

ABSTRACT
Glycosylation is a critical attribute for development and manufacturing of therapeutic monoclonal antibodies (mAbs) in the pharmaceutical industry. Conventional antibody glycan analysis is usually achieved by the 2-aminobenzamide (2-AB) hydrophilic interaction liquid chromatography (HILIC) method following the release of glycans. Although this method produces satisfactory results, it has limited use for screening a large number of samples because it requires expensive reagents and takes several hours or even days for the sample preparation. A simple and rapid glycan analysis method was not available. To overcome these constraints, we developed and compared 2 ultrafast methods for antibody glycan analysis (UMAG) that involve the rapid generation and purification of glycopeptides in either organic solvent or aqueous buffer followed by label-free quantification using matrix-assisted laser desorption/ionization-time of flight mass spectrometry. Both methods quickly yield N-glycan profiles of test antibodies similar to those obtained by the 2-AB HILIC-HPLC method. In addition, the UMAG method performed in aqueous buffer has a shorter assay time of less than 15 min, and enables high throughput analysis in 96-well PCR plates with minimal sample handling. This method, the fastest, and simplest as reported thus far, has been evaluated for glycoprofiling of mAbs expressed under various cell culture conditions, as well as for the evaluation of antibody culture clones and various production batches. Importantly the method sensitively captured changes in glycoprofiles detected by traditional 2-AB HILIC-HPLC or HILIC-UPLC. The simplicity, high speed, and low cost of this method may facilitate basic research and process development for novel mAbs and biosimilar products.
KEYWORDS: Antibody, biosimilar, high throughput, N-glycan analysis, ultrafast
Introduction
Glycosylation, which is considered one of the most important posttranslational protein modifications,1 especially for therapeutic antibodies, involves the covalent attachment of oligosaccharide molecules (glycans) to specific amino acid residues on the protein molecule. In N-glycosylation of most recombinant IgG antibodies, Asn-297, in the Fc fragment of the heavy chain, is a strictly conserved glycosylation site.2 Glycans in such IgGs belong to the bi-antennary complex type with a conserved -GlcNAc2Man3GlcNAc2- heptasaccharide core.3 Additional terminal sugars, fucose (Fuc), galactose (Gal), and N-acetylneuraminic acid (sialic acid, or NANA) residues may be attached to the core, producing a large diversity of glycan structures.4,5
The glycan moiety has a profound effect on the biological functions of a glycoprotein, such as protein cell-surface expression,6 protein quality,2 immune responses,7-10 resistance towards proteases or half-life.2,4,11-15 Glycans are essential for the antibody's effector functions because they are required for antibody binding to all Fc gamma receptors, and thus affect efficacy of the antibody if its mode of action involves antibody dependent cell-mediated cytotoxicity (ADCC).2,14,16,17 Therapeutic properties can be enhanced by glycoengineering via alteration of glycosylation sites, enzymatic glycan modification in vitro, and manipulation of cellular glycosylation machinery.4,15,18 Relating to human diseases, abnormal glycan profiles have been implicated in human genetic disorders,19,20 muscular dystrophies,21 neurological diseases,22 and cancers.23-26
During novel therapeutic mAb development, glycosylation is considered a critical quality attribute that must be closely monitored, and it has attracted increasing attention from the regulatory agencies.27 Glycans in mAbs represent an average of 2-3% of the total antibody mass, 2,4 and their expression and structure are influenced by cell culture process conditions.18,28,29 It is also particularly important to monitor and compare distribution of biosimilar antibody glycoforms with the reference product, as such comparison is mandated by regulatory agencies. Many cell culture process conditions need to be tested to ensure that biosimilar antibodies are produced with glycoprofiles consistent with the originator molecules. This creates a challenge in glycan analysis because a large numbers of samples need to be rapidly evaluated to facilitate the ongoing process optimization. Development of rapid and high throughput glycoprofiling assays is highly desired and critical to support high sample loads.30,31
Glycan analysis for N-linked glycoproteins usually involves the release of the oligosaccharide chains from the protein backbone by enzymatic cleavage with peptide N-glycosidase (PNGase F).28,32-34 Released glycans are normally labeled with a fluorescent tag such as 2-aminobenzamide (2-AB), and then quantitated by hydrophilic interaction liquid chromatography (HILIC)-HPLC. The 2-AB HILIC-HPLC method is the mainstay for glycan analysis for recombinant antibodies in the biopharmaceutical industry. However, the method is time consuming (requiring several hours with recently improved labeling chemistry or up to three days using older methods 35) and labor intensive, and becomes a bottleneck in the analysis of large numbers of samples.
As described here, we developed and evaluated 2 ultrafast methods for antibody glycan analysis (UMAG) based on glycopeptides.1,31,36-38 These methods involve the rapid generation and purification of the glycopeptides in either organic solvent or aqueous buffer, followed by detection and label-free quantification using matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF-MS), a useful method for the analysis of glycopeptides.37,39 Compared to 2-AB HILIC-HPLC, the UMAG under aqueous conditions is ultra-fast (10-15 min), robust, high throughput, and low cost.
Results
Strategy
The conventional 2-AB HILIC-HPLC method for glycan analysis requires sample preparation involving release of glycans from antibodies by PNGase F, and then labeling and separation of glycans. This procedure is lengthy and labor intensive. Our glycan analysis strategy involves ultrafast digestion of antibodies by specific proteases like trypsin, rapid purification of glycopeptides, and then label-free detection/quantification of glycopeptides by MALDI-TOF MS (Fig. 1). Our aim was to complete the analysis in minutes instead of hours to days.
Figure 1.

Strategy used in the new glycan analysis of antibodies.
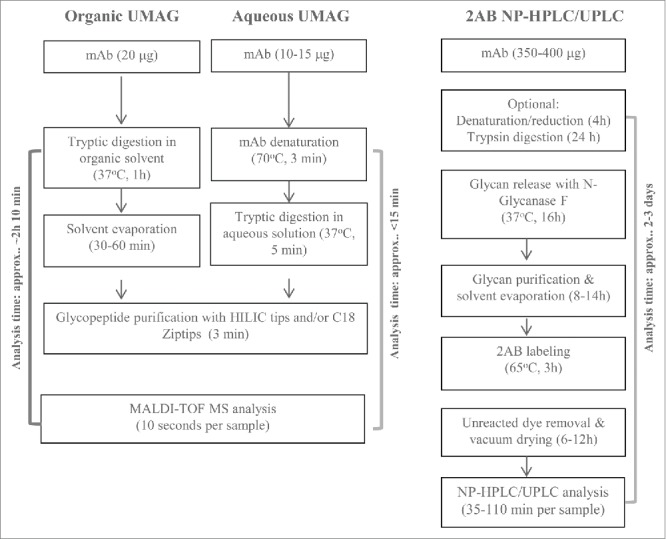
The ability to rapidly generate and purify glycopeptides is both the key and the limiting factor for establishing an ultrafast MALDI method for antibody glycan analysis (UMAG) as outlined in Fig. 1, while the time for MALDI-TOF MS analysis is relatively fast (approximately several seconds per sample). In a routine trypsin digestion, the target proteins are normally reduced, alkylated, and enzymatically digested overnight. The procedure is lengthy. Thus, a rapid digestion method in the organic solvents40 and the ultrafast digestion method in aqueous buffer we developed were tested to determine whether they achieved the goal of high throughput glycan analysis.
