

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is a signalopathy of renal tubular epithelial cells caused by naturally occurring mutations in two distinct genes, polycystic kidney disease 1 (PKD1) and 2 (PKD2). Genetic variants in PKD1, which encodes the polycystin-1 (PC-1) protein, remain the predominant factor associated with the pathogenesis of nearly two-thirds of all patients diagnosed with PKD. Although the relationship between defective PC-1 with renal cystic disease initiation and progression remains to be fully elucidated, there are numerous clinical studies that have focused upon the control of effector systems involving heterotrimeric G protein regulation. A major regulator in the activation state of heterotrimeric G proteins are G protein-coupled receptors (GPCRs), which are defined by their seven transmembrane-spanning regions. PC-1 has been considered to function as an unconventional GPCR, but the mechanisms by which PC-1 controls signal processing, magnitude, or trafficking through heterotrimeric G proteins remains to be fully known. The diversity of heterotrimeric G protein signaling in PKD is further complicated by the presence of non-GPCR proteins in the membrane or cytoplasm that also modulate the functional state of heterotrimeric G proteins within the cell. Moreover, PC-1 abnormalities promote changes in hormonal systems that ultimately interact with distinct GPCRs in the kidney to potentially amplify or antagonize signaling output from PC-1. This review will focus upon the canonical and noncanonical signaling pathways that have been described in PKD with specific emphasis on which heterotrimeric G proteins are involved in the pathological reorganization of the tubular epithelial cell architecture to exacerbate renal cystogenic pathways.
Keywords: heterotrimeric G proteins, kidney, signal transduction, polycystic kidney disease, accessory proteins
polycystic kidney disease (PKD) remains one of the most common inherited disorders, which can lead to a progressive loss in renal function (52–54). Under normal conditions, the kidney possesses a multitude of functions, including its primary roles as a filtration system to eliminate waste products from the blood, regulation in long-term blood pressure by controlling fluid and electrolyte balance, acid-base regulation, and production of endocrine hormones, such as erythropoietin, renin and active forms of vitamin D (148). In PKD, however, numerous variants in either the polycystic kidney disease 1 (PKD1) or polycystic kidney disease 2 (PKD2) gene, which are the two primary genes associated with causing the autosomal dominant form of PKD (or ADPKD), can negatively impact the normal function of the kidney by pathologically altering the architecture of the kidney through a transformative process that changes the tubular epithelial cells, largely in the collecting ducts, into fluid-filled sacs known as cysts (52, 54). The mechanisms by which cyst initiation and formation occur due to the malfunctioning gene products of PKD1 and PKD2, known as polycystin-1 (PC-1) and polycystin-2 (PC-2), respectively, remain to be fully described (60).
There is considerable evidence that this pathological switch in the tubular epithelial cell to form cysts is quite complex and involves a cascade of diverse changes, including missorting of proteins to incorrect cellular locations (5), misalignment of cell orientation (42), hypersecretion of fluids (53), reorganization of the extracellular matrix (190), apoptosis (49), and abnormal induction in cell proliferation (90). Numerous orthologous and nonorthologous rodent models of PKD have been generated to further elucidate the importance of these above described and possible other novel modes of action in vivo (55, 60, 116). At the present time, however, a major focus of many preclinical and numerous clinical trials is the importance of neutralizing heterotrimeric G protein-dependent signalopathies by either activating or inactivating cell surface G protein-coupled receptors (GPCRs) to control cystic disease progression (53, 59, 60, 174, 190).
Heterotrimeric G proteins consist of three distinct subunits, α, β, and γ, which are regulated through canonical interactions with cell surface GPCRs (189) or noncanonical associations with membrane tyrosine kinase receptors or cytoplasmic accessory proteins (127). In the classic model of GPCR signaling, ligand binding induces a conformational change in the guanine nucleotide exchange factor domain of the receptor facilitating the transfer of GTP onto the inactive Gα-GDP subunit. This exchange promoted the activation of Gα-GTP, which is the main signaling complex evaluated during cyst formation and progression in PKD. Although unbound Gβγ has been largely observed as an ancillary byproduct to Gα-subunit activation, its role as a regulator of signaling output, particularly on ion channel activity, cannot be completely ignored (31, 32, 86). In the end, G protein signaling is terminated by the reassociation of unbound Gβγ with inactivated Gα-GDP subunits.
A growing body of evidence has demonstrated that the regulation of heterotrimeric G protein activation is more diverse than previously anticipated. This was attributed to the distinct renal localization pattern for each G protein subunit expression, the combination of Gαβγ-complexes that actually exist, and the GPCR subtypes that are expressed within each cell type (127, 128). Moreover, intracellular proteins have been identified to intercede at the level of the receptor-ligand complex through unique modes of action to compete for either Gα or Gβγ binding and modulate the termination of second messenger-mediated signaling. In addition, these accessory or adaptor proteins could also perturb the magnitude of the cellular response, protein trafficking, cell orientation, and many other biological processes that would be relevant to the pathogenesis of cystic disease (127, 128). Alternatively, heterotrimeric G proteins could also be involved in an integrated cross-talk network by functioning as intermediary signal processors between traditional GPCR and other unitary transmembrane tyrosine kinase receptors (105).
In the majority of the studies describing GPCR-based signalopathies in PKD, the involvement of heterotrimeric G proteins is not generally discussed. A major reason may be that many investigators assume that the specific heterotrimeric G protein αβγ-complexes with their cognate GPCR have already been well documented. However, as new data continue to emerge regarding unexpected interactions by heterotrimeric G proteins or the identification of novel signaling pathways outside of the canonical ligand-GPCR systems, further investigations into the role of heterotrimeric G proteins are warranted to confirm their role as key signal modulators in pathways associated with cystic disease progression. With this in mind, we will provide a comprehensive overview by which heterotrimeric G proteins play a pivotal role in canonical GPCR pathways, as well as unconventional modes of action, including those regulated by PC-1, during the cystogenic process in PKD. In some cases, we will ascribe some modes of action by which heterotrimeric G protein subunits have been described in other biological systems to potentially provide important new insight into heterotrimeric G protein signal modulation in PKD.
POLYCYSTIN-1
PC-1 has been categorized as an atypical GPCR and has shown the capability to transmit physical cues from the external milieu into the cell by converting this information into a biochemical signaling output (118, 126, 192). The mechanosensation mediated by PC-1 has been postulated to be associated with its cellular localization within or at the base of the cilia (118, 119). Unlike the standard GPCR, which has a heptahelical transmembrane configuration, PC-1 has been predicted to have 11 transmembrane-spanning regions (68). Similar to all GPCRs, however, PC-1 could selectively bind multiple classes of Gα subunits at a specific stretch of 20 amino acids in the COOH terminus, known as a G protein-binding domain (GBD) (129, 130, 204). In addition, PC-1 has a highly conserved autoproteolytic site, GPCR proteolysis site (GPS), within a GPCR-Autoproteolytic INducing (GAIN) domain located on the NH2-terminal side of the first transmembrane span. The GAIN domain was found in cell adhesion GPCRs to maintain tubular epithelial cell morphology (3). In fact, loss of the GPS in PC-1 could structurally reorganize the cell toward a highly aggressive form of renal cystogenesis (202), but the importance of PC-1 targeting to the cilia remains to be resolved (19, 159). These findings provide some insight that PC-1 has the properties of a ligand-independent GPCR, yet the physiological implication to directly regulate heterotrimeric G protein signaling to maintain normal tubular architecture and function remains to be fully understood.
G Protein Subunit Binding to PC-1 COOH Terminus
As mentioned above, a number of Gα-subunits, most notably Gα12 and Gαi/o, have been shown to interact with PC1 and PC1-like proteins (31, 32, 129, 130, 203–205) (Fig. 1). Initially, Parnell et al. (129, 130) demonstrated that heterotrimeric G proteins with select Gα-subunits, namely Gαi/o, Gαs, and Gα12/13, interact with the COOH-terminal tail of PC-1. The efficiency of the Gα-subunit binding to PC-1 was dependent upon a stretch of 74 amino acids, which contained the GBD flanked by conserved tyrosine and serine phosphorylation motifs (129). Complete abolition of G protein binding was only observed when the serine phosphorylation motif was also removed in conjunction with the GBD (129). Subsequently, Yu et al. (203, 204) confirmed a selective interaction between PC-1 and Gα12, but not its closely related Gα13 (203), and that other components of the COOH-terminal region were partially required to maintain wild-type binding of Gα12 to PC-1 (204).
Fig. 1.
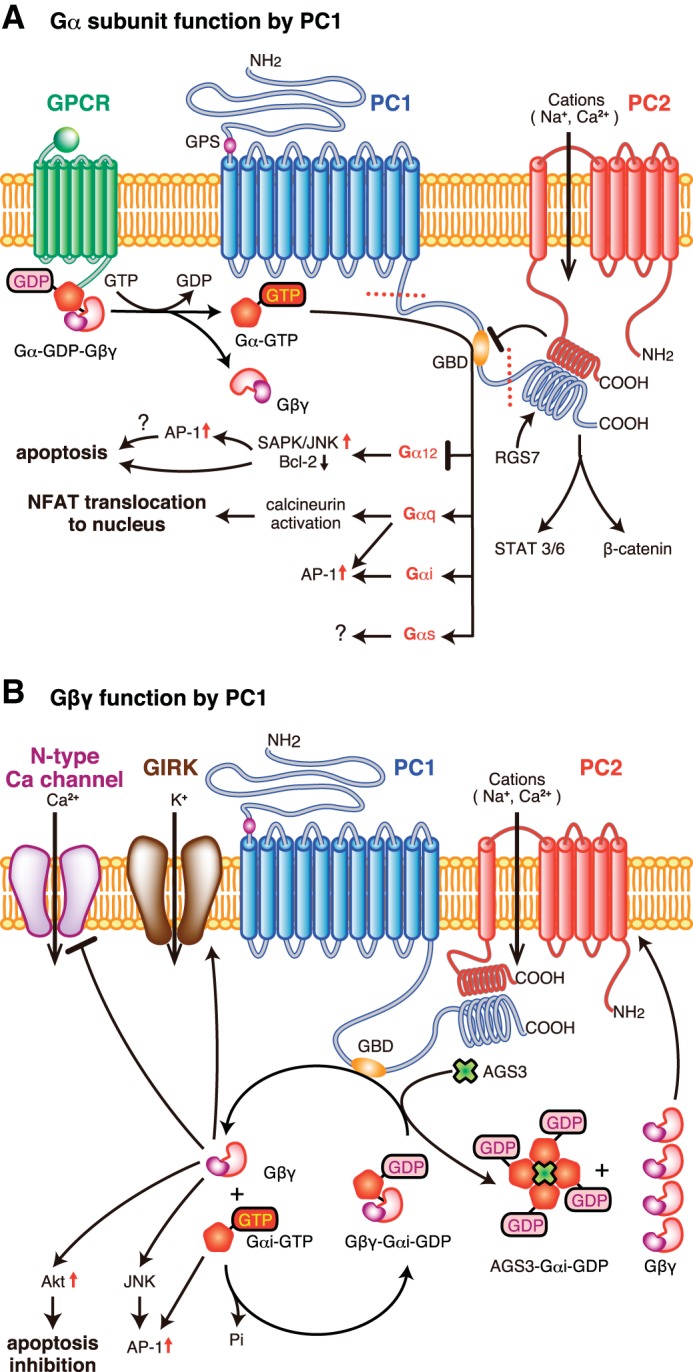
Heterotrimeric G protein regulation by polycystin-1. Polycystin-1 (PC-1; in blue) is embedded within the plasma membrane due to its 11 transmembrane-spanning regions. G protein binding and activation are regulated by a region known as GBD in the COOH terminus end of PC-1 in the cytoplasm and could regulate Gα (A) or Gβγ signaling (B). A: the GBD could interact with multiple classes of GTP-bound Gα-subunits (Gα12, Gα, Gαs, and Gα) activated by heterologous GPCRs (such as thrombin receptors) or interacting with inactivated forms of Gα-GDP. PC-1 could interact with Gα12 to block apoptosis, possibly through the activation of AP-1. Gαq signaling could activate calcineurin to promote NFAT translocation to the nucleus. Other Gα-subunits could activate AP-1 activity. The role of Gαs activation by PC-1 is not fully described but has been shown to interact directly with PC-1. PC-1 could also interact directly with other proteins, PC-2 and RGS7, at the coiled coil domain. The COOH-terminal cytoplasmic tail has 2 cleavage sites (denoted by the dotted line in the COOH-terminal tail of PC-1) to generate either a small or larger fragment that can be translocated to the nucleus to either activate STAT 3/6 or β-catenin. B: other accessory proteins in the vicinity of PC-1 may intercept G protein subunits, such as AGS3, to promote PC-2 ion channel regulation through a Gβγ-dependent mechanism. Gαi-subunit interaction with PC-1 could prevent apoptosis by activating Akt or activate JNK/AP-1 signaling. Gβγ-signaling mediated by PC-1 could negatively control N-type calcium channel and activate GIRK channels. AGS3, activator of G protein signaling 3; GBD, G protein-binding domain; GPCR, G protein-coupled receptor; GPS, G protein-coupled receptor proteolytic site; NFAT, nuclear factor of activated T cells; PC1, polycystin 1; PC2, polycystin 2; RGS7, regulator of G protein signaling 7; SAPK/JNK, stress activated protein kinase/Jun amino terminal kinase.
In addition to binding heterotrimeric G protein subunits, the GBD catalyzes the degradation of GTP to GDP nucleotides bound on Gαi-subunits (129). Although these data were suggestive that PC-1 may act similar to conventional GPCRs, it was not known whether the inactivated Gα-GDP remained bound to PC-1. A subsequent study, however, demonstrated that mutant Gα12-subunits bound to either GTP or GDP are capable of binding to PC-1 (203, 204). From these studies, PC-1 may be predicted not only to activate heterotrimeric G proteins, but also to behave as a scaffold to form protein complexes to control downstream signaling. Additional studies are clearly needed to determine whether the activation status (i.e., GTP/GDP bound form) of each distinct Gα-subunit plays a crucial role in the efficient binding to PC-1. Regardless, the COOH-terminal tail of PC-1 has shown flexibility in its ability to interact with multiple classes of Gα-subtypes, which leads to complementary or differing modes of signal processing and/or effect. To date, heterotrimeric G protein interaction with PC-1 has been described to control downstream signal pathway activation or directly modulate ion channel activity (Fig. 1).
