

ABSTRACT
The identification of functional monoclonal antibodies directed against G-protein coupled receptors (GPCRs) is challenging because of the membrane-embedded topology of these molecules. Here, we report the successful combination of llama DNA immunization with scFv-phage display and selections using virus-like particles (VLP) and the recombinant extracellular domain of the GPCR glucagon receptor (GCGR), resulting in glucagon receptor-specific antagonistic antibodies. By immunizing outbred llamas with plasmid DNA containing the human GCGR gene, we sought to provoke their immune system, which generated a high IgG1 response. Phage selections on VLPs allowed the identification of mAbs against the extracellular loop regions (ECL) of GCGR, in addition to multiple VH families interacting with the extracellular domain (ECD) of GCGR. Identifying mAbs binding to the ECL regions of GCGR is challenging because the large ECD covers the small ECLs in the energetically most favorable ‘closed conformation’ of GCGR. Comparison of Fab with scFv-phage display demonstrated that the multivalent nature of scFv display is essential for the identification of GCGR specific clones by selections on VLPs because of avid interaction. Ten different VH families that bound 5 different epitopes on the ECD of GCGR were derived from only 2 DNA-immunized llamas. Seven VH families demonstrated interference with glucagon-mediated cAMP increase. This combination of technologies proved applicable in identifying multiple functional binders in the class B GPCR context, suggesting it is a robust approach for tackling difficult membrane proteins.
KEYWORDS: DNA immunizations, glucagon receptor, llama, phage display, single-chain Fv, virus-like particles
Introduction
GPCRs, transporters and ion channels, constitute the largest family of membrane protein targets (MPTs) in drug discovery.1 To date, these are mostly targeted by small molecule compounds to modify their function, but poor drug-like properties or pharmacokinetics of the drugs are still a problem. Therapeutic monoclonal antibodies (mAbs) against these complex targets have become an interesting approach.2
While modulating antibodies against Class B and C GPCRs, and against transporters and ion channels (e.g., P2×3 and P2×7) with relatively large extracellular domains (ECD), have been reported,3-5 functional antibodies against class A GPCRs and most of the transporters and ion channels with a small extracellular domain and small extracellular loops have been more challenging, and examples of mAbs raised against these targets are rare.6,7
Hybridoma technology and phage display are 2 technologies commonly used for mAb discovery. For both technologies, antigen is required for immunization and for selection and screenings of antigen-specific clones. Identification of functional mAbs against MPTs is technically challenging because of the membrane-embedded topology of these molecules. GPCRs typically contain 7-transmembrane domains, while, for transporters and ion channels, the number of transmembrane domains can go up to 24. MPTs are dependent on a membrane environment to maintain their natural structure, and this makes it difficult to maintain the proper folding when they are expressed as soluble proteins. An additional complexity is the poor expression level of most of these MPTs, probably due to toxic effects when overexpressed, which limits accessibility of potential epitopes. MPTs are thus difficult to use for immunization, selection and screening. However, several technologies have been established to address these problems.8 Cells overexpressing the GPCR of interest or membrane fractions derived thereof are often used for immunizations. The disadvantage of immunizations with such cell-based materials is that the immune response is also directed against other membrane components, and membrane fractions lose their out-side-out orientation, directing the immune response against the intracellular epitopes as well. To prevent these off-target responses, DNA immunization, in which the host animal cells express the GPCR of interest, is an attractive approach.9
In this report, we addressed several of the complex issues with identification of functional mAbs against MPTs, using the GPCR glucagon receptor (GCGR), as a model molecule. GCGR belongs to the secretin-like type-B GPCRs, and equilibrates between an open and closed conformation. In the open conformation, the extracellular (ECD) is perpendicular to the membrane, while in the closed conformation the ECD covers the extracellular loop regions (ECL). Glucagon preferentially binds the open conformation where the C-terminal domain of glucagon binds the ECD, facilitating the penetration of the N-terminal half of glucagon into a cavity formed by the 7-TM domain.3,10,11 Active immunization of outbred llamas and phage display for the generation of potent therapeutic mAbs against membrane proteins has proven powerful.12,13 Here, we describe the successful combination of DNA immunization of outbred llamas with scFv-phage display, and selections using virus-like particles (VLP) for the identification of glucagon receptor-specific antagonistic mAbs.
Results
DNA immunization raises target-specific immune responses against GCGR
For generation of a GCGR-specific immune response, 4 llamas were immunized with pcDNA3.1 encoding the GCGR protein (aa1-477) under control of constitutively active CMV promoter. DNA injections were repeated a total of 4 times with 2-week intervals, followed by a single subcutaneous cell boost with dromedary Caki14 cells overexpressing the GCGR. Specific immune responses to GCGR were measured by flow cytometry on Caki cells expressing GCGR (Caki-GCGR), with ELISA on virus-like particles derived from GCGR overexpressing HEK293 cells (VLP-GCGR), and on recombinant extracellular domain of GCGR (ECD-GCGR, aa1-147). All four llamas showed a specific immune response to Caki-GCGR cells compared to that of the parental Caki cells (Fig. 1A). The immune responses were further evaluated on VLP-GCGR and ECD-GCGR. Three of the 4 llamas showed a response measured on VLP-GCGR (Fig. 1B). While llama 15 did not show a response to VLP-GCGR, there was a high immune titer measurable on ECD-GCGR (Fig. 1C). The anti-llama IgG mAb used to detect the immune response is directed against the conventional antibodies of camelids, which was called IgG1.15 DNA immunization without boost was already sufficient to generate a llama IgG1 response as shown in Fig. 1D for llama 73, indicating that class switch and affinity maturation had taken place even without a boost with cells. Analysis of the immune response to immobilized ECD-GCGR over time showed that all 4 llamas had a high GCGR-specific response already after the third DNA immunization (Fig. S1). Llama 73 had the highest increase with an EC50 of 0.8% after 3 DNA immunizations (3XDNA), which was improved 20-fold after the Caki-GCGR boost (Fig. 1D). A boost with Caki-GCGR cells increased the immune titers for all llamas, except llama 52. Taken together, our results suggest that DNA immunization is a very effective technology to obtain a specific immune response to membrane-embedded GCGR.
