

Abstract
We report on markedly different frequencies of genetic lesions within subsets of chronic lymphocytic leukemia patients carrying mutated or unmutated stereotyped B-cell receptor immunoglobulins in the largest cohort (n=565) studied for this purpose. By combining data on recurrent gene mutations (BIRC3, MYD88, NOTCH1, SF3B1 and TP53) and cytogenetic aberrations, we reveal a subset-biased acquisition of gene mutations. More specifically, the frequency of NOTCH1 mutations was found to be enriched in subsets expressing unmutated immunoglobulin genes, i.e. #1, #6, #8 and #59 (22–34%), often in association with trisomy 12, and was significantly different (P<0.001) to the frequency observed in subset #2 (4%, aggressive disease, variable somatic hypermutation status) and subset #4 (1%, indolent disease, mutated immunoglobulin genes). Interestingly, subsets harboring a high frequency of NOTCH1 mutations were found to carry few (if any) SF3B1 mutations. This starkly contrasts with subsets #2 and #3 where, despite their immunogenetic differences, SF3B1 mutations occurred in 45% and 46% of cases, respectively. In addition, mutations within TP53, whilst enriched in subset #1 (16%), were rare in subsets #2 and #8 (both 2%), despite all being clinically aggressive. All subsets were negative for MYD88 mutations, whereas BIRC3 mutations were infrequent. Collectively, this striking bias and skewed distribution of mutations and cytogenetic aberrations within specific chronic lymphocytic leukemia subsets implies that the mechanisms underlying clinical aggressiveness are not uniform, but rather support the existence of distinct genetic pathways of clonal evolution governed by a particular stereotyped B-cell receptor selecting a certain molecular lesion(s).
Introduction
Immunogenetic studies have been instrumental in revealing that the ontogeny of chronic lymphocytic leukemia (CLL) is not stochastic, but rather antigen-driven, through the discovery that: (i) the immunoglobulin (IG) gene repertoire of the clonotypic B-cell receptor (BcR) displays restriction and, (ii) the level of somatic hypermutations (SHM) present in rearranged IG heavy chain genes defines two disease subtypes, each associated with a different clinical course.1–5 Such studies led to the discovery of quasi-identical or stereotyped BcR IGs in more than 30% of CLL patients who can be assigned to distinct subsets, each defined by a particular BcR immunogenetic motif.6–14 Importantly, from both a biological and clinical perspective, evidence suggests that this classification of CLL based on BcR stereotypy is highly relevant and extends well beyond the SHM status of the BcR IG, thereby enabling the identification of homogeneous disease subgroups and, hence, overcoming the heterogeneity characteristic of CLL.
Indeed, studies indicate that patients with similar SHM status but assigned to different stereotyped subsets can exhibit distinct, subset-biased biological profiles and clinical behavior.10,15–25 In addition, preliminary observations in CLL, in relatively small patient series, suggest that the frequency and patterns of mutations within several genes, namely, NOTCH1, SF3B1 and TP53, may differ amongst subsets of patients carrying stereotyped BcRs, the paradigmatic example being the recently observed enrichment of SF3B1 mutations in the clinically aggressive subset #2.26–28
With this in mind, we sought to systematically evaluate the mutational status of BIRC3, MYD88, NOTCH1, SF3B1 and TP53 in 565 CLL patients assigned to one of 10 major stereotyped subsets, and representing cases with varying SHM status, i.e. cases harboring either unmutated IGHV genes (U-CLL) or mutated IGHV genes (M-CLL). We demonstrate markedly different frequencies and spectra of genomic defects amongst the various subsets. On these grounds, we speculate that common genetic aberrations, acquired and/or selected in the context of shared immune pathways originating from highly similar BcR IGs could shape the evolutionary pathway of individual CLL subsets.
Methods
Patients
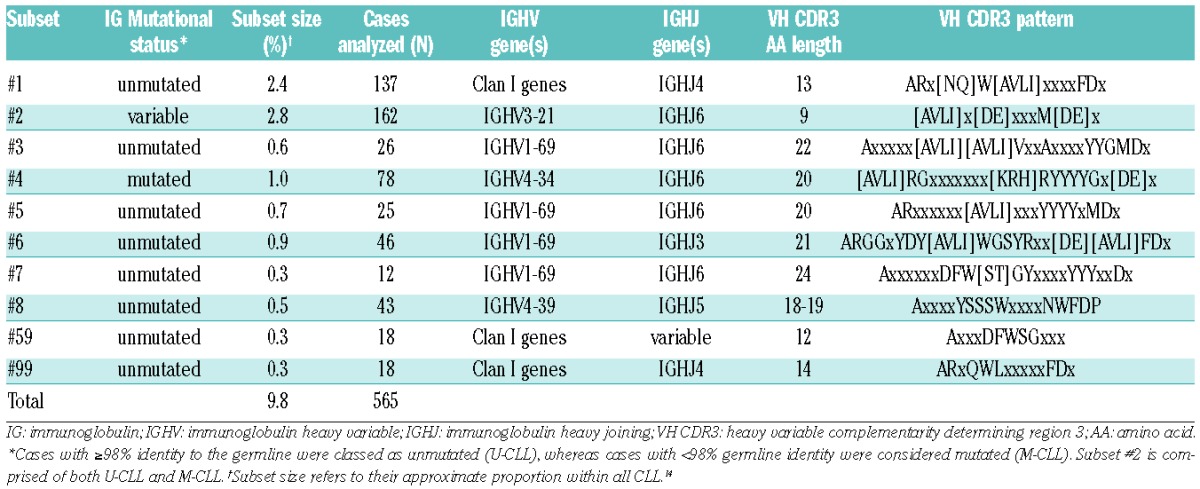
A total of 565 CLL patients, selected based on the expression of stereotyped BcR IGs leading to their assignment to a major subset,10,14 were included in this study (Table 1). A minimum requirement was that data be available for at least 10 cases/subsets to enable meaningful comparisons; this criterion resulted in 10 major subsets being evaluated. All cases were diagnosed according to the 2008 IWCLL criteria.29 Informed consent was collected according to the Declaration of Helsinki, and ethical approval was granted by local review committees.