JNK Signaling: Role of Gαi- and Gα12-Subunits
Initial experiments by Parnell et al. (130) demonstrated that c-Jun NH2-terminal kinase (JNK) activation is mediated by interaction of Gαi-subunits with the COOH-terminal tail of PC-1, but that the effector was unbound Gβγ. Subsequent experiments showed that the activation of JNK cannot be solely attributed to Gαi-subunits but involves other classes of Gα-subunits, since blockade with pertussis toxin inhibited only ∼25% of the PC-1-mediated JNK signaling (130). In tubular epithelial cells, G protein α12-subunits promote apoptosis by activating JNK and enhance Bcl-2 degradation. PC-1 interferes with the activation of apoptotic signaling by directly binding to Gα12 (204). Even under conditions of reduced by Gα12 binding to PC-1, the inhibition of apoptotic signaling is maintained. Moreover, constitutively active Gα12, QLα12, is able to bind to PC-1, and that mutant Gα12 can uncouple the interactions between QLα12 and PC-1 to prevent apoptotic signaling (204). These results suggest that Gα12 can bind to PC-1 in either the active GTP-bound or inactive GDP-bound form.
Gαi-Subunits: Ion Channels and Other Signaling Pathways
Antiapoptotic signaling was also observed following Gαi-subunit interaction with PC-1, which activates Akt through a Gβγ-PI-3K pathway (11). In addition, Gαi-subunits are capable of activating AP-1 activity, but this activation is at least twofold less potent than Gαq- or Gα12/13-subunits (130). The intermediary steps between Gα-subunit activation to the induction of AP-1 is not known, but there may be a link with JNK activation (130).
To date, however, the role of Gαi/o subunits with PC-1 has been described as a key regulator of ion channels through a Gβγ-dependent mechanism. Under basal conditions, overexpression of PC-1 exerts an inhibitory effect on N-type Ca2+ channels, while promoting G protein-coupled inward rectifying potassium channel activity (31, 32). Genetic and pharmacological approaches have demonstrated that the ion channel regulation can be attributed to the COOH-terminal region of PC-1 to bind Gαi/o-subunits to liberate unbound Gβγ (31, 32, 46). However, the role of PC-1 to control ion channel activity by Gβγ is not universal and is dependent upon heterologous proteins that bind to the coiled coil domain in the COOH-terminal tail of PC-1, which is located near the GBD (129). PC-2, which is also known as the transient receptor protein P2 ion channel (176), has been shown to have a direct relationship with PC-1 at the coiled coil domain (57, 135, 195). The role of PC-2 within the heteromeric PC-1/PC-2 complex was found to block G protein activation by PC-1, and the loss of the COOH-terminal region of PC-2 is responsible for restoring the ability of PC-1 to modulate ion channels by activating Gβγ (31, 32). The positive influence by which PC-1 activates PC-2 ion channel activity is not dependent upon G protein signaling, but rather upon the physical association between the COOH-terminal tails of PC-1 and PC-2 (31, 46, 57, 195).
Gαq-Signaling
In neurons, the role of Gαq-signaling in the presence or absence of murine PC-1 did not appear to play a role in ion channel activity, since there was no marked change in the endogenous activity of the M-type K+ channel (32). In a subsequent study, the role of the PC-1/Gαq-complex was shown to be involved in the activation of other signaling pathways unrelated to ion channel regulation. Puri et al. (134) demonstrated a synergistic role for Gαq to enhance the PC-1 activation of calcineurin and nuclear accumulation of nuclear factor of activated T cells in HEK293T cells. Gαq-subunits are also involved with promoting AP-1 activity in the presence of PC-1, similar in magnitude with Gα12/13-subunits (130). Since transgenic overexpression of PC-1 can also perturb its normal physiological function to promote renal cyst formation (168), the contrasting results regarding the role of Gαq-activation with respect to PC-1 is likely due to the differences in the experimental design (endogenous vs. transfected overexpression of Gαq), cell lines studied (sympathetic neurons vs. HEK293T), and experimental readout (electrophysiological measurement of ion channels vs. signal transduction pathway analysis). Further studies using Pkd1-deficient or hypomorph cells are needed to confirm whether Gαq signaling is actually regulated by PC-1 in renal tubular epithelial cells.
Accessory Proteins
The regulation of G protein subunit binding and downstream signaling by PC-1 may be modulated by the presence of heterologous proteins. Similar to PC-2, proteins could directly bind to specific regions near the GBD in PC-1 to interfere with G protein signaling, possibly by affecting G protein interaction. Alternatively, proteins may localize near PC-1 and interfere with G protein signaling elicited by PC-1 by binding to G protein subunits during the activation/inactivation cycle. Unlike PC-2, not all proteins that bind to the COOH-terminal end of PC-1 can alter G protein signaling. Most notably, regulator of G protein signaling 7 (RGS7), which is a member of a large family of accessory proteins that directly binds to Gα-subunits, exhibits a physical protein-protein interaction in the coiled coil domain of PC-1 (76). Other than slowing the degradation of RGS7 (76), the RGS7/PC-1 complex was not determined to alter the ability by PC-1 to control ion channel activity (32). Mutant forms of Gαi/o-subunits [i.e., GαoA (G184S) or Gαi1 (G183S)], which were designed to prevent binding to the RGS domain of RGS7, did not change PC-1-dependent regulation in Ca2+ channel activity (32). Because of these findings, the authors concluded that RGS7 is not directly involved with disrupting the PC-1/heterotrimeric G protein signaling axis (32), but further studies may be warranted to substantiate this conclusion. Several RGS proteins, including RGS7, have been shown to preferentially bind to Gβ5 rather than Gαo-GDP complexes (92). Regardless of the presence of pertussis toxin, an inhibitor of Gαi/o protein activity, RGS7 could interact with Gβ5 and the deactivation of the mutant Gαi/o could be based entirely upon its intrinsic GAP function. Therefore, the lack of regulation on ion channel activity may be masked by the preference of G protein subunits binding to RGS7 within a particular cell type. It is also important to note that the absence of the whole or even a partial fragment of the coiled coil domain is sufficient to markedly reduce G protein α-subunit binding to PC-1 (129, 204). This could suggest that heterologous protein binding at the coiled coil domain does not negatively impact, but may positively recruit, G proteins to PC-1 for activation. To date, however, there have been no published data demonstrating that protein interaction in the COOH terminus of PC-1 could modify Gα-subunit binding to regulate G protein signaling. Further studies are needed to clearly elucidate the role of G protein subunit binding and activation using mutant PC-1 with only mutations in the GBD, and not deletions in the entire PC-1 COOH-terminal region.
As mentioned above, other accessory proteins may indirectly control PC-1-dependent G protein signaling by interacting with the activated G protein subunits, and not with PC-1 itself. One example is activator of G protein signaling 3 (AGS3), which could bind multiple inactive Gαi/o-GDP-subunits and function as a guanine dinucleotide dissociation inhibitor. The liberation of free Gβγ-dimers was shown to induce heteromeric PC-1/PC-2 ion channel activity, but not with PC-2 expression alone (86). No definitive study has shown a direct protein-protein interaction between AGS3 and PC-1, but a recent study by Yeh et al. (200) clearly localized AGS3 to the basal body/centrosome region of the cilia, a common site of PC-1 expression. If this spatial localization for AGS3 is similarly observed in renal epithelial cells, then AGS3 could be in close proximity to the same pool of Gαi/o-subunits that interact with PC-1 and would be available to modulate downstream effector signaling, including the direct control of PC-2 ion channel activity through unbound Gβγ-dimers (86).
Cleavage and Nuclear Localization of PC-1 COOH-terminal Tail
In addition, the importance of G protein binding on PC-1 and its effects on the efficiency of posttranslation cleavage of its COOH-terminal tail to produce two distinct smaller protein fragments, ∼35 kDa product containing the entire intracellular COOH-terminal tail (24, 87) or ∼16 kDa product (p112) (99), remain to be determined. These truncated COOH-terminal fragments could translocate to the nucleus to promote gene expression by interacting with either β-catenin or STAT-3/-6, respectively (99, 186, 187). The extent of the cleavage of the COOH-terminal tail depends upon the expression of PC-2 and changes in intracellular calcium (11). Of these two PC-1 fragments, the larger COOH-terminal tail portion may likely contain the GBD, which could be involved in the process of cleavage efficiency, nuclear translocation, and possibly transcriptional activation.
For these reasons, to fully appreciate the physiological importance of G protein interaction with PC-1 to control normal tubular epithelial cell function, genetic constructs expressing mutant versions of PC-1 in which the GBD has been modified or removed to prevent G protein binding need to be evaluated. These additional studies would provide direct evidence regarding the functional consequences of PC-1 to bind and initiate G protein signaling to promote cardiovascular abnormalities and cystic disease development.
DYSREGULATION OF HETEROTRIMERIC G PROTEIN-DEPENDENT SIGNAL TRANSDUCTION PATHWAYS IN PKD
Loss of polycystin activity could dysregulate numerous hormonal systems that are dependent upon heterotrimeric G protein regulation in both vascular and tubular epithelial cells in the kidney (Fig. 2). To date, the gradual temporal changes in the activation or repression of signaling pathways during the early and late phases of cystic disease progression remain to be fully described. In nearly all cases, heterotrimeric G protein signaling following ligand binding to a specific GPCR has focused upon the role of the Gα-subunit, with less information known about Gβγ, in tubular epithelial and vascular cell function. In this section, we provide an overview for some of the candidate GPCR pathways that have been described in vascular and tubular epithelial cells in PKD and elucidate the cellular and/or functional changes in cardiovascular and renal function using data from animal models and humans with PKD.
Fig. 2.

Schematic overview of heterotrimeric G protein signalopathy in renal tubular epithelial and vascular cells in polycystic kidney disease. Heterotrimeric G proteins play diverse physiological roles either canonically to transmit extracellular stimuli into the cell following GPCR stimulation or noncanonically through recently identified accessory proteins. There are 4 distinct classes of Gα subunits, Gαs, Gαi, Gαq/11, and Gα12/13, which are depicted in this schematic, except for Gα12 (described in Fig. 1). 1) Cystic tubular epithelial cells. Gαs activation by multiple GPCR, including V2R, EP2/EP4, and β1/β2, could increase cAMP production and activate downstream proliferative pathways, which include Raf/MEK/ERK1/2, or other cellular functions, such as water and electrolyte balance. ANG II stimulation of the AT1R could also increase cAMP, but the mechanism of action is not known. Conversely, Gαi functions to antagonize cAMP production by activating SSTR or EP4 but could also regulate pathways mediated by Smo or FzR that control cytoskeleton remodeling, cell migration, and ion channel activity (shown in Fig. 1). Gαq activation by B2R, ETB, and AT1R could activate PLC-dependent signaling cascades, which could increase intracellular calcium or activate DAG to promote PKC function. In some cases, B2R and AT1R could also promote ERK 1/2 signaling by cross talk with EGFR. ETB inhibits cAMP production through increased production of PGE2, which is mediated by PKC. 2) Vascular cells. In smooth muscle cells, Gαq activation by the AT1R or ETA/ETB could promote vasoconstriction, which could be counteracted by opposing AT2R-Gαq vasodilation signaling pathways in the same smooth muscle cells. Alternatively, B2R or ETB stimulation of Gαq signaling in endothelial cells or neighboring tubular epithelial cells could produce paracrine factors, such as nitric oxide or prostaglandins, which could traverse to the smooth muscle cells and modulate tone. 3) Polarity and mitotic spindles. In some situations, Gαi-subunits could be involved in the process of epithelial cell polarity and mitotic spindle orientation. There is evidence that accessory proteins from the activator of G protein signaling family, namely AGS3 and AGS5/LGN, could be involved in this process. ACE, angiotensin converting enzyme; AC6, adenylate cyclase 6; AGS3, activator of G protein signaling 3; ANG I, angiotensin I; aPKC, atypical protein kinase C; AQP2, aquaporin 2; ANG II, angiotensin II; AT1R, angiotensin II receptor type 1; AT2R, angiotensin II receptor type 2; AVP, arginine vasopressin; A3AR, A3 adenosine receptor; B2R, bradykinin B2 receptor; CaMKII, calcium/calmodulin-dependent protein kinase II; CFTR, cystic fibrosis transmembrane conductance regulator; cGMP, cyclic guanosine monophosphate; COX1/2, cyclooxygenase 1/2; DAAM-1, dishevelled associated activator of morphogenesis 1; DAG, diacylglycerol; Dvl, disheveled; EGFR, epidermal growth factor receptor; Epac, exchange protein directly activated by cAMP; EP2, prostaglandin E2 receptor 2; EP4, prostaglandin E2 receptor 4; ER, endoplasmic reticulum; Erk1/2, extracellular signal regulated kinase 1/2; ET-1, endothelin-1, ETA, endothelin type A receptor; ETB, endothelin type B receptor, FzR, frizzled receptor; Gli2/3, glioma-associated oncogene 2/3; GSK-3, glycogen synthase kinase 3; IP3, inositol triphosphate; JNK, c-jun N-terminal kinase; LGN, leucine-glycine-asparagine repeat protein; MEK, MAPK/ERK kinase; mInsc, mammalian homolog of Inscuteable; NHE, Na/H exchanger; NO, nitric oxide; NuMA, nuclear mitotic apparatus; PC1, polycystin-1; PC2, polycystin-2; PGE2, prostaglandin E2; PI3-K, phosphoinositide 3 kinase; PKA, protein kinase A; PKC, protein kinase C; PLA2, phospholipase A2; PLC, phospholipase C; PTCH, Patched; Rock, Rho kinase; SST, somatostatin; SSTR, somatostatin receptor; UT, urea transporter; SMO, Smoothened; V2R, vasopressin type 2 receptor.
cAMP Regulation by Adenylyl Cyclase and Calcium: Role of Gαi-, Gαs-, and Gαq-Subunits
Renal tubular epithelial cells from animal models and humans with PKD1 or PKD2 mutations exhibit elevated production of cAMP (102); molecular strategies targeting the control of adenylyl cyclase activity have been a major focus in drug development for PKD treatment. Gαs (stimulatory) and Gαi (inhibitory) were shown to be positive and negative regulators of adenylyl cyclase activity, respectively. In the nephron, numerous isoforms of adenylyl cyclase are present, particularly adenylyl cyclase isoforms 3, 4, and 6 (AC6), but to date, only AC6 has been shown to play a crucial role in cystic disease progression (137). Under normal conditions, increased cAMP decreases the rate of tubular epithelial cell proliferation by activating the protein kinase A/c-Raf pathway (197). In cystic epithelial cells, however, increased cAMP (197) or decreased intracellular calcium (198) could switch kinase activation from c-Raf (Raf-1) to B-Raf kinase and lead to abnormal activation of the MEK/ERK1/2 signaling (Fig. 3). This would result in increased tubular epithelial cell proliferation, increased fluid secretion, and/or decreased tubulogenesis.
