

ABSTRACT
Bispecific antibodies (BsAbs) can affect multiple disease pathways, thus these types of constructs potentially provide promising approaches to improve efficacy in complex disease indications. The specific and non-specific clearance mechanisms/biology that affect monoclonal antibody (mAb) pharmacokinetics are likely involved in the disposition of BsAbs. Despite these similarities, there are a paucity of studies on the in vivo biology that influences the biodistribution and pharmacokinetics of BsAbs. The present case study evaluated the in vivo disposition of 2 IgG-fusion BsAb formats deemed IgG-ECD (extracellular domain) and IgG-scFv (single-chain Fv) in cynomolgus monkeys. These BsAb molecules displayed inferior in vivo pharmacokinetic properties, including a rapid clearance (> 0.5 mL/hr/kg) and short half-life relative to their mAb counterparts. The current work evaluated factors in vivo that result in the aberrant clearance of these BsAb constructs. Results showed the rapid clearance of the BsAbs that was not attributable to target binding, reduced neonatal Fc receptor (FcRn) interactions or poor molecular/biochemical properties. Evaluation of the cellular distribution of the constructs suggested that the major clearance mechanism was linked to binding/association with liver sinusoidal endothelial cells (LSECs) versus liver macrophages. The role of LSECs in facilitating the clearance of the IgG-ECD and IgG-scFv BsAb constructs described in these studies was consistent with the minimal influence of clodronate-mediated macrophage depletion on the pharmacokinetics of the constructs in cynomolgus monkeys The findings in this report are an important demonstration that the elucidation of clearance mechanisms for some IgG-ECD and IgG-scFv BsAb molecules can be unique and complicated, and may require increased attention due to the proliferation of these more complex mAb-like structures.
KEYWORDS: Antibody pharmacokinetics, bifunctional antibodies, bispecific antibodies, clodronate, liver sinusoidal endothelial cells, non-specific binding
Abbreviations
- BsAbs
bispecific antibodies
- mAb
monoclonal antibody
- ECD
extracellular domain
- scFv
single chain variable fragment
- LSECs
liver sinusoidal endothelial cells
- TMDD
target-mediated drug disposition
- IV
intravenous
- Gly-Ser
Glycine-Serine
- VEGF-A
vascular endothelial growth factor
- PlGF
placental growth factor (PlGF)
- SPR
surface plasmon resonance
- HC
heavy chain
- FcRn
neonatal Fc receptor
- RUs
resonance units
- KD
equilibrium dissociation constant
- pI
isoelectric
- cIEF
capillary isoelectric focusing
- Tm
thermal stability
- DSC
differential scanning calorimetry
- HIC
hydrophobic interaction chromatography
- IF
immunofluorescence
- ADA
anti-drug antibodies
- LLOQ
lower limit of quantitation
- MRD
minimum required dilution
- Cmax
maximum serum concentration
- AUC
area under the curve
- CL
clearance
- t1/2
elimination half-life
- NBF
neutral buffered formalin solution
- ddH2O
distilled deionized water
- PBS
phosphate-buffered saline
- SECs
scavenger endothelial cells
- SR
scavenger receptors
Introduction
It has been become clear that many disorders, including cancer, metabolic and autoimmune diseases, display multiple or redundant mechanisms that fuel their progression.1-3 Therapeutic strategies utilizing bispecific or multifunctional antibodies that interfere with multiple disease pathways are thus viewed as a promising way to improve or prolong efficacy in complex disease indications. Bispecific monoclonal antibodies (BsAbs) have been designed in numerous formats that take advantage of the modular nature of the basic antibody construct. In one form, a pharmacologically active peptide/protein may be fused to an agonistic/antagonistic mAb to provide additional pharmacological activities. In another, a more antibody-like bispecific molecule is composed of subunits of individual antibodies, imparting the ability to bind dual soluble or membrane-bound ligands or a combination of both, which has been described for formats such as IgG-scFv (single-chain Fv), DVD-Ig (dual-variable-domain Ig), CrossMAbs, and IgG-ECD (extracellular domain) constructs. 4,5 The IgG-ECD constructs are unique in that these formats provide the alternate ligand binding activity via addition of a non-IgG target binding domain such as the extracellular domain of a cell surface receptor to the mAb structure. As with the IgG-scFv construct, the IgG-ECD construct may position the binding partner at the N- or C-terminus of the IgG via a flexible linker region.
It is likely that some of the specific and non-specific clearance mechanisms that govern mAb pharmacokinetics are also involved in the disposition of bispecific antibodies. The expression/density/turnover of target ligands for both the mAb and binding partner fused to the mAb influence clearance through target-mediated drug disposition (TMDD).6 Non-specific mAb clearance mechanisms involving interactions with the neonatal Fc receptor (FcRn) also play an obviously critical role.7-10 Despite these similarities, an issue that has been observed during the study and development of these more complex mAb-like structures is their inferior in vivo pharmacokinetic properties relative to the mAb components, such as short-half life and reduced in vivo stability.7-10 As with mAbs, some of these observations can be ascribed to Fcγ binding in vivo or non-specific binding driven by inherently poor biophysical properties.7-11 However, there are more examples of aberrant pharmacokinetic observations for bispecific mAbs that are unexplainable, and not readily attributable to these ‘other’ mechanisms that can influence mAb disposition.11-18 Since these inferior in vivo properties can limit the potential advantages offered pharmacologically, further study of factors influencing their disposition is critical to their successful development for therapeutic application. In addition, despite the increased emphasis on developing these more complex structures, there remains a substantial gap in understanding the factors in vivo that result in the aberrant peripheral clearance of some bispecific antibodies.1-3 With these points in mind, we designed the study reported here to evaluate the in vivo mechanism(s) involved in the aberrant clearance of several IgG-ECD and an IgG-scFv construct after intravenous (IV) administration to cynomolgus monkeys.