Table 1.
Immunogenetic characteristics of the major stereotyped subsets analyzed in the present study.
Cytogenetic and SNP-array studies
Interphase fluorescence in situ hybridization (FISH) for the 13q14, 13q34, 11q22, 17p13 chromosomal regions and the centromere of chromosome 12 was performed as previously described.30 For 30 cases recurrent genomic aberration data was obtained using the Affymetrix 250K SNP Array.31
Sequence analysis of IGHV–IGHD–IGHJ rearrangements
PCR amplification, sequence analysis and interpretation of IGHV-IGHD-IGHJ rearrangements were performed following established international guidelines and using the IMGT® database and the IMGT/V-QUEST tool, as previously reported.2,7,8,10 Clonotypic IGHV gene sequences were defined as either mutated or unmutated based on the clinically relevant 98% cutoff value for identity to the closest germline gene.4,5
Assignment of cases to specific stereotyped subsets was performed following established guidelines and based on the following stringent criteria: the IG sequences must: (i) have ≥ 50% amino acid identity and 70% similarity within the variable heavy complementarity-determining region 3 (VH CDR3); (ii) have the same VH CDR3 length and the shared amino acid patterns must occur at identical codon positions; and (iii) utilize IGHV genes belonging to the same phylogenetic clan.13,14 The sole exception to these rules concerned subset #8, where the specific combination of IGHV4-39, IGHD6-13 and IGHJ5 genes resulted in a VH CDR3 motif that was shared by two subgroups of cases bearing VH CDR3s that differed in length by a single amino acid residue (18 and 19 amino acids) (Online Supplementary Table S1).10,14 For the present study, these two ‘sub-subsets’ were considered as a single entity. The immunogenetic characteristics of the subsets analyzed are provided in Table 1.
Gene mutation analysis
The date of the sample used for mutational analysis as well as the date of the first treatment was available for >80% of patients, and >80% of patients were tested prior to treatment (Online Supplementary Table S2). Mutational screening was performed for the following genes: NOTCH1, the entire exon 34 or targeted analysis for del7544_7545/p.P2514Rfs*4; TP53, exons 4–10 (depending on the medical center); SF3B1, exons 14–16; BIRC3, exons 6–9; and MYD88, exons 3 and 5 or targeted analysis for the p.L265P substitution (exon 5). Specific methodologies are detailed in the Online Supplementary Table S3.
Statistical analysis
Due to the complete absence or rarity of mutations in MYD88 and BIRC3, respectively, statistical analysis was only performed for NOTCH1, SF3B1 and TP53 gene mutations. Pearson’s Chi-squared test was used to evaluate the null hypothesis that the frequency of mutations within each of the aforementioned genes is equal among all subsets analyzed; the P value was computed by Monte Carlo simulation with 10 000 replicates. Comparisons between subsets were performed using the Fisher’s exact test and all tests were two-sided. P values were corrected for multiple comparisons using the Bonferroni method and the level of significance was set at P<0.001 (Online Supplementary Table S4). All calculations were performed using R (version 3.1.2).
Overall survival (OS) and time to first treatment (TTFT) were measured from the date of diagnosis until last follow-up/death, or date of initial treatment, respectively. Survival curves were constructed according to the Kaplan-Meier method, using Statistica Software 10.0 (Stat Soft Inc., Tulsa, OK, USA), and the log-rank test was used to determine the differences between survival proportions.
Results
Markedly different frequencies of genetic lesions amongst stereotyped CLL subsets
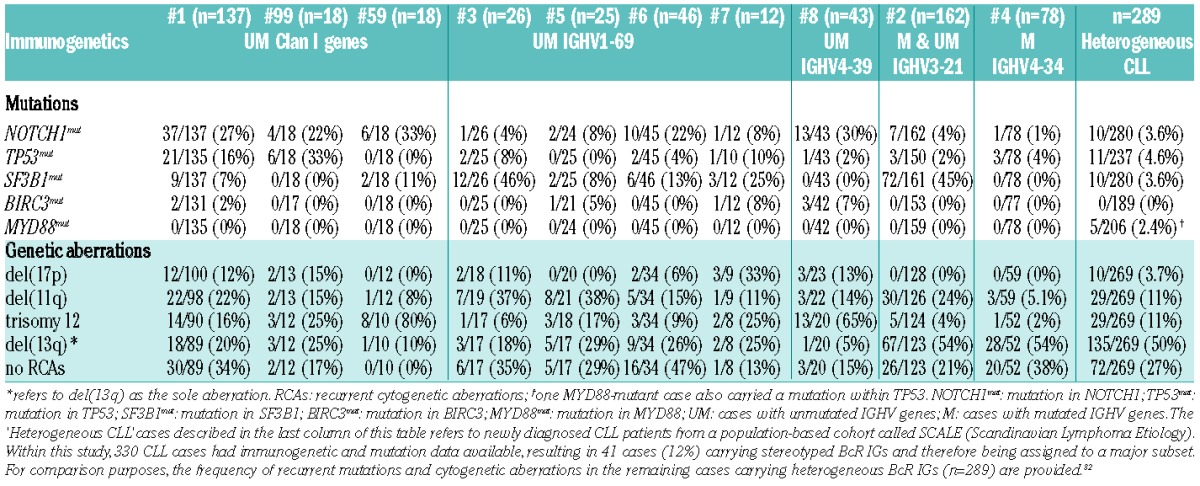
To investigate the associations between recurrent genetic lesions in CLL and BcR IG stereotypy, we profiled 565 patients assigned to one of 10 major stereotyped subsets (Table 1).10,14 Subset #4 cases (n=78) carried uniformly mutated BcR IGs (M-CLL), whereas subset #2 cases (n=162) exhibited significant heterogeneity with regard to SHM load, leading to 98 cases being considered as M-CLL and the remainder (n=64) as U-CLL (range: 93.3–99.7%; median: 97.8%). All remaining subsets concerned cases with unmutated or minimally mutated BcR IGs, hence constituting U-CLL. Mutational analysis for all 5 genes, BIRC3, MYD88, NOTCH1, SF3B1 and TP53, was performed for 520/565 (92%) cases; mutation data for one or two genes was lacking for 37 (6.5%) and 8 (1.5%) cases, respectively (Online Supplementary Table S5). For comparison purposes, data concerning the frequency of recurrent gene mutations and genetic lesions within non-stereotyped CLL is also provided in Table 2 and Online Supplementary Table S6.