Fig. 3.
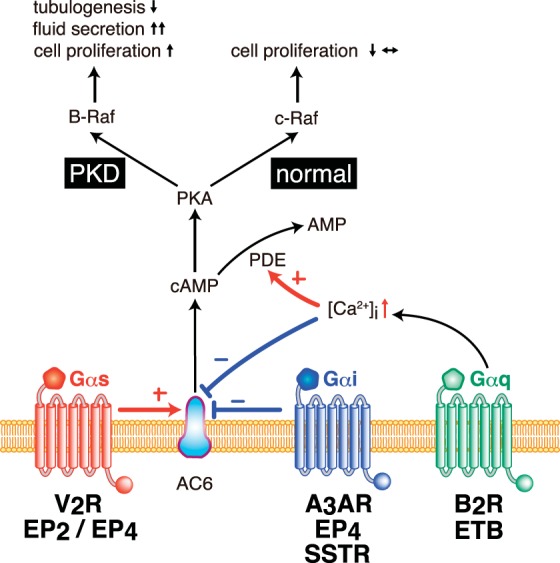
Overview of cAMP regulation by distinct heterotrimeric G protein subunits in renal tubular epithelial cells. Intracellular levels of cAMP have been shown to be regulated by an integration of numerous GPCR pathways in renal tubular epithelial cells. Adenylyl cyclase type 6 (AC6) could be either directly activated by GPCR/Gαs or inhibited by GPCR/Gαi. In addition, GPCR/Gαq signaling could stimulate intracellular calcium, which could be involved in reducing AC6 activity or accelerating cAMP catalysis by phosphodiesterase (PDE) activation. Upon changes in cAMP levels, protein kinase A (PKA)-dependent phosphorylation of Raf kinase has been shown to be involved in controlling distinct cellular functions, particularly cell proliferation. In normal renal tubular epithelial cells, c-Raf (Raf-1) kinase activates MEK/ERK1/2 signaling to prevent or reduce the activation of cell proliferative pathways. In cystic PKD epithelial cells, PKA has been shown to activate B-Raf kinase to promote increased cell proliferation, increased fluid secretion, and reduced tubulogenesis. V2R, vasopressin V2 receptor; EP2/EP4, E-prostanoid 2 or 4 receptor; A3AR, adenosine A3 receptor; SSTR, somatostatin receptor; B2R, bradykinin type 2 receptor; ETB, endothelin type B receptor; +, activate; −, inhibit.
At present, there are several GPCR systems directly associated with controlling cAMP levels through either Gαi or Gαs, including arginine vasopressin receptors, somatostatin receptor (SSTR) subtypes, β-adrenergic receptors, adenosine receptors, and E-prostanoid receptor subtypes. In addition, strategies to reduce cAMP levels by increasing intracellular calcium levels through Gαq-activation has been investigated (Figs. 2 and 3). Increased Gαq-activation by GPCR stimulation could promote either accelerated cAMP catabolism through phosphodiesterase (PDE) activation or inhibition of adenylyl cyclases (60).
Arginine vasopressin receptors.
In the kidney, arginine vasopressin (AVP) was shown to have a dual role in modulating renal blood flow distribution and control the urine-concentrating mechanism through interactions with two distinct types of receptors, vasopressin V1a (V1aR) and V2 receptors (V2R) (98, 124, 125). In the context of PKD, the primary therapeutic target of the AVP system has been to prevent or disrupt V2R activation. Mechanistically, V2R activation promotes Gαs dissociation from Gβγ to stimulate adenylyl cyclase activity and subsequently increase cAMP production. Recent data would suggest that AVP stimulation activates AC6 as the primary effector system to produce cAMP (137), and not AC3 or AC4 (77, 78). The V2R is primarily expressed in the distal tubular epithelial cells (98), and its localization is consistent with its functional role to control the urine concentration process. Changes in the cAMP levels directly alter the function of the Na+/K+/2 Cl− cotransporter in the thick ascending limb of Henle and also control the number of aquaporin-2 (AQP2) water channels and urea transporters inserted into the apical membrane in the principal cells of the collecting duct (109, 208). In cystic epithelial cells, the increased levels of cAMP are primarily through V2R activation in the basolateral membrane, but there was evidence that the V2R could also be partially misrouted to the apical membrane (144). On the basis of these findings, selective V2R antagonists, such as Tolvaptan, have been developed as a treatment strategy to attenuate cystic disease progression by preventing the activation of the Gαs subunits (61, 171, 173).
As described earlier, alterations in intracellular calcium levels could also play a role in regulating cAMP levels. In terms of the AVP system, the V1aR has been shown to activate Ca2+-dependent pathways through Gαq-subunits, and this change in calcium modulates the V2R-dependent cAMP production in renal tubular epithelial cells. Moreover, the V1aR could selectively reduce renal medullary blood flow by vasoconstriction of the renal vasculature. This would minimize solute wash-out in the renal medulla and, thereby, maximize urine concentration (43, 44). Since polydipsia has been observed as a side effect due to chronic V2R blockade in patients with ADPKD (61, 171), there may be some potential benefit if a partial antagonist to the V2R is employed to block cAMP production through the Gαs pathway, which also has modest V1aR agonist ability to activate Ca2+ pathways in both the tubules and blood vessels. However, there are caveats to this proposed logic, since the vascular sensitivity to AVP in PKD kidneys remains largely unknown and selective V1aR stimulation has been shown to promote persistent elevations in systemic blood pressure (28, 82) or lead to inappropriate activation of procystic hormones, such as PGE2 (see next section). It is also important to note that intracellular calcium levels in PKD epithelial cells are lower than in noncystic epithelial cells, so the counterregulatory systems that would be involved in reducing cAMP through V1aR are likely inferior to the AVP/V2R/Gαs axis. To date, however, the role of this V1aR pathway in PKD remains to be investigated, and the primary mode of action by AVP is through the actions of the V2R.
Prostaglandin E2 receptors.
Prostaglandin E2 (PGE2) has been implicated as a causative factor in cystic disease by a number of observations, including its accumulated levels in cyst fluid, ability to promote cAMP production and chloride secretion, and induction of cyst growth in Madin-Darby canine kidney (MDCK) epithelial cells (199). PGE2 exerts its biological effects by binding to one of four distinct GPCR subtypes known as the E-prostanoid receptors (EP1-4) (201), but the primary receptors are likely EP4 (40, 95) and possibly EP2 (41). PGE2 binding to EP4 activates Gαs-subunits to induce cAMP production in murine IMCD3 and also PC-1-deficient collecting duct cell lines (40, 95). There is also evidence in human cystic epithelial cells that EP2 may also play a role in the cAMP response (41). In some cells, PGE2 could also activate Gαi-subunits to inhibit adenylyl cyclase through EP4 stimulation (201). Alternatively, there is evidence that PGE2 synthesis could be positively regulated by AVP through the V1aR in either the interstitial medullary cells or cortical collecting ducts (15), and this could synergize the production of cAMP. These latter findings may partially explain why the magnitude of cAMP induction by PGE2 in cultured cells from ADPKD and ARPKD cysts is markedly less robust compared with AVP (8). Consistent with other hormones activating cAMP/PKA in cystic epithelial cells, PGE2 activates the conventional B-Raf/MEK/ERK1/2 pathway. In addition, PGE2 could mediate cAMP-dependent activation of membrane-associated exchange protein 1/2 (Epac1/2) (201). Although the role of Epac1/2 in the cystic kidney needs further study, there is evidence that the cAMP-Epac pathway promotes cyst growth by hyperproliferation in liver cells (6).
These studies demonstrate that there may be a synergistic role to synthesize PGE2 by other procystic hormones and that PGE2 could subsequently promote increased cAMP-dependent procystic signaling through the activation of distinct G protein subunits.
Adrenergic receptors.
In general, adrenergic receptor activation controls long-term blood pressure through a number of distinct modes, including fluid and electrolyte balance, renal blood flow distribution, and renin production (27, 35). Hyperactivity of the sympathetic nervous system has been shown to be associated with high blood pressure in humans with ADPKD (79). Selective loss of renal nerve activity reduces the kidney-to-body weight ratio, an index of cystic disease progression, in the Han:Sprague-Dawley (SPRD) rat, a nonorthologous model of ADPKD (45). The adrenergic receptor subtypes associated with the sympathetic nervous system are categorized as α and β and are distributed in various vascular and tubular epithelial cells. Blockade of the β1/β2-receptor subtypes, which are involved in Gαs-activation of adenylyl cyclase, does not improve renal function as measured by microalbuminuria and glomerular filtration rate in ADPKD patients (180, 206). Renal tubular epithelial cell expression of β1- and β2-receptor subtypes are localized throughout the nephron from the proximal tubules to the collecting ducts (161, 196). Incubation with isoproterenol, a nonselective β1/β2-receptor agonist, could induce cAMP production in microdissected distal tubules and cortical collecting ducts (111, 149) and correlate with mitogenesis in renal tubular epithelial cells (193). The adrenergic α2-receptor has been shown to inhibit adenylyl cyclase through Gαi-activation, but its role has not been evaluated in PKD.
At this time, the role of the adrenergic receptor system as it relates to the pathogenesis of cyst progression in PKD remains to be fully determined. In fact, there is recent evidence that deficits in renal function in ADPKD patients may be preceded by extrarenal pathologies to the heart, since the β-adrenergic system has been shown to be perturbed in terms of its ability to express calcium handling proteins (84). Further studies are needed to better understand the importance of the adrenergic receptors in PKD.
Somatostatin receptors.
The somatostatin receptors (SSTR) represent another set of GPCR that could activate the inhibitory Gαi-subunits to reduce adenylyl cyclase activity and decrease cAMP production. In mice, rat and human kidneys, there are five known types of SSTR, and their distribution could be localized to each segment of the nephron, although the collecting duct is the most conserved segment to express SSTR2 (7, 12). Under normal conditions, somatostatin could inhibit renal tubular phosphate transports in both humans and rats (136, 177, 182) and partially antagonize the vasopressin V2 receptor-mediated changes in water transport by reducing cAMP production (191). In the cystic epithelia of PKD patients, this pathway is believed to be involved in the dysregulation of tubular epithelial cell function and promotes the transformation to a cystic phenotype. There is evidence that SSTR3 is associated with the cilia (71), but for therapeutic purposes, short peptide somatostatin analogs with longer circulating half-lives have been developed, such as octreotide, lanreotide, and pasireotide, which could have their own distinct SSTR-binding affinity (58, 103). Currently, these SSTR agonists are widely evaluated in clinical trials as potential anticystic agents to attenuate cystic disease progression in PKD (21, 67). Although there have been some promising results to slow cystic disease in the liver, the kidney has exhibited somewhat varied responses to SSTR agonists (20, 21, 66, 67, 181). To further clarify the role of the SSTR system as an anticystic therapy for the kidney, there is an on-going clinical trial to focus on the effects of lanreotide on improving renal function in ADPKD patients (104).
However, the differences in the biological effect of somatostatin agonists in the liver vs. kidney need further investigation but may be associated with the differential abundance and pattern of expression of SSTR and its associated adenylyl cyclase partners within the nephron. In cystic cholangiocytes, SSTR1 and SSTR2 expression is markedly reduced in cystic cholangiocytes compared with normal cells and that chronic stimulation with either octreotide or pasireotide results in a compensatory increase in the receptor levels for SSTR1 and SSTR2 similar to levels observed in normal cells (103). Similar types of studies will need to be performed in PKD kidneys to confirm whether the use of SSTR agonists could augment the magnitude of adenylyl cyclase inhibition by upregulating receptor expression and/or localization to slow renal cystic disease progression.
Adenosine receptors.
Adenosine is a nucleoside that has been shown to play an important role in the control of renal function by balancing blood flow with the constant energy demands in the kidney (156, 179). Catabolism of cAMP, ATP, and AMP could lead to conversion into adenosine, which could activate one of four distinct GPCR, known as adenosine receptor A1 (A1AR), A2AAR, A2BAR, and A3AR (179). In PC-1-deficient cells, a selective induction of the A3AR relative to the other AR isoforms has been detected (1). A3AR reduces cAMP production by activating Gαi-subunits and coincides with reduced cystic epithelial cell numbers following stimulation of the A3AR using a selective agonist (1). Similar to other pathways that control adenylyl cyclase, phosphorylation of ERK1/2 is decreased in the presence of an A3AR agonist, which suggests that PKA is involved in stimulating Raf/MEK/ERK signaling. Alternatively, A3AR may stimulate phospholipase Cβ (PLC) to reduce ERK1/2 activation via PI3K/Akt signaling (106), which could also involve either Gαq or possibly unbound Gβγ-dimers. Further studies will be required to determine the relative importance of adenosine on cAMP production compared with the other adenylyl cyclase-regulating hormonal systems.
In all, these studies demonstrate that multiple hormonal systems integrate into the regulation of the cAMP system and that the use of a single inhibitor to control cAMP production would be compromised by the multifactorial input of numerous hormones.
Gαq and Calcium Regulation
Although the list of interacting signaling pathways expands for each G protein subunit, Gαq-subunits are classically known to be directly involved in regulating intracellular calcium levels by activating PLC (147). The pathways involved in Gαq-signaling are illustrated in Fig. 2. PLC catabolized phosphatidylinositol-4,5-bisphosphate (PIP2) into two products, inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 traverses to the sarcoplasmic reticulum to stimulate calcium channel activity and promote the movement of the calcium into the cytoplasm. DAG activates protein kinase C to negatively affect the activation of Gαi-subunits. Unconventionally, Gαq could also promote signaling cross talk between GPCR and the phosphorylation of growth factor receptors to activate downstream signaling pathways (29). In this section, we focus upon the role of Gαq in the renin-angiotensin, kallikrein-kinin, endothelin, and EGFR systems.