The IgG-scFv construct was made by fusing the scFv derived from a mAb (sc2) to the C-terminal end of the heavy chain (HC) of a different mAb (GB) via a flexible Glycine-Serine (Gly-Ser) linker (yielding GB-sc2) (Fig. 1). The IgG-ECD construct used the D2 domain of vascular endothelial growth factor receptor 1 (VEGFR1, reported previously 19), an ∼11.5 kDa protein, as the ECD fusion partner to 3 different IgGs with similar orientation and linker (yielding G1-ECD, G2-ECD, G3-ECD). The IgG-scFv construct was made with a scFv that was targeted to a soluble ligand having minimal peripheral concentrations in normal animals. Similarly, peripheral concentrations of the ECD antigens, including vascular endothelial growth factor (VEGF-A) and placental growth factor (PlGF) are in the low ng/mL and pg/mL range in normal animals, and these would not be predicted to influence the clearance of the D2 domain ECD used to construct the IgG-ECD constructs at the doses administered.20,21 Each of the IgG components of the bispecific molecules were also targeted to soluble ligands having low/no significant peripheral levels in cynomolgus monkeys, with the exception of G2 used to make G2-ECD. The mAb portion of G2-ECD had interactions with a cell surface receptor and the shed soluble domain of the same receptor, and thus its pharmacokinetics were examined at doses known to saturate TMDD. We observed rapid clearance (> 0.5 mL/hr/kg) of all constructs in cynomolgus monkeys. Characterizing the cellular distribution of the constructs after IV administration to cynomolgus monkeys suggested that the major clearance mechanism was linked to binding/association with liver sinusoidal endothelial cells (LSECs) vs. liver macrophages. The role of LSECs in facilitating the clearance of one of the IgG-ECD BsAbs studied was further supported by the depletion of macrophages, which did not significantly improve this molecule's pharmacokinetics in cynomolgus monkeys. While the exact biochemical interaction has not been defined, this mechanism of clearance is consistent with the physiological role of these cell types in the liver.
Figure 1.
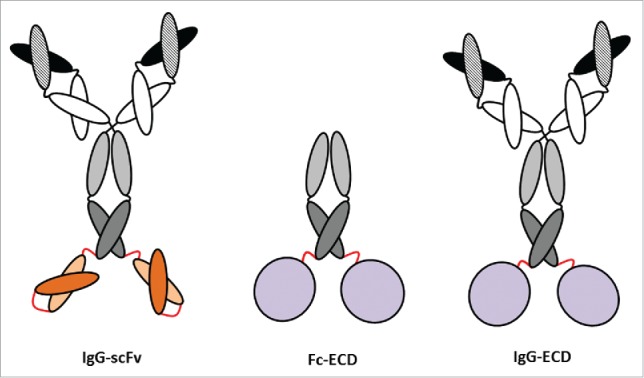
Cartoon representations of the BsAbs, IgG-ECD, IgG-scFv and the Fc-ECD molecules examined in the current study. The Glycine-Serine linkers shown in red represent the positions at which the linkers are used to covalently link the ECD or scFv to the IgG and Fc components of the bispecific molecules. In the cartoon representation of the IgG-scFv, the dark and light orange structures represent antigen-binding regions of the scFv. The gray circles show the position of the ECD fusion in the cartoon representation of the IgG-ECD and Fc-ECD.
Results
Design rational for BsAbs
The IgG-ECD bispecific constructs used 3 well-characterized humanized IgG4 molecules deemed G1, G2 and G3, which have different antigen-binding properties. A diagram of an IgG-ECD construct representative of the G1-ECD, G2-ECD and G3-ECD molecules is shown in Fig. 1. G1 binds a soluble ligand having minimal peripheral concentrations in normal cynomolgus monkeys. In contrast, G2 targets a cell surface receptor and the soluble shed version of the same receptor, which are present in normal cynomolgus monkeys. The G3 mAb has no known binding target in cynomolgus monkeys. Each of these molecules also included modifications: 1) 2 mutations were made in the Fc region to eliminate any potential for effector function; and 2) each IgG also had a serine mutated to a proline in the hinge region to eliminate in vivo arm exchange with endogenous IgGs.22,23 Our previous studies with these mAbs showed each has well behaved in vivo pharmacokinetics and stability in cynomolgus monkeys (Figs. 2A-C and 3). As the fusion partner to mAbs G1, G2 and G3 for the BsAb model system, we used the well-folded ∼11.5 kDa D2 domain of VEGFR1 described previously19 as the ECD fusion partner to the 3 different IgGs with the same fusion position orientation and linker region. The ECD binds its soluble targets VEGF-A and PlGF, which are known to have low ng/mL and pg/mL circulating concentrations in the blood of normal animals, respectively.20,21 The ECD was fused via a flexible glycine-serine linker of the same length to the C-terminal end of the HC each of the 3 mAb partners. The C-terminal HC position was initially selected for the fusion configuration of the ECD protein domain for each of the mAbs because this theoretically allowed the bispecific constructs increased target binding accessibility such that the mAbs could engage their targets concomitantly with protein binding its antigen.