Figure 1.
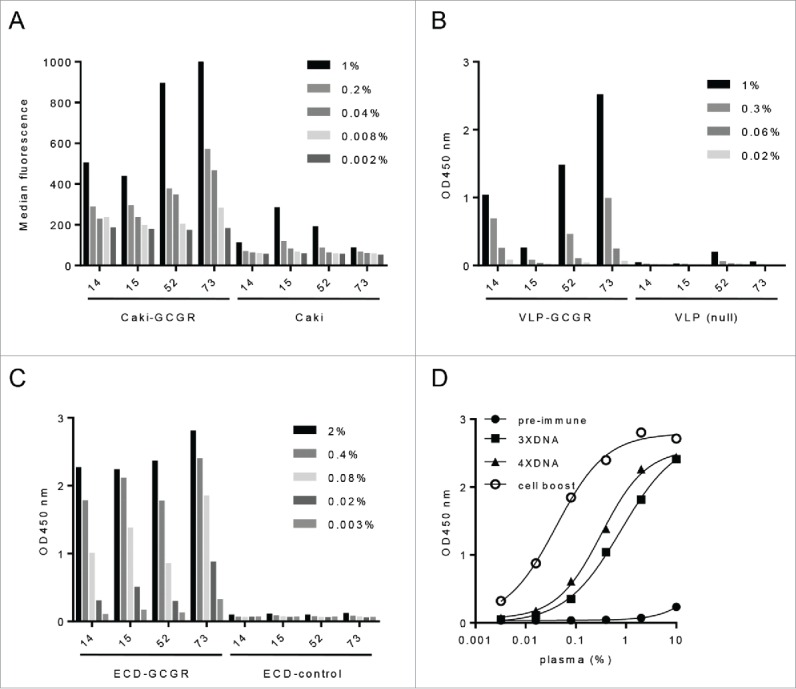
GCGR-specific IgG1 immune responses from DNA immunized llamas. (A) Immune response (% plasma) after Caki-GCGR boost against Caki-GCGR compared to Caki parental, measured by flow cytometry and binding expressed as median fluorescence. (B) Immune response (% plasma) against VLP-GCGR after Caki-GCGR boost compared to VLP-null measured by ELISA at OD 450 nm. (C) Immune response (% plasma) against ECD-GCGR compared to control after Caki-GCGR boost measured by ELISA at OD 450 nm. (D) Immune response (% plasma) against ECD-GCGR over time (pre-immune, 3XDNA, 4XDNA and cell boost) for llama 73 measured by ELISA at OD 450 nm.
Identification of specific antibodies binding to the extracellular domain and extracellular loops of GCGR
Fab phage selections on recombinant ECD-GCGR identify specific clones binding to different epitopes on the extracellular domain. To identify GCGR-specific antibodies, Fab expressing phage libraries were constructed from peripheral blood lymphocytes (PBL) of the GCGR positive llama 14 and 73 after the boost with Caki-GCGR cells. Phage selections showed 1,000-10,000-fold enrichment of binding phage over the control on immobilized recombinant ECD-GCGR after 3 rounds of selection. Individual clones were screened as Fab in periplasmic extracts for binding to ECD-GCGR using surface plasmon resonance (SPR) with 56% (158 of 280 clones) having off rates in the low, 10−3 (s−1), range correlating with low nanomolar affinities. Sequencing revealed a total of 10 different VH families based on HCDR3 sequences and lengths (Fig. S2).16 One representative clone of each VH family was reformatted into human IgG1. GCGR binding on cells was confirmed with flow cytometry with EC50 of 2–20 nM (Table 1). Affinities of the mAbs to cells expressing GCGR were in the range of affinities (KD) measured by SPR on ECD-GCGR (Table 1).
Table 1.
Binding characteristics of GCGR specific mAbs (defined by VH family) selected from Fab libraries on ECD-GCGR by SPR (KD) and flow cytometry on CHO-GCGR (EC50).
| Clone | VH family (based on HCDR3 seq*) | KD(nM) | CHO-GCGR (EC50 nM) |
|---|---|---|---|
| 1C3 | 1 | 4.7 | 5.2 |
| 9H3 | 2 | 33 | 11 |
| 1G7 | 3 | 5.2 | 9.4 |
| 1G3 | 4 | 1.4 | 13 |
| 6C6 | 5 | 4.5 | 19 |
| 7B3 | 6 | 13 | 4.1 |
| 8E2 | 7 | 4.9 | 2.6 |
| 8G5 | 8 | 3.5 | 9.4 |
| 8B1 | 9 | 7.9 | 1.7 |
| 9D7 | 10 | 2.2 | 4.5 |
complete sequences (heavy chain and light chain) of the clones of the different VH families are shown in Fig. S2.
scFv phage display identifies additional ECD-GCGR specific clones. To further expand the panel of GCGR specific clones, we performed selections on full-length native GCGR on VLP derived from GCGR overexpressing HEK293 cells (VLP-GCGR). After three rounds of selection with Fab phage libraries 14 and 73 (Vκ and Vλ), only the 73Vκ library showed limited enrichment, but screening on ECD-GCGR did not yield GCGR-specific clones. The background on VLP-null was relatively high for all libraries, and screening on VLP-GCGR and recombinant ECD-GCGR did not yield any specific GCGR clones. ECD-GCGR selections were included as control, and gave 100-fold enrichments over the irrelevant control, but no new VH families were identified.
