

Abstract
Novel drugs are required to shorten the duration of treatment for tuberculosis (TB) and to combat the emergence of drug resistance. One approach has been to identify and target Mycobacterium tuberculosis (Mtb) virulence factors, which promote the establishment of TB infection and pathogenesis. Mtb produces a number of virulence factors, including two protein tyrosine phosphatases (PTPs), mPTPA and mPTPB, to evade the antimicrobial functions of host macrophages. To assess the therapeutic potential of targeting the virulent Mtb PTPs, we developed highly potent and selective inhibitors of mPTPA (L335-M34) and mPTPB (L01-Z08) with drug-like properties. We tested the bactericidal activity of L335-M34 and L01-Z08 alone or together in combination with the standard antitubercular regimen of isoniazid-rifampicin-pyrazinamide (HRZ) in the guinea pig model of chronic TB infection, which faithfully recapitulates some of the key histological features of human TB lesions. Following a single dose of L335-M34 50mg/kg and L01-Z08 20 mg/kg, plasma levels were maintained at levels 10-fold greater than the biochemical IC50 for 12–24 hours. Although neither PTP inhibitor alone significantly enhanced the antibacterial activity of HRZ, dual inhibition of mPTPA and mPTPB in combination with HRZ showed modest synergy, even after 2 weeks of treatment. After 6 weeks of treatment, the degree of lung inflammation correlated with the bactericidal activity of each drug regimen. This study highlights the potential utility of targeting Mtb virulence factors, and specifically the Mtb PTPs, as a strategy for enhancing the activity of standard anti-TB treatment.
Keywords: Mycobacterium tuberculosis, protein tyrosine phosphatases, inhibitors, pharmacokinetics, rifampin, isoniazid, pyrazinamide, chemotherapy, bactericidal, sterilizing, activity, guinea pig, virulence factors
INTRODUCTION
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis (TB), which infects a third of the world’s population, causing between 1.2–2 million deaths annually 1. Although curative drug regimens are available, such therapy is onerous and the emergence of HIV/AIDS has triggered a resurgence of TB 2. A major obstacle to TB eradication efforts is antibiotic resistance, due primarily to inadequate adherence to the treatment regimen, which is complex, requiring multiple drugs for a minimum of 6 months. Multidrug-resistant (MDR) TB now affects over 50 million people, with an increasing number of cases of extensively drug-resistant (XDR) TB, which carries high mortality rates due to limited treatment options 3. The prevalence of MDR and XDR TB and the ongoing AIDS epidemic highlight the need to identify new drug targets and develop innovative strategies to combat drug-susceptible and drug-resistant TB 4. Recent work has focused on identifying and targeting pathogen virulence factors, which promote the establishment of infection and TB-related pathogenesis 5, 6.
Protein tyrosine phosphatases (PTPs) constitute a large family of signaling enzymes that, together with protein tyrosine kinases (PTKs), modulate the proper cellular level of protein tyrosine phosphorylation 7, 8. Malfunction of either PTKs or PTPs results in aberrant protein tyrosine phosphorylation, which has been linked to the etiology of many human diseases, including cancer, diabetes, and immune dysfunction 9. The importance of PTPs in cellular physiology is further underscored by the fact that they are often exploited and subverted by pathogenic bacteria to cause infection. The PTPs mPTPA and mPTPB from Mtb are required for optimal bacillary survival within host macrophages 10–14 and in animal models 10, 15. Although Mtb itself lacks endogenous protein tyrosine phosphorylation, mPTPA and mPTPB support Mtb infection by acting on macrophage proteins to modulate host-pathogen interactions. Specifically, mPTPA prevents phagolysosome acidification by dephosphorylation of its substrate, Human Vacuolar Protein Sorting 33B 16, resulting in the exclusion of the macrophage vacuolar-H+-ATPase (V-ATPase) from the vesicle 17. We previously reported that once inside the macrophage, mPTPB activates Akt signaling and simultaneously blocks ERK1/2 and p38 activation to prevent host macrophage apoptosis and cytokine production (12). Importantly, deletion of mPTPA or mPTPB decreases Mtb survival within interferon-γ (IFN-γ)-activated macrophages and severely reduces the Mtb bacillary load in the lungs of chronically guinea pigs 10, 18. Moreover, Mtb recombinant strains deficient in PTP activity were found to protect guinea pigs against challenge with virulent Mtb 15. The finding that mPTPA and mPTPB mediate Mtb survival within macrophages by targeting host cell processes 12, 14, 15 led to the hypothesis that specific inhibition of their phosphatase activity may augment intrinsic host signaling pathways to eradicate TB infection. To this end, we and others have shown that small molecule mPTPB inhibitors are capable of reversing the altered host immune responses induced by the bacterial phosphatase and impairing Mtb survival in macrophages, validating the concept that chemical inhibition of mPTPB may be useful for TB treatment 19, 20.
In the current study, we describe the design, synthesis and characterization of the most potent and selective inhibitor for mPTPA. We then report the evaluation of the bactericidal activity of specific mPTPA and mPTPB inhibitors, either alone or as a cocktail, in combination with the standard regimen of isoniazid-rifampicin-pyrazinamide (HRZ) in a well-validated chronic TB infection model in guinea pigs. Pharmacokinetic studies were performed to establish clinically relevant doses of each inhibitor. Therapeutic efficacy was assessed by the change in lung mycobacterial load and pathology. The public health significance of this research lies in the development of a novel class of agents for more effective treatment of drug-susceptible and drug-resistant TB.
RESULTS AND DISCUSSION
Development of potent and selective mPTPA and mPTPB inhibitors
Therapeutic targeting of PTPs has historically been stalled by difficulties in achieving inhibitor selectivity and bioavailability. The highly conserved PTP active site presents considerable challenges in obtaining compounds that can selectively inhibit the target of interest without adversely hitting other PTPs. In order to accommodate phospho-substrates, the PTP active site is positively charged, which favors negatively charged molecules in high throughput screening campaigns that suffer from poor cell membrane permeability. To address the selectivity issue, we have pioneered a novel paradigm for the acquisition of potent and selective PTP inhibitors by targeting both the PTP active site and unique pockets in the vicinity of the active site 21, 22. To address the bioavailability issue, we sought to explore the existing natural product and FDA-approved drug space for previously unknown PTP inhibitory activities, since these molecules already possess acceptable pharmacological properties. We previously identified benzofuran salicylic acid as a privileged pharmacophore for mPTPB (12). Using a fragment-based medicinal chemistry approach, we transformed the benzofuran salicylic acid core into a highly potent and selective mPTPB inhibitor (L01-Z08, Table 1) with excellent in vivo efficacy 20.