Table 2.
Frequency of recurrent mutations and cytogenetic aberrations within each subset included in the present study.
MYD88 mutations
All cases analyzed (n=557) were devoid of MYD88 mutations, which, bearing in mind that our cohort was predominantly composed of U-CLL, was not surprising, since existing evidence indicates that MYD88 mutations exclusively occur in M-CLL.32–37 That said, 32% (176/557) of cases analyzed concerned M-CLL, subset #2 (n=98) (mixed SHM profile) and subset #4 (n=78), and the complete absence of MYD88 mutations amongst these cases implies that mutations within MYD88 are absent from M-CLL assigned to major stereotyped subsets.
BIRC3 mutations
Mutations within BIRC3 were infrequent (7/541 cases, 1.3%) and primarily concerned truncating mutations i.e. small frameshift deletions, duplications or insertions, as opposed to single nucleotide variants (Table 2; Online Supplementary Tables S6 and S7). BIRC3-mutated cases lacked del(17p), however a single subset #1 case did coexist with a TP53 mutation within the DNA-binding domain (Online Supplementary Table S7); the remaining cases harbored del(11q) (n=1), trisomy 12 (n=3) or both del(11q) and trisomy 12 (n=2). Five out of 7 (71%) BIRC3-mutant cases carried mutations within one of the other genes analyzed; 4/5 cases had concurrent mutations within NOTCH1. Irrespective of subset assignment, BIRC3 mutations never coincided with SF3B1 mutations (Online Supplementary Table S7).
NOTCH1 mutations
Mutations within exon 34 of the NOTCH1 gene were detected in 15% (82/563) of cases. The frequency of NOTCH1 mutations varied considerably among subsets and can be summarized as follows: (i) subsets #1, #59 and #99, all concerning U-CLL, exhibited high frequencies of NOTCH1 mutations (22%, 30% and 33%, respectively); (ii) NOTCH1 mutations were also enriched in subset #6 (22%), but relatively infrequent in subsets #3, #5 and #7 (4%, 8% and 8%, respectively), despite the fact that all these subsets utilize the IGHV1-69 gene and concern U-CLL; (iii) NOTCH1 mutations were prevalent in the clinically aggressive subset #8 (IGHV4-39/IGKV1(D)-39), ranging from 21–34% depending on the VH CDR3 length and 18 or 19 amino acids, respectively; (iv) aberrations within NOTCH1 were uncommon in subset #2, which comprises both U- and M-CLL, being present in only 4% of analyzed cases (7/162; 5/7 (71%) NOTCH1-mutant subset #2 cases concerned M-CLL while the remaining 2 cases were U-CLL); and, finally, (v) defects within NOTCH1 were rare in subset #4 (1/78; 1.3%) (Figure 1; Table 2; Online Supplementary Tables S1 and S6). To account for multiple testing, Bonferroni correction was performed and pairwise comparisons between subsets was checked at a level of significance of P<0.001, indicating that the frequencies of NOTCH1 mutations in subsets #2 and #4 were statistically significant compared to the frequencies observed in subsets #1, #6, #8 and #59 (Online Supplementary Table S4A). Although significance was not reached for other subset comparisons, this could in part be explained by the small number of cases included within each of these subsets, thus underscoring an inherent limitation when analyzing stereotyped subsets due to the fact that even the largest subsets account for less than 5% of all CLL cases.
Figure 1.
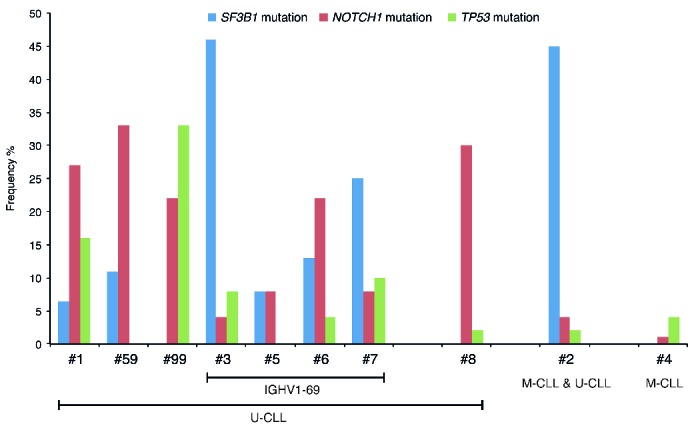
Recurrent gene mutations in stereotyped CLL subsets. The distribution of mutations within NOTCH1, SF3B1 and TP53 varied considerably between the major stereotyped subsets included in the present study. All cases were devoid of MYD88 mutations and BIRC3 mutations were rare, with no clear bias to any subset (this data is not shown in the graph). M-CLL: mutated IGHV gene; U-CLL: unmutated IGHV gene.
Although NOTCH1 mutations tended to coincide with trisomy 12, their co-occurrence differed among subsets (Table 2; Online Supplementary Table S6). Concurrent mutations were uncommon in NOTCH1-mutated cases, with the vast majority (79%; 65/82) devoid of mutations in the other four genes included in the study. The combinatorial patterns of mutations occurring in the remaining 21% of cases (17/82) were as follows: (i) NOTCH1 and TP53 mutations coexisted in 7/17 (41%) cases comprising subset #1 (n=4), subset #99 (n=2) and subset #6 (n=1); (ii) mutations in NOTCH1 and either SF3B1 or BIRC3 occurred at similar frequencies, 5/17 (29%) and 4/17 (24%), respectively, with no bias to any particular subset; and (iii) a single subset #1 case carried mutations in NOTCH1, TP53 and SF3B1. Notably, subsets harboring NOTCH1 mutations at high frequencies were either absent for (subsets #8 and #99) or concerned a single case with a co-occurring SF3B1 mutation (subsets #1, #6 and #59).