At this point, we had shown that Fab selections on ECD-GCGR generated specific clones binding to native GCGR on cells, but we could not identify Fab binding to GCGR on VLP. As previously identified, specific VHH can be readily selected against GPCR (CXCR4) expressed on VLP (Fig. S3), which may be due to their multivalent display on phage, resulting in avid binding to its immobilized target. We therefore hypothesized that converting the Fab libraries into scFv libraries would display the antibody fragments multivalent by the phage17-19 and possibly allow selection of additional GCGR binders, including the ones directed against the extracellular loops. Fig. S4, shows 100-fold higher phage titers of a GCGR-ECD-binding phage clone, 1C3 when coated with GCGR-VLP compared to recombinant GCGR-ECD. This indicates that there are fewer target molecules available on the GCGR-VLP compared to recombinant ECD-GCGR. ScFv display of 1C3 did not show much advantage over Fab display when selected on a 2 µg/ml coating of recombinant ECD-GCGR. However, when we selected on a coating of 5 U/well GCGR-VLP, the phage output titer was 100-fold higher with scFv display. This demonstrates that multivalent display of 1C3 scFv has an advantage over monovalent Fab display when there are low target densities.
ScFv libraries (Vλ and Vκ) based on the RNA samples of the DNA immunized and Caki-GCGR boosted llama 73 were constructed, and phage selections on the recombinant ECD-GCGR and VLP-GCGR were performed with Fab and scFv libraries in parallel. After a first round on VLP-GCGR, a second-round selection was performed on ECD-GCGR to investigate if we had enriched for ECD-GCGR binders in the first round. Both the Fab and scFv libraries from the first round on ECD-GCGR showed phage enrichments over the control, whereas only scFv display showed clear enrichment (±1000 -fold) on ECD-GCGR after a first round on VLP-GCGRs (Fig. 2A and 2B). Screening for binding to ECD-GCGR after 2 rounds of selections revealed 77% positive binders (32/45). Most of the selected scFv clones after the second round on GCGR-VLPs showed the full-length scFv-encoding sequence as compared to the Fab fragments, which were truncated.
Figure 2.

Enrichment of GCGR specific phage after 3 rounds of VLP selections, scFv ELISA and mAb binding to GCGR expressing HEK293 cells. (A) GCGR-specific enrichments after 3 rounds of selection with libraries Fab and scFv 73Vκ and (B) libraries Fab and scFv 73Vλ. (C) GCGR-specific phage enrichments with and without counter selections with VLP-null. (D) scFv from library scFv 73Vλ tested for binding in ELISA with immobilized GCGR-VLP or negative control CXCR4-VLP or (E) immobilized ECD-GCGR. Detected with anti-c-myc and read at OD 450 nm. (F) FACS binding of mAbs to GCGR- (dark blue) or GCGRΔECD- (green) expressing HEK293 cells with and without ECD-GCGR competition (light blue). MOCK was included as a negative control (red). mAbs were detected with anti-human Fc-PE.
Positive clones from the scFv libraries belonged to previously identified VH families 1, 2 and 10 from 73 FabVλ. In addition, 6 new VH families were identified from 73 FabVλ (VH families 11–16), also binding to ECD-GCGR. ScFv recognizing ECD-GCGR was measured using SPR and revealed off rates (kd) of 3.3-0.3×10−3 (s−1) (Fig. S5). No clones were screened from the Fab libraries.
Counter selection with VLP-null and ECD-GCGR removes background binders and identifies ECD and ECL GCGR binders. To reduce the background binding to VLP-null (not containing GCGR), input phage from scFv libraries 73Vκ and Vλ, were pre-incubated with a 10-fold excess of VLP-null in suspension, and selected on immobilized VLP-GCGR in a first round. Selections without VLP-null counter selections were included as control. In second and third selection rounds, ECD-GCGR was included with and without VLP-null counter selections. After the second selection round, there was only enrichment on the ECD-GCGR and no difference in enrichment over the VLP-null control, indicating enrichment for specific binders in the first round on VLP-GCGR with VLP-null counter selections. Continuing with the third round, we observed a 300-fold enrichment on the VLP-GCGR and the ECD-GCGR after VLP-null counter selections in all 3 rounds, indicating more enrichment of GCGR-specific clones during the previous rounds including the counter selections (Fig. 2C). While the enrichments for VLP-GCGR were similar with and without counter selection; the background on the VLP-null was strongly reduced by counter selection.
Clones originating from round 3, counter selected with excess of VLP-null and selected on the VLP-GCGR, were investigated for specific binding to full-length GCGR and to ECD-GCGR in ELISA. Binding ELISA revealed that most GCGR-specific clones bound recombinant ECD-GCGR. One new large family (9 clones, family 19) originating from library 73Vλ bound full-length GCGR on VLP-GCGR, without binding to ECD-GCGR, indicating recognition of a different part of GCGR than the ECD (Fig. 2D and 2E). Two different clones from VH family 19 with the same CDR3 sequence, but containing amino acid differences in the remnant of the VH and different VL (6C6 and 6A5), and one clone from VH family 20 (6B3, control ECD-GCGR binder) were converted to IgG1 and further characterized for binding to GCGR expressed on Chinese hamster ovary (CHO) cells and VLP. Sequences of 6C6 and 6A5 are shown in Fig. S6.
To demonstrate that mAbs 6A5 and 6C6 did not require ECD for GCGR binding, a truncated GCGR that lacked the ECD (GCGRΔECD, aa146-447) was constructed. HEK293E cells were transfected with constructs encoding GCGR and GCGRΔECD. Binding analysis of mAbs to HEK293E cells overexpressing GCGR and GCGRΔECD confirmed previous results, with 6C6 and 6A5 binding to the GCGR and GCGRΔECD, whereas 6B3 only bound GCGR (Fig. 2F). After pre-incubation with an excess of recombinant ECD-GCGR, the binding of 6B3 to 293E cells overexpressing GCGR was lost, whereas mAbs 6C6 and 6A5 still showed binding to GCGR (Fig. 2F).