Table 1.
Molecular and Cellular Properties of lead mPTPA and mPTPB inhibitors
| Structure | Name | Target | Biochemical potency against target (IC50, nM) | Fold selectivity | In vitro anti-Mtb activity (uM) | |||
|---|---|---|---|---|---|---|---|---|
| mPTPA | mPTPB | vs. mPTPA/B | vs. PTP panela | MABA-MICb H37Rv Erdman |
Mtb-infected macro phagesc | |||

|
L335M34 | mPTPA | 160 | >3200 | >20 | >20 | >10 >10 |
1.38 |

|
L01Z08 | mPTPB | 2500 | 38 | 66 | >37 | >10 >10 |
<5 |
Human PTP panel: PTP1B, TC-PTP, SHP1, SHP2, FAp1, Lyp, Meg2, HePTP, laforin, VHX, VHR, LMWPTP, Cdc14A, PTPα, LAR, CD45, PTPRG
MABA-MIC = Microplate Alamar Blue assay for Minimum Inhibitory Concentration
IC90 in macrophages activated with γ-interferon
More recently, we discovered that cefsulodin, a third generation cephalosporin β-lactam antibiotic, exhibits inhibitory activity against a number of PTPs 23. Fragmentation analysis of cefsulodin identified α-sulfophenylacetic amide (SPAA) as an mPTP-inhibiting pharmacophore and a novel pTyr mimetic. Structure-guided and fragment-based optimization of SPAA led to compound L335-M34, which displayed an IC50 value of 160 nM for mPTPA (Table 1). Kinetic analysis revealed that L335-M34 is a reversible and competitive inhibitor of mPTPA with a Ki of 56 ± 2.0 nM (Figure 1). To determine the specificity of L335-M34, we measured its inhibitory activity toward mPTPB and a panel of mammalian PTPs, including cytosolic PTPs, PTP1B, TC-PTP, SHP1, SHP2, FAP1, Lyp, PTP-Meg2, and HePTP, the receptor-like PTPs, PTPα, LAR, CD45 and PTPRG, the dual specificity phosphatases VHR, Laforin, VHX, and Cdc14A, and the low molecular weight PTP. As shown in Table 1, L335-M34 is highly selective for mPTPA, exhibiting greater than 20-fold selectivity over all PTPs examined. To the best of our knowledge, L335-M34 represents the most potent and specific mPTPA inhibitor reported to date 24–27.
Figure 1. Compound L335-M34 is a reversible and competitive inhibitor of mPTPA with pNPP as a substrate.

Line weaver-Burk plot for L335-M34 -mediated mPTPA inhibition. Compound L335-M34 concentrations were 0 (●), 50 (○), 100 (▽), 150 (▼), and 200 nM (■), respectively. The Ki value of 56 ± 2.0 nM was determined from three independent measurements.
Cellular activity of mPTPA and mPTPB inhibitors L335-M34 and L01Z08
The mPTPA inhibitor L335-M34 is highly selective for its target, with an IC50 of 160 nM against mPTPA but no significant activity against mPTPB or a panel of human PTPs at concentrations below 3 μM. Because mPTPA is a secreted virulence factor that regulates host antibacterial responses rather than Mtb physiology 28, it was unsurprising that L335-M34 was devoid of activity in standard MIC assays; however, the compound was able to markedly decrease bacterial load in Mtb-infected macrophages at low micromolar concentrations (Table 1).
The L01 family comprises three highly active and selective mPTPB inhibitors 20. The selected lead compound from this series, L01-Z08, displayed a potency of 38 nM against mPTPB and was 66-fold less potent against mPTPA and at least 37-fold selective when screened against a panel of 17 human PTPs. Like the mPTPA inhibitor L335-M34, L01-Z08 was inactive in the MIC assay but displayed potent anti-Mtb activity in J774A.1 macrophages (Table 1).
mPTPA and mPTPB inhibitors are bioavailable and well tolerated in guinea pigs following oral dosing
As shown in Figure 2, L01-Z08 and L335-M34 showed good oral bioavailability and half-life in guinea pigs (Table 2). Both drugs were rapidly absorbed, reaching peak concentrations within a few hours with a typical biphasic plasma clearance curve. Because the drugs were to be co-formulated for oral delivery in the therapy study, a combination PK study was performed to confirm that they were amenable to co-administration. Co-formulating L01-Z08 with L335-M34 in a single dosing solution did not negatively affect uptake or clearance of either drug. In fact, the bioavailability of L335-M34 was affected only moderately; a slight increase in uptake rate led to a greater peak concentration, which was offset by somewhat more rapid clearance, so that the overall exposure (AUCALL) was essentially unchanged. By contrast, L01-Z08 exposure was enhanced by co-administration (both Cmax and beta-phase T1/2 were elevated), but a reduction in the volume of distribution suggested that drug delivery to the tissues was probably not improved (Table 2). The PK study strongly indicated that the two compounds could be delivered with adequate efficiency in the guinea pig by the oral route. As shown in the bottom panel of Figure 2, at the doses selected for use in the efficacy study, L335-M34 and L01-Z08 were detected at concentrations 10-fold in excess of their biochemical IC50 values for 12–14 and 20–24 hours, respectively, suggesting that once daily oral dosing was an appropriate schedule for each drug. However, it should be noted that a higher degree of selectivity for these compounds was observed in the biochemical assays (IC50) than in the whole-cell assays in macrophages (growth inhibition/IC90), perhaps due to cell permeability issues29.
Figure 2. Pharmacokinetic profile of lead mPTP inhibitors in guinea pig plasma.

Concentration of mPTP inhibitors in the plasma over time. L335-M34 (mPTPA) 50 mg/kg or L01-Z08 (mPTPB) 20 mg/kg was given orally once alone or in combination and blood samples were collected at the indicated intervals relative to dosing. N=3 per time point; graph represents mean ± SD values.
Table 2.