Renin-angiotensin system.
The renin-angiotensin system (RAS) remains one of the best-characterized hormonal systems in the field of cardio-renal biology, and its role in blood pressure regulation and renal cystic disease progression in PKD continues to be an area of intense investigation (88).
The effector hormone angiotensin II (ANG II) is generated by a proteolytic cascade initiated by the cleavage of the precursor protein, angiotensinogen, which is an α2-globulin formed primarily in the liver and, to a lesser extent, in the kidney. Angiotensinogen is subsequently catabolized by the endopeptidase renin to form angiotensin I (ANG I). Renin is synthesized and released into the circulation from the juxtaglomerular apparatus in the kidney and can be regulated in part by the adrenergic β1/Gαs/cAMP pathway (35). ANG I is predominantly converted to ANG II by angiotensin-converting enzyme (ACE), but chymase can also perform this function to a lesser degree. At this point, ANG II exhibits multifunctional effects due to its ability to interact with two distinct GPCR subtypes, AT1 (AT1R) and AT2 (AT2R), which activate a diverse set of Gα-subunits throughout the kidney, including the vasculature, tubular epithelial cells of the nephron, and interstitial cells (96, 97, 108). In fact, all of the other components of RAS, including angiotensinogen, renin, ACE, and ANG II, are produced in the kidney (96, 97). Moreover, there is evidence in polycystic kidney disease (PCK) rat kidneys and biopsied human ADPKD kidneys that various components of the RAS, particularly renin, are elevated compared with their noncystic counterparts (50, 51, 172).
In PKD, investigators studying the biological functions of ANG II have focused almost entirely upon elucidating the mechanisms involved in blood pressure regulation and its impact on renal cyst progression. ANG II has potent vasoconstrictor effects in the systemic circulation via the AT1R/Gαq/11 pathways (69, 108). In the kidney, ANG II could also promote Na+/H+ exchange to enhance reabsorption of sodium and water by activating apical AT1R in the proximal tubules (69). Since the etiology associated with hypertension in humans with PKD remains largely undetermined (75, 88, 96), the primary treatment modality has been to administer inhibitors to either angiotensin converting enzymes (ACEI) or antagonists to angiotensin receptors (ARB), particularly the AT1R (23). Unfortunately, the recently completed HALT-PKD trial was unable to prove definitively that cystic disease progression was slowed in either early (150) and late-stage (170) ADPKD patients following treatment of either ACEI (Lisinopril) alone or in combination with an ARB (telmistartan). Similarly, rodent models of PKD have shown only modest to little efficacy on cystic disease progression following blockade of the RAS with Lisinopril, even with reduced blood pressures (70, 143). Interestingly, a selective reduction in angiotensinogen levels using antisense oligonucleotide technology did markedly attenuate the increase in cyst area within the Pkd1 null kidneys with only a mild reduction in systolic blood pressure compared with the effects of Lisinopril (143). From these studies, it appears the RAS may play a direct mitogenic role in tubular epithelial cells, but additional studies are needed to dissect the direct ANG II-mediated tubular effects from those changes attributed to physical factors, such as blood pressure and renal blood and tubular luminal flow. These future studies will likely require the evaluation of subpressor doses of ANG II or servo-controlled renal perfusion pressure in the presence of ANG II in rodent models of PKD to determine whether renal cystic disease progression can be modulated by RAS.
Regardless, there are other recent studies that have described an effect by the RAS on tubular epithelial cells with defective polycystin or ciliary proteins. In human ADPKD cystic epithelial cells, defective PC-1 function negatively affected the shear stress-increased transient response to cytoplasmic Ca2+ concentration. However, the effect of ANG II was essentially retained at near normal Ca2+ responses as control normal kidney cells (194), which would suggest that the receptor localization and signaling pathways associated with the ANG II/AT1R axis is essentially intact during cystic disease progression. In cilia-deficient collecting duct epithelial cells, ANG II stimulation of the apical AT1R was also able to increase cAMP production significantly, similar in magnitude as the costimulation of both apical and basolateral AT1R activation (142). Previous studies focusing on urine-concentrating mechanisms have observed that there is a cooperative effect on cAMP production between ANG II and AVP on their respective AT1R and V2R, respectively (89, 157). A genetic loss of the AT1R in collecting duct reduces the maximal urine-concentrating capacity through decreased production of AQP2 water channels (157) and insertion of AQP2 into the apical membrane (89). To date, it is unclear how ANG II/AT1R activates a calcium-dependent pathway to increase cAMP production, but it may involve the activation of alternate subtypes of adenylyl cyclase, such as AC3 and AC5. However, neither of those AC subtypes in the collecting ducts affects urine concentration under basal conditions (78, 138). Alternatively, AT1R may also activate other downstream signaling pathways by interacting with Gα12/α13 (48, 141, 162, 178) or even directly stimulating the Raf/MEK/ERK1/2 pathway (169, 185). It may be possible that the AT2R, which is not well described in PKD, may need to be evaluated before we fully comprehend the importance of AT1R and its associated heterotrimeric G protein signaling in PKD. The AT2R has been shown to oppose the AT1R-mediated vasoconstrictor effects exerted by promoting the production of bradykinin and exaggerate the vasodilator response of smooth muscle cells (153). Additionally, AT2R could modulate other AT1R functions by regulating sodium excretion (153), cell growth, and anti-inflammatory pathways (33, 34).
These studies clearly demonstrate that the RAS could activate a diverse signaling network, which interacts with a number of other hormonal systems to dysregulate blood pressure and potentially regulate a network of signaling pathways that could lead to changes in tubular epithelial cell architecture and function. Further studies are needed to isolate systematically each potential signaling partner and identify how they alter the expression profile, localization, and functional effects of both AT1R and AT2R in renal cells during the cystogenic process.
Kallikrein-kinin system.
Under normal conditions, the primary functions of bradykinin are involved in regulating vascular tone and sodium and water balance in the kidney (72). The biological role of the kallikrein-kinin system (KKS) needs further exploration, especially since the catabolism of active bradykinin is regulated by ACE (also known as kininase II), which is the same enzyme involved in the production of ANG II (72). The KKS in PKD has not aroused much interest, since one study found was no detectable change in the KKS (16), although this finding was observed only in a small group of human ADPKD patients. However, in the heterozygous Han:SPRD rats, a nonorthologous model of ADPKD, urinary kallikrein and bradykinin levels were actually detected at higher levels compared with age-matched wild-type controls (16, 17).
Bradykinin elicits its biological function through the activation of one or both receptor subtypes, B1 (B1R) and B2 (B2R) (110). Both receptor subtypes have been identified in collecting duct epithelial cells in embryonic (39) and postnatal Oak Ridge polycystic kidney (Orpk) mouse kidneys (18). Functionally, blockade of the B2R reduces proteinuria and albuminuria in the Han:SPRD rat (17), similar to the findings observed with ACE inhibition (17, 37, 38, 75). In mice, the loss of the B2R did not lead to abnormal renal architecture or function, but high salt intake during the period of kidney development led to tubular dilation. In some cases, the formation of cystic cells was described in the kidneys (39). The signaling pathways perturbed in KKS during cyst formation remain largely unknown. Generally, the B2R interacts with Gαq-subunits to promote PLC signaling and increase intracellular Ca2+, which could reduce adenylyl cyclase activity or increase production of nitric oxide/cGMP second messengers to reduce vascular tone (110). Alternatively, the B2R could be involved in the control of proliferation in collecting duct cells through the transactivation of the EGFR/ERK1/2 signaling axis by Gαq-subunits (112).
Although there is a paucity of data regarding the dysregulation of the KKS in PKD, further exploration using mice that are genetically deficient of bradykinin receptor subtypes would be essential to obtain an unequivocal answer to the question of whether the KKS is a fundamentally important hormone that modulates cystic signaling pathways.
Endothelin receptors.
Endothelin (ET)-1 exerts potent vasoconstriction in the systemic and renal circulation and promotes natriuresis and diuresis by the kidneys (30). Over the years, evidence has accumulated that endothelin, specifically ET-1, plays a role in blood pressure regulation and cystic disease progression in PKD (63–65, 107, 113, 117, 123, 183). In hypertensive ADPKD patients, plasma ET-1 levels are elevated compared with healthy patients (107, 183), and endothelin has been detected in cyst fluid (113). Moreover, renal ET-1 mRNA (117) and protein content (64, 65) has been detected at a higher level in rodent models of ADPKD, and ET-1 production is localized to renal cystic epithelial cells in mice, rats, and humans (63, 65, 123) and in human vascular smooth muscle cells (123). Intriguingly, the transgenic overexpression of ET-1 in mice results in the formation of cysts in the kidney (63).
The biological actions by ET-1 are elicited by its ability to bind and activate distinct receptor subtypes, ETA and ETB (4, 145). ETA mRNA and protein are localized to vascular smooth muscle cells in renal blood vessels and glomeruli (167, 188) and protein expression in the distal tubules and collecting duct cells (188). ETB is expressed in the endothelial cell part of the blood vessels (188) and epithelial cells in the proximal tubule (188) and inner medullary collecting ducts (167, 188). The renal ETA and ETB mRNA (117), protein levels (123), or ligand binding (64) has been shown to be either higher or lower depending upon the PKD species being investigated. Stimulation of ETA and ETB could activate a number of G protein α-subunit partners, including Gαq, Gα12/13, and Gαi, depending on the cell type and the context of its activation (30). As part of the global signaling network in Fig. 2, the most common ETA/ETB signaling is illustrated in epithelial and vascular cells. More descriptive signaling pathways for renal ETA/ETB can be found elsewhere (80). Functionally, receptor blockade of the endothelin receptors to attenuate cystic disease progression has resulted in conflicting results. Pharmacological blockade of ETB increased renal cAMP levels, which was associated with accelerated cystic disease progression (22). In addition, ETB blockade enhanced urine concentration, which suggested that there may be an integration of signaling pathways with the V2R/Gαs-axis. This observation may be consistent with an earlier study that showed ETB inhibited cAMP production by a Gαi-dependent mechanism, possibly through increased production of PGE2 (81). Interestingly, concomitant blockade of ETA receptors could prevent the pathogenic effects mediated by the ETB receptor (22). Conversely, receptor antagonism of the ETA, and not ETB, was instrumental in exaggerating tubular epithelial cell proliferation and cystic disease progression in Han:SPRD (cy/+) rats. Similar to the mouse study, coantagonism of ETA and ETB receptors attenuated the effects by ET-1 on cystic disease progression (62). These somewhat contradictory results have suggested that a critical balance between ETA and ETB function may be required to maintain normal kidney structure and function. However, further development in endothelin receptor blockers and also their use in orthologous models of PKD is needed to clarify whether ETA or ETB is the crucial receptor that is involved in the cystic disease process.
Tyrosine kinase receptors.
In PKD, growth factors, including epidermal growth factor (EGF), are well established to interact with single transmembrane-spanning tyrosine kinase receptors, which initiate a process in which homo- or heterodimerization occurs leading to the recruitment of a number of adaptor proteins (13). This results in the subsequent activation of downstream proliferative signaling pathways, including the canonical Raf/MEK/ERK1/2 pathway (164). In this signaling paradigm, EGFR functions as a downstream signaling effector system, which could be transactivated by heterotrimeric G protein subunits, namely Gαq-subunits (29). As described earlier, bradykinin and ANG II are bound to their respective GPCR to promote EGFR phosphorylation (112), and this may be the mode of action mediated by Gαq. It remains to be determined whether the cross talk mediated by Gαq between various GPCR with the EGFR system occurs with basal membrane receptors or the receptors that are mistargeted to the apical membrane, which are known to have a procystic role in PKD (163, 165). In the end, ligand-activated GPCR with the EGFR system likely provides additional pleiotropic signal diversity within the cell to further complicate the therapeutic development to treat PKD.
EMERGING AREAS OF HETEROTRIMERIC G PROTEIN SIGNALING
In this section, we will describe GPCR-dependent and -independent signaling pathways that have been implicated more recently in PKD (Fig. 2). At present, the role of the heterotrimeric G proteins in these pathways has not been fully described other than in nonrenal systems, which provide potential mechanisms of action that may be relevant in the pathogenesis of PKD.
Hedgehog Signaling
Recent evidence has suggested that the Hedgehog (Hh) signaling pathway may function through the regulation of G protein signaling and control cystic disease progression (174, 175). Multiple components of the Hh system, including Patched (Ptch), the putative receptor for Hh ligands, are localized to the cilia (114). In the absence of Hh, ciliary Ptch is inactivated. Upon interaction with Hh ligands, Ptch exits the cilia and removes its repressive effects on Smoothened (Smo), a seven-transmembrane GPCR associated with Gαi-subunits (152). Smo facilitates the transmission of signaling effects initiated by Hh by accumulating in the cilia. This activates a series of signaling events converting full-length glioblastoma (Gli) transcription factors, Gli2 and Gli3, from transcriptional repressors to transcriptional activators for cellular proliferation (192).
In multiple rodent models of PKD, including a Pkd1 mutant mouse, aberrantly high expression of all three Gli subtypes has been detected (175). Blockade of Hh signaling reduces cystic disease progression, but the noncanonical role of Smo-Gαi-signaling has not been studied. In some invertebrate and mammalian cells, the activation of Gli2/3 may (56, 122, 139) or may not (100) be dependent upon the availability of Gαi-dependent signaling. There is evidence that Smo can activate effector targets normally associated with Gαi activation, including PI3-K dependent cell migration (133) and inhibition of adenylyl cyclase to reduce cAMP levels (152). However, blockade of Hh signaling did not affect the activity of CREB, which is a marker for cAMP production, in explanted cystic kidneys, suggesting that the inhibition of Gαi signaling may not be involved (175). Further studies will be required to confirm whether trimeric G protein signaling is a crucial player in Hh signaling in orthologous model of PKD. These findings would suggest that Hh signaling may provide a relationship between ciliary dysfunction with cyst formation.