To select for more ECL binders, VLP-null and ECD-GCGR were combined for counter selections in the second and third round of selections. VLP-null was included in all 3 selection rounds. The combined counter selection resulted in decreased numbers of ECD binding phage and 10-300-fold enrichment after 3 rounds of selections on VLP-GCGR from both 73 scFv libraries (Fig. 3A).
Figure 3.

Selections for GCGR-ECL-specific phage after 2 and 3 rounds VLP selections and binding to GCGRΔECD and GCGR in competition with ECD-GCGR by flow cytometry. (A) Phage output titers from scFv libraries 73Vκ and 73Vλ after selections on VLP and counter selections with VLP-null and ECD-GCGR after 2 (upper panel) and 3 (lower panel) rounds. (B) FACS binding of mAbs to GCGR- (dark blue) or GCGRΔECD- (green) expressing HEK293 cells with and without ECD-GCGR competition (light blue). MOCK was included as a negative control (red). mAbs were detected with anti-human Fc-PE.
ScFv binding ELISA showed that counter selection with VLP-null and ECD-GCGR only revealed 20% ECD-GCGR binders from library 73Vκ and none from library 73Vλ. Sequence analysis showed many clones belonging to VH family 19. In addition, one new family was identified: family 29 (clones 9C8 and 9A4; sequences are shown in Fig. S5), which specifically bound to VLP-GCGR, and not to ECD-GCGR. After conversion to IgG1, binding to the ECL regions of GCGR was confirmed as both mAbs showed binding to GCGRΔECD and GCGR in competition with ECD-GCGR by flow cytometry (Fig. 3B). In conclusion, by counter selection with a combination of VLP-null and ECD-GCGR, we were able to identify a second family (family 29) of clones binding to the ECL regions of GCGR.
Antibodies binding to different epitopes on ECD-GCGR inhibit glucagon-induced activity
Representative mAbs of the 10 different VH families were investigated for functional activity in a glucagon-induced cAMP assay using stable CHO-GCGR cells. Seven of these mAbs were able to interfere with glucagon-mediated cAMP increase (Fig. 4A). They were all ECD-GCGR- binders, whereas the mAbs representing both ECL-binding families did not show any activity in the cAMP assay.
Figure 4.
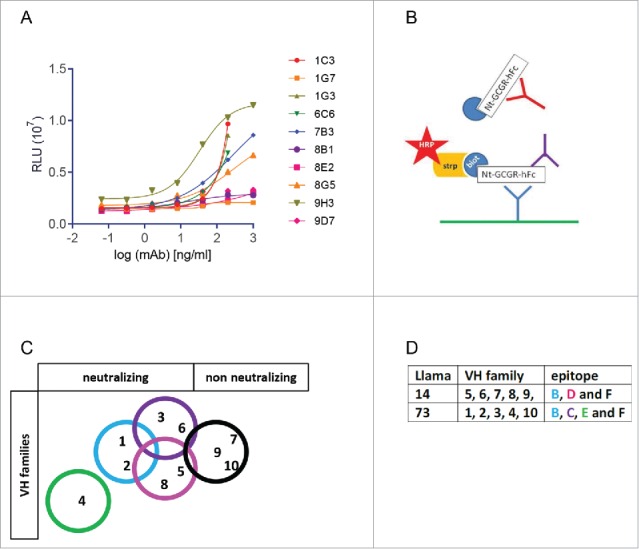
Glucagon-induced cAMP activity assay and epitope mapping of mAbs. (A) Inhibition of glucagon-induced cAMP activity by serially diluted mAbs, expressed as Relative Luminescence Units (107 RLU). Binding of GCGR-specific mAbs on GCGR-overexpressing CHO cells analyzed by fluorescence-activated cell sorting. (B) Schematic epitope overview, analyzed by competition assay. Biotinylated mAbs were allowed to bind coated ECD-GCGR in competition with a 50-fold excess of unbiotinylated mAb. (C) Overview of epitope coverage by the VH families in a Venn diagram and (D) in a table.
The 10 antibodies were tested against one another in a competitive binding ELISA using one mAb in solid phase (6.7 nM), binding the 67 pM biotinylated ECD-GCGR in the presence of 6.7 μM (100-fold access) of the competing mAb. Fig. 4B shows a schematic representation of the assay. Epitope mapping using competition ELISA revealed binding to 5 different epitopes on the ECD of the GCGR by the 10 different mAbs (Fig. 4C), indicating that the 2 outbred llamas 14 and 73, give different epitope coverage. The three mAbs binding to the ECD-GCGR that did not show any interference with glucagon-mediated cAMP increase bind to the same epitope (Fig. 4D). Taken together, selections on recombinant ECD-GCGR yield high affinity antibodies, binding to native GCGR and covering different epitopes on the extracellular domain.
Discussion
Finding mAbs against MPTs is difficult, and identification of mAbs that modulate the activity of these targets is even more challenging. Functional mAbs against GPCRs have been reported, but antibody discovery against this class of targets is still difficult. In this report, we demonstrate that DNA immunization of llamas effectively elicited a target-specific immune response against a MPT, without any off-target responses. Boosts with cells overexpressing GCGR resulted in higher target-specific plasma titers. We choose Caki cells overexpressing GCGR because they originate from dromedaries, which are evolutionarily related to llamas, aiming for less off-target responses. One immunization with Caki-GCGR cells was sufficient to boost the response against GCGR without raising a high response against other antigens on the cells, whereas a full cell immunization campaign with 6 repetitive injections of Caki-GCGR cells resulted in a high off-target response as observed in a separate study (unpublished data).