UMAG with digestion of antibody in organic solvent
Recombinant mAb A (IgG1) was first digested with trypsin at 37°C for 1 hour in 10 mM Tris, pH 8.0 containing 80% acetonitrile (ACN) in the presence of 2 mM CaCl2. ACN acts as a denaturing agent to unfold and denature proteins, and allows the protease more access to the cleavage sites on the target proteins, and thus facilitates digestion.40 The digest was lyophilized and resuspended in 0.1% trifluoroacetic acid (TFA), and analyzed by MALDI-TOF MS.
Multiple peptide peaks were detected in trypsin digests of mAb A in the presence of 80% ACN (Fig. 2A). Glycopeptides were not detected due to ionization suppression and their lower molar concentration compared to non-glycosylated peptides. Thus, glycopeptides were enriched with a C18 Ziptip followed by a HILIC tip. The glycopeptides were clearly detected by MALDI within a mass range of 2200-3000, although some non-glycosylated peptides were also observed (Fig. 2B). Detected glycopeptides correspond to the tryptic peptide (EEQYNSTYR) with different glycans attached (such as G0, G0F, G1, G1F, and G2F) (Table 1). Some glycopeptides, such as G0F-GlcNAC and G0-GlcNAC that were not identified by the 2-AB HILIC-HPLC method were also observed (Fig. 2C).
Figure 2.
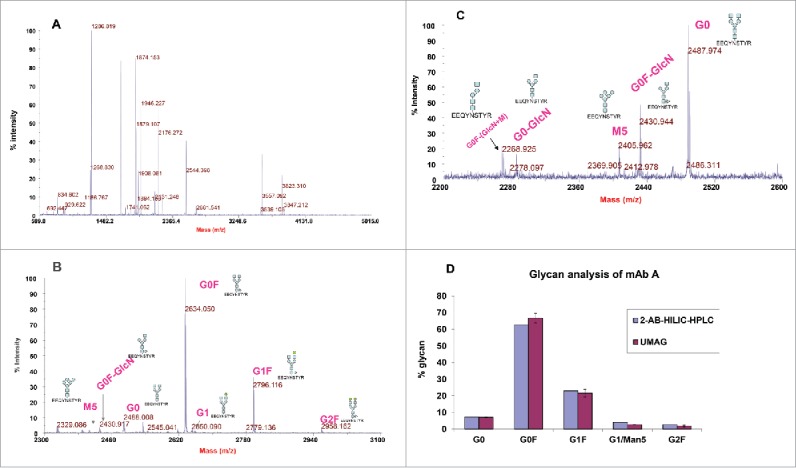
Detection of mAb A glycopeptides by MALDI-TOF MS. (A) MALDI-TOF mass spectrum of trypsin digests of mAb A. mAb A was digested with trypsin in the presence of 80% acetonitrile for 1 hour at 37°C. The digest (1 μl) was directly spotted and analyzed by MALDI-TOF MS. (B) Enrichment of glycopeptides with a C18 Ziptip and then with a HILIC tip and detection by MALDI-TOF. The glycosylated peptides were indicated. (C) Detection of minor glycopeptides G0F-GlcN, G0-GlcN, and G0F-GlcN-Man. The sugar symbols: blue square = N-acetyl glucosamine, blue circle=mannose, green circle = galactose, triangle = fucose. (D) Comparison of mAb A glycoprofiles analyzed by ultrafast MALDI-TOF antibody glycan analysis (UMAG) (n = 2) and 2-AB HILIC-HPLC. The antibody was digested under the organic conditions, purified by C18 Ziptips/HILIC tips and detected by MALDI-TOF.
Table 1.
Glycopeptide masses (Da) of mAb A by ultrafast MALDI-TOF antibody glycan analysis (UMAG) with trypsin digestion in organic solvent.
| Glycopeptide | Structure | Theoretical (M+H)+ | Measured (M+H)+ |
|---|---|---|---|
| Peptide alone | EEQYNSTYR | 1189.512 | not detected |
| G0 | Man3(glcNAc)4 | 2487.988 | 2488.008 |
| G0F | Man3(glcNAc)4(Fuc)1 | 2634.046 | 2634.050 |
| G1 | (Gal)1Man3(glcNAc)4 | 2650.041 | 2650.090 |
| G1F | (Gal)1Man3(glcNAc)4(Fuc)1 | 2796.099 | 2796.116 |
| G2F | (Gal)2Man3(glcNAc)4(Fuc)1 | 2958.152 | 2958.162 |
| Man5 | Man5(glcNAc)2 | 2405.935 | 2405.934 |
| G0F-GlcNAc | Man3(glcNAc)3(Fuc)1 | 2430.967 | 2430.917 |
| G0-GlcNAc | Man3(glcNAc)3 | 2284.909 | 2284.924 |
| G0F-GlcNAc-Man | Man2(glcNAc)3(Fuc)1 | 2268.914 | 2268.925 |
For comparison, mAb A was analyzed with UMAG and the conventional 2-AB HILIC- HPLC glycan method. In UMAG, the cluster peak area for each glycopeptide in the MALDI-TOF spectrum was integrated and used to determine their percentage. The glycoprofile of the antibody determined by these 2 methods was similar (Fig. 2D).
The feasibility of UMAG to assess the glycan changes was also performed with a set of mAb B samples derived from cell culture that were grown in the presence of different amounts of galactose. mAb B purified from these cultures displayed decreasing % of G0F and increasing % of G1F in the presence of increasing amounts of galactose, as determined by both UMAG and 2-AB HILIC-HPLC method (Fig. 3). Although the experiment was not repeated due to the limited amount of material available, the finding was consistent with the previous observation that the galactosylation of the antibody, i.e., the conversion of glycoform G0F into G1F, was greatly affected by galactose in the cell media.41,42 Therefore this UMAG method performed with trypsin in organic solvent (referred to as organic UMAG) was able to detect changes in mAb N-glycan profiles.
Figure 3.
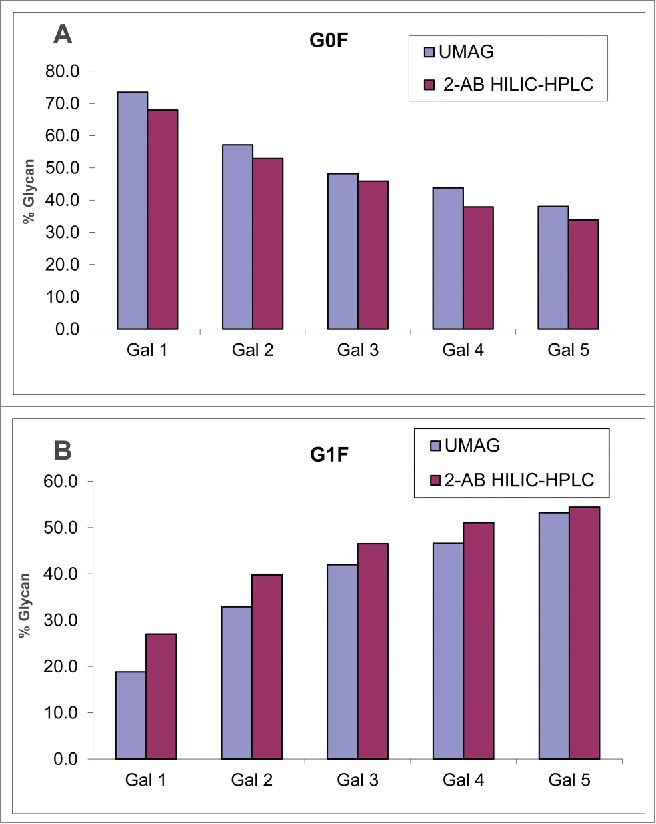
Analysis of mAb B samples purified from different cell cultures by organic UMAG and 2-AB HILIC-HPLC. G0F (A) and G1F (B) were listed for comparison. Samples Gal1-5 correspond to antibody from cell cultures containing increasing amounts of galactose.