Wingless/Int1 and Frizzled Receptor
Frizzled (Fz) receptors, which have the structure-function relationship similar to a GPCR, are an integral component for the wingless/int1 (Wnt) signaling pathway (36). At present, 10 different Fz receptors have been identified to bind and process the signaling of various Wnt ligands into the cell, and their general biological activity has been reviewed in greater detail elsewhere (47, 74). In brief, Wnt signaling is generally categorized as either β-catenin dependent (canonical) or β-catenin independent (noncanonical). In canonical Wnt signaling, β-catenin is a major signaling effector, which accumulates and induces transcriptional activation of Wnt target genes. In the noncanonical pathway, Wnt/Fz signaling could stimulate one of two pathways, which are involved in calcium signaling or planar cell polarity pathways (36). The role of heterotrimeric G proteins in the Wnt signaling pathways has not been well described, particularly in the context of PKD, but there is an accumulating body of evidence in nonrenal cells that both canonical and noncanonical Wnt signaling regulates the activation of multiple classes of Gα-subunits, including Gαi, Gαq, Gαs, and Gα12 (93, 120, 154), by interacting with Fz receptors and other Wnt signaling components, disshevelled and axin (2, 94, 158). These G protein-dependent pathways are subsequently involved in various pathways associated with proliferation, cytoskeletal changes, and cell motility (36).
At present, there are an increasing number of studies demonstrating perturbed regulation in Wnt signaling pathways in renal cystic disease (47, 73, 74, 184). Loss of Wnt9b has been shown to disrupt planar cell polarity to promote renal cyst formation through noncanonical signaling (73), whereas a loss of aquaporin-1 expression accelerated cystic disease progression in a mouse model of ADPKD by promoting canonical Wnt/β-catenin signaling (184). However, the role of Wnt signaling to regulate cystogenesis is not universal (160) and may be attributed to the use of different genetic strains in these studies. Alternatively, there is a plethora of Wnt ligands that could interact with numerous types of Fz receptors, so the potential network of diverse intertwining signaling branches could be quite expansive. Therefore, it will be extremely challenging to unravel, particularly if one or more subtypes of heterotrimeric G protein complexes, which have yet to be identified, which are involved in the process of the various extrinsic cues.
Gαi-Subunits in Mitotic Spindle Regulation
Proper orientation of mitotic spindles is a central mechanism that enables tubular epithelial cells to maintain their complement of polarity proteins and complete a round of symmetric cell division (10). Oriented cell division involves tight regulation of mitotic spindle alignment using protein complexes controlling either apico-basal polarity or planar cell polarity. Defective control of the mitotic spindles was believed to be a causative factor in the initiation and progression of cystic kidney disease, but the findings are not universal (42, 73, 101, 121, 131). Moreover, the signaling pathways involved in the misalignment of the mitotic spindles are not well established, even though it was believed to be attributed to dysfunctional planar cell polarity pathway. There are, however, compelling data in MDCK cells that alterations in the localization and composition of apico-basolateral polarity protein complexes could be a critical factor in the orientation of the mitotic spindles (207). In tubular epithelial cells, the alignment of the mitotic spindles is perpendicular to the apico-basal direction (207). To achieve this proper alignment during mitosis, it is essential for the formation of a core tripartite complex composed of: 1) Gαi-subunits of heterotrimeric inhibitory G proteins; 2) Leu-Gly-Asn (LGN), a cytosolic scaffold protein containing GoLoco/G protein regulatory (GPR) motifs; and 3) nuclear mitotic apparatus (NuMA), a nuclear coiled coil microtubule-binding protein. During mitosis, Gαi-subunits are localized to the basolateral cortex, and LGN captures the inactive forms of Gαi-GDP by direct protein interaction at the COOH-terminal GoLoco/GPR motifs in LGN. At this point, nuclear dissolution allows for NuMA translocation to the lateral cortex by binding to the NH2-terminal TPR domains in LGN. Subsequently, dynein is recruited and accumulates at the cell cortex by binding to NuMA, which promotes spindle elongation. LGN plays a central role in maintaining proper epithelial cell architecture following cell division. Loss of LGN expression or a lack of lateral cortex targeting of LGN leads to increased epithelial cells with an abnormal number of lumens that exhibit aberrant tilting of the mitotic spindles in the apico-basal direction (207).
More recent studies demonstrate that LGN could be a pivotal switch between the apico-basal and planar cell polarity complexes to control the orientation of the mitotic spindles (9, 140). LGN has the capability to shift its binding affinity from NuMA in the apico-basal complex to Disks large homolog 1 (Dlg1), which leads to the regulation of the mitotic spindle in the PCP direction. From these studies, it would be difficult to conclude which pathways would be disrupted during the changes in morphogenetic programming in the normal tubular epithelial cell as it transitions to a cystic phenotype.
At present, the localization of LGN in normal and PKD kidneys is thought to be exclusively in distal tubular epithelial cells (91), but its biological role remains largely undefined in vivo. However, a close homolog of LGN, known as activator of G protein signaling 3 (AGS3), has a beneficial role in attenuating cystic disease progression (86). Similar to LGN, AGS3 is normally distributed throughout the cytoplasm but can localize to the spindle poles in the presence of Gαi (115). It remains to be determined whether the same complement of polarity proteins can interchangeably bind to AGS3 as they do for LGN. However, it is clear that heterotrimeric Gαi-subunits could play a pivotal role in the absence of GPCR activation to control mitotic spindle orientation through their interaction with accessory proteins, such as LGN/AGS3.
Gβγ-Signaling
To date, Gβγ-dimers, the native binding partner of Gα-subunits, have not been as well characterized compared with Gα-subunits as regulators of signal transduction pathways in PKD. Gβγ has been shown to have a diverse array of functions within the cell, including the ability to regulate ion channel activity and PI3K activation (26, 155). As described earlier and illustrated in Fig. 1B, full-length PC1 bound to Gαi/o-subunits leads to the regulation of Ca2+ and K+ channels through a Gβγ-dependent mechanism (31, 32). Similarly, AGS3 functions to bind Gαi-subunits and release Gβγ-dimers that increase the heteromeric PC1/PC2 ion channel activity (86). In other studies, the availability of Gβγ-dimers mediated by AGS3 could control mitotic spindle orientation (146). The pool of Gβγ-dimers by AGS3 could be regulated near the cilia (200), but further studies are needed to confirm whether Gβγ has a key role in the transformation of normal tubular into cystic epithelial cells. In vertebrate and mammalian cells, Wnt ligand activation of selective Fz, in particular Fz2 and Fz7, elicits induction of calcium transients through a Gαi-dependent mechanism. Downstream signaling is controlled by the release of Gβγ-dimers, resulting in the activation of PI3-K activity (154), protein kinase C translocation (151), calmodulin-dependent protein-kinase II (83), and also cdc42 activity (132). Further studies are needed to confirm the role of Wnt/Fz and the activation of Gβγ-dependent pathways in PKD, but there is emerging evidence that cdc42 knockout in mice can produce focal cysts in the kidney (25). As mentioned earlier, PC-1 could also induce Akt function to prevent apoptotic signaling by interacting with active forms of Gαi-subunits, which ultimately leads to an increased pool of unbound Gβγ-subunits (14). The Gβγ would subsequently activate the p110β subunit of PI-3K (85, 166), which would result in the activation of Akt.
Although these downstream signaling pathways may seem distinct, a common theme that has arisen appears to be that the sequestration of Gαi-subunits promotes the hyperactivity of Gβγ. In this scenario, the Gαi-subunit functions more like an entity that is designed to inactivate Gβγ, rather than pursue its own functional role to regulate other effector systems, such as inhibiting adenylyl cyclase.
PERSPECTIVES AND CONCLUSIONS
Heterotrimeric G protein signaling remains an integral part of the cell signaling network during the establishment of normal tubular epithelial cell homeostasis but also in the adaptive process by the cell in response to continual extrinsic stressors. To date, the main method to control G protein activation has been predominantly through the design and development of pharmacological drugs targeted to specific GPCR, including those abnormally active in PKD. This type of control mechanism is primarily used to regulate the activity of the Gα-subunits. As described in this review, there is increased diversity by which heterotrimeric G protein signaling can occur within the cell, especially with the accumulating evidence of nonconventional mechanisms to control Gα-subunit function. Moreover, changes in Gβγ function within the cell could have a fundamentally important role in the control of tubular epithelial cell biology by either complementing Gα-directed signaling or, in some cases, being the major driving force depending upon the specific Gα-subunit to regulate downstream signaling cascades.
It has been nearly two decades since the initial observation that PC-1 could bind to heterotrimeric G proteins (129), and nearly two-thirds of all humans diagnosed with ADPKD have mutations in the PKD1 gene. At present, there are >60 clinically identified variants in the germline and somatic cells within the G protein-binding domain (amino acids 4110–4183) (http://pkdb.mayo.edu/). There is yet a paucity of information regarding the importance of how mutant forms of PC-1 can dysregulate tubular epithelial cell biology. Moreover, the role of heterotrimeric G protein binding and function in PKD has not yet been fully resolved and likely requires additional areas of investigation. First, remove the G protein-binding domain in PC-1 to assess whether this region is an essential part of the pathogenic process to either initiate or exacerbate tubular epithelial cell transformation into cystic cells. Second, determine the Gα-subtype and its preferred state of activity (i.e., GTP- or GDP-bound) for efficient binding to PC-1. Third, define whether the Gα or Gβγ is the primary signaling effector following interaction with PC-1 and whether there is a positive or negative influence on signaling output depending upon the activation of each individual G protein subunit. Fourth, determine whether subcellular targeting of the COOH-terminal fragment of PC-1 to the nucleus can be influenced by G protein binding, which could modify transcriptional profiling. Last, identify the composition of the Gαβγ-complex within the cystic epithelial cell to determine whether selective interactions occur to drive signal processing toward cyst progression.
In all, heterotrimeric G proteins are clearly an important part of the cellular repertoire to control signal transduction within the tubular epithelial cell. However, the regulation by which heterotrimeric G proteins are controlled continues to expand far beyond the membrane-associated GPCRs. In fact, heterotrimeric G proteins may function, albeit through unconventional modes of action, by altering its normal subcellular location (i.e., the plasma membrane) to other sites, such as the mitotic spindle organizing center, through novel interactions with cytoplasmic proteins. As the data continue to accumulate regarding the role of G proteins in PKD, the design and development of selective Gα and/or Gβγ modulators will enable researchers to determine whether specific G protein subunits can be therapeutically targeted to attenuate the procystic signaling pathways in vascular and tubular epithelial cells in the kidney.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-90123 (to F. Park).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
T.H. and F.P. prepared figures; T.H. and F.P. drafted manuscript; T.H. and F.P. edited and revised manuscript; T.H. and F.P. approved final version of manuscript.
ACKNOWLEDGMENTS
Current address for T. Hama: Dept. of Pediatrics, Wakayama Medical Univ., Wakayama, Japan.