SF3B1 mutations
Mutations within the SF3B1 gene (hotspot exons 14–16) were detected in 19% (106/564) of cases. A very high frequency of SF3B1 mutations was not only observed in subset #2 (45%; 72/161) but also in subset #3 (46%; 12/26), which collectively accounted for 79% (84/106) of all SF3B1 mutated cases. The frequency of SF3B1 mutations in both subsets #2 and #3 sharply contrasted the frequency observed in all other subsets, and reached statistical significance when compared to subsets #1, #4, #8 and #99 (P<0.001, the frequency of SF3B1 mutations in subset #2 also reached significance when compared to subsets #5 and #6) (Figure 1; Table 2; Online Supplementary Tables S4B, S6 and S8). No significant difference in the distribution of SF3B1 mutations was observed between subset #2 M-CLL cases (47/98; 48%) versus subset #2 U-CLL cases (25/64; 39%) (P=0.27).
The SF3B1 mutation distribution within subset #2 was remarkably skewed, with 77% (58/75; 3 subset #2 cases carried 2 SF3B1 mutations) of mutations localized to two codons (p.K700E: n=43/75, 57%; p.G742D: n=15/75, 20%; Figure 2A; Online Supplementary Table S8). Similar to subset #2, the p.K700E substitution accounted for a high proportion of SF3B1 mutations in subset #3 (33%; 4/12), while the p.G742D mutation was absent (Figure 2A; Online Supplementary Table S8).
Figure 2.
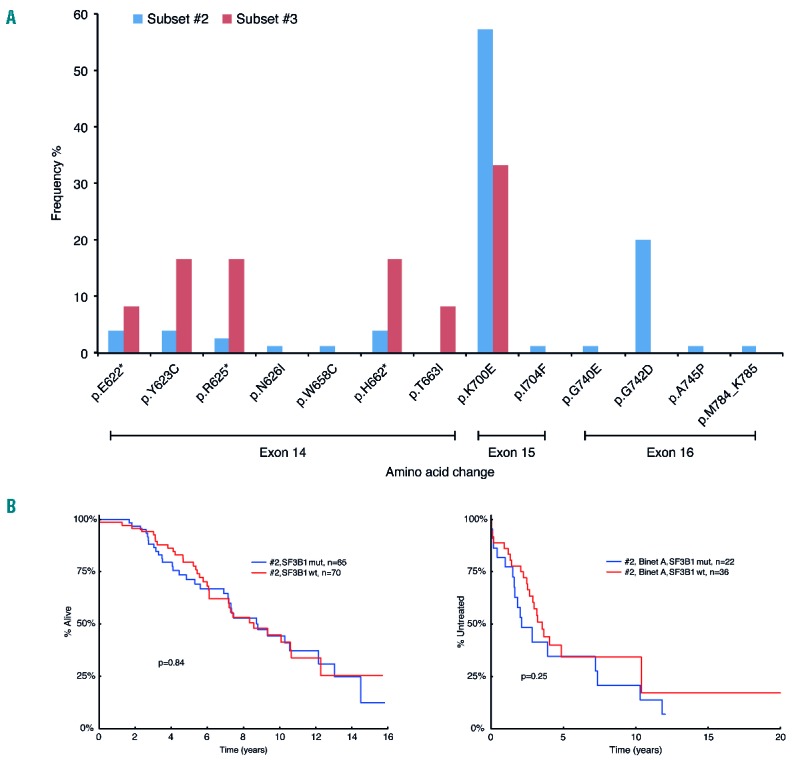
SF3B1 mutations in subsets #2 and #3. (A) Distribution of SF3B1 mutations in subsets #2 and #3. Overall, 45% (72/161) of subset #2 and 46% (12/26) of subset #3 cases were found to carry mutations within SF3B1. While the majority of subset #2 cases (69/72; 96%) carried a single SF3B1 mutation, 3 cases had 2 mutations within SF3B1 (2/3 cases carried the p.K700E change, with one case carrying both p.K700E and p.G742D). Although the most frequent amino acid change in both subsets involved a lysine to glutamic acid substitution at codon 700 (exon 15), representing 57% (43/75) and 33% (4/12) of all SF3B1 mutations in subsets #2 and #3, respectively, the overall distribution of mutations varied. To elaborate, in subset #3, all remaining SF3B1 mutations (excluding p.K700E) occur within exon 14. This is in contrast to the SF3B1 mutational profile in subset #2 where only 17% of mutations are found within exon 14 and where two particular substitutions account for 96% and 88% of the alterations observed in exon 15 and exon 16 (p.K700E and p.G742D, respectively). *indicates that more than one amino acid change occurred at this codon (detailed in Online Supplementary Table S8). (B) Prognostic implications of SF3B1 mutations within subset #2 on overall survival (OS) and time to first treatment (TTFT). (Binet A cases).
When considering cytogenetic aberrations, subset #2 was enriched for del(11q) (30/126; 24%) and isolated del(13q) (67/123; 54%), in line with previous reports.16,23,38 SF3B1-mutated subset #2 cases showed a negative association with isolated del(13q) (25/57 vs. 42/66; P=0.028). Within subset #2 cases, del(11q) was associated with shorter TTFT (Online Supplementary Figure S1A), whereas SF3B1 mutations had no significant impact on either OS or TTFT (for Binet A patients) (Figure 2B; Online Supplementary Figure S1B-S1F). Despite their immunogenetic differences, similar to subset #2, subset #3 was also enriched for del(11q) (7/19; 37%), with both SF3B1-mutant and wild-type subset #3 cases carrying del(11q) in similar proportions (Table 2).