Pharmacokinetics of mPTPA and mPTPB inhibitors in guinea pigsa
| Compound | Dose type | AUCALL hr*ng/ml | Clearance ml/hr/kg | CMAX ng/ml | TMAXb hr | Half-lifehr | VDc |
|---|---|---|---|---|---|---|---|
| L335M34 | Single | 54406.61 | 917.462 | 5142.47 | 2.5 | 5.197 | 6878.467 |
| L335M34 | Co-adminc | 52752.61 | 950.034 | 7064.956 | 2.5 | 4.16 | 5702.19 |
| L01Z08 | Single | 13166.89 | 1518.474 | 1870.43 | 1.587 | 5.512 | 12074.939 |
| L01Z08 | Co-adminc | 28161.76 | 701.564 | 3059.668 | 1.587 | 6.141 | 6215.132 |
Data represent mean values for 2–3 animals
TMAX is the time required to achieve the maximal concentration (CMAX)
VD = Volume of distribution
Both compounds L335M34 (50mg/kg) and L01Z08 (20 mg/kg) were co-administered orally
Guinea pigs receiving L01-Z08 20 mg/kg and L335-M34 50 mg/kg once daily alone or in combination for 6 weeks showed no overt signs of toxicity and displayed similar mean weight gain to those receiving HRZ (Figure S1). All guinea pigs receiving L01-Z08 and L335-M34 survived and gained weight throughout the course of the efficacy study.
Dual inhibition of mPTPA and mPTPB significantly reduces guinea pig lung bacillary burden relative to HRZ alone
Since each mPTP modulates distinct Mtb clearance pathways in macrophages, we hypothesized that dual inhibition of mPTPA and mPTPB would enhance the bactericidal activity of the standard antitubercular regimen in guinea pigs more than adjunctive therapy with either agent alone. Following aerosol infection of guinea pigs with Mtb H37Rv, 2.06 ± 0.15 log10 bacilli were deposited in the lungs on Day -27, and the organisms multiplied to a peak burden of 6.11 ± 0.15 log10 CFU on Day 0 (time of treatment initiation). Thereafter, bacillary growth was controlled in the lungs of untreated guinea pigs, which had 5.82 ± 0.17 log10 CFU in the lungs at the conclusion of the study.
Following two weeks of treatment, all guinea pigs in the HRZ, HRZ+L335-M34 (A), HRZ+L01-Z08 (B), and HRZ+AB groups were able to contain Mtb multiplication in the lungs, resulting in mean bacillary burdens of 4.44 ± 0.31, 4.07 ± 0.15, 4.15 ± 0.17, and 3.77±0.21 log10, respectively. After 2 weeks of treatment, lung CFU counts in animals treated with HRZ+AB were significantly (p < 0.01) lower than those treated with HRZ (Figure 3A). However, no significant differences were observed in lung CFU between HRZ+A, HRZ+B and HRZ+AB.
Figure 3. Activity of adjunctive mPTPA and mPTPB inhibitors against chronic TB infection in guinea pigs.

Animals were infected via aerosol with ~102 colony-forming units (CFU) of M. tuberculosis H37Rv and were either left untreated or were treated with drugs beginning 4 weeks after infection. Log10 colony-forming units (CFU) in the lungs are shown after 2 (A), 4 (B), and 6 (C) weeks of treatment. No drug= untreated, HRZ: Isoniazid (H), 60/Rifampin (R), 100/Pyrazinamide (Z), 300; mPTPA/A: L01-Z08, 20; mPTPB/B: L335-M34, 50; Numbers after each drug refer to doses (mg/kg). n=4 Guinea pigs per time point. *p<0.05, **p<0.01, ***p<0.001, HRZ versus HRZ+A+B.
At four weeks post-treatment, a similar trend was seen and the hierarchy of bactericidal activities of the various regimens was: HRZ+AB (1.15 ± 0.17 log10 CFU)> HRZ+B (1.64 ± 0.19 log10 CFU)> HRZ+A (1.65 ± 0.2 log10 CFU)>HRZ (1.70 ± 0.12 log10 CFU) (Figure 3B). HRZ+AB was significantly more active than HRZ+B, HRZ+A, and HRZ alone (p < 0.001).
After 6 weeks of treatment, HRZ reduced mean lung CFU by 4.27 log10 compared to untreated controls (Figure 2C). The addition of L01-Z08 (A) or L335-M34 (B) to the standard regimen further reduced mean lung CFU by 0.14 log10 and 0.17 log10, respectively, and the combination (AB) lowered mean lung CFU by 0.45 log10 (p<0.0001) relative to HRZ (Figure 3C).
The gross pathology of guinea pig lungs (data not shown), as well as mean guinea pig lung and spleen weights (Figure S2) correlated with the efficacy of the various chemotherapy regimens. Interestingly, the mean lung surface area involved by inflammation after 6 weeks of treatment was significantly lower in the HRZ+AB (9.23%) group relative to the HRZ group (36.28%) (p=0.028). Our results suggest that this effect on improved histopathology is primarily conferred by inhibition of mPTPA (HRZ+A vs. HRZ+AB, p=0.68) (Figure 4, Figures S3).
Figure 4. Lung inflammation 6 weeks after initiation of treatment.

Results are represented as percentage of lung surface area involved, calculated using imageJ software. Compared to RHZ, RHZ+A/RHZ+A+B are more effective in reducing lung lesion size and number in M. tuberculosis-infected guinea pigs at Month 1.5 after treatment. *p<0.05, HRZ versus HRZ+A/HRZ+A+B.
Our fragment-based lead optimization strategies have yielded two compounds, L01-Z08 and L335-M34, with potent activity against intracellular Mtb, as well as favorable PK and toxicity profiles. L01-Z08 and L335-M34 are inhibitors of the key secreted Mtb enzymes, mPTPB and mPTPA, respectively, and thus provide a novel mechanism of action for the treatment of TB. Both phosphatases are secreted by Mtb into the cytoplasm of the macrophage and are important for persistence of mycobacterial infection 10, 30. In order to determine the potential for translation of our findings to the clinical arena, we evaluated whether mPTP inhibitors could be beneficial as an adjunctive treatment when combined with the standard first-line regimen against drug-susceptible TB in guinea pigs. The two mPTPA and mPTPB inhibitor lead compounds showed promising oral bioavailability and tolerability in this model. Although each inhibitor alone added little bactericidal activity to the standard regimen, dual inhibition of mPTPA and mPTPB significantly reduced the lung bacillary burden relative to HRZ at each time point studied.
PTKs are the molecular targets for a growing number of anticancer agents 31, however there is a notable absence of drugs targeting the PTPs. Although many disease-relevant pathways are also controlled by PTPs 32–34, the latter have proven to be exceptionally challenging targets for the development of new therapeutic agents 34, due primarily to the poor bioavailability of existing PTP inhibitory compounds. The observed oral bioavailability and in vivo efficacy of L01-Z08 and L335-M34 are promising and further demonstrate that it is feasible to obtain PTP inhibitors that are sufficiently polar to bind the active site and yet still possess favorable pharmacological properties for therapeutic development.