Figure 2.

The IgG-ECD BsAbs show faster clearance than their respective parental mAbs, but the clearance of a Fc-EDC fusion protein is similar to Fc following administration to cynomolgus monkeys. The mean pharmacokinetic profiles of (A) G1 and G1-ECD; (B) G2 and G2-ECD; (C) G3 and G3-ECD; and (D) Fc and Fc-ECD following a single IV administration to cynomolgus monkeys. Data are the mean for 2 animals/timepoint for all molecules.
Figure 3.
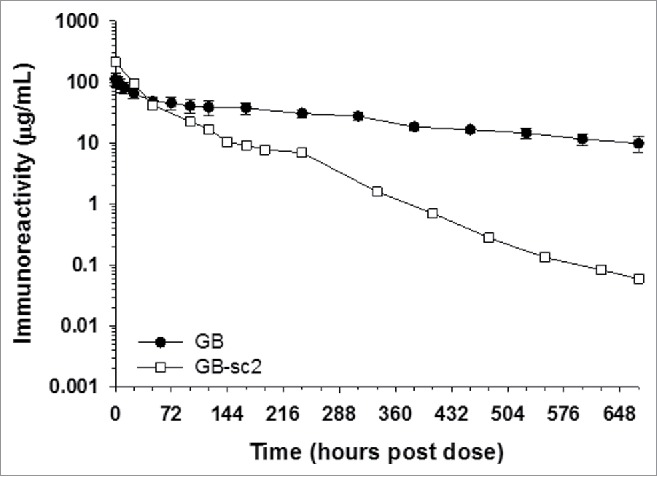
IgG-scFv BsAbs show rapid clearance in vivo. The mean pharmacokinetic profiles GB and GB-sc2 following a single IV administration to cynomolgus monkeys. Data for GB are the mean of 3 animals/timepoint, whereas PK data for GB-sc2 are for 2 animals/timepoint.
The IgG-scFv bispecific molecule was constructed with a similar orientation (i.e., C-terminal HC) and linker region as the IgG-ECD molecules (Fig. 1). In the first step, a scFv element was constructed from a separate humanized IgG4 mAb by linking the variable HC and light chain domains via a flexible Gly-Ser linker. Similar to the molecules described above, the hinge and Fc regions contain the same mutations to reduce any potential for effector function and arm exchange.22,23 Secondly, fusion of this stabilized scFv element to the C-terminus of GB to form GB-sc2 was facilitated by an additional flexible Gly-Ser linker to generate GB-sc2. Similar to G1, GB binds soluble ligands that both have minimal peripheral concentrations in normal cynomolgus monkeys. When examined as a mAb, GB also showed well-behaved in vivo pharmacokinetics and stability in cynomolgus monkeys (Fig. 3). With respect to sc2, the molecule interacts with soluble antigens that have low circulating levels in the blood in normal animals (data not shown).
In vitro characterization of the IgG-ECD and IgG-scFv BsAbs
Following the construction of the IgG-ECD and IgG-scFv BsAbs, we characterized biochemical and biophysical molecular properties in vitro that have been shown to have an influence on mAb and protein clearance in vivo.13-15, 24-26 Specifically, we evaluated their isoelectric points (pI), thermal stabilities (Tm), as well as their propensity for electrostatic (i.e., charge) and hydrophobic-related interactions using heparin or hydrophobic interaction chromatography (HIC), respectively. Additionally, each variant was evaluated for its cynomolgus monkey FcRn-binding properties. The data from these analyses is summarized in Table 1.
Table 1.
Biophysical and cynomolgus monkey FcRn binding properties of the IgG-ECD and IgG-scFv BsAbs.
| Tm (°C) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Molecules | Cynomolgus Monkey FcRn Kd (nM) at pH 6 Kd1 Kd2 | Heparin Binding ([NaCl] at main peak elution apex mM) | HIC Retention (min) | Predicted pI (VNTI) | Measured pI | CH2 | CH3/FAB | Fusion | |
| G1 | 623 | 33 | No Binding | 9.9 | 6.86 | 7.55 | 70.4 | 75.9 | N/A |
| G1-ECD | 339 | 57 | No Binding | 13.3/14.0 | 6.72 | 7.51 | 67.9 | 75.4 | 51.9 |
| G2 | 1342 | 54 | 160 | 7.5 | 8.03 | 8.45 | 69.6 | 73.9 | N/A |
| G2-ECD | 1013 | 68 | 105 | 7.8/9.4 | 7.87 | 8.29 | 68.2 | 73.9 | 50.5 |
| G3 | 801 | 45 | No Binding | No Retention | 7.82 | 7.97 | 70.6 | 78.6 | N/A |
| G3-ECD | 759 | 56 | No Binding | 9.1 | 7.21 | 7.8 | 67.9 | 77.5 | 51.9 |
| Fc | 195 | 96 | No Binding | No Retention | 5.78 | 5.82 | 68.1 | 70.2 | N/A |
| Fc-ECD | 300 | 43 | No Binding | 7.8 | 6.29 | 6.41 | 63.3 | 66.9 | 54.3 |
| GB* | 1030 | 66 | No Binding | No Retention | 7.57 | 8.03 | 66.8 | 71.8 | 71.8 |
| GB-sc2 | 989 | 63 | 150 | No Retention | 6.78 | 7.55 | 66.6 | 72.1 | 66.6 |
Kd1 and Kd2 are the low and high affinity FcRn equilibrium binding constants; electrostatic and hydrophobic-related interactions were determined using heparin or hydrophobic interaction chromatography, respectively; N/A = not applicable; ND = not determined.