UMAG with digestion of antibodies in aqueous solution
Although the organic UMAG worked quite well, it required a time-consuming step (30 min-1 hour) for solvent evaporation and sample rehydration. To further increase the throughput, ultrafast digestion of antibodies under aqueous conditions was developed. After testing many conditions, the following procedure was found to work the best. Since the intact antibody is resistant to cleavage by trypsin in aqueous solutions, mAb A was denatured at a high temperature (70°C) by mixing with 8 M urea and 10 mM DTT for 3 min. The denatured antibody was then diluted with 20 mM (NH4)2HCO3 to reduce the urea concentration, and digested for a short period of time by trypsin at an enzyme/substrate ratio of 1:30. The kinetic study of the digestion suggested that the majority of cleavages occurred at 37°C in a short time period (5 min), and almost no intact antibody remained as demonstrated by RP-HPLC analysis of the trypsin digest of mAb A (Fig. 4A), and denaturation at 4-8 M urea worked equally well (data not shown).
Figure 4.
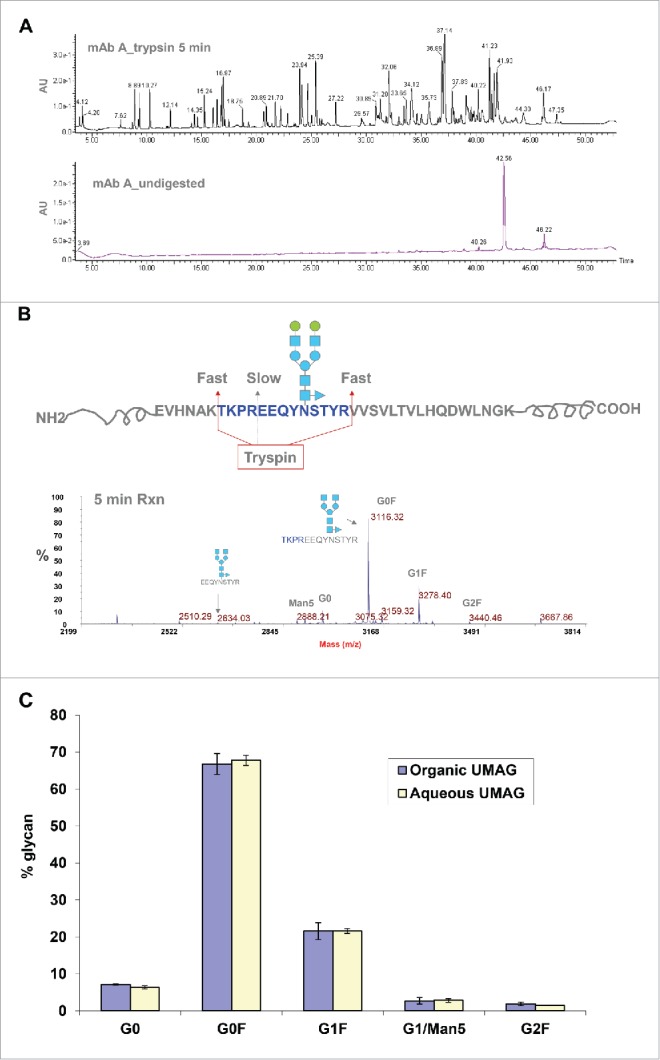
Analysis of mAb A glycan profiles by UMAG in aqueous solution. (A) Analysis of mAb A digested by trypsin for 5 min (top) and undigested mAb A (bottom) by RP-HPLC. Undigested mAb A was injected at a 1/6 amount of digested antibody. (B) Different rates of cleavage by trypsin under aqueous conditions near the glycosylation site. The majority of glycopeptides were missed cleavage peptides with an internal trypsin site. (C) Comparison of glycoprofiles analyzed by organic UMAG under organic conditions (blue) (n = 2) and aqueous UMAG under aqueous conditions (yellow) (n = 4).
Bypassing the solvent evaporation step required for organic UMAG, glycopeptides were directly purified from the aqueous trypsin digest of mAb A in the aqueous solution using C18 Ziptips and detected by MALDI-TOF MS. We found that the purification step with HILIC tips adding little to the procedure. The majority of purified glycopeptides contained the peptide (TKPREEQYNSTYR) with a missed cleavage site in the middle, and only a very small percentage of glycopeptides contained EEQYNSTYR (Fig. 4B), likely due to the steric hindrance of the glycans or the interference of proline near the cleavage site. A kinetic study showed that 24 hours were required to complete digestion in the missed cleavage site (data not shown). Expected and measured masses of different glycopeptides containing the longer peptide (TKPREEQYNSTYR) generated under the aqueous conditions are listed in Table 2. Longer glycopeptides (TKPREEQYNSTYR), yielded similar glycoprofiles as short glycopeptides (EEQYNSTYR) obtained under organic digesting conditions (Fig. 4C). Compared to the organic UMAG, the aqueous UMAG was much simpler and drastically reduced the total analysis time from 1 hour 50 min to less than 15 min by use of a faster trypsin digestion step and elimination of the drying step (Fig. 10).
Table 2.
Glycopeptide masses (Da) of mAb A by ultrafast MALDI-TOF antibody glycan analysis (UMAG) with trypsin digestion in the aqueous solution.
| Glycopeptide | Structure | Theoretical (M+H)+ | Measured (M+H)+ |
|---|---|---|---|
| Peptide alone | TKPREEQYNSTYR | 1671.8019 | Not detected |
| G0 | Man3(glcNAc)4 | 2970.2779 | 2970.28 |
| G0F | Man3(glcNAc)4(Fuc)1 | 3116.3358 | 3116.32 |
| G1 | (Gal)1Man3(glcNAc)4 | 3132.3307 | 3132.36 |
| G1F | (Gal)1Man3(glcNAc)4(Fuc)1 | 3278.3886 | 3278.40 |
| G2F | (Gal)2Man3(glcNAc)4(Fuc)1 | 3440.4414 | 3440.45 |
| Man5 | Man5(glcNAc)2 | 2888.2247 | 2888.21 |
| G0F-GlcNAc | Man3(glcNAc)3(Fuc)1 | 2913.2564 | 2913.23 |
| G0-GlcNAc | Man3(glcNAc)3 | 2767.1985 | 2767.17 |
| G0F-GlcNAc-Man | Man2(glcNAc)3(Fuc)1 | 2751.2036 | 2751.19 |
Figure 10.
Comparison of organic UMAG, aqueous UMAG, and 2-AB HILIC-HPLC/ UPLC methods.