Finally, across all subsets, SF3B1 mutations tended to occur in isolation, e.g. for SF3B1-mutant cases, 94% (68/72) of subset #2, 75% (9/12) of subset #3 and 83% (5/6) of subset #6 had no coincident mutations within any other gene analyzed. The sole exception concerned subset #1, within which 56% (5/9) of SF3B1-mutant cases coexisted with mutations in TP53 (one case also carried a mutation within NOTCH1).
TP53 mutations
Overall, the majority of cases analyzed had no mutations within TP53 (93%; 508/547). That said, we noted differences in the frequency of TP53 mutations amongst specific subsets, ranging from 0% to 33% (Figure 1; Table 2; Online Supplementary Tables S4C, S6 and S9). In particular, we found that TP53 mutations were: (i) absent in subset #59; (ii) rare in subsets #2 (3/150; 2%), #4 (3/78; 4%) and #8 (1/43; 2%); (iii) had varying, albeit low, frequencies in U-CLL subsets utilizing the IGHV1-69 gene (Subsets #3: 2/25 (8%); #5: 0/25 (0%); #6: 2/45 (4%) and #7: 1/10 (10%)); and (iv) were enriched in subsets #1 (21/135; 16%) and #99 (6/18; 33%); the latter representing a less populated subset that immunogenetically bears a high similarity to subset #1. The high frequency of TP53 mutations in these latter two subsets reached statistical significance compared to the frequency observed in subset #2 (Online Supplementary Table S4C). Of note, when incorporating del(17p) into our analysis, the frequency of TP53 aberrant cases in subsets #1 and #99 increased to 22% and 40%, respectively (Online Supplementary Table S9). Subsets #5 and #59 remained unaffected by any TP53 lesions, while the number of cases inflicted with TP53 defects increased to 3/8 (37.5%) for subset #7 and 4/19 (21%) for subset #3 (Online Supplementary Table S9).
As mentioned above, a significant proportion of TP53-mutated cases carried concurrent mutations in other genes analyzed. More specifically, almost 50% of TP53-mutant subset #1 cases were positive for either SF3B1 or NOTCH1 mutations (both 4/21, 19%), whereas one case was positive for all three mutations; an additional case carried a mutation within BIRC3. Regarding subset #99, 2/6 (33%) of TP53-mutant cases carried double mutations, with both cases carrying the recurrent 2 base pair deletion in NOTCH1.
Discussion
Within this study, by combining data concerning both recurrent gene mutations and cytogenetic aberrations in the largest series of stereotyped subset CLL cases studied to date for this purpose, we were able to offer important novel insights into the molecular mechanisms driving the pathogenesis and evolution of each subset. Overall, we document that the genetic makeup of individual stereotyped subsets is remarkably distinct, which alludes to the subset-biased acquisition and/or selection of genetic aberrations likely in the context of particular BcR signaling initiated by the subset-specific IG.
Beginning with U-CLL subsets, it is noteworthy that even when comparing subsets with BcR IGs utilizing the same IGHV gene, their genomic profiles are distinct, as exemplified by the subsets utilizing IGHV1-69 (subsets #3, #5, #6 and #7), which displayed markedly different spectra of genomic aberrations. For instance, SF3B1 mutations were detected in an impressive 46% of subset #3 cases versus only 8% of subset #5 cases. Similar observations regarding the skewed distribution of particular recurrent gene mutations were also apparent for NOTCH1 mutations, which were detected at a much higher frequency in subset #6 (22%) versus all other IGHV1-69 subsets (frequencies 4–8%); interestingly, within subset #6, NOTCH1 mutations rarely co-occurred with trisomy 12. Finally, TP53 defects due to mutations within TP53 and/or del(17p) ranged from 37.5% in subset #7 to 0% in subset #5.
Switching our focus to U-CLL subsets #8 and #59, two clinically aggressive subsets, with the former exhibiting the highest risk for Richter’s transformation amongst all CLL,15 revealed a very high frequency of trisomy 12 (65% and 80%, respectively) and an enrichment in NOTCH1 mutations (30% and 33%, respectively), with a relatively low frequency or complete absence of all other genomic aberrations evaluated.23
A similarly skewed distribution of genomic aberrations was identified for another very aggressive CLL subset, subset #2, which is the largest subset overall.14,23 Indeed, we herein confirm and significantly extend previous observations that subset #2 is remarkably enriched for mutations within the SF3B1 gene.26,27 Of note, the targeting profile of mutations in the hotspot regions of SF3B1 (exons 14–16) clearly differed in subset #2 from that in subset #3. Although the actual significance of these observations is currently unknown, the high frequency and striking bias of SF3B1 mutations to subsets #2 and #3 bodes strongly for their critical role in the pathobiology of these subsets. Furthermore, it supports the argument that the mechanisms underlying clinical aggressiveness in CLL are not uniform, but rather differ among the various disease subgroups.
Along these lines, the relative paucity of TP53 defects in subset #2 (2%) implies that the poor prognosis of subset #2 is attributable to other factors, with SF3B1 seemingly appearing as a top candidate. That said, when comparing subset #2 cases with/without SF3B1 mutations, no difference in OS or TTFT (Binet A cases) were observed, suggesting that SF3B1 dysregulation alone does not explain the clinical aggressiveness of subset #2. Since SF3B1 gene mutations are also found in other malignancies, most notably myelodysplastic syndrome, within which their presence appears to confer a more favorable prognosis,39 it is conceivable that the functional impact of SF3B1 mutations may be influenced by the specific microenvironment and/or differ in distinct blood cell lineages, hence producing context-dependent effects.