NCR = no column retention observed.
Fusion = ECD or scFv.
The pIs of the molecules calculated in silico based on their primary amino acid sequence using Vector NTI software were comparable to those determined through empirical isoelectric focusing methods (Table 1). For the majority of these molecules, we observed that the experimentally determined pI values fell within a narrow pH range from 7.5 to 8.5, indicating that, at physiological pH, these molecules would be expected to have a very weak to weak positive charge. Additionally, it was noted that fusion of the parental mAb did not grossly alter the molecules apparent charge properties (Table 1).
Given that the glycocalyx of endothelial cells creates a negatively charged surface that can promote non-productive charge-based interactions, we leveraged a heparin-based surrogate column assay to determine the degree of charge-based interaction for each of the molecules evaluated in this study. In this experiment, samples were injected over a column of heparin sepharose, and then eluted with a linear gradient of increasing ionic strength. Retention of compound and elution time was then used to determine the potential for charge-based interactions. Despite the slight positive charge present on all of the molecules tested, we observed that the majority of the molecules were not retained by the column. For the few molecules that did show retention, all were noted to elute at sodium chloride concentrations that approximate those found under physiological conditions (Table 1). The relative comparison of the parental mAbs with their BsAb counterparts indicated no gross change in column association, with the noted exception of GB and GB-sc2.
Non-specific interactions driven by hydrophobic association were evaluated using a HIC-based column assay in which molecules were formulated in high ionic strength (1 M ammonium sulfate solution at pH 6.7), and then injected onto a solid phase hydrophobic resin. Retained materials were eluted from the column with a linear gradient of low ionic strength and hydrophobicity was assessed as a function of retention time. Fusion of the ECD component to the 3 mAbs generally resulted in an increased hydrophobicity, relative to parent mAb, as evidenced by increased retention time. However, evaluation of GB and GB-sc2 did not show any specific increase in hydrophobicity as both proteins were not retained under the conditions tested.
Perturbations to the structural integrity (or thermal stability) of the mAbs and BsAbs were measured by differential scanning calorimetry (DSC; Table 1). Analysis of G1 and G1-ECD indicated that fusion of ECD to the C-terminal end of G1 lead to a slight decrease in the overall thermal stability of the CH2 domain present in G1 (∼3°C), while no changes in CH3 or Fab domains were measured. In the context of G2 or G3, no gross changes in any domain of the IgG were found to be perturbed. Furthermore, the Tm of the ECD was found to be invariant regardless of the antibody to which it was fused. With respect to GB and GB-sc2, compared to each other, no gross alteration of the thermal stability of any domains within in the molecules were found to be altered.
The cynomolgus monkey FcRn binding was measured using a previously reported surface plasmon resonance (SPR) approach.27 Resultant data was fit to a biphasic model that reported 2 affinities for FcRn binding (Table 1). The low affinity binding values ranged from 339 to 1342 nM; the higher affinity Kd values ranged from 33 to 68 nM (Table 1). A relative comparison of parental mAbs with their corresponding bispecific forms did not reveal any substantial alteration of measured binding affinities of either the weak or strong affinities, indicating a conserved engagement profile with cynomolgus monkey FcRn.
Pharmacokinetics of the parental and IgG-ECD bispecific mAbs in cynomolgus monkeys
Given the IgG-ECD constructs displayed in vitro characteristics that were reasonably similar to their respective parental mAbs (Table 1), we were prompted to determine their in vivo pharmacokinetic properties in cynomolgus monkeys. We had previously established (data not shown) that there were insignificant peripheral levels of antigen for the ECD protein domain component to bind in normal cynomolgus monkeys. As a result, the influence of target binding due to the fusion partner component on the IgG-ECD bispecific construct kinetics is limited/negligible.