REFERENCES
- 1.Aguiari G, Varani K, Bogo M, Mangolini A, Vincenzi F, Durante C, Gessi S, Sacchetto V, Catizone L, Harris P, Rizzuto R, Borea PA, Del Senno L. Deficiency of polycystic kidney disease-1 gene (PKD1) expression increases A(3) adenosine receptors in human renal cells: implications for cAMP-dependent signalling and proliferation of PKD1-mutated cystic cells. Biochim Biophys Acta 1792: 531–540, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Angers S, Thorpe CJ, Biechele TL, Goldenberg SJ, Zheng N, MacCoss MJ, Moon RT. The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation. Nat Cell Biol 8: 348–357, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Arac D, Boucard AA, Bolliger MF, Nguyen J, Soltis SM, Sudhof TC, Brunger AT. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J 31: 1364–1378, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arai H, Hori S, Aramori I, Ohkubo H, Nakanishi S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature 348: 730–732, 1990. [DOI] [PubMed] [Google Scholar]
- 5.Avner ED. Epithelial polarity and differentiation in polycystic kidney disease. J Cell Sci Suppl 17: 217–222, 1993. [DOI] [PubMed] [Google Scholar]
- 6.Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD). Hepatology 49: 160–174, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bates CM, Kegg H, Grady S. Expression of somatostatin in the adult and developing mouse kidney. Kidney Int 66: 1785–1793, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM Jr, Grantham JJ. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int 66: 964–973, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Bell GP, Fletcher GC, Brain R, Thompson BJ. Aurora kinases phosphorylate Lgl to induce mitotic spindle orientation in Drosophila epithelia. Curr Biol 25: 61–68, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergstralh DT, St Johnston D. Spindle orientation: what if it goes wrong? Sem Cell Dev Biol 34: 140–145, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertuccio CA, Chapin HC, Cai Y, Mistry K, Chauvet V, Somlo S, Caplan MJ. Polycystin-1 C-terminal cleavage is modulated by polycystin-2 expression. J Biol Chem 284: 21011–21026, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhandari S, Watson N, Long E, Sharpe S, Zhong W, Xu SZ, Atkin SL. Expression of somatostatin and somatostatin receptor subtypes 1–5 in human normal and diseased kidney. J Histochem Cytochem 56: 733–743, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birtwistle MR, Hatakeyama M, Yumoto N, Ogunnaike BA, Hoek JB, Kholodenko BN. Ligand-dependent responses of the ErbB signaling network: experimental and modeling analyses. Mol Syst Biol 3: 144, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boca M, Distefano G, Qian F, Bhunia AK, Germino GG, Boletta A. Polycystin-1 induces resistance to apoptosis through the phosphatidylinositol 3-kinase/Akt signaling pathway. J Am Soc Nephrol 17: 637–647, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonvalet JP, Pradelles P, Farman N. Segmental synthesis and actions of prostaglandins along the nephron. Am J Physiol Renal Fluid Electrolyte Physiol 253: F377–F387, 1987. [DOI] [PubMed] [Google Scholar]
- 16.Braun C, Kleemann T, Birck R, Hilgenfeldt U, Riester U, Tschope C, van der Woude FJ, Rohmeiss P. Increased activity of the renal kallikrein-kinin system in autosomal dominant polycystic kidney disease in rats, but not in humans. Int Immunopharmacol 2: 1949–1956, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Braun C, Kleemann T, Hilgenfeldt U, Riester U, Rohmeiss P, van der Woude FJ. Activity and functional significance of the renal kallikrein-kinin-system in polycystic kidney disease of the rat. Kidney Int 61: 2149–2156, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Bunni MA, Morinelli TA, Bell PD, Raymond JR, Garnovskaya MN. Bradykinin B2 receptor activates extracellular signal regulated kinase in renal cells from a mouse model of polycystic kidney disease. FASEB J 24, Suppl: lb703, 2010. [Google Scholar]
- 19.Cai Y, Fedeles SV, Dong K, Anyatonwu G, Onoe T, Mitobe M, Gao JD, Okuhara D, Tian X, Gallagher AR, Tang Z, Xie X, Lalioti MD, Lee AH, Ehrlich BE, Somlo S. Altered trafficking and stability of polycystins underlie polycystic kidney disease. J Clin Invest 124: 5129–5144, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caroli A, Antiga L, Cafaro M, Fasolini G, Remuzzi A, Remuzzi G, Ruggenenti P. Reducing polycystic liver volume in ADPKD: effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol 5: 783–789, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caroli A, Perico N, Perna A, Antiga L, Brambilla P, Pisani A, Visciano B, Imbriaco M, Messa P, Cerutti R, Dugo M, Cancian L, Buongiorno E, De Pascalis A, Gaspari F, Carrara F, Rubis N, Prandini S, Remuzzi A, Remuzzi G, Ruggenenti P, ALADIN study group. Effect of longacting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet 382: 1485–1495, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Chang MY, Parker E, El Nahas M, Haylor JL, Ong AC. Endothelin B receptor blockade accelerates disease progression in a murine model of autosomal dominant polycystic kidney disease. J Am Soc Nephrol 18: 560–569, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 323: 1091–1096, 1990. [DOI] [PubMed] [Google Scholar]
- 24.Chauvet V, Tian X, Husson H, Grimm DH, Wang T, Hiesberger T, Igarashi P, Bennett AM, Ibraghimov-Beskrovnaya O, Somlo S, Caplan MJ. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J Clin Invest 114: 1433–1443, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi SY, Chacon-Heszele MF, Huang L, McKenna S, Wilson FP, Zuo X, Lipschutz JH. Cdc42 deficiency causes ciliary abnormalities and cystic kidneys. J Am Soc Nephrol 24: 1435–1450, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clapham DE, Neer EJ. G protein beta gamma subunits. Ann Rev Pharmacol Toxicol 37: 167–203, 1997. [DOI] [PubMed] [Google Scholar]
- 27.Cowley AW., Jr Long-term control of arterial blood pressure. Physiol Rev 72: 231–300, 1992. [DOI] [PubMed] [Google Scholar]
- 28.Cowley AW Jr, Szczepanska-Sadowska E, Stepniakowski K, Mattson D. Chronic intravenous administration of V1 arginine vasopressin agonist results in sustained hypertension. Am J Physiol Heart Circ Physiol 267: H751–H756, 1994. [DOI] [PubMed] [Google Scholar]
- 29.Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J 16: 7032–7044, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, Pollock DM, Webb DJ, Maguire JJ. Endothelin. Pharmacol Rev 68: 357–418, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delmas P, Nauli SM, Li X, Coste B, Osorio N, Crest M, Brown DA, Zhou J. Gating of the polycystin ion channel signaling complex in neurons and kidney cells. FASEB J 18: 740–742, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Delmas P, Nomura H, Li X, Lakkis M, Luo Y, Segal Y, Fernandez-Fernandez JM, Harris P, Frischauf AM, Brown DA, Zhou J. Constitutive activation of G-proteins by polycystin-1 is antagonized by polycystin-2. J Biol Chem 277: 11276–11283, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Dhande I, Ali Q, Hussain T. Proximal tubule angiotensin AT2 receptors mediate an anti-inflammatory response via interleukin-10: role in renoprotection in obese rats. Hypertension 61: 1218–1226, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dhande I, Ma W, Hussain T. Angiotensin AT2 receptor stimulation is anti-inflammatory in lipopolysaccharide-activated THP-1 macrophages via increased interleukin-10 production. Hypertens Res 38: 21–29, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DiBona GF, Kopp UC. Neural control of renal function. Physiol Rev 77: 75–197, 1997. [DOI] [PubMed] [Google Scholar]
- 36.Dijksterhuis JP, Petersen J, Schulte G. WNT/Frizzled signalling: receptor-ligand selectivity with focus on FZD-G protein signalling and its physiological relevance: IUPHAR Review 3. Br J Pharmacol 171: 1195–1209, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ecder T, Chapman AB, Brosnahan GM, Edelstein CL, Johnson AM, Schrier RW. Effect of antihypertensive therapy on renal function and urinary albumin excretion in hypertensive patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis 35: 427–432, 2000. [DOI] [PubMed] [Google Scholar]
- 38.Ecder T, Edelstein CL, Fick-Brosnahan GM, Johnson AM, Chapman AB, Gabow PA, Schrier RW. Diuretics versus angiotensin-converting enzyme inhibitors in autosomal dominant polycystic kidney disease. Am J Nephrol 21: 98–103, 2001. [DOI] [PubMed] [Google Scholar]
- 39.El-Dahr SS, Harrison-Bernard LM, Dipp S, Yosipiv IV, Meleg-Smith S. Bradykinin B2 null mice are prone to renal dysplasia: gene-environment interactions in kidney development. Physiol Genomics 3: 121–131, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Elberg D, Turman MA, Pullen N, Elberg G. Prostaglandin E2 stimulates cystogenesis through EP4 receptor in IMCD-3 cells. Prostaglandins Other Lipid Mediat 98: 11–16, 2012. [DOI] [PubMed] [Google Scholar]
- 41.Elberg G, Elberg D, Lewis TV, Guruswamy S, Chen L, Logan CJ, Chan MD, Turman MA. EP2 receptor mediates PGE2-induced cystogenesis of human renal epithelial cells. Am J Physiol Renal Physiol 293: F1622–F1632, 2007. [DOI] [PubMed] [Google Scholar]
- 42.Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M. Defective planar cell polarity in polycystic kidney disease. Nat Genet 38: 21–23, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Franchini KG, Cowley AW Jr. Renal cortical and medullary blood flow responses during water restriction: role of vasopressin. Am J Physiol Regul Integr Comp Physiol 270: R1257–R1264, 1996. [DOI] [PubMed] [Google Scholar]
- 44.Franchini KG, Cowley AW Jr. Sensitivity of the renal medullary circulation to plasma vasopressin. Am J Physiol Regul Integr Comp Physiol 271: R647–R653, 1996. [DOI] [PubMed] [Google Scholar]
- 45.Gattone VH 2nd, Siqueira TM Jr, Powell CR, Trambaugh CM, Lingeman JE, Shalhav AL. Contribution of renal innervation to hypertension in rat autosomal dominant polycystic kidney disease. Exp Biol Med (Maywood) 233: 952–957, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Giamarchi A, Feng S, Rodat-Despoix L, Xu Y, Bubenshchikova E, Newby LJ, Hao J, Gaudioso C, Crest M, Lupas AN, Honore E, Williamson MP, Obara T, Ong AC, Delmas P. A polycystin-2 (TRPP2) dimerization domain essential for the function of heteromeric polycystin complexes. EMBO J 29: 1176–1191, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goggolidou P. Wnt and planar cell polarity signaling in cystic renal disease. Organogenesis 10: 86–95, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gohla A, Schultz G, Offermanns S. Role for G(12)/G(13) in agonist-induced vascular smooth muscle cell contraction. Circ Res 87: 221–227, 2000. [DOI] [PubMed] [Google Scholar]
- 49.Goilav B. Apoptosis in polycystic kidney disease. Biochim Biophys Acta 1812: 1272–1280, 2011. [DOI] [PubMed] [Google Scholar]
- 50.Goto M, Hoxha N, Osman R, Dell KM. The renin-angiotensin system and hypertension in autosomal recessive polycystic kidney disease. Pediatr Nephrol 25: 2449–2457, 2010. [DOI] [PubMed] [Google Scholar]
- 51.Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int 33: 1084–1090, 1988. [DOI] [PubMed] [Google Scholar]
- 52.Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 359: 1477–1485, 2008. [DOI] [PubMed] [Google Scholar]
- 53.Grantham JJ. Mechanisms of progression in autosomal dominant polycystic kidney disease. Kidney Int Suppl 63: S93–S97, 1997. [PubMed] [Google Scholar]
- 54.Grantham JJ, Mulamalla S, Swenson-Fields KI. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol 7: 556–566, 2011. [DOI] [PubMed] [Google Scholar]
- 55.Guay-Woodford LM. Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol 285: F1034–F1049, 2003. [DOI] [PubMed] [Google Scholar]
- 56.Hammerschmidt M, McMahon AP. The effect of pertussis toxin on zebrafish development: a possible role for inhibitory G-proteins in hedgehog signaling. Dev Biol 194: 166–171, 1998. [DOI] [PubMed] [Google Scholar]
- 57.Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408: 990–994, 2000. [DOI] [PubMed] [Google Scholar]
- 58.Hannon JP, Nunn C, Stolz B, Bruns C, Weckbecker G, Lewis I, Troxler T, Hurth K, Hoyer D. Drug design at peptide receptors: somatostatin receptor ligands. J Mol Neurosci 18: 15–27, 2002. [DOI] [PubMed] [Google Scholar]
- 59.Happe H, de Heer E, Peters DJ. Polycystic kidney disease: the complexity of planar cell polarity and signaling during tissue regeneration and cyst formation. Biochim Biophys Acta 1812: 1249–1255, 2011. [DOI] [PubMed] [Google Scholar]
- 60.Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest 124: 2315–2324, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Higashihara E, Torres VE, Chapman AB, Grantham JJ, Bae K, Watnick TJ, Horie S, Nutahara K, Ouyang J, Krasa HB, Czerwiec FS, TEMPOFormula and 156-05-002 Study Investigators. Tolvaptan in autosomal dominant polycystic kidney disease: three years' experience. Clin J Am Soc Nephrol 6: 2499–2507, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hocher B, Kalk P, Slowinski T, Godes M, Mach A, Herzfeld S, Wiesner D, Arck PC, Neumayer HH, Nafz B. ETA receptor blockade induces tubular cell proliferation and cyst growth in rats with polycystic kidney disease. J Am Soc Nephrol 14: 367–376, 2003. [DOI] [PubMed] [Google Scholar]
- 63.Hocher B, Thone-Reineke C, Rohmeiss P, Schmager F, Slowinski T, Burst V, Siegmund F, Quertermous T, Bauer C, Neumayer HH, Schleuning WD, Theuring F. Endothelin-1 transgenic mice develop glomerulosclerosis, interstitial fibrosis, and renal cysts but not hypertension. J Clin Invest 99: 1380–1389, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hocher B, Zart R, Braun N, Schwarz A, van der Woude F, Rohmeiss P, Koppenhagen K. The endothelin system in polycystic kidneys of Han:SPRD rats. J Cardiovasc Pharmacol 31, Suppl 1: S342–S344, 1998. [DOI] [PubMed] [Google Scholar]
- 65.Hocher B, Zart R, Schwarz A, Vogt V, Braun C, Thone-Reineke C, Braun N, Neumayer HH, Koppenhagen K, Bauer C, Rohmeiss P. Renal endothelin system in polycystic kidney disease. J Am Soc Nephrol 9: 1169–1177, 1998. [DOI] [PubMed] [Google Scholar]
- 66.Hogan MC, Masyuk TV, Page L, Holmes DR 3rd, Li X, Bergstralh EJ, Irazabal MV, Kim B, King BF, Glockner JF, Larusso NF, Torres VE. Somatostatin analog therapy for severe polycystic liver disease: results after 2 years. Nephrol Dial Transplant 27: 3532–3539, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, Kim B, King BF, Glockner J, Holmes DR 3rd Rossetti S, Harris PC, LaRusso NF, Torres VE. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol 21: 1052–1061, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 10: 151–160, 1995. [DOI] [PubMed] [Google Scholar]