Confirmation of glycopeptides produced under aqueous conditions
PNGase F digestion experiments were performed to prove that the MALDI peaks detected were glycopeptides. After cleavage with PNGase F, the original asparagine in the peptide is converted into an Asp, and therefore the mass for the generated peptide increases by 1 Da. A peak with mass of 1672.8 Da corresponding to TKPREEQYNSTYR was observed following PNGase treatment, while the glycopeptide peaks disappeared (Fig. 5). These data indicate that the detected peaks are glycopeptides containing N-glycans.
Figure 5.

Treatment of glycopeptides with PNGase F to confirm the composition of glycans. Top graph represents glycopeptides before PNGase F treatment, and bottom graph shows peptides after PNGase F treatment. The peptide (TKPREEQYNSTYR) after removal of glycans with PNGase F showed the expected mass of 1672.8 Da.
Adaption to the high throughput assay in 96-well PCR plates
High throughput assessment of the aqueous UMAG procedure was conducted by analyzing a set of mAb H samples in a 96-well PCR plate using a multichannel pipette for sample handling. The antibody samples were first purified by Protein A chromatography from the cell cultures grown under 11 different conditions. Samples were also analyzed by 2-AB HILIC-UPLC. The changes of glycan profiles with differing cell culture conditions were very similar whether determined by either the high throughput aqueous UMAG or 2-AB HILIC-HPLC method (Fig. 6). For example, the trend of changes in major glycopeptides G0, G0F, and G1F due to the cell culture conditions was very similar by both methods, in spite of some differences in the absolute percentages of some glycan species. These data also suggest that the antibody glycosylation varies frequently in response to altered physiological conditions that lead to changes in the complex glycosylation machinery in the cells.28,29 Therefore, the high throughput nature of this UMAG is an efficient way to rapidly analyze a large number of samples or cell culture test conditions in parallel. This is particularly important in the development of process conditions for the optimization of biosimilar glycoprofiles, and in the support of process comparability studies for late stage programs.
Figure 6.
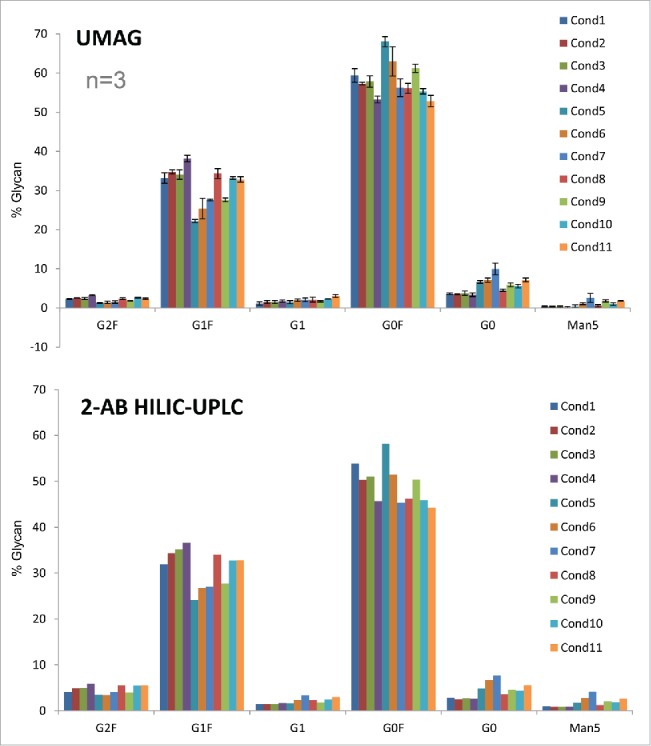
Glycan analysis of mAb H samples produced under different culture conditions by the high throughput aqueous UMAG performed on a PCR plate and 2-AB HILIC-UPLC.
In comparison to 2-AB HILIC HPLC/UPLC, the high throughput aqueous UMAG was further evaluated by analyzing the glycoprofiles of several mAbs from different production batches (Figs. 7A and 7B). Again, both methods demonstrated similar levels of each glycoform among production batches.
Figure 7.
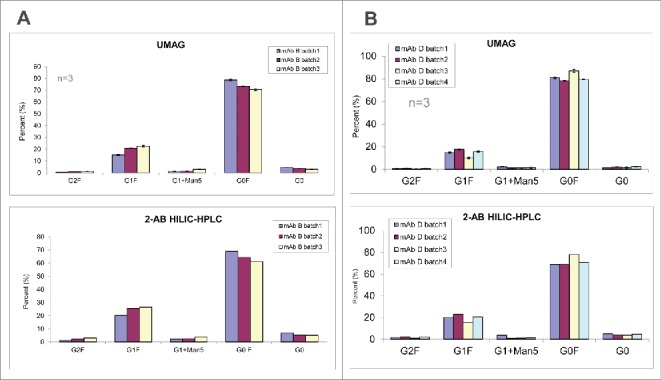
Glycan analysis of mAb B (A) and mAb D (B) samples different production batches by the high throughput aqueous UMAG and 2-AB HILIC-UPLC.
UMAG was also applied to evaluate and compare commercial originator antibody batches and biosimilar clones/batches (Fig. 8A). Several originator mAb H batches exhibited significant variation in the glycoprofiles by both methods, and some biosimilar clones or batches showed comparable glycan profiles as originator's material.
Figure 8.

(A) Glycan analysis of mAb H samples from different originator production lots and biosimilar samples from different clones/production batches by the high throughput aqueous UMAG and 2-AB HILIC-UPLC. (B) Fast characterization of the glycosylation pathways using mannosidase inhibitor kifunensine in the cell culture by the aqueous UMAG. Top graph represents glycan profile of mAb H from the control cell culture, and bottom graph shows glycan profiles when kifunensine was present in the culture. The inset plot is included to show mannose 7. Kifunensine completely blocked the process of the normal glycans, and resulted in accumulation of high mannose glycans (Man7-9).
Additionally, UMAG was used to assess the effect of glycosidase inhibitors on the antibody glycoforms (Fig 8B). A typical profile containing G0, G0F, G1F, G1, and G2F was seen for mAb H produced from the control culture. However, these typical glycoforms were absent from the antibody produced in the culture with inhibitor kifunensine, which is a specific inhibitor for mannosidase. Instead, glycoforms containing high mannose glycans (Man7, Man8, and Man9) were detected, indicating that the processing of high mannose glycan precursors was totally inhibited by blocking mannosidase activity.43 Therefore, aqueous UMAG, coupled with mass spectrometry, is useful for rapid detection and characterization of new glycan structures and glycosylation sites that traditional 2-AB labeling HILIC-HPLC method might fail to identify or detect.
Reproducibility and intermediate precision of the high throughput UMAG
The inter-day precision and assay reproducibility is shown in Fig. 9. Two antibodies, mAb A and mAb H were independently analyzed by 2 analysts on 3 different dates. Similar results were obtained by these analysts and the assay inter-day precision was high with low variability (Fig. 9). The differences of glycoprofiles between the 2 different antibodies were clearly demonstrated by both of the analysts.
Figure 9.