With respect to the mAbs used in the IgG-ECD molecules, earlier studies showed mAb G1 displayed nearly dose proportional increases in exposure over a broad dose range in cynomolgus monkeys (1–10 mg/kg; data not shown). Thus the kinetics of G1-ECD was evaluated following a single 2 mg/kg IV administration to cynomolgus monkeys. In contrast, mAb G2 had displayed evidence of non-linear kinetics consistent with TMDD influencing the clearance (data not shown). To evaluate the kinetics of G2-ECD, a higher single IV dose of 15 mg/kg, known to saturate target using mAb G2 in cynomolgus monkeys, was used with the intent of eliminating/limiting the influence of TMDD on the interpretation of the kinetics of G2-ECD. Following administration to cynomolgus monkeys, both the G1-ECD and G2-ECD constructs, showed rapid blood clearance and shorter half-life relative to their corresponding parent mAb (Fig. 2 and Table 2). The G1-ECD and G2-ECD constructs were characterized by ∼10- and ∼7-fold faster elimination, respectively, and ∼3- and ∼5-fold shorter half-life, respectively, relative to their corresponding parent mAb (Fig. 2 and Table 2).
Table 2.
Mean cynomolgus monkey pharmacokinetic parameters of the IgG-ECD and Fc-ECD molecules.
| Molecules | Dose (mg/kg) | Cmax (μg/mL) | AUC0-∞(hr*μg/mL) | Vss (mL/kg) | CL (mL/hr/kg) | T1/2 (hr) |
|---|---|---|---|---|---|---|
| G1 | 2 (n = 2) | 69.7 | 6615 | 40.9 | 0.30 | 37.2 |
| G1-ECD | 2 (n = 2) | 51.4 | 606 | 60.3 | 3.32 | 12.0 |
| G2 | 10 (n = 2) | 327.0 | 42344 | 39.4 | 0.24 | 143.3 |
| G2-ECD | 15 (n = 2) | 480.0 | 12532 | 56.7 | 1.23 | 39.9 |
| G3 | 2 (n = 2) | 68.6 | 7645 | 34.7 | 0.26 | 91.1 |
| G3-ECD | 2 (n = 2) | 78.4 | 1509 | 75.3 | 1.35 | 134.6 |
| Fc | 2 (n = 2) | 56.1 | 2841 | 86.3 | 0.70 | 91.6 |
| Fc-ECD | 2 (n = 2) | 61.0 | 2034 | 105.4 | 0.99 | 78.4 |
Cmax, maximal observed plasma or serum concentration; AUC0-∞, area under the plasma or serum concentration curve from zero to infinity; Vss, volume of distribution at steady state; CL, clearance; T1/2, elimination half-life. Time points included in the estimation of terminal T1/2: G1 336–600 hours post dose; G1-ECD 24–120 hours post dose; G2 576–648 hours post dose; G2-ECD 96–504 hours post dose; G3 432–672 hours post dose; G3-ECD 240–672 hours post dose; Fc and Fc-ECD 360–648 hours post dose.
Anti-drug antibodies (ADA) were observed for the parental G1 and G2 mAbs later in the concentration versus time profile (>336 hours post dose) (ADA titer data not shown). Given the ADA was evident >336 hours after administration, there was enough concentration vs. time data to assess the clearance of the molecules, and thus the ADA did not influence the interpretation of the kinetic data (Fig. 2). However, ADA titers were not detected for the G1-ECD and G2-ECD BsAb groups, and thus did not explain the aberrant clearance of these BsAbs (ADA titer data not shown). In addition, concentrations of the ECD-based BsAbs measured using a VEGF-based antigen capture and Fc (i.e., mAb) detection ELISA also showed the BsAbs had rapid clearance relative to their parental mAbs, indicating that functional ECD is present on the BsAbs and not clipped from the mAb in vivo (data not shown). In an effort to more definitively rule out the possibility that target binding by the D2 domain of VEGFR1 ECD protein domain component or TMDD due to antigen binding by the G1 and G2 mAb component were causing the rapid clearance when combined in the context of the G1-ECD and G2-ECD bispecific molecules, we conducted 2 additional cynomolgus monkey pharmacokinetic experiments.
TMDD was obviated in the cases of G1-ECD and G2-ECD as stated above; however, in our first effort, we decided to extend our characterization of the fusion of our ECD protein domain to the C-terminal HC of another well characterized, internally developed humanized IgG4 (mAb G3) (Eli Lilly and Company proprietary antibody) with no known binding target in cynomolgus monkeys. Similar to the other 2 IgG-ECD constructs, G3-ECD showed rapid blood clearance characterized by ∼5-fold faster elimination relative to its parent mAb ‘C’ following a single 2 mg/kg IV administration to cynomolgus monkeys (Fig. 2 and Table 2). Given the kinetic observations for all 3 IgG-ECD constructs were similar, these findings suggested that non-specific clearance factor(s) were likely responsible for the in vivo disposition observations.
In our second follow-up experiment, we were prompted to examine the pharmacokinetics of the ECD protein domain fused to the C-terminal end of an IgG4-derived human Fc fragment to generate Fc-ECD (Fig. 1). No statistically significant differences in blood clearance of Fc-ECD relative to the parental Fc were noted in cynomolgus monkeys after a single 2 mg/kg IV administration of each (Fig. 2 and Table 2). The data suggested that there were factors specifically related to the context of fusion format (i.e., Fc versus IgG) with the ECD protein domain that influenced the in vivo pharmacokinetics.