- 69.Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol 20: 953–970, 2006. [DOI] [PubMed] [Google Scholar]
- 70.Jia G, Kwon M, Liang HL, Mortensen J, Nilakantan V, Sweeney WE, Park F. Chronic treatment with lisinopril decreases proliferative and apoptotic pathways in autosomal recessive polycystic kidney disease. Pediatr Nephrol 25: 1139–1146, 2010. [DOI] [PubMed] [Google Scholar]
- 71.Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141: 1208–1219, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kakoki M, Smithies O. The kallikrein-kinin system in health and in diseases of the kidney. Kidney Int 75: 1019–1030, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, Carroll TJ. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat Genet 41: 793–799, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kawakami T, Ren S, Duffield JS. Wnt signalling in kidney diseases: dual roles in renal injury and repair. J Pathol 229: 221–231, 2013. [DOI] [PubMed] [Google Scholar]
- 75.Kennefick TM, Al-Nimri MA, Oyama TT, Thompson MM, Kelly FJ, Chapman JG, Anderson S. Hypertension and renal injury in experimental polycystic kidney disease. Kidney Int 56: 2181–2190, 1999. [DOI] [PubMed] [Google Scholar]
- 76.Kim E, Arnould T, Sellin L, Benzing T, Comella N, Kocher O, Tsiokas L, Sukhatme VP, Walz G. Interaction between RGS7 and polycystin. Proc Natl Acad Sci USA 96: 6371–6376, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kittikulsuth W, Stuart D, Kohan DE. Adenylyl cyclase 4 does not regulate collecting duct water and sodium handling. Physiol Rep 2: e00277, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kittikulsuth W, Stuart D, Van Hoek AN, Stockand JD, Bugaj V, Mironova E, Blount MA, Kohan DE. Lack of an effect of collecting duct-specific deletion of adenylyl cyclase 3 on renal Na+ and water excretion or arterial pressure. Am J Physiol Renal Physiol 306: F597–F607, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Klein IH, Ligtenberg G, Oey PL, Koomans HA, Blankestijn PJ. Sympathetic activity is increased in polycystic kidney disease and is associated with hypertension. J Am Soc Nephrol 12: 2427–2433, 2001. [DOI] [PubMed] [Google Scholar]
- 80.Kohan DE, Inscho EW, Wesson D, Pollock DM. Physiology of endothelin and the kidney. Compr Physiol 1: 883–919, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kohan DE, Padilla E, Hughes AK. Endothelin B receptor mediates ET-1 effects on cAMP and PGE2 accumulation in rat IMCD. Am J Physiol Renal Fluid Electrolyte Physiol 265: F670–F676, 1993. [DOI] [PubMed] [Google Scholar]
- 82.Koshimizu TA, Nasa Y, Tanoue A, Oikawa R, Kawahara Y, Kiyono Y, Adachi T, Tanaka T, Kuwaki T, Mori T, Takeo S, Okamura H, Tsujimoto G. V1a vasopressin receptors maintain normal blood pressure by regulating circulating blood volume and baroreflex sensitivity. Proc Natl Acad Sci USA 103: 7807–7812, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kuhl M, Sheldahl LC, Malbon CC, Moon RT. Ca2+/calmodulin-dependent protein kinase II is stimulated by Wnt and Frizzled homologs and promotes ventral cell fates in Xenopus. J Biol Chem 275: 12701–12711, 2000. [DOI] [PubMed] [Google Scholar]
- 84.Kuo IY, Kwaczala AT, Nguyen L, Russell KS, Campbell SG, Ehrlich BE. Decreased polycystin 2 expression alters calcium-contraction coupling and changes beta-adrenergic signaling pathways. Proc Natl Acad Sci USA 111: 16604–16609, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kurosu H, Maehama T, Okada T, Yamamoto T, Hoshino S, Fukui Y, Ui M, Hazeki O, Katada T. Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110beta is synergistically activated by the betagamma subunits of G proteins and phosphotyrosyl peptide. J Biol Chem 272: 24252–24256, 1997. [DOI] [PubMed] [Google Scholar]
- 86.Kwon M, Pavlov TS, Nozu K, Rasmussen SA, Ilatovskaya DV, Lerch-Gaggl A, North LM, Kim H, Qian F, Sweeney WE Jr, Avner ED, Blumer JB, Staruschenko A, Park F. G-protein signaling modulator 1 deficiency accelerates cystic disease in an orthologous mouse model of autosomal dominant polycystic kidney disease. Proc Natl Acad Sci USA 109: 21462–21467, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lal M, Song X, Pluznick JL, Di Giovanni V, Merrick DM, Rosenblum ND, Chauvet V, Gottardi CJ, Pei Y, Caplan MJ. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum Mol Genet 17: 3105–3117, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lawson CR, Doulton TW, MacGregor GA. Autosomal dominant polycystic kidney disease: role of the renin-angiotensin system in raised blood pressure in progression of renal and cardiovascular disease. J Renin-Angiotensin-Aldosterone Syst 7: 139–145, 2006. [DOI] [PubMed] [Google Scholar]
- 89.Lee BH, Kwon TH. Regulation of AQP2 in collecting duct: an emphasis on the effects of angiotensin II or aldosterone. Electrolyte Blood Press 5: 15–22, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee K, Battini L, Gusella GL. Cilium, centrosome and cell cycle regulation in polycystic kidney disease. Biochim Biophys Acta 1812: 1263–1271, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lenarczyk M, Pressly JD, Arnett J, Regner KR, Park F. Localization and expression profile of group I and II activators of G-protein signaling in the kidney. J Mol Histol 46: 123–136, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Levay K, Cabrera JL, Satpaev DK, Slepak VZ. Gbeta5 prevents the RGS7-Galphao interaction through binding to a distinct Ggamma-like domain found in RGS7 and other RGS proteins. Proc Natl Acad Sci USA 96: 2503–2507, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu T, DeCostanzo AJ, Liu X, Wang H, Hallagan S, Moon RT, Malbon CC. G protein signaling from activated rat frizzled-1 to the beta-catenin-Lef-Tcf pathway. Science 292: 1718–1722, 2001. [DOI] [PubMed] [Google Scholar]
- 94.Liu X, Rubin JS, Kimmel AR. Rapid, Wnt-induced changes in GSK3beta associations that regulate beta-catenin stabilization are mediated by Galpha proteins. Curr Biol 15: 1989–1997, 2005. [DOI] [PubMed] [Google Scholar]
- 95.Liu Y, Rajagopal M, Lee K, Battini L, Flores D, Gusella GL, Pao AC, Rohatgi R. Prostaglandin E2 mediates proliferation and chloride secretion in ADPKD cystic renal epithelia. Am J Physiol Renal Physiol 303: F1425–F1434, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Loghman-Adham M, Soto CE, Inagami T, Cassis L. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 287: F775–F788, 2004. [DOI] [PubMed] [Google Scholar]
- 97.Loghman-Adham M, Soto CE, Inagami T, Sotelo-Avila C. Expression of components of the renin-angiotensin system in autosomal recessive polycystic kidney disease. J Histochem Cytochem 53: 979–988, 2005. [DOI] [PubMed] [Google Scholar]
- 98.Lolait SJ, O'Carroll AM, McBride OW, Konig M, Morel A, Brownstein MJ. Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature 357: 336–339, 1992. [DOI] [PubMed] [Google Scholar]
- 99.Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, Kane ME, Obara T, Weimbs T. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev Cell 10: 57–69, 2006. [DOI] [PubMed] [Google Scholar]
- 100.Low WC, Wang C, Pan Y, Huang XY, Chen JK, Wang B. The decoupling of Smoothened from Galphai proteins has little effect on Gli3 protein processing and Hedgehog-regulated chick neural tube patterning. Dev Biol 321: 188–196, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Luyten A, Su X, Gondela S, Chen Y, Rompani S, Takakura A, Zhou J. Aberrant regulation of planar cell polarity in polycystic kidney disease. J Am Soc Nephrol 21: 1521–1532, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mangoo-Karim R, Uchic M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci USA 86: 6007–6011, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Gradilone SA, Huang B, Masyuk AI, Hogan MC, Torres VE, Larusso NF. Pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in rodents with polycystic kidney and liver diseases. Hepatology 58: 409–421, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Meijer E, Drenth JP, d'Agnolo H, Casteleijn NF, de Fijter JW, Gevers TJ, Kappert P, Peters DJ, Salih M, Soonawala D, Spithoven EM, Torres VE, Visser FW, Wetzels JF, Zietse R, Gansevoort RT, DIPAK Consortium. Rationale and design of the DIPAK 1 study: a randomized controlled clinical trial assessing the efficacy of lanreotide to Halt disease progression in autosomal dominant polycystic kidney disease. Am J Kidney Dis 63: 446–455, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Melenhorst WB, Mulder GM, Xi Q, Hoenderop JG, Kimura K, Eguchi S, van Goor H. Epidermal growth factor receptor signaling in the kidney: key roles in physiology and disease. Hypertension 52: 987–993, 2008. [DOI] [PubMed] [Google Scholar]
- 106.Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Baraldi PG, Borea PA. Modulation of the Akt/Ras/Raf/MEK/ERK pathway by A(3) adenosine receptor. Purinergic Signal 2: 627–632, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Merta M, Reiterova J, Rysava R, Tesar V, Zavada J, Jachymova M, Zima T. Role of endothelin and nitric oxide in the pathogenesis of arterial hypertension in autosomal dominant polycystic kidney disease. Physiol Res 52: 433–437, 2003. [PubMed] [Google Scholar]
- 108.Miyata N, Park F, Li XF, Cowley AW Jr. Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am J Physiol Renal Physiol 277: F437–F446, 1999. [DOI] [PubMed] [Google Scholar]
- 109.Moeller HB, Olesen ET, Fenton RA. Regulation of the water channel aquaporin-2 by posttranslational modification. Am J Physiol Renal Physiol 300: F1062–F1073, 2011. [DOI] [PubMed] [Google Scholar]
- 110.Moreau ME, Garbacki N, Molinaro G, Brown NJ, Marceau F, Adam A. The kallikrein-kinin system: current and future pharmacological targets. J Pharmacol Sci 99: 6–38, 2005. [DOI] [PubMed] [Google Scholar]
- 111.Morel F. Sites of hormone action in the mammalian nephron. Am J Physiol Renal Fluid Electrolyte Physiol 240: F159–F164, 1981. [DOI] [PubMed] [Google Scholar]
- 112.Mukhin YV, Garnovsky EA, Ullian ME, Garnovskaya MN. Bradykinin B2 receptor activates extracellular signal-regulated protein kinase in mIMCD-3 cells via epidermal growth factor receptor transactivation. J Pharmacol Exp Therapeut 304: 968–977, 2003. [DOI] [PubMed] [Google Scholar]
- 113.Munemura C, Uemasu J, Kawasaki H. Epidermal growth factor and endothelin in cyst fluid from autosomal dominant polycystic kidney disease cases: possible evidence of heterogeneity in cystogenesis. Am J Kidney Dis 24: 561–568, 1994. [DOI] [PubMed] [Google Scholar]
- 114.Murdoch JN, Copp AJ. The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res A Clin Mol Teratol 88: 633–652, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nadella R, Blumer JB, Jia G, Kwon M, Akbulut T, Qian F, Sedlic F, Wakatsuki T, Sweeney WE Jr, Wilson PD, Lanier SM, Park F. Activator of G protein signaling 3 promotes epithelial cell proliferation in PKD. J Am Soc Nephrol 21: 1275–1280, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nagao S, Kugita M, Yoshihara D, Yamaguchi T. Animal models for human polycystic kidney disease. Exp Anim 61: 477–488, 2012. [DOI] [PubMed] [Google Scholar]
- 117.Nakamura T, Ebihara I, Fukui M, Osada S, Tomino Y, Masaki T, Goto K, Furuichi Y, Koide H. Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol 4: 1064–1072, 1993. [DOI] [PubMed] [Google Scholar]
- 118.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33: 129–137, 2003. [DOI] [PubMed] [Google Scholar]
- 119.Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 117: 1161–1171, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nichols AS, Floyd DH, Bruinsma SP, Narzinski K, Baranski TJ. Frizzled receptors signal through G proteins. Cell Signal 25: 1468–1475, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nishio S, Tian X, Gallagher AR, Yu Z, Patel V, Igarashi P, Somlo S. Loss of oriented cell division does not initiate cyst formation. J Am Soc Nephrol 21: 295–302, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ogden SK, Fei DL, Schilling NS, Ahmed YF, Hwa J, Robbins DJ. G protein Galphai functions immediately downstream of Smoothened in Hedgehog signalling. Nature 456: 967–970, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exp Nephrol 93: e80, 2003. [DOI] [PubMed] [Google Scholar]
- 124.Ostrowski NL, Lolait SJ, Bradley DJ, O'Carroll AM, Brownstein MJ, Young WS 3rd. Distribution of V1a and V2 vasopressin receptor messenger ribonucleic acids in rat liver, kidney, pituitary and brain. Endocrinology 131: 533–535, 1992. [DOI] [PubMed] [Google Scholar]
- 125.Ostrowski NL, Young WS 3rd, Knepper MA, Lolait SJ. Expression of vasopressin V1a and V2 receptor messenger ribonucleic acid in the liver and kidney of embryonic, developing, and adult rats. Endocrinology 133: 1849–1859, 1993. [DOI] [PubMed] [Google Scholar]
- 126.Pan J, Seeger-Nukpezah T, Golemis EA. The role of the cilium in normal and abnormal cell cycles: emphasis on renal cystic pathologies. Cell Mol Life Sci 70: 1849–1874, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Park F. Accessory proteins for heterotrimeric G-proteins in the kidney. Front Physiol 6: 219, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Park F. Activators of G protein signaling in the kidney. J Pharmacol Exp Therapeut 353: 235–245, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Parnell SC, Magenheimer BS, Maser RL, Rankin CA, Smine A, Okamoto T, Calvet JP. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem Biophys Res Commun 251: 625–631, 1998. [DOI] [PubMed] [Google Scholar]
- 130.Parnell SC, Magenheimer BS, Maser RL, Zien CA, Frischauf AM, Calvet JP. Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins. J Biol Chem 277: 19566–19572, 2002. [DOI] [PubMed] [Google Scholar]
- 131.Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet 17: 1578–1590, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Penzo-Mendez A, Umbhauer M, Djiane A, Boucaut JC, Riou JF. Activation of Gbetagamma signaling downstream of Wnt-11/Xfz7 regulates Cdc42 activity during Xenopus gastrulation. Dev Biol 257: 302–314, 2003. [DOI] [PubMed] [Google Scholar]
- 133.Polizio AH, Chinchilla P, Chen X, Manning DR, Riobo NA. Sonic Hedgehog activates the GTPases Rac1 and RhoA in a Gli-independent manner through coupling of smoothened to Gi proteins. Sci Signal 4: pt7, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Puri S, Magenheimer BS, Maser RL, Ryan EM, Zien CA, Walker DD, Wallace DP, Hempson SJ, Calvet JP. Polycystin-1 activates the calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway. J Biol Chem 279: 55455–55464, 2004. [DOI] [PubMed] [Google Scholar]
- 135.Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet 16: 179–183, 1997. [DOI] [PubMed] [Google Scholar]