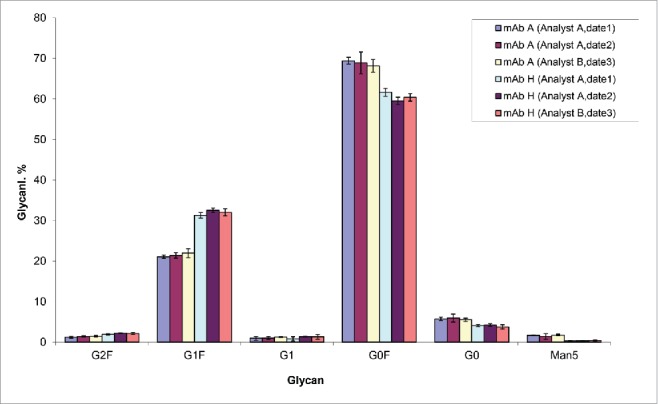
Reproducibility and intermediate precision of the high throughput UMAG. mAb A and mAb H biosimilar samples were analyzed. The test (n=5) was performed by 2 analysts on 3 different dates.
Discussion
A high throughput strategy is particularly desirable for assays that are needed to assess the properties vital for the molecule's function, but require a long assay time.30,31 Glycoprofiling is critical to understand antibody structure-activity relationships, to assess novel drug candidates in early development, and to ensure product comparability and consistency during the commercial product manufacture. For biosimilar antibodies, demonstration that the glycan profiles are equivalent or similar to the reference product is required for regulatory approval. A large number of cell culture process conditions need to be screened, and the antibody glycoprofiles must be analyzed prior to establishing the manufacture process. However, due to the long sample preparation time and high reagent cost, the conventional 2-AB glycan analytical methods are not ideal for handling large numbers of cell culture samples (Fig. 10).
We thus developed two ultrafast MALDI methods for analysis of glycan profile (UMAG) based on glycopeptides that were derived from the antibody following trypsin digestion in either organic solvent or aqueous buffer. Rather than requiring the purified glycan moieties, as is the case for the traditional 2-AB labeling HILIC-HPLC method, both UMAG methods utilize a protocol for the rapid generation and purification of glycopeptides, followed by detection with MALDI-TOF MS. The methods were ultrafast and able to generate similar glycan profiles as the conventional 2-AB HILIC-HPLC/UPLC method, and both can potentially be adapted to high throughput analysis in 96-well plates.
Of the two UMAG methods, we considered the aqueous UMAG to be superior to the organic UMAG because it has several advantages (Fig. 10). This procedure incorporates an ultrafast trypsin digestion protocol that we developed, and can be completed readily within 15 minutes, compared to 130 minutes for the organic UMAG. In addition, the aqueous UMAG allows easier adaption to high throughput analysis in a 96-well PCR plate than the organic UMAG. A plate of 96 samples can be analyzed within 2.5 hours by a proficient analyst, with an average of less than 2 min per sample. This reflects a significant reduction of the assay time compared to the conventional 2-AB HILIC-HPLC. Since the digestion efficiency could vary with different substrate proteins, the incubation time with trypsin might need to be determined case-by-case in UMAG. However, 5 min was sufficient to generate consistent results for most mAbs we tested. In comparing reagent costs, we found the UMAG to be highly cost effective (<$0.30 per sample vs ∼$150 per sample for 2-AB HILIC-HPLC). Also, the procedure can be readily adapted to automation by using TECAN or other automatic liquid handlers, and it is particularly useful for analyzing low volume cell culture samples because a small amount of the antibody is required compared to the 2-AB HILIC-HPLC method. The aqueous UMAG procedure was tested and worked equally well on antibodies with different glycopeptides, e.g., TKPREEQYNSTYR, TKPREEQFNSTYR, and TKPREEQFNSTFR, representing glycosylation sites for IgG1, IgG2, and IgG4, respectively.
In UMAG, the ionization efficiency of different glycopeptides generated from the identical glycosylation site is largely dependent on the same peptide and less on the attached glycans. Low ionization/detection bias for glycopeptides in MALDI-TOF MS was observed in comparison to electrospray ionization MS.44 Therefore, the ion peak area of glycopeptides can be used to establish a relative abundance profile of N-glycans present in a mAb digest. The glycoprofiles from our UMAG are thus generally in good agreement with those determined by 2-AB HILIC-HPLC/UPLC methods, although there are minor differences in the absolute percentage of certain glycan species. We evaluated the high throughput aqueous UMAG by analyzing a variety of antibody samples, from different mAbs, clones, production batches, and originator antibody batches and biosimilar samples. The method, albeit ultrafast and simple, is able to capture the same trends in the changes (differences) among different samples as those detected by the 2-AB HILIC-HPLC/UPLC method. Moreover, the quantitation of the glycans in mAbs by UMAG is reproducible, with high intermediate precision by different analysts. Therefore, we believe that our method is highly valuable for screening cell culture conditions and batch variation, in particular when large numbers of samples are involved. In addition, the method allows fast identification of unknown peaks that the conventional method cannot identify. Aqueous UMAG was able to quickly detect and identify the new forms of high mannose glycans in mAbs, when a mannosidase inhibitor was present in the cell culture medium. UMAG was also successfully used to analyze the glycan profiles on Fc fragment of the Fc-fusion proteins (data not shown).
The major shortcoming of UMAG is that glycopeptides with sialylated glycans cannot quantitatively be analyzed since these acidic glycans may dissociate in-source and post-source during the reflector mode in TOF detection,45 and are also chemically labile under the acidic conditions used during the purification of glycopeptides, a common characteristic associated with MALDI-TOF MS-based methods. As the sialylated glycans are present in most recombinant mAbs at a very low level (<1 %), this shortcoming of UMAG is expected to have a minimal impact. In summary, the UMAG method provides a valuable addition to the tool kit for glycan characterization and profiling of mAbs for cell culture process development, in-process sample analysis, production batch comparability studies, and biosimilar drug evaluation. This method is also applicable for characterization of glycoproteins other than mAbs.
Materials and methods
Chemicals and materials
Sequence grade modified trypsin (cat # V5113) was purchased from Promega.
Ammonia bicarbonate, urea, and dithiothreitol (DTT) were purchased from Sigma. ACN, TFA, formic acid, CaCl2 and a stock solution of Tris (1M, pH 8.0 at room temperature) were purchased from Fisher Scientific. The MALDI matrix α-cyano-4-hydroxycinnamic acid (CHCA) was from Sigma/Fluka. Matrix dihydrobenzoic acid (DHB) was from Applied Biosystems. PNGase F (cat# P0705L) was from New England Biolabs. External calibration standards were from Sigma including angiotensin II (1,045.5423 Da), Bradykinin fragment 1-7(756.3997 Da), P14R (1,532.8582 Da), ACTH fragment 18-39 (2,464.1989 Da), insulin chain B oxidized (3,493.6513 Da).
Ziptips (C18, cat ZTC18S096) was purchased from Millipore, and ZIC-HILIC Proteastips (Cat# SP120-24) was purchased from Proteas BioScience.
Purification of antibodies
mAbs A, B, D, and H were produced in Chinese hamster ovary cells and purified by Protein A chromatography or purified by the 2 or 3 column purification processes. mAb A was in 10 mM citrate, pH 5.6, mAb B was provided in Tris and acetate, pH 5.6. mAbs D and H were kept in His/Arg buffer, pH 5.6. The concentrations of antibodies were determined by UV absorbance at 280 nm.
