
- Termeer C.; Benedix F.; Sleeman J.; Fieber C.; Voith U.; Ahrens T.; Miyake K.; Freudenberg M.; Galanos C.; Simon J. C. Oligosaccharides of Hyaluronan Activate Dendritic Cells Via Toll-Like Receptor 4. J. Exp. Med. 2002, 195, 99–111. 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorko J. I.; Hess K. L.; Pineault K. G.; Jewell C. M. Intrinsic Immunogenicity of Rapidly-Degradable Polymers Evolves During Degradation. Acta Biomater. 2016, 32, 24–34. 10.1016/j.actbio.2015.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyano D. F.; Goldsmith M.; Solfiell D. J.; Landesman-Milo D.; Miranda O. R.; Peer D.; Rotello V. M. Nanoparticle Hydrophobicity Dictates Immune Response. J. Am. Chem. Soc. 2012, 134, 3965–3967. 10.1021/ja2108905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunshine J. C.; Perica K.; Schneck J. P.; Green J. J. Particle Shape Dependence of CD8+ T Cell Activation by Artificial Antigen Presenting Cells. Biomaterials 2014, 35, 269–277. 10.1016/j.biomaterials.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou P. M.; Babensee J. E. Macrophage and Dendritic Cell Phenotypic Diversity in the Context of Biomaterials. J. Biomed. Mater. Res., Part A 2011, 96A, 239–260. 10.1002/jbm.a.32971. [DOI] [PubMed] [Google Scholar]
- Stein E. W.; Volodkin D. V.; McShane M. J.; Sukhorukov G. B. Real-Time Assessment of Spatial and Temporal Coupled Catalysis within Polyelectrolyte Microcapsules Containing Coimmobilized Glucose Oxidase and Peroxidase. Biomacromolecules 2006, 7, 710–719. 10.1021/bm050304j. [DOI] [PubMed] [Google Scholar]
- Jewell C. M.; Lynn D. M. Multilayered Polyelectrolyte Assemblies as Platforms for the Delivery of DNA and Other Nucleic Acid-Based Therapeutics. Adv. Drug Delivery Rev. 2008, 60, 979–999. 10.1016/j.addr.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. Y.; Moskowitz J. S.; Veselinovic J.; Rosario R. A.; Timachova K.; Blaisse M. R.; Fuller R. C.; Klibanov A. M.; Hammond P. T. Dual Functional Polyelectrolyte Multilayer Coatings for Implants: Permanent Microbicidal Base with Controlled Release of Therapeutic Agents. J. Am. Chem. Soc. 2010, 132, 17840–17848. 10.1021/ja106288c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Bjornmalm M.; Caruso F. Assembly of Layer-by-Layer Particles and Their Interactions with Biological Systems. Chem. Mater. 2014, 26, 452–460. 10.1021/cm402126n. [DOI] [Google Scholar]
- Johnston A. P.; Kamphuis M. M.; Such G. K.; Scott A. M.; Nice E. C.; Heath J. K.; Caruso F. Targeting Cancer Cells: Controlling the Binding and Internalization of Antibody-Functionalized Capsules. ACS Nano 2012, 6, 6667–6674. 10.1021/nn3010476. [DOI] [PubMed] [Google Scholar]
- DeMuth P. C.; Min Y.; Irvine D. J.; Hammond P. T. Implantable Silk Composite Microneedles for Programmable Vaccine Release Kinetics and Enhanced Immunogenicity in Transcutaneous Immunization. Adv. Healthcare Mater. 2014, 3, 47–58. 10.1002/adhm.201300139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu B. B.; Park M. H.; Hagerman S. R.; Hammond P. T. Multimonth Controlled Small Molecule Release from Biodegradable Thin Films. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 12175–12180. 10.1073/pnas.1323829111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath E.; Sukhorukov G. B.; Caruso F.; Davis S. A.; Mohwald H. Novel Hollow Polymer Shells by Colloid-Templated Assembly of Polyelectrolytes. Angew. Chem., Int. Ed. 1998, 37, 2201–2205. . [DOI] [PubMed] [Google Scholar]
- De Koker S.; De Geest B. G.; Singh S. K.; De Rycke R.; Naessens T.; Van Kooyk Y.; Demeester J.; De Smedt S. C.; Grooten J. Polyelectrolyte Microcapsules as Antigen Delivery Vehicles to Dendritic Cells: Uptake, Processing, and Cross-Presentation of Encapsulated Antigens. Angew. Chem., Int. Ed. 2009, 48, 8485–8489. 10.1002/anie.200903769. [DOI] [PubMed] [Google Scholar]
- De Geest B. G.; Willart M. A.; Hammad H.; Lambrecht B. N.; Pollard C.; Bogaert P.; De Filette M.; Saelens X.; Vervaet C.; Remon J. P.; Grooten J.; De Koker S. Polymeric Multilayer Capsule-Mediated Vaccination Induces Protective Immunity against Cancer and Viral Infection. ACS Nano 2012, 6, 2136–2149. 10.1021/nn205099c. [DOI] [PubMed] [Google Scholar]
- De Cock L. J.; De Koker S.; De Geest B. G.; Grooten J.; Vervaet C.; Remon J. P.; Sukhorukov G. B.; Antipina M. N. Polymeric Multilayer Capsules in Drug Delivery. Angew. Chem., Int. Ed. 2010, 49, 6954–6973. 10.1002/anie.200906266. [DOI] [PubMed] [Google Scholar]