To identify mAbs against MPTs, multiple bait materials, including recombinant extracellular loops, cells, membrane fractions, proteoliposomes, VLPs or purified recombinant membrane protein, have been used.20 Recombinant extracellular loops are pure, but only represent a part of the whole target, and therefore may not elicit clones against discontinuous epitopes. One approach to solve that problem are recombinant loops fused to each other in so-called CLIPS.21 Cells and membrane fractions contain many other proteins, and therefore give high background outputs during phage selections. VLP are membrane derived and contain other cell surface proteins as well, which might explain why we observed some background binding to VLP-null during our phage selections on VLPs. This could easily be diminished by counter selection with a high excess of null VLP. VLP appeared to be the bait material with the highest molecular density of full-length GCGR that was commercially available. VLP have the advantage that they have a much higher density of the target GPCR compared to cells and membrane fractions and can easily be coated onto an ELISA plate.22 The molecular density of target molecules on VLPs is still much lower compared to a coating of recombinant protein. In this report, we demonstrated that, with these ‘relatively low densities’ of coated target molecules, phage selections benefit from multivalent display of antibody fragments due to avid binding. With Fab display, which is strictly monovalent,23 we were able to identify 5 VH families by selections on the recombinant ECD of GCGR, whereas scFv-display with selections on full-length GCGR on VLP allowed us to successfully identify a much higher diversity of VH families, including 2 families that bound to the ECL regions. This is, to our knowledge, the first report describing mAbs against the ECL regions of a type-B GPCR, which is challenging because the closed conformation, where the ECD of GCGR covers the ECLs, is energetically more favorable.10,11
Phage display in antibody discovery allows scientists to easily focus on a particular epitope using different approaches. Most of the GCGR-specific clones found by panning on GCGR VLPs bound to the ECD of GCGR. By performing counter selections with a large excess of recombinant ECD of GCGR during our selections on VLP-GCGR, we were able to identify additional ECL binding clones. In conclusion, by combining DNA immunization with scFv-display and selections on VLPs, we identified 23 different VH families of GCGR-specific clones derived from only 2 outbred llamas.
Most of the clones binding to the ECD of GCGR were able to inhibit glucagon-mediated cAMP increase. Antagonistic mAb against the ECD of GCGR, reported by Mukund et al,11 either block residues of the glucagon-binding cleft or allosterically prevent the conformational positioning of the ECD that is essential for GCGR activation.3 The clones binding to the small ECL regions of GCGR were not able to inhibit glucagon-mediated cAMP increase, which does not exclude antagonistic GCGR-ECL-binding mAbs from being found. The identification of antagonistic mAb directed against the ECD of GCGR may represent a more general approach for identification of antagonistic mAbs against other secretin-like type B GPCRs, which share structural features and ligand interaction hotspots, and therefore most likely similar conformational changes of the ECD.10,11
In summary, we demonstrated that DNA immunization is a very efficient way of immunization to generate specific immune responses to an MPT like GCGR. VLPs are the source of full-length GCGR material with a high amount of full-length GCGR. ScFv-display on VLP in combination with counter selections removing the ECD-GCGR binders, allows the identification of clones that bind to ECL regions. The advantage of scFv-display over Fab-display is that multiple copies of scFv are displayed per phage,17-19 resulting in avid binding. Avid phage binding is essential when panning on relatively low target density coatings.
Materials and methods
Constructs and cell lines
pCDNA3.1-hGCGR (aa1-477, Genbank accession number: NM_000160.4) was provided by Magali Waelbroeck (University of Brussels, Belgium). It was transformed in chemically competent E. coli Top 10 cells and plasmid DNA was isolated from a culture in 12L LB medium (supplemented with 2% glucose (w/v) and 100 µg/ml ampicillin) using the EndoFree Plasmid Giga Kit (Qiagen #12391).
Camelid Caki cells (dromedary renal fibroblasts, a kind gift from Serge Muyldermans, University of Brussels, Belgium), as well as CHO cells, were transfected with pCDNA3.1-hGCGR (same construct as for immunizations) and made stable by minimal dilution and culture in the presence of 200 µg/ml neomycin in 50% Dulbecco's Modified Eagle's Medium (DMEM, Gibco #31331) + 50% F12 medium (Sigma-Aldrich #51651C) supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich #F7524) and penicillin-streptomycin (Sigma-Aldrich #P4333). CHO cells stably transfected with CXCR4 (a kind gift from John Wijdenes, Diaclone, France) were cultured in the presence of 200 µg/ml hygromycin B. Caki cells were cultured in DMEM medium and CHO in Ham's F12 nutrient mixture with 10% FBS.
HEK293E cells were transiently transfected with pCDNA3.1-hGCGR (aa1-477), pCDNA3.1-hGCGRΔECD (aa146-447) or MOCK using pCDNA3.1-CXCR4 and cultured in DMEM with 10% FBS.
Recombinant extracellular domain of GCGR aa1-147 (ECD-GCGR) was PCR amplified from pCDNA3.1-hGCGR using T7 primer and Nt hGCGR 2 (AS) primer (ACTGCGTCTCCTCGA TCTGGAAGCTGCTGTACATC), and cloned by PmeI-BsmBI restriction cloning in pUPE-Fc vector. ECD-GCGR-Fc was produced by HEK293E cells (ATCC #CRL-10852) and purified using Protein A (GE Healthcare #17-5138-07) according to standard protocols.
pCDNA3.1-hGCGRΔECD was generated by splice overlap extension PCR on pCDNA3.1-hGCGR using the primers dNt GCGR-S (GACTACAACGACGACGACGACAACAGTGGGCTACAGCCTGTC), BGH reverse, and T7 primer Flag dNt GCGR-AS (GTTGTCGTCGTCGTCGTTGTAGTCAGCGGAGGGGACCTGTG).