Given the unique mechanisms of action of the mPTPA and mPTPB inhibitors, these compounds are expected to provide additive bactericidal activity to the standard regimen for drug-susceptible TB, as well as to novel regimens for drug-resistant TB. Moreover, concomitant treatment with such inhibitors may reduce the risk for selection of strains resistant to currently available anti-TB drugs during treatment 8. Previous work has shown that small molecule inhibitors of both mPTPA and mPTPB are capable of reducing intracellular mycobacteria in infected macrophages 8, 13, 35. It is interesting to note that adjunctive inhibition of mPTPA led to improved lung histopathology relative to standard treatment alone. A recent study showed that mPTPA dephosphorylates a second substrate, glycogen synthase kinase-α (GSK-α), causing its activation and the subsequent inhibition of the cell death program in infected macrophages 35. Based on available data, dual inhibition of mPTPA and mPTPB appears to undermine Mtb infection by: (i) increasing intracellular destruction of bacteria 36; (ii) promoting host-beneficial apoptosis of infected macrophages 13; and (iii) increasing host immunologic awareness of, and responsiveness to, Mtb infection 8, 35.
Previous studies37 have indicated that mPTPA is not essential for Mtb survival in mice, implying that the murine model fails to recapitulate the phenotypes reported in human macrophages 16. Although the mouse model has long been used to evaluate TB drugs38, it has been increasingly recognized in the TB field that observations made in mice are not predictive of treatment outcomes in human clinical trials, nor is early “sterilization” a predictor of cure 39,40. In the current study, we used the well-characterized guinea pig model of TB chemotherapy41–48. Compared to mice, guinea pig TB granulomas more closely approximate their human counterparts with respect to cellular composition, granuloma architecture, and the presence of caseation necrosis 49. In addition, tissue hypoxia is present in guinea pig TB granulomas 46, 50, but absent in mouse TB lung lesions 51, 52. These histological and microenvironmental factors, which may be biological determinants of Mtb persistence 53, as well as concordance of treatment outcomes with those of recent human studies 39, 54, make the guinea pig model an attractive one for testing the activity of novel antitubercular agents 41–44, 46, 47. However, the antitubercular activity of these agents could be further characterized in other clinically relevant models, such as the rabbit and nonhuman primate.
Our study contains several limitations. Considering the robust antitubercular activity of the mPTP inhibitors in macrophages, the corresponding activity of these agents against Mtb in guinea pig lungs was relatively modest. One potential explanation for this is reduced drug bioavailability at the site of infection. Although plasma levels of each inhibitor were determined after oral dosing, we did not directly measure drug concentrations in the lung lesions. Using MALDI-TOF, Prideaux et al. showed that the antitubercular activity of moxifloxacin may be limited due to inadequate penetration into necrotic granulomas in rabbits 55,56. As suggested by the discrepancy between the biochemical IC50 values of each agent and their corresponding IC90 values against intracellular Mtb, the entry of these compounds into macrophages may be somewhat limited. In addition, we have shown previously that a large population of bacilli is found in the extracellular compartment of necrotic lung granulomas in guinea pigs 43. Based on their mechanism of action, mPTP inhibitors would likely have no activity against such organisms. Finally, it is possible that functional deficiency of mPTPA and/or mPTPB may be compensated by alternative Mtb phosphatases, such as SapM15. Recently, Chauhan et al. generated an Mtb mutant (MtbΔmms) by disrupting the genes encoding PtpA, PtpB and SapM. After ten weeks of infection, guinea pigs infected with MtbΔmms showed a 4.69 log10 reduction in lung CFU relative to those infected with the isogenic wild-type strain 15. This mutant was more attenuated in the guinea pig model of chronic TB relative to an Mtb mutant lacking only PtpB 10. Future studies will focus on dose optimization, taking into account both plasma and tissue concentrations of mPTP inhibitors. In addition, we will attempt to identify potent inhibitors of Mtb SapM, which can be co-formulated with mPTPA and mPTPB inhibitors.
Our data support the further development of the Mtb PTP inhibitor class of drugs. We showed that PTP inhibitors lack direct antimicrobial activity but promote intracellular Mtb killing in vitro. Our findings suggest a modest increase in killing by the standard regimen with dual mPTPA/B inhibition, as well as a favorable PK interaction between the agents. Significantly, our data suggest that PTP inhibitors may improve clinical outcomes by ameliorating lung pathology. Further studies are needed to better characterize the sterilizing activity of these agents and their potential for improving TB-induced lung pathology.
MATERIALS AND METHODS
General procedures for the preparation of inhibitors
Reagents were used as purchased from Sigma-Aldrich and Fisher Scientific. 1H and 13C NMR spectra were obtained on a BrukerAvance II 500 MHz NMR spectrometer with TMS or residual solvent as standard. Mass spectra were obtained using an Agilent Technologies 6130 quadrupole LC/MS. HPLC purification was carried out on a Waters Delta 600 equipped with a Sunfire Prep C18 OBD column (30 mm/150 mm, 5 μm) with methanol water (both containing 0.1% TFA) as mobile phase (gradient: 50–100% methanol, flow 10 mL/min). The purity of all final tested compounds was established to be >95% by Agilent Technologies 6130 quadrupole LC/MS by using methanol water (both containing 0.1% TFA) as the mobile phase (gradient: 0–100% methanol, flow 1.0 mL/min), with UV monitoring at the fixed wavelength of 254 nm.
Synthesis of L335-M34 (mPTPA)
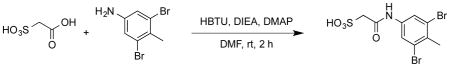
To a round-bottom flask was added sulfoacetic acid (0.14g, 1.0 mmol), DMF (5 ml), HBTU (0.379g, 1 mmol), 3, 5-dibromo-4-methyl aniline (0.265g, 1 mmol), DIEA (0.52 mL, 3 mmol), DMAP (0.012g, 0.1 mmol). The mixture was stirred at rt for 2 h, then it was subjected to reversed phase HPLC purification to give product L335-M34 as a white solid (0.385g, 99% yield). 1H NMR (500 MHz, DMSO) δ 10.08 (s, 1H), 7.87 (s, 2H), 3.52 (s, 2H), 2.42 (s, 3H); 13C NMR (DMSO) δ 164.6, 138.9, 130.5, 124.1, 121.8, 59.1, 22.6. ESI-HRMS Cacld. for C9H10Br2NO4S (M+H+): 385.8692, found 385.8696.