Characterization of the pharmacokinetics of G1-ECD following macrophage depletion in cynomolgus monkeys
The lack of specific target binding in cynomolgus monkeys of the G1-, G2- and G3- ECD molecules, as well as the general observation of rapid clearance in the context of 3 IgG-ECD molecules, suggested that a high-capacity, non-specific clearance mechanism(s) may be contributing to the aberrant kinetic findings. Macrophages are found essentially in all tissues and are known to engulf and proteolyze circulating species non-specifically. In an effort to characterize the role of macrophages in the rapid peripheral clearance of the IgG-ECD, the kinetics of one of the constructs (G1-ECD) was evaluated in cynomolgus monkeys pretreated with liposomes containing clodronate to deplete these cells in vivo. Only G1-ECD was evaluated under these conditions since the goal was to simply profile the role of macrophages as a clearance mechanism for the bispecific molecules.
Immunohistochemistry showed pretreatment with the clodronate liposomes reduced the number of liver macrophages 48 hours post pretreatment to a severe grade level (Table 3). The findings also showed liver macrophage numbers partially recovered to marked and moderate grade levels at 120 and 192 hours post pretreatment, respectively (Table 3). Upon liver macrophage depletion, it is well known that monocytes are recruited to the tissue where they can differentiate into tissue macrophages or dendritic cells.28 In response to the decreased number of liver macrophages, hematology results showed a delayed increase in circulating absolute monocytes in the blood (Table 3). Pretreatment with control liposomes (empty), however, had no effect on either of these parameters since these contained no clodronate (Table 3).
Table 3.
Circulating absolute monocytes in the blood (macrophage precursors) and incidence and severity of liposomal clodronate-related reduction in liver macrophages following liposome treatment.
| Treatment | Parameters | Prior to Pretreatment | Predose | 48 hr post pretreatmenta | 120 hr post pretreatmenta | 192 hr post pretreatmentb |
|---|---|---|---|---|---|---|
| Clodronate Liposomes + G1-ECD (n = 2) | Absolute Monocytes | NA | 0.39 | 0.30 | 1.00 | 1.14 |
| Reduction in Liver Macrophages | NA | NA | NA | NA | Grade 3 | |
| Empty Liposomes + G1-ECD (n = 2) | Absolute Monocytes | NA | 0.13 | 0.20 | 0.18 | 0.16 |
| Reduction in Liver Macrophages | NA | NA | NA | NA | None | |
| Clodronate Liposomes (n = 1) | Absolute Monocytes | 0.67 | NA | 0.22 | 1.03 | 1.62 |
| Reduction in Liver Macrophages | NA | NA | Grade 5 | Grade 4 | Grade 3 | |
| EmptyLiposomes (n = 1) | Absolute Monocytes | 0.37 | NA | 0.29 | 0.21 | 0.32 |
| Reduction in Liver Macrophages | NA | NA | None | None | None |
Reduction in liver macrophages based on microscopic evaluation conducted on biopsy samples collected from males treated with clodronate liposomes and empty liposomes.
Reduction in liver macrophages based on microscopic evaluation conducted on liver samples collected at necropsy from all males in all groups.
Absolute monocyte units = E3/µL
NA = Not Applicable
Grade 5 = Severe = approximately 95% reduction in macrophage numbers from the baseline
Grade 4 = Marked = approximately 70% reduction in macrophage numbers from the baseline
Grade 3 = Moderate = approximately 40% reduction in macrophage numbers from the baseline
None = No reduction in macrophage numbers from the baseline
The G1-ECD showed a slightly slower clearance (∼30%) following a single 2 mg/kg IV administration to cynomolgus monkeys pretreated with clodronate-containing liposomes relative to those treated with empty liposomes (Fig. 4 and Table 4). Although G1-ECD exposure in the clodronate pretreated animals shows trends of improved exposure along the entire concentration vs. time profile relative to control liposome pretreated animals, the clearance observations did not appear different enough to suggest that macrophages contribute meaningfully to the aberrant BsAb pharmacokinetics. Thus, the findings suggest macrophages likely contribute marginally to the rapid peripheral clearance of the IgG-ECD disposition, but do not account for the major clearance mechanism(s).
Figure 4.
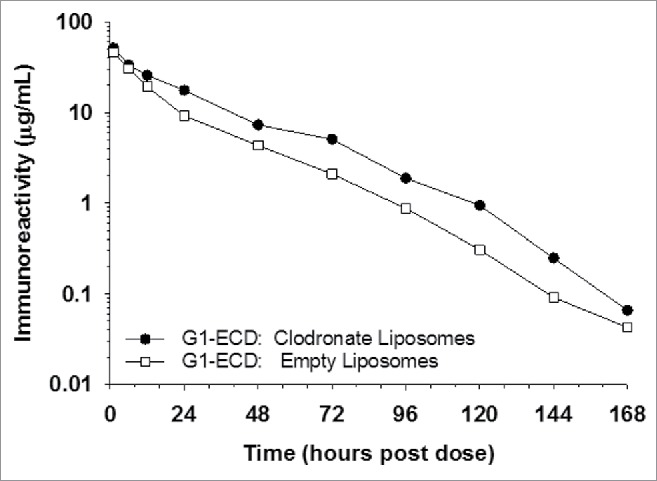
Macrophage depletion using clodronate containing liposomes marginally changes the pharmacokinetics of G1-ECD in cynomolgus monkeys. The mean pharmacokinetic profiles of G1-ECD following a single IV dose to cynomolgus monkeys treated with clodronate-containing liposomes (depletes macrophages) or control liposomes (empty) prior to G1-ECD administration. Data are the mean for 2 animals/timepoint for all molecules.