Flow cytometry
100,000 Caki or HEK293E cells expressing hGCGR, hGCGRΔECD or MOCK were incubated with mAbs (1 µg/ml) for 1 h at 4°C, washed and incubated with phycoerythrin-conjugated goat anti-human antibody for 1 h at 4°C before reading using fluorescence-activated cell sorting (FACScount, BD Biosciences). For competition experiments, a 100-fold excess of ECD-GCGR (over the anti-GCGR mAb) was incubated with the anti-GCGR mAb.
Immunization and library construction
Four llamas were immunized with DNA, repeated a total of 4 times with 2-week intervals, followed by a single subcutaneous cell boost with dromedary Caki cells overexpressing the GCGR. They were housed with water and food ad libitum. All animal studies were conducted in accordance with European directive 2010/63/EU and with national legislative regulations after local ethical approval by the Ethical Committee for Animal Testing, Antwerp University. Llamas were anesthetized for approximately 30 min with an intramuscular injection of 1.5 ml Hella-Brunner mix (500 mg Xylazine and 150 mg ketamine 100) and then injected with 1 ml pCDNA3.1-hGCGR (2 mg/ml) intradermally, divided over at least 8 injection spots. Directly after injection, an electric pulse of 450 V with a resistance below 3000 Ω was given, using the Agile Pulse In Vivo system (BTX #47-0400N) with 4×6, 2 mm needle (BTX #47–0050). Cell boost was performed with 10×106 stable transfected Caki-hGCGR cells, stored frozen in DMEM + 10%FBS + 10% DMSO, washed in phosphate-buffered saline (PBS) twice and transported on ice before injected subcutaneously.
Plasma was prepared from 10 ml blood, collected before the start of the immunizations (pre-immune), a week after the third and the fourth DNA immunizations (3XDNA and 4XDNA) and a week after the cell boost in EDTA tubes. PBLs were isolated from 400 ml blood collected 4 d after completion of all immunizations; RNA was isolated, cDNA amplified and 4 Fab libraries (llamas 14 and 73) were constructed as previously described.12 Two scFv libraries were constructed from llama 73 by amplification of the heavy chain (VH) and the light chain (Vλ and Vκ) from the primary Fab libraries, digestion with restriction enzymes SfiI/NotI for VH and ApaLI/AscI for Vλ and Vκ, extracted and ligated into the pSc vector. Vector pSc was derived from the pCB3 phagemid vector and has the LacZ promoter, RBS, gene3 leader, SfiI/NotI restriction sites, (Gly4Ser)3 linker, ApaLI/AscI restriction sites, His6, c-myc-tag, amber stop and gene3. The E. coli strain TG1 (Netherland Culture Collection of Bacteria, The Netherlands) was transformed using recombinant phagemids to generate 4 different Fab-expressing and 2 scFv-expressing phage libraries (one λ and one κ library per immunized llama).
CXCR4 DNA immunizations of 2 llamas were performed as described for GCGR; VHH libraries were prepared as previously described.24
Phage selection
Phage were produced as previously described17 and selections for GCGR specific binders were performed using HEK293 derived virus-like particles (VLP, Integral Molecular) expressing GCGR (#RR-0999), CXCR4 (#RR-0830) or empty (null), ECD-GCGR, ECLs of GCGR and irrelevant recombinant protein. For CXCR4 selections, VLP expressing CXCR4 were used in the same way as for GCGR. VLPs were immobilized in maxisorb plates (Nunc #442404) at 20 and 2 U/well and the recombinant proteins at 10 and 1 µg/ml in PBS overnight (ON) at 4°C. VLPs were washed with PBS containing 0.01% Tween 80 and the recombinant proteins with PBS containing 0.05% Tween 80. Blocking was performed with 2% Marvel skimmed milk solution (Chivers Ireland LTD, Dublin, Ireland) in PBS for 1 h, then 1011 phage/well were added and incubated for 1 h at room temperature (RT) with shaking. Elution was performed with 10 mg/ml trypsin (Sigma-Aldrich #T1426-5G) for 30 min with shaking before the reaction was neutralized by addition of 4 mg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (Sigma-Aldrich #A8456). Exponentially grown (OD600 0.5) TG1 cells were infected with eluted phage (phage rescue) for 30 min at 37°C without shaking, followed by addition of 2TY with 100 µg/ml ampicillin and 2% glucose (w/v), then ON incubation at 37°C with shaking at 100 rpm. The outputs after TG1 infection were titrated on LB agar plates containing 100 µg/ml ampicillin, and the number of eluted phage from the different conditions was calculated and compared with a background control (PBS only, irrelevant protein or null VLP) after ON incubation at 37°C.
For counter selections, phage were incubated with 100 U null VLPs or 50 µg/ml ECD-GCGR for 1 h before being adding to the immobilized material.
Second or third round selections were performed using the rescued phage from the previous round. Briefly, the ON rescues were diluted 1/100 and grown in LB containing ampicillin and 2% glucose until OD600 reached 0.5. Helper phage M13-KO7 (Thermo Fisher #18311019) were added (phage: bact ratio 10:1) and allowed to infect for 30 min without shaking at 37°C. The medium was exchanged by centrifugation and resuspension in 50 ml 2TY/Amp/Kan (50 µg/ml) and incubation ON at 28°C for phage production. Phage were precipitated using PEG precipitation as previously described.17 Selections were performed as described for the first round, but with decreased amounts of phage (1010 phage/well).
ELISA and surface plasmon resonance binding assays
The immune response was investigated using ELISA with 1 µg/ml immobilized ECD-GCGR, blocking in 1% casein in PBS and incubation with a dilution series of the plasma, starting at 10%, detected with mouse anti-llama IgG1 (in-house antibody) and horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (1/5000, Jackson ImmunoResearch #715-035-150). A positive signal was revealed with 3,3′,5,5′-tetramethylbenzidine (TMB) and H2SO4 and read at optical density (OD) 450 nm.
Phage ELISA was performed using the same set-up as for the immune response or with immobilized VLP-CXCR4, but the phage were detected with a HRP-conjugated M13 antibody (GE Healthcare #27–9421).