Kinetic analysis of mPTPA inhibition
The phosphatase activity of mPTPA was assayed using p-nitrophenyl phosphate (pNPP) as a substrate at 25°C in 50 mM 3,3-dimethylglutarate buffer, pH 7.0, containing 1 mM EDTA with an ionic strength of 0.15 M adjusted by NaCl. The reaction was started by the addition of 50 μl of the enzyme to 150 μl of reaction mixture containing pNPP and various concentrations of the inhibitor, in a 96-well plate. The final concentration for mPTPA was 5 nM. The final concentration for pNPP was 1 mM, which was the Km value for mPTPA). The reaction was quenched after 15 min by the addition of 50 μl of 5N NaOH, and then 200 μl of reaction mixture was transferred to a 96-well plate. The nonenzymatic hydrolysis of pNPP was corrected by measuring the control without the addition of enzyme. The amount of product p-nitrophenol was determined from the absorbance at 405 nm detected by a SpectraMax 384PLUS microplate spectrophotometer (Molecular Devices) using a molar extinction coefficient of 18,000 M−1cm−1. IC50 values were calculated by fitting the absorbance at 405 nm versus inhibitor concentration to the following equation:
where AI is the absorbance at 405 nm of the sample in the presence of inhibitor; A0 is the absorbance at 405 nm in the absence of inhibitor; and [I] is the concentration of the inhibitor.
For selectivity studies, the PTPs, including mPTPB, PTP1B, TC-PTP, SHP1, SHP2, FAP1, Lyp, PTP-MEG2, HePTP, PTPα, LAR, CD45, PTPγ, VHR, Laforin, VHX, Cdc14A, and the low molecular weight PTP were expressed and purified from E. coli. The final concentration for all PTPs was 5 nM. The inhibition assay for these PTPs were performed under the same conditions as mPTPA except using a different pNPP concentration corresponding to the Km of the PTP studied. Inhibitor concentrations used for IC50 measurements cover the range from 0.2 to 5× of the IC50 value.
Mycobacterium tuberculosis strains
The Johns Hopkins Center for Tuberculosis Research laboratory reference strain Mtb H37Rv 41 was passaged twice through mice and frozen in aliquots at –80°C before use. Aliquots were thawed and grown to logarithmic phase (optical density at 600nm = 0.6) in Middlebrook 7H9 broth (Difco Laboratories Detroit, MI) supplemented with 10% OADC (Becton Dickinson), 0.05% Tween, and 0.1% glycerol prior to aerosol infection.
Animals
Female guinea pigs (273±20.91g) with and without jugular vein catheters were purchased from Charles River Labs (Wilmington, MA). The animals were maintained under specific pathogen-free conditions and fed water and chow ad libitum. All procedures 57 followed protocols approved by the Institutional Animal Care and Use Committee at the Johns Hopkins University School of Medicine.
In vitro anti-Mtb assays
Alamar Blue assay
A colorimetric, microplate-based Alamar Blue assay (MABA) method was used to determine the MICs of mPTPA/B against M. tuberculosis isolates, as described earlier 58. Briefly, Cultures were incubated at 37°C without shaking in 96-well plates. AlamarBlue reagent (Invitrogen) was added at 1:5 v/v prior to readout 24 hours later using a Fluostar Optima fluorescence plate reader (BMG Labtech), equipped with a 544 nm excitation filter and a 590 nm emission filter.
Macrophage assays
Inhibition of growth of M. tuberculosis (Erdman and H37Rv) in a macrophage cell culture was assessed as previously described 13. Following activation with 50 U/ml IFN-γ (Sigma, 087k1288), J774 macrophages were infected with M. tuberculosis strain at a multiplicity of infection of 1:1 for 1 hr, washed and incubated with 20 μg/ml amikacin containing DMEM before adding the test compounds. Cells were washed; lysed and different dilutions were plated on 7H11 agar plates. Colonies were counted after 3 weeks of incubation at 37°C.
Pharmacokinetics and bioavailability studies
Separate groups of three catheterized guinea pigs each were given: (i) a single dose of L01-Z08 at 20 mg/kg, or L335-M34 at 50mg/kg of body weight; (ii) L01-Z08 at 20 mg/kg and L335-M34 at 50 mg/kg were given together to test for possible drug-drug interactions that might alter the uptake and/or clearance of one or both of the compounds. All drugs were suspended in 1% DMSO, 0.5% DEA, 48.5% PEG 400, 50% water. Blood was collected for analysis of these drugs in plasma pre-dose, and at 1, 4, 8, 24, and 48 hours post-dose. Plasma was separated and stored at −70°C until analysis. Plasma drug concentrations were determined by liquid chromatography–mass spectrometry and liquid chromatography–tandem mass spectrometry over the concentration range of 0.005–1 mg/L with dilution to 10 mg/L. Pharmacokinetic variables were calculated from individual drug concentration–time data using non-compartmental methods as implemented in WinNonlin version 5.0 (Pharsight, Mountain View, CA, USA) as described earlier46, 47.
Aerosol infections
Log-phase cultures of Mtb H37Rv were diluted 500-fold (to ~105 bacilli/mL) in 1× PBS for aerosol infection of guinea pigs. A total of 73 guinea pigs were aerosol-infected with a Madison chamber aerosol generation device (College of Engineering Shops, University of Wisconsin, Madison, WI) calibrated to deliver approximately 102 bacilli into guinea pig lungs, as previously described 43.
Antibiotic treatment
Beginning 28 days after aerosol infection, guinea pigs were randomized to different treatment groups control groups. Guinea pigs were treated 5 days per week for 6 weeks at different dose ranges, as indicated in Table S1. Isoniazid (INH, H; Sigma), rifampicin (RIF, R; Sigma), pyrazinamide (PZA, Z; Sigma) were dissolved in sterile distilled water. A cocktail solution of HRZ was prepared weekly and kept at 4°C. Aarden compounds (L01-Z08 and L335-M34) were suspended in formulation and stored at room temperature for up to 1 week.
All animals were treated with a formulation consisting of 20% pumpkin (wt/vol) (Libby’s 100% pure pumpkin) mixture supplemented with Vitamin C (50 mg/kg mean body weight) and commercial lactobacillus (BD lactinex) (all purchased from Walmart, Towson, MD) to improve palatability and help stabilize the cecal flora, thereby preventing gastrointestinal dysbacteriosis or antibiotic-associated enteritis, as previously described 46, 47. Drug doses were administered in a final volume of 0.5ml and were delivered in the posterior oropharynx by an automatic pipette with disposable tip.
Study end points
Guinea pigs were sacrificed on the day after aerosol infection (Day-27), on the day of treatment initiation (Day 0) and at the indicated time points after treatment to determine the numbers of colony-forming units (CFU) implanted in the lungs, pretreatment baseline CFU counts, and the post-treatment CFU counts, respectively.