The discussion about aggressive CLL subsets culminates with subset #1, the biggest within U-CLL and second largest overall after subset #2.14 In contrast to the subsets mentioned above, the pattern of cytogenetic aberrations and recurrent gene mutations in subset #1 is quite heterogeneous (Figure 3A and 3B; Table 2). Of note, TP53 disruption (due to del(17p) and/or TP53 mutations), NOTCH1 mutations and trisomy 12 were all frequent in subset #1 (22%, 27% and 16% of cases, respectively); although lesions within both TP53 and NOTCH1 co-occurred in only 5 cases, all were negative for trisomy 12. Altogether, these findings support the existence of distinct genetic pathways of clonal evolution in subset #1, one influenced and dictated by TP53 disruption, while the other is dependent on trisomy 12 and/or NOTCH1 mutations. Regarding the latter, given the very recent discovery of recurrent mutations in the 3’ UTR of NOTCH1 in approximately 3% of CLL patients,40 it is plausible that their frequency in both subset #1 and CLL at large has been underestimated to date, nevertheless, this does not detract from the apparent non-random association of the p.P2514Rfs*4 NOTCH1 mutation reported herein with certain stereotyped CLL subsets.
Figure 3.

Main biological associations within subset #1. (A) Concurrent mutations within subset #1. Only cases for which the mutational status of all 3 genes (NOTCH1, SF3B1 and TP53) was available were included in the figure (n=135). Fifty-seven cases had a mutation in at least one of the gene hotspots whereas 78 cases were wild-type for these genes. (B) Spectrum of mutations and genomic aberrations within subset #1. Despite a high frequency of NOTCH1 mutations, a large proportion (58%) of subset #1 cases carried no mutations within the 5 genes analyzed. Specifically, considering the subset #1 cases lacking any recurrent gene mutations, 35% also lacked any recurrent genetic aberrations. Collectively, this resulted in the absence of any recurrent gene mutation or cytogenetic aberration in approximately 20% of subset #1 cases, thereby implying that additional mechanisms must account for the clinically aggressive nature of this subset. Only cases for which the mutational status of all 3 genes (NOTCH1, SF3B1 and TP53) was available were included in the figure (n=135). *indicates that none of the known recurrent genomic aberrations were present; NOTCH1mut: mutation in NOTCH1 only; TP53mut: mutation in TP53 only; SF3B1mut: mutation in SF3B1 only. Concurrentmut refers to the presence of mutations in more than one of the genes analyzed. Absolute numbers and percentages are provided in brackets. For del(17p), 2/53 correspond to the 4% indicated in the figure. (C) The frequency of NOTCH1 mutations in subset #1 cases varies depending on specific IGHV gene usage. Only the top 3 utilized IGHV genes within subset #1 patients in our cohort were included in the graph, collectively accounting for 73% (99/136) subset #1 cases. Mutations within NOTCH1 were found to be particularly frequent in subset #1 cases expressing IGHV1-2*02 (17/39; 44%).
We next integrated these findings with the specific immunogenetic nature of subset #1, taking into consideration the fact that cases assigned to this subset differ from most major CLL subsets, in that they do not all express the same IGHV gene but rather carry IGHV genes that share common ancestry and thus, belong to the same IGHV phylogenetic clan, clan I (comprising IGHV1, IGHV5 and IGHV7 genes).10,14 Focusing on the IGHV1-2*02 and IGHV1-3*01 genes, which accounted for 64% of all subset #1 cases (28.7% and 35.3%, respectively), it was interesting to note that 17/39 (43.6%) IGHV1-2*02 expressing cases carried mutations within NOTCH1 compared to only 7/48 (14.6%) IGHV1-3*01 expressing subset #1 cases (P=0.003) (Figure 3C). This finding further exemplifies how the expression of a particular stereotyped immunoglobulin may be linked to a distinct evolutionary pathway through the acquisition of specific genomic aberrations. Whether these genomic differences may translate into different clinical outcomes remains to be elucidated.
Another important observation relates to the finding that approximately 20% of subset #1 cases were negative for any of the genetic lesions tested for (mutations or aberrations). Thus, for at least a proportion of subset #1 cases, it appears that the full extent of their genomic complexity has yet to be revealed, and additional mechanisms must underlie their clinical aggressiveness. This claim is supported by recent high-throughput studies which indicate that mutations within the NFKBIE or RPS15 gene may serve as novel pathogenic mechanisms linked to a more aggressive disease.41,42 While highlighting an inherent limitation of the present study, which was restricted to the analysis of 5 recurrently mutated genes, these results, together with the recent finding of non-coding recurrent mutations in CLL, emphasize the need for more comprehensive approaches, such as whole-genome sequencing, in order to obtain a complete picture of the genomic landscape of CLL subsets.40
Finally, we looked to subset #4, the largest subset within M-CLL and now considered as a prototype for indolent disease.14,23 Previous studies have reported that subset #4 is virtually devoid of cytogenetic aberrations associated with adverse prognosis [del(11q) and, especially, del(17p)].16 Herein, we not only confirm and significantly extend these observations, but also take a decisive step further by showing that subset #4 essentially lacks recurrent gene mutations, at least amongst the five genes analyzed. These results provide insights into the ‘mild’ genomic background of subset #4 CLL, which is reflected in the indolent clinical course experienced by these patients.
Although our understanding of the genetic basis, clonal architecture and evolution in CLL pathogenesis is rapidly advancing, the CLL cell of origin and the precise timeline of events leading to leukemogenesis remain elusive. It has recently been proposed that hematopoietic stem cells (HSCs) may be involved in CLL pathogenesis, since they have been shown to harbor mutations within potential CLL oncogenes.43,44 These cells could undergo antigen-driven clonal selection and progressively acquire additional genetic abnormalities giving rise to a clonal B cell expansion, termed monoclonal B-cell lymphocytosis (MBL), which we know invariably precedes CLL, and ultimately may progress to CLL. However, in trying to reconcile the above scenario with the biology of CLL, this model fails to account for the fact that none of the recurrent CLL chromosomal abnormalities were found in the HSC compartment; how could such clones be selected and subsequently expand into CLL¿ In addition, CLL patients who achieve remissions do not relapse with polyclonal disease or distinct clones. Further studies are therefore required to both confirm and clarify the precise role of HSCs in CLL pathogenesis.