For screening of binding clones, TG1 E. coli was infected with selected phages and individual colonies were isolated. Secretion of Fab or scFv fragments was induced using IPTG (Thermo Fisher #R0391), and the Fab-containing periplasmic fraction of bacteria was collected. Binding of Fab or scFv to ECD-GCGR was determined by ELISA using ECD-GCGR in solid phase and periplasmic crude extract in solution. Binding was revealed using HRP-conjugated anti-MYC antibody (Bethyl Laboratories #A190-105P). Fab or scFv that scored positive in ELISA were further investigated using SPR (BIACORE 3000 apparatus, GE Healthcare). ECD-GCGR was immobilized on a CM5 chip using amine coupling in sodium acetate buffer (GE Healthcare #BR100012). The periplasmic extracts with the Fab or scFv fragments were loaded with a flow rate of 30 µl/min. The Fab off-rates (koff) were measured over a 2-minute period. Regeneration was performed with 10 mM glycine pH1.5 for 10 sec. Binding clones were sent out for sequencing (LGC Genomics), and divided into families based on VH CDR3 sequence length and homology.16 VH families were given an internal number not based on IMGT (International Immunogenetics Information System) nomenclature.
Production and purification of IgG1
Antibody (mAb) recloning to IgG1, transfection, production in HEK293E cells (ATCC #CRL-10852) and Protein A purification were performed as previously described.12,25
Epitope mapping with competitive binding ELISA
Competitive binding ELISA was used for epitope mapping. One mAb was immobilized on maxisorb plates at 6.7 nM ON at 4°C in PBS. After blocking with 1% casein-PBS for 1 h at RT, 67 pM N-terminal biotinylated ECD-GCGR in combination with 6.7 μM (100-fold access) of the second mAb were added and incubated 1 h at RT. After incubation with HRP-conjugated streptavidin (1/5000) and washing with PBS-T, TMB was added and the reaction was stopped with H2SO4 and read at optical density 450 nm. Biotinylated ECD-GCGR was then detected by the antibodies where competition decreased the signal.
cAMP assay
mAbs were investigated for agonism and antagonism using cAMP-Glo max assay according to the manufacturer's protocol (Promega #V1681). Briefly, serial dilutions of the antibodies (starting at 10 µM) were pre-incubated with CHO cells overexpressing human GCGR for 30 min at 37°C before addition of 0.3 µM glucagon to induce cAMP for another 30 min at 37°C. cAMP-Glo ONE and PKA enzyme from the kit were added, and after 20 min at RT, Kinase Glo substrate in Kinase Glo-Buffer from the kit was added and incubated for 10 min at RT. cAMP was detected using a Glomax multi-detection system (Promega), the RLU were plotted against the concentration of antibodies and EC50 calculated using Graphpad prism v.6.
Supplementary Material
Disclosure of potential conflicts of interest
Bas van der Woning, Gitte De Boeck, Christophe Blanchetot, Vladimir Bobkov, Michael Saunders, Anna Hultberg and Hans De Haard are all employees of argenx. The other authors declare no conflict of interest.
Acknowledgments
We would like to thank Jo Leroy, veterinarian at the Faculty of Biomedical, Pharmaceutical and Veterinary Sciences, University of Antwerp, for their help with the DNA immunizations.
Funding
This work has been funded by the Flemish Agency for Innovation by Science and Technology (project number: IWT110484) and European Union's Horizon2020 MSCA Program under grant agreement 641833 (ONCORNET).
References
- 1.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov 2002; 1:727-30; PMID:12209152; http://dx.doi.org/ 10.1038/nrd892 [DOI] [PubMed] [Google Scholar]
- 2.Hutchings CJ, Koglin M, Marshall FH. Therapeutic antibodies directed at G protein-coupled receptors. MAbs 2010; 594-606; PMID:20864805; http://dx.doi.org/ 10.4161/mabs.2.6.13420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koth CM, Murray JM, Mukund S, Madjidi A, Minn A, Clarke HJ, Wong T, Chiang V, Luis E, Estevez A, et al.. Molecular basis for negative regulation of the glucagon receptor. Proc Natl Acad Sci U S A 2012; 109:14393-8; PMID:22908259; http://dx.doi.org/ 10.1073/pnas.1206734109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kemp EH, Gavalas NG, Akhtar S, Krohn KJ, Pallais JC, Brown EM, Watson PF, Weetman AP. Mapping of human autoantibody binding sites on the calcium-sensing receptor. J Bone Miner Res 2010; 25:132-40; PMID:19580466; http://dx.doi.org/ 10.1359/jbmr.090703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravn P, Madhurantakam C, Kunze S, Matthews E, Priest C, O'Brien S, Collinson A, Papworth M, Fritsch-Fredin M, Jermutus L, et al.. Structural and pharmacological characterization of novel potent and selective monoclonal antibody antagonists of glucose-dependent insulinotropic polypeptide receptor. J Biol Chem 2013; 288:19760-72; PMID:23689510; http://dx.doi.org/ 10.1074/jbc.M112.426288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossant CJ, Carroll D, Huang L, Elvin J, Neal F, Walker E, Benschop JJ, Kim EE, Barry ST, Vaughan TJ. Phage display and hybridoma generation of antibodies to human CXCR2 yields antibodies with distinct mechanisms and epitopes. MAbs 2014; 6:1425-38; PMID:25484064; http://dx.doi.org/ 10.4161/mabs.34376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagemann UB, Gunnarsson L, Geraudie S, Scheffler U, Griep RA, Reiersen H, Duncan AR, Kiprijanov SM. Fully human antagonistic antibodies against CCR4 potently inhibit cell signaling and chemotaxis. PLoS One 2014; 9:e103776; PMID:25080123; http://dx.doi.org/ 10.1371/journal.pone.0103776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Webb DR, Handel TM, Kretz-Rommel A, Stevens RC. Opportunities for functional selectivity in GPCR antibodies. Biochem Pharmacol 2013; 85:147-52; PMID:22975405; http://dx.doi.org/ 10.1016/j.bcp.2012.