Animal body weights were recorded on a weekly basis and lung and spleen weights were recorded at the time of necropsy. The lungs of each animal were examined at necropsy for grossly visible lesions, and random samples from the left caudal lung lobe were dissected, placed into 10% buffered formaldehyde, and paraffin embedded for histopathological staining with hematoxylin and eosin (H&E). At least one entire H&E-stained cross section per animal lung (4 animals/group) was analyzed for degree of inflammation. The surface area occupied by granulomatous inflammation was determined by ImageJ software-based morphometry of digitized images of lung sections and results are represented as percentage of lung surface area involved 47, 59.
The remaining lungs were homogenized in 10 ml PBS and homogenates were plated on 7H11 plates containing cycloheximide (50 mg/L), carbenicillin (100 mg/L), polymyxin B (200000 U/L), and trimethoprim (20 mg/L) and incubated for 28 days at 37°C for CFU enumeration.
Statistical analysis
CFU data were derived from 4 to 5 animals per group. Log-transformed CFU were used to calculate means and standard deviations. Comparisons of data among experimental groups were performed by Student’s t-test. Group means were compared by one-way analysis of variance (ANOVA) with Dunnett’s post-test (D0 or untreated controls vs. treatment groups) or Bonferroni comparison (all treatment groups), using GraphPad Prism version 4 (GraphPad, San Diego, CA). Values of p < 0.05 were considered to be statistically significant.
Acknowledgments
The authors wish to thank Gregg Timony, Receptos, Inc., for WinNonlin analysis of pharmacokinetic data.
Financial support: This work was supported by National Institutes of Health and the National Institute of Allergy and Infectious Diseases, Phase I STTR No. 1R41I106123-01 to FB, RO1 CA69202 to ZYZ; ACTG grant 110007 and NIAID grant UH2AI122309 to PCK, respectively.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Potential conflicts of interest: NKD, RH, MLP, YH and PCK: No conflicts. FB and ZYZ have ownership interests in Aarden Pharmaceuticals Inc.
Author contributions: Experimental conception and design was by NKD, ZYZ, FB and PCK. Data acquisition, analysis, and/or interpretation was by all authors. The manuscript was drafted by NKD, ZYZ and PCK and approved by all authors.
Disclaimer: The funding sources had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
References
- 1.Herbert N, George A, Sharma V, Oliver M, Oxley A, Raviglione M, Zumla AI. World TB Day 2014: finding the missing 3 million. Lancet. 2014;383:1016–1018. doi: 10.1016/S0140-6736(14)60422-0. [DOI] [PubMed] [Google Scholar]
- 2.Lienhardt C, Glaziou P, Uplekar M, Lonnroth K, Getahun H, Raviglione M Baroness Masham of I. Global tuberculosis control: lessons learnt and future prospects. Nature reviews Microbiology. 2012;10:407–416. doi: 10.1038/nrmicro2797. [DOI] [PubMed] [Google Scholar]
- 3.Kempker RR, Kipiani M, Mirtskhulava V, Tukvadze N, Magee MJ, Blumberg HM. Acquired Drug Resistance in Mycobacterium tuberculosis and Poor Outcomes among Patients with Multidrug-Resistant Tuberculosis. Emerging infectious diseases. 2015;21:992–1001. doi: 10.3201/eid2106.141873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y. The magic bullets and tuberculosis drug targets. Annual review of pharmacology and toxicology. 2005;45:529–564. doi: 10.1146/annurev.pharmtox.45.120403.100120. [DOI] [PubMed] [Google Scholar]
- 5.Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nature chemical biology. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 6.Silva AP, Tabernero L. New strategies in fighting TB: targeting Mycobacterium tuberculosis-secreted phosphatases MptpA & MptpB. Future medicinal chemistry. 2010;2:1325–1337. doi: 10.4155/fmc.10.214. [DOI] [PubMed] [Google Scholar]
- 7.Hunter T. The Croonian Lecture 1997. The phosphorylation of proteins on tyrosine: its role in cell growth and disease. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 1998;353:583–605. doi: 10.1098/rstb.1998.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beresford NJ, Mulhearn D, Szczepankiewicz B, Liu G, Johnson ME, Fordham-Skelton A, Abad-Zapatero C, Cavet JS, Tabernero L. Inhibition of MptpB phosphatase from Mycobacterium tuberculosis impairs mycobacterial survival in macrophages. The Journal of antimicrobial chemotherapy. 2009;63:928–936. doi: 10.1093/jac/dkp031. [DOI] [PubMed] [Google Scholar]
- 9.He RJ, Yu ZH, Zhang RY, Zhang ZY. Protein tyrosine phosphatases as potential therapeutic targets. Acta pharmacologica Sinica. 2014;35:1227–1246. doi: 10.1038/aps.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh R, Rao V, Shakila H, Gupta R, Khera A, Dhar N, Singh A, Koul A, Singh Y, Naseema M, Narayanan PR, Paramasivan CN, Ramanathan VD, Tyagi AK. Disruption of mptpB impairs the ability of Mycobacterium tuberculosis to survive in guinea pigs. Molecular microbiology. 2003;50:751–762. doi: 10.1046/j.1365-2958.2003.03712.x. [DOI] [PubMed] [Google Scholar]
- 11.Singh R, Singh A, Tyagi AK. Deciphering the genes involved in pathogenesis of Mycobacterium tuberculosis. Tuberculosis. 2005;85:325–335. doi: 10.1016/j.tube.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 12.Koul A, Herget T, Klebl B, Ullrich A. Interplay between mycobacteria and host signalling pathways. Nature reviews Microbiology. 2004;2:189–202. doi: 10.1038/nrmicro840. [DOI] [PubMed] [Google Scholar]
- 13.Zhou B, He Y, Zhang X, Xu J, Luo Y, Wang Y, Franzblau SG, Yang Z, Chan RJ, Liu Y, Zheng J, Zhang ZY. Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4573–4578. doi: 10.1073/pnas.0909133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castandet J, Prost JF, Peyron P, Astarie-Dequeker C, Anes E, Cozzone AJ, Griffiths G, Maridonneau-Parini I. Tyrosine phosphatase MptpA of Mycobacterium tuberculosis inhibits phagocytosis and increases actin polymerization in macrophages. Research in microbiology. 2005;156:1005–1013. doi: 10.1016/j.resmic.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 15.Chauhan P, Reddy PV, Singh R, Jaisinghani N, Gandotra S, Tyagi AK. Secretory phosphatases deficient mutant of Mycobacterium tuberculosis imparts protection at the primary site of infection in guinea pigs. PloS one. 2013;8:e77930. doi: 10.1371/journal.pone.0077930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bach H, Papavinasasundaram KG, Wong D, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell host & microbe. 2008;3:316–322. doi: 10.1016/j.chom.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 17.Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A. 2011;108:19371–19376. doi: 10.1073/pnas.1109201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cowley SC, Babakaiff R, Av-Gay Y. Expression and localization of the Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Research in microbiology. 2002;153:233–241. doi: 10.1016/s0923-2508(02)01309-8. [DOI] [PubMed] [Google Scholar]