Table 4.
Mean cynomolgus monkey pharmacokinetic parameters of the G1-ECD molecule following treatment with empty liposomes or liposomes containing clodronate.
| Treatment | Dose (mg/kg) | Cmax (μg/mL) | AUC0-∞ (hr*μg/mL) | Vss (mL/kg) | CL (mL/hr/kg) | T1/2 (hr) |
|---|---|---|---|---|---|---|
| Empty Liposomes | 2 (n = 2) | 45.9 | 828 | 57.4 | 2.42 | 17.9 |
| Clodronate Liposomes | 2 (n = 2) | 51.7 | 1250 | 47.8 | 1.60 | 18.5 |
Cmax, maximal observed plasma concentration; AUC0-∞, area under the plasma concentration curve from zero to infinity; Vss, volume of distribution at steady-state; CL, clearance; T1/2, elimination half-life. Time points included in the estimation of terminal T1/2: 24–168 hours post dose.
Pharmacokinetics of the parental and IgG-scFv bispecific mAbs in cynomolgus monkeys
The pharmacokinetic properties of our IgG-scFv constructs was also examined in cynomolgus monkeys. Similar to some of our IgG-ECD constructs, earlier studies showed that there were low/negligible concentrations of the target antigens for both the IgG and scFv components of GB-sc2 in normal cynomolgus monkeys to influence the pharmacokinetics of the molecules. Thus, the pharmacokinetics of GB-sc2 were evaluated following a single IV administration of 5 mg/kg each to cynomolgus monkeys. The in vivo evaluation showed the GB-sc2 construct had rapid blood clearance and a short terminal phase half-life compared to its parental mAb GB (Fig. 3 and Table 5).
Table 5.
Mean (± SD when applicable) cynomolgus monkey pharmacokinetic parameters of the IgG and IgG-scFv molecule.
| Molecules | Dose (mg/kg) | Cmax (μg/mL) | AUC0-∞ (hr*μg/mL) | Vss (mL/kg) | CL(mL/hr/kg) | T1/2 (hr) |
|---|---|---|---|---|---|---|
| GB | 5 (n = 3) | 111.2 ± 24.3 | 25,333 ± 8128 | 89.9 ± 14.7 | 0.21 ± 0.071 | 313.60 ± 54.0 |
| GB-sc2 | 5 (n = 2) | 212.0 | 8609 | 40.0 | 0.59 | 72.3 |
Cmax, maximal observed plasma or serum concentration; AUC0-∞, area under the plasma or serum concentration curve from zero to infinity; Vss, volume of distribution at steady state; CL, clearance; T1/2, elimination half-life. Time points included in the estimation of terminal T1/2: GB 48–168 hours post dose; GB-sc2 336–672 hours post dose.
Distribution of IgG-ECD and IgG-scFv constructs in various cynomolgus monkey tissues by immunofluorescence
Biodistribution studies with exogenously administered mAbs have shown that there are generally 5 highly vascularized tissues in which IgGs distribute following dosing.29 These tissues include the liver, kidneys, spleen, skin and muscle. In an effort to delineate the types of cells the BsAb molecules were binding within these tissues, immunofluorescence (IF) was conducted on samples collected from cynomolgus monkeys. Since G1-ECD was more extensively studied in the previous pharmacokinetic studies of IgG-ECD constructs, the cellular distribution of G1-ECD and G1 were studied in greater detail. In addition, limited distribution of GB-sc2 within liver tissue was also studied following administration to cynomolgus monkeys.
For G1 and G1-ECD, the distribution within each of the 5 tissues named above was evaluated at 6 and 24 hours following a single IV administration of the constructs. The timepoints were chosen based on the cynomolgus monkey pharmacokinetic profiles displaying a reasonable divergence in the peripheral exposure of the compounds (Fig. 2). At both 6 and 24 hours post administration, G1 was not detected in any of the tissue samples. G1-ECD was detected only at 6 hours post administration in the liver and kidney, with all the other tissue samples showing no appreciable signal over background (kidney data not shown; Fig. 5). Under the tissue staining conditions used, the intensity of the staining for IgG-ECD and IgG-scFv constructs was qualitatively greater in the liver tissue than the kidney, thus liver tissue became the focus of additional cellular staining. Figs. 5 and 6 show the IF of G1 and G1-ECD in the liver, respectively, at 6 hours post administration. Fig. 7 shows the GB-sc2 in the liver following a single IV administration of the construct to cynomolgus monkeys at 6 hours post administration. All the bispecific constructs show multifocal labeling of the sinusoidal lining around the hepatocytes consistent with endothelial cell association (Figs. 6–7).
Figure 5.