A minute proportion of MBL transform to overt CLL requiring treatment (1–2%/year), thus significant somatic changes coupled with microenvironmental factors must occur.45 The existence of stereotyped BcRs together with the tendency of CLL cells to express poly- and auto-reactive BcRs are indicative of selective pressures, such as autoantigens or microbial pathogens, that favor specific IG gene rearrangements. Such persistent stimulation through the surface IG may be the central event that drives the evolution from a preleukemic state to overt leukemia, thereafter chronic BcR engagement may favor the selection of a monoclonal population which subsequently acquires genetic alterations, which in some instances may provide a survival and growth advantage.45
In conclusion, our present findings imply that distinctive modes of microenvironmental interactions, mediated by certain stereotyped BcRs, may be associated with the selection or occurrence of particular genetic aberrations, with the combined effect determining both clonal and clinical evolution, and ultimately disease outcome. Our study further serves to highlight the fact that even findings within smaller subsets help to dissect the pathophysiology of CLL, and may eventually have clinical usability akin to the known prognostication value of TP53 gene mutations, despite being present in only 5–10% of cases at diagnosis or the association between subset #8 (0.6% of all CLL) and Richter’s transformation.15,46 Overall, since the distinct genomic profiles evidenced amongst stereotyped CLL subsets are potentially linked to varying mechanisms of clinical aggressiveness based on a reliance on specific intracellular signaling pathways, our findings may have important implications for patient monitoring and therapeutic management in the era of targeted pathway inhibition.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/8/959
Funding
The authors would like to thank the Swedish Cancer Society, the Swedish Research Council, The Lion’s Cancer Research Foundation and Selander’s Foundation, Uppsala; the ENosAI project (code 09SYN-13-880) co-funded by the EU and the General Secretariat for Research and Technology of Greece; the KRIPIS action, funded by the General Secretariat for Research and Technology of Greece; the AIRC Special Program Molecular Clinical Oncology, 5 × 1000, #9965 and 10007 and Investigator grant, Milano, Italy); Ricerca Finalizzata 2010 - Ministero della Salute, Roma, Italy; Leukaemia and Lymphoma Research, Kay Kendall Leukaemia Fund, UK; the Spanish ICGC-CLL Genome Project funded by the Instituto de Salud Carlos III (ISCIII); The Danish Cancer Society; the research grants IGA MZ CR NT13493-4/2012 and MSMT CR VaVPI project CZ.1.05/1.1.00/02.0068 (CEITEC); H2020 “AEGLE, An analytics framework for integrated and personalized healthcare services in Europe”, and H2020 “MEDGENET, Medical Genomics and Epigenomics Network” (No.692298), both funded by the European Commission, for their financial support; and also Barbara Kantorova and Veronika Navrkalova for their technical help with NOTCH1 and SF3B1 sequencing analysis of the Czech cohort.
References
- 1.Hashimoto S, Dono M, Wakai M, et al. Somatic diversification and selection of immunoglobulin heavy and light chain variable region genes in IgG+ CD5+ chronic lymphocytic leukemia B cells. J Exp Med. 1995;181(4):1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fais F, Ghiotto F, Hashimoto S, et al. Chronic lymphocytic leukemia B cells express restricted sets of mutated and unmutated antigen receptors. J Clin Invest. 1998;102(8):1515–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dighiero G, Hamblin TJ. Chronic lymphocytic leukaemia. Lancet. 2008; 371(9617): 1017–1029. [DOI] [PubMed] [Google Scholar]
- 4.Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–1847. [PubMed] [Google Scholar]
- 5.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–1854. [PubMed] [Google Scholar]
- 6.Tobin G, Thunberg U, Johnson A, et al. Chronic lymphocytic leukemias utilizing the VH3-21 gene display highly restricted Vlambda2-14 gene use and homologous CDR3s: implicating recognition of a common antigen epitope. Blood. 2003;101(12): 4952–4957. [DOI] [PubMed] [Google Scholar]
- 7.Ghiotto F, Fais F, Valetto A, et al. Remarkably similar antigen receptors among a subset of patients with chronic lymphocytic leukemia. J Clin Invest. 2004;113(7):1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Messmer BT, Albesiano E, Efremov DG, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med. 2004;200(4):519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tobin G, Thunberg U, Karlsson K, et al. Subsets with restricted immunoglobulin gene rearrangement features indicate a role for antigen selection in the development of chronic lymphocytic leukemia. Blood. 2004;104(9):2879–2885. [DOI] [PubMed] [Google Scholar]
- 10.Stamatopoulos K, Belessi C, Moreno C, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood. 2007;109(1):259–270. [DOI] [PubMed] [Google Scholar]
- 11.Murray F, Darzentas N, Hadzidimitriou A, et al. Stereotyped patterns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood. 2008;111(3):1524–1533. [DOI] [PubMed] [Google Scholar]
- 12.Bomben R, Dal Bo M, Capello D, et al. Molecular and clinical features of chronic lymphocytic leukaemia with stereotyped B cell receptors: results from an Italian multicentre study. Br J Haematol. 2009;144(4): 492–506. [DOI] [PubMed] [Google Scholar]
- 13.Darzentas N, Hadzidimitriou A, Murray F, et al. A different ontogenesis for chronic lymphocytic leukemia cases carrying stereotyped antigen receptors: molecular and computational evidence. Leukemia. 2010;24(1):125–132. [DOI] [PubMed] [Google Scholar]
- 14.Agathangelidis A, Darzentas N, Hadzidimitriou A, et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood. 2012;119(19):4467–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossi D, Spina V, Cerri M, et al. Stereotyped B-cell receptor is an independent risk factor of chronic lymphocytic leukemia transformation to Richter syndrome. Clin Cancer Res. 2009;15(13):4415–4422. [DOI] [PubMed] [Google Scholar]