Digestion of mAbs in organic solvents
Twenty μg of antibody in 2 μl was mixed with 398 μl of the organic trypsin digestion buffer (80% ACN) in 10 mM Tris, and 2 mM CaCl2, pH 8.0; trypsin (0.4 μg) at a 1:40 enzyme/substrate ratio was then added and the resulting solution was incubated at 37°C for 1 hour. The digest was dried in a speed Vac for 30 min to 1 hour. The dried residue was dissolved in 10 μl 0.1% TFA and subjected to purification as below.
Digestion of mAbs in aqueous solutions
To compare the efficiency of trypsin digestion, and the ensuing purification of the glycopeptides, the antibody was also subjected to trypsin digestion in aqueous buffer. First, 10-15 μg of the antibody in a total volume of 1-10 μl, minimally purified by Protein A column, was fully mixed, and denatured in 10 μl of 8 M urea in 20 mM ammonium bicarbonate (NH4)2HCO3, pH 7.8, containing 10 mM DTT at 70°C for 3 min. After denaturing, an aliquot (100 μl) of trypsin in 20 mM (NH4)2HCO3, pH 7.8, was added at 1:30 enzyme/substrate ratio to the antibody solution, fully mixed, and incubated at 37°C for 5 min. The enzymatic reaction was terminated by adding 1.0 μL of 10% TFA and glycopeptides were purified as follows. For high throughput analysis, samples were treated as above in 96-well PCR plates (ABI optical PCR plates) using a PCR cycler (Gene Amp PCR) or a water bath.
RP-HPLC
The digested sample was separated on a HALO ES-C18, 2.1X150 mm, 2.7 μm (Part No.: 92122-702) column at 0.4 mL/min at 60°C. The peptides were eluted by following gradients: 98% mobile phase A (0.1% TFA in H2O) and 2% mobile phase B (100% ACN and 0.1% TFA) for 1 min, then by increasing mobile phase B 10% to 18% in 14 min, then from 18% to 40% in 30 min, and then to 45% in 5 min. In final wash step, mobile phase B was increased from 45% to 88% in 3 min and then held at 88% for 4 min. The column was equilibrated by running 2% mobile phase B for 8 min. The auto-sampler was kept at 5°C. UV absorbance was set at 214 nm.
Purification of glycopeptides with C18 Ziptips
Glycopeptides from the trypsin digestion in organic or aqueous solution were enriched with C18 Ziptips. The tips were preconditioned by pipetting 3 times with 20 μl 100% ACN /0.1% TFA, followed by washing with 3 × 20 ul of 0.1% TFA. Peptides were loaded onto the Ziptips by pipetting the peptide solution up and down for at least 3 times. The tips were washed 3 times with 20 μl of 0.1% TFA. The glycopeptides were then eluted with 10 μl of 9% ACN/0.1% TFA by pipetting up and down 3 times. For samples in PCR plates, a multichannel pipette was used for manual sample handling or a liquid handling system can be incorporated for automated sample handling.
Purification of glycopeptides with HILIC tips
The enrichment of glycopeptides was also assessed with HILIC tips (from Proteas) alone or together with C18 Ziptips. The peptide mixtures after digestion or after C18 Ziptip purification were loaded onto the conditioned HILIC tips by pipetting up and down at least 5 times, and washed 3 times with 80% ACN/0.5% TFA or 2% formic acid, and then glycopeptides were eluted with 10 μl of 2% formic acid.
Treatment of glycopeptides with PNGase F
The purified glycopeptides were dried by air flow, and resuspended in 10 μl of 1x PNGase F buffer (50 mM sodium phosphate, pH 7.5) and treated with 0.5 μl PNGase F (500,000 units/ml) at 37°C overnight. PNGase-treated glycopeptides were analyzed by MALDI-TOF MS as described below.
MALDI-TOF MS of peptides and glycopeptides
Following elution, 2 μl of purified glycopeptides were directly spotted onto the MALDI metal plate (Opti-TOF® 384 well MALDI plate,123x81mm, or 96-well 123x81 mm plate from Applied Biosystems), and air-dried at room temperature. Then 1.5 μl of matrix mixture (10 mg/ml each of CHCA and DHB in 70% ACN and 0.1% TFA) was spotted onto each sample well. After crystallization at room temperature, MS analysis was performed with ABI 4800 Plus MALDI-TOF/TOF analyzer (Applied Biosystems, Foster City, USA), equipped with on-axis laser radiation. Measurements were made in the positive ion reflector mode with the target voltage at 20 kV, first grid voltage at 80% of the target voltage, and delayed extraction at 600 ns. The MS spectra over a range of m/z 2,200–4,200 for different N-glycopeptides were acquired by submitting each spot to multiple laser shots (500-625) at an instrument voltage of 4150 V. All shots were summed per spectrum. Spectra are calibrated externally with calibration standards or internally with glycopeptides of known masses.
Processing of MS data and quantification using mass software
The quantification of the glycopeptides was done with integrated cluster mass peak areas for each glycopeptide using the analysis tool of the Spot Manager provided with the MS instrument. The mass of each glycopeptide was entered in the analysis tool, and the cluster peak area for every sample in the spot set was extracted and output into an Excel spreadsheet. The percentage of glycopeptides was calculated from the sum of cluster peak areas using the automated Excel template.
Glycan analysis by 2-AB HILIC-HPLC or HILIC-UPLC
For comparison, the antibody samples were also analyzed by 2-AB HILIC-HPLC or HILIC-UPLC. The basic method is described as below. The materials required for the assay, including N-Glycanase-Plus, GlycoClean R cartridge column, 2-AB- labeling kit, S cartridge column, and GlycoSep N column were purchased from Prozyme.
Release of glycans from monoclonal antibodies
An aliquot (10 μl) of sample (˜35-40 mg/ml) was deglycosylated in 80 μl of Tris Reaction buffer by incubating with 2 μl of N-Glycanase-Plus at 37°C overnight. Released glycans were purified by chromatography on GlycoClean R cartridge column. The R cartridge was primed with methanol (2 ml) and equilibrated with 5% acetic acid (6 ml). The sample was loaded onto the cartridge, washed with 5% acetic acid 3 times (1 ml each), and the eluent was collected. The collected eluent was dried in a vacuum centrifuge for ∼6 hours and stored at 4°C until used for labeling.
Labeling of glycan with 2-AB- labeling kit
Purified dry glycan samples were labeled with 2-AB- labeling kit from Prozyme. The acetic acid/DMSO/2-AB dye mixture was prepared according to manufacturer's instructions, and added to a vial of reductant (sodium cyanoborohydride, 6 mg). Each dried glycan sample was dissolved in an aliquot (5 μl) of labeling reagent and incubated at 65°C in a heating block for 3 hours. Labeled glycans were cleaned to remove unreacted dye and salts by gravity-fed chromatography on the S cartridge column. The eluates were collected and, dried by vacuum centrifugation for ∼6 hours, re-suspended in purified water (100 μl) and transferred to HPLC vials for analysis.