- 136.Ray C, Carney S, Morgan T, Gillies A. Somatostatin as a modulator of distal nephron water permeability. Clin Sci 84: 455–460, 1993. [DOI] [PubMed] [Google Scholar]
- 137.Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J Am Soc Nephrol 25: 232–237, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rieg T, Kohan DE. Regulation of nephron water and electrolyte transport by adenylyl cyclases. Am J Physiol Renal Physiol 306: F701–F709, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Riobo NA, Saucy B, Dilizio C, Manning DR. Activation of heterotrimeric G proteins by Smoothened. Proc Natl Acad Sci USA 103: 12607–12612, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Saadaoui M, Machicoane M, di Pietro F, Etoc F, Echard A, Morin X. Dlg1 controls planar spindle orientation in the neuroepithelium through direct interaction with LGN. J Cell Biol 206: 707–717, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sagara Y, Hirooka Y, Nozoe M, Ito K, Kimura Y, Sunagawa K. Pressor response induced by central angiotensin II is mediated by activation of Rho/Rho-kinase pathway via AT1 receptors. J Hypertens 25: 399–406, 2007. [DOI] [PubMed] [Google Scholar]
- 142.Saigusa T, Dang Y, Bunni MA, Amria MY, Steele SL, Fitzgibbon WR, Bell PD. Activation of the intrarenal renin-angiotensin-system in murine polycystic kidney disease. Physiol Rep 3: e12405, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Saigusa T, Dang Y, Mullick AE, Yeh ST, Zile MR, Baicu CF, Bell PD. Suppressing angiotensinogen synthesis attenuates kidney cyst formation in a Pkd1 mouse model. FASEB J 30: 370–379, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Saigusa T, Reichert R, Guare J, Siroky BJ, Gooz M, Steele S, Fenton RA, Bell PD, Kolb RJ. Collecting duct cells that lack normal cilia have mislocalized vasopressin-2 receptors. Am J Physiol Renal Physiol 302: F801–F808, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sakurai T, Yanagisawa M, Takuwa Y, Miyazaki H, Kimura S, Goto K, Masaki T. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 348: 732–735, 1990. [DOI] [PubMed] [Google Scholar]
- 146.Sanada K, Tsai LH. G protein betagamma subunits and AGS3 control spindle orientation and asymmetric cell fate of cerebral cortical progenitors. Cell 122: 119–131, 2005. [DOI] [PubMed] [Google Scholar]
- 147.Sanchez-Fernandez G, Cabezudo S, Garcia-Hoz C, Beninca C, Aragay AM, Mayor F Jr, Ribas C. Galphaq signalling: the new and the old. Cell Signal 26: 833–848, 2014. [DOI] [PubMed] [Google Scholar]
- 148.Schauf CL, Moffett DF, Moffett SB. Human Physiology: Foundations & Frontiers. St. Louis, MO: Times Mirror/Mosby College Pub, 1990. [Google Scholar]
- 149.Schramm M, Selinger Z. Message transmission: receptor controlled adenylate cyclase system. Science 225: 1350–1356, 1984. [DOI] [PubMed] [Google Scholar]
- 150.Schrier RW, Abebe KZ, Perrone RD, Torres VE, Braun WE, Steinman TI, Winklhofer FT, Brosnahan G, Czarnecki PG, Hogan MC, Miskulin DC, Rahbari-Oskoui FF, Grantham JJ, Harris PC, Flessner MF, Bae KT, Moore CG, Chapman AB, HALT-PKD Trial Investigators. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 371: 2255–2266, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sheldahl LC, Park M, Malbon CC, Moon RT. Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr Biol 9: 695–698, 1999. [DOI] [PubMed] [Google Scholar]
- 152.Shen F, Cheng L, Douglas AE, Riobo NA, Manning DR. Smoothened is a fully competent activator of the heterotrimeric G protein G(i). Mol Pharmacol 83: 691–697, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Siragy HM, Carey RM. The subtype 2 (AT2) angiotensin receptor mediates renal production of nitric oxide in conscious rats. J Clin Invest 100: 264–269, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Slusarski DC, Corces VG, Moon RT. Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nature 390: 410–413, 1997. [DOI] [PubMed] [Google Scholar]
- 155.Smrcka AV. G protein betagamma subunits: central mediators of G protein-coupled receptor signaling. Cell Mol Life Sci 65: 2191–2214, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Spielman WS, Arend LJ. Adenosine receptors and signaling in the kidney. Hypertension 17: 117–130, 1991. [DOI] [PubMed] [Google Scholar]
- 157.Stegbauer J, Gurley SB, Sparks MA, Woznowski M, Kohan DE, Yan M, Lehrich RW, Coffman TM. AT1 receptors in the collecting duct directly modulate the concentration of urine. J Am Soc Nephrol 22: 2237–2246, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Stemmle LN, Fields TA, Casey PJ. The regulator of G protein signaling domain of axin selectively interacts with Galpha12 but not Galpha13. Mol Pharmacol 70: 1461–1468, 2006. [DOI] [PubMed] [Google Scholar]
- 159.Su X, Wu M, Yao G, El-Jouni W, Luo C, Tabari A, Zhou J. Regulation of polycystin-1 ciliary trafficking by motifs at its C-terminus and polycystin-2 but not by cleavage at the GPS site. J Cell Sci 128: 4063–4073, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sugiyama N, Tsukiyama T, Yamaguchi TP, Yokoyama T. The canonical Wnt signaling pathway is not involved in renal cyst development in the kidneys of inv mutant mice. Kidney Int 79: 957–965, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sundaresan PR, Fortin TL, Kelvie SL. Alpha- and beta-adrenergic receptors in proximal tubules of rat kidney. Am J Physiol Renal Fluid Electrolyte Physiol 253: F848–F856, 1987. [DOI] [PubMed] [Google Scholar]
- 162.Suzuki H, Kimura K, Shirai H, Eguchi K, Higuchi S, Hinoki A, Ishimaru K, Brailoiu E, Dhanasekaran DN, Stemmle LN, Fields TA, Frank GD, Autieri MV, Eguchi S. Endothelial nitric oxide synthase inhibits G12/13 and rho-kinase activated by the angiotensin II type-1 receptor: implication in vascular migration. Arterioscler Thromb Vasc Biol 29: 217–224, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int 57: 33–40, 2000. [DOI] [PubMed] [Google Scholar]
- 164.Sweeney WE Jr, Avner ED. Pathophysiology of childhood polycystic kidney diseases: new insights into disease-specific therapy. Pediatr Res 75: 148–157, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sweeney WE Jr, Hamahira K, Sweeney J, Garcia-Gatrell M, Frost P, Avner ED. Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability. Kidney Int 64: 1310–1319, 2003. [DOI] [PubMed] [Google Scholar]
- 166.Taussig R, Gilman AG. Mammalian membrane-bound adenylyl cyclases. J Biol Chem 270: 1–4, 1995. [DOI] [PubMed] [Google Scholar]
- 167.Terada Y, Tomita K, Nonoguchi H, Marumo F. Different localization of two types of endothelin receptor mRNA in microdissected rat nephron segments using reverse transcription and polymerase chain reaction assay. J Clin Invest 90: 107–112, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Thivierge C, Kurbegovic A, Couillard M, Guillaume R, Cote O, Trudel M. Overexpression of PKD1 causes polycystic kidney disease. Mol Cell Biol 26: 1538–1548, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tian Y, Smith RD, Balla T, Catt KJ. Angiotensin II activates mitogen-activated protein kinase via protein kinase C and Ras/Raf-1 kinase in bovine adrenal glomerulosa cells. Endocrinology 139: 1801–1809, 1998. [DOI] [PubMed] [Google Scholar]
- 170.Torres VE, Abebe KZ, Chapman AB, Schrier RW, Braun WE, Steinman TI, Winklhofer FT, Brosnahan G, Czarnecki PG, Hogan MC, Miskulin DC, Rahbari-Oskoui FF, Grantham JJ, Harris PC, Flessner MF, Moore CG, Perrone RD, HALT-PKD Trial Investigators. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 371: 2267–2276, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS, Investigators TT . Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367: 2407–2418, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Torres VE, Donovan KA, Scicli G, Holley KE, Thibodeau SN, Carretero OA, Inagami T, McAteer JA, Johnson CM. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 42: 364–373, 1992. [DOI] [PubMed] [Google Scholar]
- 173.Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH 2nd. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med 10: 363–364, 2004. [DOI] [PubMed] [Google Scholar]
- 174.Tran PV, Sharma M, Li X, Calvet JP. Developmental signaling: does it bridge the gap between cilia dysfunction and renal cystogenesis? Birth Defects Res C Embryo Today 102: 159–173, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tran PV, Talbott GC, Turbe-Doan A, Jacobs DT, Schonfeld MP, Silva LM, Chatterjee A, Prysak M, Allard BA, Beier DR. Downregulating hedgehog signaling reduces renal cystogenic potential of mouse models. J Am Soc Nephrol 25: 2201–2212, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tsiokas L. Function and regulation of TRPP2 at the plasma membrane. Am J Physiol Renal Physiol 297: F1–F9, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tulassay T, Tulassay Z, Rascher W, Szucs L, Seyberth HW, Nagy I. Effect of somatostatin on kidney function and vasoactive hormone systems in health subjects. Klin Wochenschr 69: 486–490, 1991. [DOI] [PubMed] [Google Scholar]
- 178.Ushio-Fukai M, Griendling KK, Akers M, Lyons PR, Alexander RW. Temporal dispersion of activation of phospholipase C-beta1 and -gamma isoforms by angiotensin II in vascular smooth muscle cells. Role of alphaq/11, alpha12, and beta gamma G protein subunits. J Biol Chem 273: 19772–19777, 1998. [DOI] [PubMed] [Google Scholar]
- 179.Vallon V, Muhlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev 86: 901–940, 2006. [DOI] [PubMed] [Google Scholar]
- 180.van Dijk MA, Breuning MH, Duiser R, van Es LA, Westendorp RG. No effect of enalapril on progression in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 18: 2314–2320, 2003. [DOI] [PubMed] [Google Scholar]
- 181.van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MG, Hoffmann AL, Dekker HM, de Man RA, Drenth JP. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology 137: 1661–1668, 2009. [DOI] [PubMed] [Google Scholar]
- 182.Vora JP, Owens DR, Ryder R, Atiea J, Luzio S, Hayes TM. Effect of somatostatin on renal function. Br Med J 292: 1701–1702, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wang D, Strandgaard S. The pathogenesis of hypertension in autosomal dominant polycystic kidney disease. J Hypertens 15: 925–933, 1997. [DOI] [PubMed] [Google Scholar]
- 184.Wang W, Li F, Sun Y, Lei L, Zhou H, Lei T, Xia Y, Verkman AS, Yang B. Aquaporin-1 retards renal cyst development in polycystic kidney disease by inhibition of Wnt signaling. FASEB J 29: 1551–1563, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA 100: 10782–10787, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Weimbs T, Olsan EE, Talbot JJ. Regulation of STATs by polycystin-1 and their role in polycystic kidney disease. JAKSTAT 2: e23650, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Weimbs T, Talbot JJ. STAT3 signaling in polycystic kidney disease. Drug Discov Today Dis Mech 10: e113–e118, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wendel M, Knels L, Kummer W, Koch T. Distribution of endothelin receptor subtypes ETA and ETB in the rat kidney. J Histochem Cytochem 54: 1193–1203, 2006. [DOI] [PubMed] [Google Scholar]
- 189.Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev 85: 1159–1204, 2005. [DOI] [PubMed] [Google Scholar]
- 190.Wilson PD. Polycystic kidney disease: new understanding in the pathogenesis. Int J Biochem Cell Biol 36: 1868–1873, 2004. [DOI] [PubMed] [Google Scholar]
- 191.Winkler SN, Torikai S, Levine BS, Kurokawa K. Effect of somatostatin on vasopressin-induced antidiuresis and renal cyclic AMP of rats. Miner Electrolyte Metab 7: 8–14, 1982. [PubMed] [Google Scholar]
- 192.Winyard P, Jenkins D. Putative roles of cilia in polycystic kidney disease. Biochim Biophys Acta 1812: 1256–1262, 2011. [DOI] [PubMed] [Google Scholar]
- 193.Wolf G, Neilson EG. Isoproterenol induces mitogenesis in MCT and LLC-PK1 tubular cells. J Am Soc Nephrol 4: 1995–2002, 1994. [DOI] [PubMed] [Google Scholar]
- 194.Xu C, Rossetti S, Jiang L, Harris PC, Brown-Glaberman U, Wandinger-Ness A, Bacallao R, Alper SL. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am J Physiol Renal Physiol 292: F930–F945, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Xu GM, Gonzalez-Perrett S, Essafi M, Timpanaro GA, Montalbetti N, Arnaout MA, Cantiello HF. Polycystin-1 activates and stabilizes the polycystin-2 channel. J Biol Chem 278: 1457–1462, 2003. [DOI] [PubMed] [Google Scholar]
- 196.Yamaguchi I. Evidence for beta-adrenoceptors in rat proximal convoluted tubules. Kurume Med J 40: 159–167, 1993. [DOI] [PubMed] [Google Scholar]
- 197.Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int 63: 1983–1994, 2003. [DOI] [PubMed] [Google Scholar]
- 198.Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 279: 40419–40430, 2004. [DOI] [PubMed] [Google Scholar]
- 199.Ye M, Grant M, Sharma M, Elzinga L, Swan S, Torres VE, Grantham JJ. Cyst fluid from human autosomal dominant polycystic kidneys promotes cyst formation and expansion by renal epithelial cells in vitro. J Am Soc Nephrol 3: 984–994, 1992. [DOI] [PubMed] [Google Scholar]
- 200.Yeh C, Li A, Chuang JZ, Saito M, Caceres A, Sung CH. IGF-1 activates a cilium-localized noncanonical Gbetagamma signaling pathway that regulates cell-cycle progression. Dev Cell 26: 358–368, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev 65: 1010–1052, 2013. [DOI] [PubMed] [Google Scholar]
- 202.Yu S, Hackmann K, Gao J, He X, Piontek K, Garcia-Gonzalez MA, Menezes LF, Xu H, Germino GG, Zuo J, Qian F. Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc Natl Acad Sci USA 104: 18688–18693, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yu W, Kong T, Beaudry S, Tran M, Negoro H, Yanamadala V, Denker BM. Polycystin-1 protein level determines activity of the Galpha12/JNK apoptosis pathway. J Biol Chem 285: 10243–10251, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Yu W, Ritchie BJ, Su X, Zhou J, Meigs TE, Denker BM. Identification of polycystin-1 and Galpha12 binding regions necessary for regulation of apoptosis. Cell Signal 23: 213–221, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yuasa T, Takakura A, Denker BM, Venugopal B, Zhou J. Polycystin-1L2 is a novel G-protein-binding protein. Genomics 84: 126–138, 2004. [DOI] [PubMed] [Google Scholar]
- 206.Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 23: 573–579, 2008. [DOI] [PubMed] [Google Scholar]
- 207.Zheng Z, Zhu H, Wan Q, Liu J, Xiao Z, Siderovski DP, Du Q. LGN regulates mitotic spindle orientation during epithelial morphogenesis. J Cell Biol 189: 275–288, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zittema D, Boertien WE, van Beek AP, Dullaart RP, Franssen CF, de Jong PE, Meijer E, Gansevoort RT. Vasopressin, copeptin, and renal concentrating capacity in patients with autosomal dominant polycystic kidney disease without renal impairment. Clin J Am Soc Nephrol 7: 906–913, 2012. [DOI] [PubMed] [Google Scholar]