- 16.Marincevic M, Cahill N, Gunnarsson R, et al. High-density screening reveals a different spectrum of genomic aberrations in chronic lymphocytic leukemia patients with ‘stereotyped’ IGHV3-21 and IGHV4-34 B-cell receptors. Haematologica. 2010;95(9):1519–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marincevi3c M, Mansouri M, Kanduri M, et al. Distinct gene expression profiles in subsets of chronic lymphocytic leukemia expressing stereotyped IGHV4-34 B-cell receptors. Haematologica. 2010;95(12): 2072–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arvaniti E, Ntoufa S, Papakonstantinou N, et al. Toll-like receptor signaling pathway in chronic lymphocytic leukemia: distinct gene expression profiles of potential pathogenic significance in specific subsets of patients. Haematologica. 2011;96(11):1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maura F, Cutrona G, Fabris S, et al. Relevance of stereotyped B-cell receptors in the context of the molecular, cytogenetic and clinical features of chronic lymphocytic leukemia. PloS one. 2011;6(8):e24313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ntoufa S, Vardi A, Papakonstantinou N, et al. Distinct innate immunity pathways to activation and tolerance in subgroups of chronic lymphocytic leukemia with distinct immunoglobulin receptors. Mol Med. 2012;18:1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanduri M, Marincevic M, Halldorsdottir AM, et al. Distinct transcriptional control in major immunogenetic subsets of chronic lymphocytic leukemia exhibiting subset-biased global DNA methylation profiles. Epigenetics. 2012;7(12):1435–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papakonstantinou N, Ntoufa S, Chartomatsidou E, et al. Differential microRNA profiles and their functional implications in different immunogenetic subsets of chronic lymphocytic leukemia. Mol Med. 2013;19:115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baliakas P, Hadzidimitriou A, Sutton L, et al. Clinical effect of stereotyped B-cell receptor immunoglobulins in chronic lymphocytic leukaemia: a retrospective multicentre study. Lancet Haematol. 2014;1(2): 74–84. [DOI] [PubMed] [Google Scholar]
- 24.Del Giudice I, Chiaretti S, Santangelo S, et al. Stereotyped subset #1 chronic lymphocytic leukemia: a direct link between B-cell receptor structure, function, and patients’ prognosis. Am J Hematol. 2014;89(1):74–82. [DOI] [PubMed] [Google Scholar]
- 25.Gounari M, Ntoufa S, Apollonio B, et al. Excessive antigen reactivity may underlie the clinical aggressiveness of chronic lymphocytic leukemia stereotyped subset #8. Blood. 2015;125(23):3580–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strefford JC, Sutton LA, Baliakas P, et al. Distinct patterns of novel gene mutations in poor-prognostic stereotyped subsets of chronic lymphocytic leukemia: the case of SF3B1 and subset #2. Leukemia. 2013;27(11):2196–2199. [DOI] [PubMed] [Google Scholar]
- 27.Rossi D, Spina V, Bomben R, et al. Association between molecular lesions and specific B-cell receptor subsets in chronic lymphocytic leukemia. Blood. 2013;121(24):4902–4905. [DOI] [PubMed] [Google Scholar]
- 28.Malcikova J, Stalika E, Davis Z, et al. The frequency of TP53 gene defects differs between chronic lymphocytic leukaemia subgroups harbouring distinct antigen receptors. Br J Haematol. 2014;166(4):621–625. [DOI] [PubMed] [Google Scholar]
- 29.Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baliakas P, Iskas M, Gardiner A, et al. Chromosomal translocations and karyotype complexity in chronic lymphocytic leukemia: a systematic reappraisal of classic cytogenetic data. Am J Hematol. 2014; 89(3):249–255. [DOI] [PubMed] [Google Scholar]
- 31.Gunnarsson R, Isaksson A, Mansouri M, et al. Large but not small copy-number alterations correlate to high-risk genomic aberrations and survival in chronic lymphocytic leukemia: a high-resolution genomic screening of newly diagnosed patients. Leukemia. 2010;24(1):211–215. [DOI] [PubMed] [Google Scholar]
- 32.Cortese D, Sutton LA, Cahill N, et al. On the way towards a ‘CLL prognostic index’: focus on TP53, BIRC3, SF3B1, NOTCH1 and MYD88 in a population-based cohort. Leukemia. 2014;28(3):710–713. [DOI] [PubMed] [Google Scholar]
- 33.Baliakas P, Hadzidimitriou A, Sutton LA, et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia. 2015;29(2):329–336. [DOI] [PubMed] [Google Scholar]
- 34.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez-Trillos A, Pinyol M, Navarro A, et al. Mutations in TLR/MYD88 pathway identify a subset of young chronic lymphocytic leukemia patients with favorable outcome. Blood. 2014;123(24):3790–3796. [DOI] [PubMed] [Google Scholar]
- 37.Baliakas P, Hadzidimitriou A, Agathangelidis A, et al. Prognostic relevance of MYD88 mutations in CLL: the jury is still out. Blood. 2015;126(8):1043–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. [DOI] [PubMed] [Google Scholar]
- 39.Malcovati L, Papaemmanuil E, Bowen DT, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011;118(24):6239–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puente XS, Bea S, Valdes-Mas R, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519–524. [DOI] [PubMed] [Google Scholar]
- 41.Mansouri L, Sutton LA, Ljungstrom V, et al. Functional loss of IkappaBepsilon leads to NF-kappaB deregulation in aggressive chronic lymphocytic leukemia. J Exp Med. 2015;212(6):833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ljungstrom V, Cortese D, Young E, et al. Whole-exome sequencing in relapsing chronic lymphocytic leukemia: clinical impact of recurrent RPS15 mutations. Blood. 2016;127(8):1007–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kikushige Y, Ishikawa F, Miyamoto T, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20(2):246–259. [DOI] [PubMed] [Google Scholar]
- 44.Damm F, Mylonas E, Cosson A, et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014;4(9):1088–1101. [DOI] [PubMed] [Google Scholar]
- 45.Sutton LA, Rosenquist R. Deciphering the molecular landscape in chronic lymphocytic leukemia: time frame of disease evolution. Haematologica. 2015;100(1):7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pospisilova S, Sutton LA, Malcikova J, et al. Innovation in the prognostication of chronic lymphocytic leukemia: how far beyond TP53 gene analysis can we go¿ Haematologica. 2016;101(3):263–265. [DOI] [PMC free article] [PubMed] [Google Scholar]