Analysis of glycans with HILIC HPLC or UPLC
Purified 2-AB labeled glycans were resolved by HILIC HPLC on a GlycoSep N Column using a Waters 2695 HPLC system with a Waters 2475 Multi-fluorescence detector (excitation at 330 nm and emission at 420 nm), or on Acquity UPLC Glycan BEH Amide Column (130À 17 um, 21x100mm) by a Waters UPLC Acquity system. Solvent A was ACN, and solvent B was 50 mM ammonium formate at pH 4.4. In HILIC-HPLC, the labeled glycans were eluted at 0.4 ml/min with a gradient of 35% to 50% solvent B within 70 min, followed by 100% solvent B for 10 min. The column was then equilibrated with 35% solvent B for 30 min before next injection. In HILIC-UPLC, the glycans were eluted at 0.4 ml/min with a gradient of 28-38% solvent B within 30 min, followed by 100% solvent B for 5 min.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
Authors want to thank Dr. Steve Farrand and Dr. Susan Cannon-Carlson for their support, and Dr. Robin Ehrick and Dr. Yan-Hui Liu for reviewing and discussion of the paper.
References
- 1.Mirgorodskaya E, Krogh TN, Roepstorff P. Characterization of protein glycosylation by MALDI-TOFMS. Methods Mol Biol 2000; 146:273-92; PMID:10948508; http://dx.doi.org/ 10.1385/1-59259-045-4:273 [DOI] [PubMed] [Google Scholar]
- 2.Janin-Bussat M-C, Wagner-Rousset E, Klinguer-Hamour C, Corvaia N, van Dorsselaer A, Beck A. Antibody Glycans Characterization In: Kontermann R, Du··bel S, eds. Antibody Enigineering Springer-Verlag; Berlin Heidelberg, 2010; 635-56. [Google Scholar]
- 3.Corfield AP, Berry M. Glycan variation and evolution in the eukaryotes. Trends Biochem Sci 2015; 40:351-9; PMID:26002999; http://dx.doi.org/ 10.1016/j.tibs.2015.04.004 [DOI] [PubMed] [Google Scholar]
- 4.Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol 2007; 25:21-50; PMID:17029568; http://dx.doi.org/ 10.1146/annurev.immunol.25.022106.141702 [DOI] [PubMed] [Google Scholar]
- 5.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem 1985; 54:631-64; PMID:3896128; http://dx.doi.org/ 10.1146/annurev.bi.54.070185.003215 [DOI] [PubMed] [Google Scholar]
- 6.Shakin-Eshleman SH, Remaley AT, Eshleman JR, Wunner WH, Spitalnik SL. N-linked glycosylation of rabies virus glycoprotein. Individual sequons differ in their glycosylation efficiencies and influence on cell surface expression. J Biol Chem 1992; 267:10690-8; PMID:1587845 [PubMed] [Google Scholar]
- 7.van Kooyk Y, Rabinovich GA. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol 2008; 9:593-601; PMID:18490910; http://dx.doi.org/ 10.1038/ni.f.203 [DOI] [PubMed] [Google Scholar]
- 8.Crispin M. Breaking the allergic response by disrupting antibody glycosylation. J Exp Med 2015; 212:433; PMID:25847971; http://dx.doi.org/ 10.1084/jem.2124insight2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyons JJ, Milner JD, Rosenzweig SD. Glycans Instructing Immunity: The Emerging Role of Altered Glycosylation in Clinical Immunology. Front Pediatr 2015; 3:54; PMID:26125015; http://dx.doi.org/ 10.3389/fped.2015.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB. Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: a critical review. J Autoimmun 2015; 57:1-13; PMID:25578468; http://dx.doi.org/ 10.1016/j.jaut.2014.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog 2005; 21:11-6; PMID:15903235; http://dx.doi.org/ 10.1021/bp040016j [DOI] [PubMed] [Google Scholar]
- 12.Beck A, Wagner-Rousset E, Bussat MC, Lokteff M, Klinguer-Hamour C, Haeuw JF, Goetsch L, Wurch T, Van Dorsselaer A, Corvaïa N. Trends in glycosylation, glycoanalysis and glycoengineering of therapeutic antibodies and Fc-fusion proteins. Curr Pharm Biotechnol 2008; 9:482-501; PMID:19075687; http://dx.doi.org/ 10.2174/138920108786786411 [DOI] [PubMed] [Google Scholar]
- 13.Raju TS, Scallon BJ. Glycosylation in the Fc domain of IgG increases resistance to proteolytic cleavage by papain. Biochem Biophys Res Commun 2006; 341:797-803; PMID:16442075; http://dx.doi.org/ 10.1016/j.bbrc.2006.01.030 [DOI] [PubMed] [Google Scholar]
- 14.Liu L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and fc-fusion proteins. J Pharm Sci 2015; 104:1866-84; PMID:25872915; http://dx.doi.org/ 10.1002/jps.24444 [DOI] [PubMed] [Google Scholar]
- 15.Elliott S, Lorenzini T, Asher S, Aoki K, Brankow D, Buck L, Busse L, Chang D, Fuller J, Grant J, et al.. Enhancement of therapeutic protein in vivo activities through glycoengineering. Nat Biotechnol 2003; 21:414-21; PMID:12612588; http://dx.doi.org/ 10.1038/nbt799 [DOI] [PubMed] [Google Scholar]
- 16.Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nature reviews Drug discovery 2009; 8:226-34; PMID:19247305; http://dx.doi.org/ 10.1038/nrd2804 [DOI] [PubMed] [Google Scholar]
- 17.Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol 2008; 20:471-8; PMID:18606225; http://dx.doi.org/ 10.1016/j.coi.2008.06.007 [DOI] [PubMed] [Google Scholar]
- 18.Hossler P, Khattak SF, Li ZJ. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology 2009; 19:936-49; PMID:19494347; http://dx.doi.org/ 10.1093/glycob/cwp079 [DOI] [PubMed] [Google Scholar]
- 19.Freeze HH, Aebi M. Altered glycan structures: the molecular basis of congenital disorders of glycosylation. Curr Opin Struct Biol 2005; 15:490-8; PMID:16154350; http://dx.doi.org/ 10.1016/j.sbi.2005.08.010 [DOI] [PubMed] [Google Scholar]
- 20.Marquardt T, Denecke J. Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 2003; 162:359-79; PMID:12756558; http://dx.doi.org/ 10.1007/s00431-002-1136-0 [DOI] [PubMed] [Google Scholar]
- 21.Muntoni F, Torelli S, Brockington M. Muscular dystrophies due to glycosylation defects. Neurotherapeutics 2008; 5:627-32; PMID:19019316; http://dx.doi.org/ 10.1016/j.nurt.2008.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lefebvre T, Guinez C, Dehennaut V, Beseme-Dekeyser O, Morelle W, Michalski JC. Does O-GlcNAc play a role in neurodegenerative diseases? Expert review of proteomics 2005; 2:265-75; PMID:15892570; http://dx.doi.org/ 10.1586/14789450.2.2.265 [DOI] [PubMed] [Google Scholar]
- 23.Dennis JW, Granovsky M, Warren CE. Protein glycosylation in development and disease. Bioessays 1999; 21:412-21; PMID:10376012; http://dx.doi.org/ 10.1002/(SICI)1521-1878(199905)21:5%3c412::AID-BIES8%3e3.0.CO;2-5 [DOI] [PubMed] [Google Scholar]