- 19.Zeng LF, Xu J, He Y, He R, Wu L, Gunawan AM, Zhang ZY. A facile hydroxyindole carboxylic acid based focused library approach for potent and selective inhibitors of Mycobacterium protein tyrosine phosphatase B. ChemMedChem. 2013;8:904–908. doi: 10.1002/cmdc.201300115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He Y, Xu J, Yu ZH, Gunawan AM, Wu L, Wang L, Zhang ZY. Discovery and evaluation of novel inhibitors of mycobacterium protein tyrosine phosphatase B from the 6-Hydroxy-benzofuran-5-carboxylic acid scaffold. Journal of medicinal chemistry. 2013;56:832–842. doi: 10.1021/jm301781p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He R, Zeng LF, He Y, Zhang S, Zhang ZY. Small molecule tools for functional interrogation of protein tyrosine phosphatases. The FEBS journal. 2013;280:731–750. doi: 10.1111/j.1742-4658.2012.08718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puius YA, Zhao Y, Sullivan M, Lawrence DS, Almo SC, Zhang ZY. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: a paradigm for inhibitor design. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13420–13425. doi: 10.1073/pnas.94.25.13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He R, Yu ZH, Zhang RY, Wu L, Gunawan AM, Lane BS, Shim JS, Zeng LF, He Y, Chen L, Wells CD, Liu JO, Zhang ZY. Exploring the Existing Drug Space for Novel pTyr Mimetic and SHP2 Inhibitors. ACS medicinal chemistry letters. 2015;6:782–786. doi: 10.1021/acsmedchemlett.5b00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manger M, Scheck M, Prinz H, von Kries JP, Langer T, Saxena K, Schwalbe H, Furstner A, Rademann J, Waldmann H. Discovery of Mycobacterium tuberculosis protein tyrosine phosphatase A (MptpA) inhibitors based on natural products and a fragment-based approach. Chembiochem: a European journal of chemical biology. 2005;6:1749–1753. doi: 10.1002/cbic.200500171. [DOI] [PubMed] [Google Scholar]
- 25.Rawls KA, Lang PT, Takeuchi J, Imamura S, Baguley TD, Grundner C, Alber T, Ellman JA. Fragment-based discovery of selective inhibitors of the Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Bioorganic & medicinal chemistry letters. 2009;19:6851–6854. doi: 10.1016/j.bmcl.2009.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandra K, Dutta D, Das AK, Basak A. Design, synthesis and inhibition activity of novel cyclic peptides against protein tyrosine phosphatase A from Mycobacterium tuberculosis. Bioorganic & medicinal chemistry. 2010;18:8365–8373. doi: 10.1016/j.bmc.2010.09.052. [DOI] [PubMed] [Google Scholar]
- 27.Chiaradia LD, Martins PG, Cordeiro MN, Guido RV, Ecco G, Andricopulo AD, Yunes RA, Vernal J, Nunes RJ, Terenzi H. Synthesis, biological evaluation, and molecular modeling of chalcone derivatives as potent inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatases (PtpA and PtpB) Journal of medicinal chemistry. 2012;55:390–402. doi: 10.1021/jm2012062. [DOI] [PubMed] [Google Scholar]
- 28.He Y, Zeng LF, Yu ZH, He R, Liu S, Zhang ZY. Bicyclic benzofuran and indole-based salicylic acids as protein tyrosine phosphatase inhibitors. Bioorganic & medicinal chemistry. 2012;20:1940–1946. doi: 10.1016/j.bmc.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moreira W, Ngan GJ, Low JL, Poulsen A, Chia BC, Ang MJ, Yap A, Fulwood J, Lakshmanan U, Lim J, Khoo AY, Flotow H, Hill J, Raju RM, Rubin EJ, Dick T. Target mechanism-based whole-cell screening identifies bortezomib as an inhibitor of caseinolytic protease in mycobacteria. mBio. 2015;6:e00253–00215. doi: 10.1128/mBio.00253-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 31.Janne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nature reviews Drug discovery. 2009;8:709–723. doi: 10.1038/nrd2871. [DOI] [PubMed] [Google Scholar]
- 32.Andersen JN, Jansen PG, Echwald SM, Mortensen OH, Fukada T, Del Vecchio R, Tonks NK, Moller NP. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2004;18:8–30. doi: 10.1096/fj.02-1212rev. [DOI] [PubMed] [Google Scholar]
- 33.Arena S, Benvenuti S, Bardelli A. Genetic analysis of the kinome and phosphatome in cancer. Cellular and molecular life sciences: CMLS. 2005;62:2092–2099. doi: 10.1007/s00018-005-5205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barr AJ. Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development. Future medicinal chemistry. 2010;2:1563–1576. doi: 10.4155/fmc.10.241. [DOI] [PubMed] [Google Scholar]
- 35.Poirier V, Bach H, Av-Gay Y. Mycobacterium tuberculosis promotes anti-apoptotic activity of the macrophage by PtpA protein-dependent dephosphorylation of host GSK3alpha. The Journal of biological chemistry. 2014;289:29376–29385. doi: 10.1074/jbc.M114.582502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pizarro-Cerda J, Cossart P. Subversion of phosphoinositide metabolism by intracellular bacterial pathogens. Nature cell biology. 2004;6:1026–1033. doi: 10.1038/ncb1104-1026. [DOI] [PubMed] [Google Scholar]
- 37.Grundner C, Cox JS, Alber T. Protein tyrosine phosphatase PtpA is not required for Mycobacterium tuberculosis growth in mice. FEMS microbiology letters. 2008;287:181–184. doi: 10.1111/j.1574-6968.2008.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Groote MA, Gilliland JC, Wells CL, Brooks EJ, Woolhiser LK, Gruppo V, Peloquin CA, Orme IM, Lenaerts AJ. Comparative studies evaluating mouse models used for efficacy testing of experimental drugs against Mycobacterium tuberculosis. Antimicrobial agents and chemotherapy. 2011;55:1237–1247. doi: 10.1128/AAC.00595-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, Pappas F, Phillips PP, Nunn AJ. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. The New England journal of medicine. 2014;371:1577–1587. doi: 10.1056/NEJMoa1407426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitchison DA, Chang KC. Experimental Models of Tuberculosis: Can We Trust the Mouse? American journal of respiratory and critical care medicine. 2009;180:201–202. doi: 10.1164/rccm.200905-0708ED. [DOI] [PubMed] [Google Scholar]