- De Koker S.; Naessens T.; De Geest B. G.; Bogaert P.; Demeester J.; De Smedt S.; Grooten J. Biodegradable Polyelectrolyte Microcapsules: Antigen Delivery Tools with Th17 Skewing Activity after Pulmonary Delivery. J. Immunol. 2010, 184, 203–211. 10.4049/jimmunol.0803591. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Chiu Y. C.; Tostanoski L. H.; Jewell C. M. Polyelectrolyte Multilayers Assembled Entirely from Immune Signals on Gold Nanoparticle Templates Promote Antigen-Specific T Cell Response. ACS Nano 2015, 9, 6465–6477. 10.1021/acsnano.5b02153. [DOI] [PubMed] [Google Scholar]
- Jewell C. M.; Zhang J. T.; Fredin N. J.; Wolff M. R.; Hacker T. A.; Lynn D. M. Release of Plasmid DNA from Intravascular Stents Coated with Ultrathin Multilayered Polyelectrolyte Films. Biomacromolecules 2006, 7, 2483–2491. 10.1021/bm0604808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volodkin D. V.; Petrov A. I.; Prevot M.; Sukhorukov G. B. Matrix Polyelectrolyte Microcapsules: New System for Macromolecule Encapsulation. Langmuir 2004, 20, 3398–3406. 10.1021/la036177z. [DOI] [PubMed] [Google Scholar]
- Getts D. R.; Martin A. J.; McCarthy D. P.; Terry R. L.; Hunter Z. N.; Yap W. T.; Getts M. T.; Pleiss M.; Luo X.; King N. J.; Shea L. D.; Miller S. D. Microparticles Bearing Encephalitogenic Peptides Induce T-Cell Tolerance and Ameliorate Experimental Autoimmune Encephalomyelitis. Nat. Biotechnol. 2012, 30, 1217–1224. 10.1038/nbt.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Haynie D. T. Multilayer Biomimetics: Reversible Covalent Stabilization of a Nanostructured Biofilm. Biomacromolecules 2004, 5, 1667–1670. 10.1021/bm0496155. [DOI] [PubMed] [Google Scholar]
- Zhong Y.; Li B.; Haynie D. T. Fine Tuning of Physical Properties of Designed Polypeptide Multilayer Films by Control of Ph. Biotechnol. Prog. 2006, 22, 126–132. 10.1021/bp050130h. [DOI] [PubMed] [Google Scholar]
- Jewell C. M.; Fuchs S. M.; Flessner R. M.; Raines R. T.; Lynn D. M. Multilayered Films Fabricated from an Oligoarginine-Conjugated Protein Promote Efficient Surface-Mediated Protein Transduction. Biomacromolecules 2007, 8, 857–863. 10.1021/bm0609442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddohi S.; Killingsworth C. E.; Kipper M. J. Polyelectrolyte Multilayer Assembly as a Function of Ph and Ionic Strength Using the Polysaccharides Chitosan and Heparin. Biomacromolecules 2008, 9, 2021–2028. 10.1021/bm8002573. [DOI] [PubMed] [Google Scholar]
- Flessner R. M.; Jewell C. M.; Anderson D. G.; Lynn D. M. Degradable Polyelectrolyte Multilayers That Promote the Release of Sirna. Langmuir 2011, 27, 7868–7876. 10.1021/la200815t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherbet G. V.; Lakshmi M. S.; Cajone F. Isoelectric Characteristics and the Secondary Structure of Some Nucleic Acids. Biophys. Struct. Mech. 1983, 10, 121–128. 10.1007/BF00537554. [DOI] [PubMed] [Google Scholar]
- Tang C. L.; Alexov E.; Pyle A. M.; Honig B. Calculation of Pkas in RNA: On the Structural Origins and Functional Roles of Protonated Nucleotides. J. Mol. Biol. 2007, 366, 1475–1496. 10.1016/j.jmb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Itano K.; Choi J.; Rubner M. F. Mechanism of the Ph-Induced Discontinuous Swelling/Deswelling Transitions of Poly(Allylamine Hydrochloride)-Containing Polyelectrolyte Multilayer Films. Macromolecules 2005, 38, 3450–3460. 10.1021/ma047667g. [DOI] [Google Scholar]
- Schoeler B.; Poptoshev E.; Caruso F. Growth of Multilayer Films of Fixed and Variable Charge Density Polyelectrolytes: Effect of Mutual Charge and Secondary Interactions. Macromolecules 2003, 36, 5258–5264. 10.1021/ma034018g. [DOI] [Google Scholar]
- Deshayes S.; Morris M. C.; Divita G.; Heitz F. Cell-Penetrating Peptides: Tools for Intracellular Delivery of Therapeutics. Cell. Mol. Life Sci. 2005, 62, 1839–1849. 10.1007/s00018-005-5109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walrant A.; Correia I.; Jiao C. Y.; Lequin O.; Bent E. H.; Goasdoue N.; Lacombe C.; Chassaing G.; Sagan S.; Alves I. D. Different Membrane Behaviour and Cellular Uptake of Three Basic Arginine-Rich Peptides. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 382–393. 10.1016/j.bbamem.2010.09.009. [DOI] [PubMed] [Google Scholar]
- Copolovici D. M.; Langel K.; Eriste E.; Langel U. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8, 1972–1994. 10.1021/nn4057269. [DOI] [PubMed] [Google Scholar]
- Nembrini C.; Stano A.; Dane K. Y.; Ballester M.; van der Vlies A. J.; Marsland B. J.; Swartz M. A.; Hubbell J. A. Nanoparticle Conjugation of Antigen Enhances Cytotoxic T-Cell Responses in Pulmonary Vaccination. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, E989–997. 10.1073/pnas.1104264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudra J. S.; Tian Y. F.; Jung J. P.; Collier J. H. A Self-Assembling Peptide Acting as an Immune Adjuvant. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 622–627. 10.1073/pnas.0912124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.