- 24.Lowe JB, Marth JD. A genetic approach to Mammalian glycan function. Annu Rev Biochem 2003; 72:643-91; PMID:12676797; http://dx.doi.org/ 10.1146/annurev.biochem.72.121801.161809 [DOI] [PubMed] [Google Scholar]
- 25.Chaiyawat P, Netsirisawan P, Svasti J, Champattanachai V. Aberrant O-GlcNAcylated Proteins: New Perspectives in Breast and Colorectal Cancer. Front Endocrinol (Lausanne) 2014; 5:193; PMID:25426101; http://dx.doi.org/ 10.3389/fendo.2014.00193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dingjan T, Spendlove I, Durrant LG, Scott AM, Yuriev E, Ramsland PA. Structural biology of antibody recognition of carbohydrate epitopes and potential uses for targeted cancer immunotherapies. Mol Immunol 2015; 67(2 Pt A):75-88; PMID:25757815 [DOI] [PubMed] [Google Scholar]
- 27.Reusch D, Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology 2015; 25:1325-34; PMID:26263923; http://dx.doi.org/ 10.1093/glycob/cwv065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolarich D, Lepenies B, Seeberger PH. Glycomics, glycoproteomics and the immune system. Curr Opin Chem Biol 2012; 16:214-20; PMID:22221852; http://dx.doi.org/ 10.1016/j.cbpa.2011.12.006 [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Balog CI, Stavenhagen K, Koeleman CA, Scherer HU, Selman MH, Deelder AM, Huizinga TW, Toes RE, Wuhrer M. Fc-glycosylation of IgG1 is modulated by B-cell stimuli. Mol Cell Proteomics 2011; 10:M110 004655; PMID:AMBIGUOUS; http"//dx.doi.org/ 10.1074/mcp.M110.004655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shubhakar A, Reiding KR, Gardner RA, Spencer DI, Fernandes DL, Wuhrer M. High-Throughput Analysis and Automation for Glycomics Studies. Chromatographia 2015; 78:321-33; PMID:25814696; http://dx.doi.org/ 10.1007/s10337-014-2803-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reusch D, Haberger M, Selman MH, Bulau P, Deelder AM, Wuhrer M, Engler N. High-throughput work flow for IgG Fc-glycosylation analysis of technological samples. Anal Biochem 2013; 432:82-9; PMID:23026777; http://dx.doi.org/ 10.1016/j.ab.2012.09.032 [DOI] [PubMed] [Google Scholar]
- 32.O'Neill RA. Enzymatic release of oligosaccharides from glycoproteins for chromatographic and electrophoretic analysis. J Chromatogr A 1996; 720:201-15; PMID:8601190; http://dx.doi.org/ 10.1016/0021-9673(95)00502-1 [DOI] [PubMed] [Google Scholar]
- 33.Tarentino AL, Plummer TH Jr. Enzymatic deglycosylation of asparagine-linked glycans: purification, properties, and specificity of oligosaccharide-cleaving enzymes from Flavobacterium meningosepticum. Methods Enzymol 1994; 230:44-57; PMID:8139511; http://dx.doi.org/ 10.1016/0076-6879(94)30006-2 [DOI] [PubMed] [Google Scholar]
- 34.Huhn C, Selman MH, Ruhaak LR, Deelder AM, Wuhrer M. IgG glycosylation analysis. Proteomics 2009; 9:882-913; PMID:19212958; http://dx.doi.org/ 10.1002/pmic.200800715 [DOI] [PubMed] [Google Scholar]
- 35.Reusch D, Haberger M, Maier B, Maier M, Kloseck R, Zimmermann B, Hook M, Szabo Z, Tep S, Wegstein J, et al.. Comparison of methods for the analysis of therapeutic immunoglobulin G Fc-glycosylation profiles–part 1: separation-based methods. mAbs 2015; 7:167-79; PMID:25524468; http://dx.doi.org/ 10.4161/19420862.2014.986000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woo CM, Iavarone AT, Spiciarich DR, Palaniappan KK, Bertozzi CR. Isotope-targeted glycoproteomics (IsoTaG): a mass-independent platform for intact N- and O-glycopeptide discovery and analysis. Nat Methods 2015; 12:561-7; PMID:25894945; http://dx.doi.org/ 10.1038/nmeth.3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huberty MC, Vath JE, Yu W, Martin SA. Site-specific carbohydrate identification in recombinant proteins using MALD-TOF MS. Anal Chem 1993; 65:2791-800; PMID:8250262; http://dx.doi.org/ 10.1021/ac00068a015 [DOI] [PubMed] [Google Scholar]
- 38.Thaysen-Andersen M, Mysling S, Hojrup P. Site-specific glycoprofiling of N-linked glycopeptides using MALDI-TOF MS: strong correlation between signal strength and glycoform quantities. Anal Chem 2009; 81:3933-43; PMID:19358553; http://dx.doi.org/ 10.1021/ac900231w [DOI] [PubMed] [Google Scholar]
- 39.Papac DI, Wong A, Jones AJ. Analysis of acidic oligosaccharides and glycopeptides by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Chem 1996; 68:3215-23; PMID:8797382; http://dx.doi.org/ 10.1021/ac960324z [DOI] [PubMed] [Google Scholar]
- 40.Strader MB, Tabb DL, Hervey WJ, Pan C, Hurst GB. Efficient and specific trypsin digestion of microgram to nanogram quantities of proteins in organic-aqueous solvent systems. Anal Chem 2006; 78:125-34; PMID:16383319; http://dx.doi.org/ 10.1021/ac051348l [DOI] [PubMed] [Google Scholar]
- 41.Gramer MJ, Eckblad JJ, Donahue R, Brown J, Shultz C, Vickerman K, Priem P, van den Bremer ET, Gerritsen J, van Berkel PH. Modulation of antibody galactosylation through feeding of uridine, manganese chloride, and galactose. Biotechnol Bioeng 2011; 108:1591-602; PMID:21328321; http://dx.doi.org/ 10.1002/bit.23075 [DOI] [PubMed] [Google Scholar]
- 42.Hills AE, Patel A, Boyd P, James DC. Metabolic control of recombinant monoclonal antibody N-glycosylation in GS-NS0 cells. Biotechnol Bioeng 2001; 75:239-51; PMID:11536148; http://dx.doi.org/ 10.1002/bit.10022 [DOI] [PubMed] [Google Scholar]
- 43.Elbein AD, Tropea JE, Mitchell M, Kaushal GP. Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J Biol Chem 1990; 265:15599-605; PMID:2144287 [PubMed] [Google Scholar]
- 44.Stavenhagen K, Hinneburg H, Thaysen-Andersen M, Hartmann L, Varon Silva D, Fuchser J, Kaspar S, Rapp E, Seeberger PH, Kolarich D. Quantitative mapping of glycoprotein micro-heterogeneity and macro-heterogeneity: an evaluation of mass spectrometry signal strengths using synthetic peptides and glycopeptides. J Mass Spectrom 2013; 48:627-39; PMID:23722953; http://dx.doi.org/ 10.1002/jms.3210 [DOI] [PubMed] [Google Scholar]
- 45.Harvey DJ. Structural determination of N-linked glycans by matrix-assisted laser desorption/ionization and electrospray ionization mass spectrometry. Proteomics 2005; 5:1774-86; PMID:15832364; http://dx.doi.org/ 10.1002/pmic.200401248 [DOI] [PubMed] [Google Scholar]