- 41.Ahmad Z, Fraig MM, Bisson GP, Nuermberger EL, Grosset JH, Karakousis PC. Dose-dependent activity of pyrazinamide in animal models of intracellular and extracellular tuberculosis infections. Antimicrobial agents and chemotherapy. 2011;55:1527–1532. doi: 10.1128/AAC.01524-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahmad Z, Fraig MM, Pinn ML, Tyagi S, Nuermberger EL, Grosset JH, Karakousis PC. Effectiveness of tuberculosis chemotherapy correlates with resistance to Mycobacterium tuberculosis infection in animal models. The Journal of antimicrobial chemotherapy. 2011;66:1560–1566. doi: 10.1093/jac/dkr188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmad Z, Klinkenberg LG, Pinn ML, Fraig MM, Peloquin CA, Bishai WR, Nuermberger EL, Grosset JH, Karakousis PC. Biphasic kill curve of isoniazid reveals the presence of drug-tolerant, not drug-resistant, Mycobacterium tuberculosis in the guinea pig. The Journal of infectious diseases. 2009;200:1136–1143. doi: 10.1086/605605. [DOI] [PubMed] [Google Scholar]
- 44.Ahmad Z, Nuermberger EL, Tasneen R, Pinn ML, Williams KN, Peloquin CA, Grosset JH, Karakousis PC. Comparison of the ‘Denver regimen’ against acute tuberculosis in the mouse and guinea pig. The Journal of antimicrobial chemotherapy. 2010;65:729–734. doi: 10.1093/jac/dkq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmad Z, Pinn ML, Nuermberger EL, Peloquin CA, Grosset JH, Karakousis PC. The potent bactericidal activity of streptomycin in the guinea pig model of tuberculosis ceases due to the presence of persisters. The Journal of antimicrobial chemotherapy. 2010;65:2172–2175. doi: 10.1093/jac/dkq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dutta NK, Alsultan A, Gniadek TJ, Belchis DA, Pinn ML, Mdluli KE, Nuermberger EL, Peloquin CA, Karakousis PC. Potent rifamycin-sparing regimen cures guinea pig tuberculosis as rapidly as the standard regimen. Antimicrobial agents and chemotherapy. 2013;57:3910–3916. doi: 10.1128/AAC.00761-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dutta NK, Illei PB, Peloquin CA, Pinn ML, Mdluli KE, Nuermberger EL, Grosset JH, Karakousis PC. Rifapentine is not more active than rifampin against chronic tuberculosis in guinea pigs. Antimicrobial agents and chemotherapy. 2012;56:3726–3731. doi: 10.1128/AAC.00500-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dutta NK, Pinn ML, Zhao M, Rudek MA, Karakousis PC. Thioridazine lacks bactericidal activity in an animal model of extracellular tuberculosis. Journal of Antimicrobial Chemotherapy. 2013;68:1327–1330. doi: 10.1093/jac/dkt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flynn J, Chan J. Animal models of tuberculosis. In: Rom W, Garay S, editors. Tuberculosis. 2. Lippincott Williams & Wilkins; Philadelphia: 2004. pp. 237–250. [Google Scholar]
- 50.Lenaerts AJ, Hoff D, Aly S, Ehlers S, Andries K, Cantarero L, Orme IM, Basaraba RJ. Location of persisting mycobacteria in a Guinea pig model of tuberculosis revealed by r207910. Antimicrob Agents Chemother. 2007;51:3338–3345. doi: 10.1128/AAC.00276-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai MC, Chakravarty S, Zhu G, Xu J, Tanaka K, Koch C, Tufariello J, Flynn J, Chan J. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cellular microbiology. 2006;8:218–232. doi: 10.1111/j.1462-5822.2005.00612.x. [DOI] [PubMed] [Google Scholar]
- 52.Aly S, Wagner K, Keller C, Malm S, Malzan A, Brandau S, Bange FC, Ehlers S. Oxygen status of lung granulomas in Mycobacterium tuberculosis-infected mice. J Pathol. 2006;210:298–305. doi: 10.1002/path.2055. [DOI] [PubMed] [Google Scholar]
- 53.Dutta NK, Karakousis PC. Latent tuberculosis infection: myths, models, and molecular mechanisms. Microbiology and molecular biology reviews: MMBR. 2014;78:343–371. doi: 10.1128/MMBR.00010-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dorman SE, Goldberg S, Stout JE, Muzanyi G, Johnson JL, Weiner M, Bozeman L, Heilig CM, Feng PJ, Moro R, Narita M, Nahid P, Ray S, Bates E, Haile B, Nuermberger EL, Vernon A, Schluger NW. Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: study 29 of the tuberculosis trials consortium. The Journal of infectious diseases. 2012;206:1030–1040. doi: 10.1093/infdis/jis461. [DOI] [PubMed] [Google Scholar]
- 55.Prideaux B, Via LE, Zimmerman MD, Eum S, Sarathy J, O’Brien P, Chen C, Kaya F, Weiner DM, Chen PY, Song T, Lee M, Shim TS, Cho JS, Kim W, Cho SN, Olivier KN, Barry CE, 3rd, Dartois V. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nature medicine. 2015;21:1223–1227. doi: 10.1038/nm.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Via LE, Savic R, Weiner DM, Zimmerman MD, Prideaux B, Irwin SM, Lyon E, O’Brien P, Gopal P, Eum S, Lee M, Lanoix JP, Dutta NK, Shim T, Cho JS, Kim W, Karakousis PC, Lenaerts A, Nuermberger E, Barry CE, 3rd, Dartois V. Host-Mediated Bioactivation of Pyrazinamide: Implications for Efficacy, Resistance, and Therapeutic Alternatives. ACS infectious diseases. 2015;1:203–214. doi: 10.1021/id500028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klinkenberg LG, Sutherland LA, Bishai WR, Karakousis PC. Metronidazole lacks activity against Mycobacterium tuberculosis in an in vivo hypoxic granuloma model of latency. J Infect Dis. 2008;198:275–283. doi: 10.1086/589515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, Gilman RH. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. Journal of clinical microbiology. 1998;36:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dutta NK, Alsultan A, Peloquin CA, Karakousis PC. Preliminary pharmacokinetic study of repeated doses of rifampin and rifapentine in guinea pigs. Antimicrobial agents and chemotherapy. 2013;57:1535–1537. doi: 10.1128/AAC.01933-12. [DOI] [PMC free article] [PubMed] [Google Scholar]