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hutchings CJ, Cseke G, Osborne G, Woolard J, Zhukov A, Koglin M, Jazayeri A, Pandya-Pathak J, Langmead CJ, Hill SJ, et al.. Monoclonal anti-beta1-adrenergic receptor antibodies activate G protein signaling in the absence of β-arrestin recruitment. MAbs 2014; 6:246-61; PMID:24253107; http://dx.doi.org/ 10.4161/mabs.27226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L, Yang D, de Graaf C, Moeller A, West GM, Dharmarajan V, Wang C, Siu FY, Song G, Reedtz-Runge S, et al.. Conformational states of the full-length glucagon receptor. Nat Commun 2015; 6:7859; PMID:26227798; http://dx.doi.org/ 10.1038/ncomms8859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukund S, Shang Y, Clarke HJ, Madjidi A, Corn JE, Kates L, Kolumam G, Chiang V, Luis E, Murray J, et al.. Inhibitory mechanism of an allosteric antibody targeting the glucagon receptor. J Biol Chem 2013; 288:36168-78; PMID:24189067; http://dx.doi.org/ 10.1074/jbc.M113.496984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basilico C, Hultberg A, Blanchetot C, de Jonge N, Festjens E, Hanssens V, Osepa S-I, De Boeck G, et al.. Four individually druggable MET hotspots mediate HGF-driven tumor progression. J Clin Invest 2014; 124:3172-86; PMID:24865428; http://dx.doi.org/ 10.1172/JCI72316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dreier T, Moshier M, Gabriels S, Wajant H, Brouckaert P, Huyghe L, Van Hauwermeiren T, Silence K. ARGX-110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. MAbs 2014; 2:523-32; PMID:24492296; http://dx.doi.org/ 10.4161/mabs.27398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wernery U, Knowles NJ, Hamblin C, Wernery R, Joseph S, Kinne J, Nagy P. Abortions in dromedaries (Camelus dromedarius) caused by equine rhinitis A virus. J Gen Virol 2008; 89:660-66; PMID:18272756; http://dx.doi.org/ 10.1099/vir.0.82215-0 [DOI] [PubMed] [Google Scholar]
- 15.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R. Naturally occurring antibodies devoid of light chains. Nature 1993; 363:446-8; PMID:8502296; http://dx.doi.org/ 10.1038/363446a0 [DOI] [PubMed] [Google Scholar]
- 16.Barbas CF, Björling E, Chiodi F, Dunlop N, Cababa D, Jones TM, Zebedee SL, Persson M, Nara PL, Norrby E. Recombinant human Fab fragments neutralize human type 1 immunodeficiency virus in vitro. Proc Natl Acad Sci U S A. 1992; 89:9339-43; PMID:1384050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Haard HJ, van Neer N, Reurs A, Hufton SE, Roovers RC, Henderikx P, de Bruïne AP, Arends J-W, Hoogenboom HR. A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J Biol Chem 1999; 274:18218-30; PMID:10373423; http://dx.doi.org/ 10.1074/jbc.274.26.18218 [DOI] [PubMed] [Google Scholar]
- 18.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization: Human antibodies from V-gene libraries displayed on phage. J Mol Biol 1991; 222:581-97; PMID:1748994; http://dx.doi.org/ 10.1016/0022-2836(91)90498-U [DOI] [PubMed] [Google Scholar]
- 19.Griffiths AD, Malmqvist M, Marks JD, Bye JM, Embleton MJ, McCaffert J, Baierl M, Holligerl KP, Gorick BD, Hughes-Jones N, Hoogenboom HR, Winter G. Human anti-self antibodies with high specificity from phage display libraries. EMBO J 1993; 12:725-34; PMID:7679990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klarenbeek A, Maussang D, Blanchetot C, Saunders M, van der Woning S, Smit M, de Haard H, Hofman E. Targeting chemokines and chemokine receptors with antibodies. Drug Discov Today Technol 2012; 9:237-44; http://dx.doi.org/ 10.1016/j.ddtec.2012.05.003 [DOI] [PubMed] [Google Scholar]
- 21.Boshuizen RS, Marsden C, Turkstra J, Rossant CJ, Slootstra J, Copley C, Schwamborn K. A combination of in vitro techniques for efficient discovery of functional monoclonal antibodies against human CXC chemokine receptor-2 (CXCR2). MAbs 2014; 6:1415-24; PMID:25484047; http://dx.doi.org/ 10.4161/mabs.36237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fong RH, Banik SS, Mattia K, Barnes T, Tucker D, Liss N, Lu K, Selvarajah S, Srinivasan S, Mabila M, et al.. Exposure of epitope residues on the outer face of the chikungunya virus envelope trimer determines antibody neutralizing efficacy. J Virol 2014; 88:14364-79; PMID:25275138; http://dx.doi.org/ 10.1128/JVI.01943-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baca M, Presta LG, O'Connor SJ, Wells JA. Antibody humanization using monovalent phage display. J Biol Chem 1997; 272:10678-84; PMID:9099717; http://dx.doi.org/ 10.1074/jbc.272.16.10678 [DOI] [PubMed] [Google Scholar]
- 24.De Haard HJ, Bezemer S, Ledeboer AM, Müller WH, Boender PJ, Moineau S, Coppelmans M-C, Verkleij AJ, Frenken LG, Verrips CT. Llama antibodies against a lactococcal protein located at the tip of the phage tail prevent phage infection. J Bacteriol 2005; 187:4531-41; PMID:15968064; http://dx.doi.org/ 10.1128/JB.187.13.4531-4541.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res 2002; 30:E9; PMID:11788735 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.