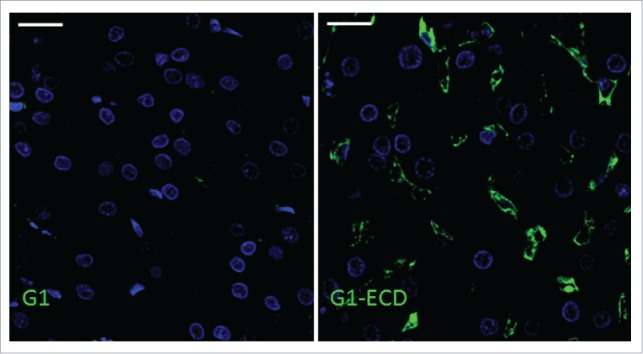
Immunofluorescence shows the presence of BsAb G1-ECD in the liver but not mAb G1. In panels A and B, green and blue represent detection of BsAb or mAb and nuclei, respectively. Immunofluorescence detection of (A) G1 and (B) G1-ECD in cynomolgus monkey liver following a single IV administration of each molecule at 6 hours post administration. Data shown are from representative liver sections from a single cynomolgus monkey for each compound. The scale bars represent 20 μm.
Figure 6.

Immunohistochemistry analysis of BsAb G1-ECD in cynomolgus monkey liver shows co-localization with LSEC marker but less with liver macrophages (Kupffer cells). In panels A-G, green, blue, red and magenta represent detection of BsAb, hepatocyte nuclei, LSECs and macrophages (Kupffer cells), respectively. (A) Multi-channel immunofluorescence image with detection of G1-ECD, LSECs, Kupffer cells and hepatocyte nuclei. The boxed area in Panel A shows the area from which the higher magnification insets B-G are derived. (B) G1-ECD detection using an anti-human IgG (Bethyl Laboratories A80–319A); (C) detection of the endothelial cell marker CD31/PECAM1 (R&D Systems AF806); (D) overlay of the G1-ECD and CD31 detection signals; (E) detection of the resident macrophage marker CD68 (ThermoFischerScientific MA5–13324); (F) overlay of the G1-ECD and CD68 detection signals; (G) multi-channel overlay image of all the insets. Data are from representative liver sections from a single cynomolgus monkey at 6 hours post administration. The scale bar for Panel A represents 50 μm, whereas the scale bars for the inset Panels B-G represents 20 μm.
Figure 7.

GB-sc2 shows co-localization with LSEC marker but not liver macrophages (Kupffer cells) in cynomolgus monkeys. In panels A-G, green, blue, red and magenta represent detection of GB-sc2, hepatocyte nuclei, LSECs and macrophages (Kupffer cells), respectively. (A) Multi-channel immunofluorescence image with detection of GB-sc2, LSECs, Kupffer cells and hepatocyte nuclei. The boxed area in Panel A shows the area from which the higher magnification insets B-G are derived. (B) GB-sc2 detection using an anti-human IgG (Bethyl Laboratories A80–319A); (C) detection of the endothelial cell marker CD31/PECAM1 (R&D Systems AF806); (D) overlay of the GB-sc2 and CD31 detection signals; (E) detection of the resident macrophage marker CD68 (ThermoFischerScientific MA5–13324); (F) overlay of the G1-ECD and CD68 detection signals; (G) multi-channel overlay image of all the insets. Data are from representative liver sections from a single cynomolgus monkey at 6 hours post administration. The scale bar for Panel A represents 50 μm, whereas the scale bars for the inset Panels B-G represents 20 μm.
Additional identification of the cell types where G1-ECD and GB-sc2 colocalized within the liver was pursued by IF detection of both CD31 and CD68, which are commonly used to identify endothelial cells and Kupffer cell (liver macrophages), respectively, in liver. The G1-ECD and GB-sc2 showed a similar staining pattern as the CD31 marker, indicating the bispecific constructs are binding predominately to liver sinusoidal endothelial cells (LSECs) relative to Kupffer cells (Figs. 6–7). Colocalization calculations of the BsAbs with CD31 were not reported since CD31 was being used as an approach to specifically identify LSECs. Taken together, these data suggest the LSECs are linked to the aberrant clearance of the IgG-ECD/scFv constructs.
Discussion
In the course of our pharmacokinetic characterization of several IgG-scFv and IgG-ECD fusion constructs, we observed unusually rapid clearance of these molecules when IV administered to cynomolgus monkeys. The C-terminal HC IgG-ECD constructs elicited rapid clearance in vivo with a variety of several different parental mAbs. In addition, an IgG-scFv (C-terminal HC fusion) construct also led to rapid clearance. With the exception of one IgG-ECD molecule (G2-ECD), constructs were made with IgGs/ECD/scFv targeted to soluble ligands having minimal peripheral concentrations in normal animals and having no interaction with cell surface receptors, thus eliminating these as potential mechanisms for the observed clearance. In the case of G2-ECD, usually rapid clearance was still observed at a dose level known to saturate TMDD for the parental G2 molecule in cynomolgus monkeys, similar to the other IgG/ECD/scFv BsAb molecules. While previous studies have also reported aberrant clearance of bispecific mAbs, exploration into the physiological mechanisms involved was not extensively pursued.9,11, 18 Mechanisms that have been previously characterized to influence the pharmacokinetics of mAbs, such as interaction with FcRn, TMDD and biophysical characteristics, did not appear to account for the observations with these constructs and are not implicated as causative in this report.14,30,31 FcRn binding of our constructs show receptor binding affinities at pH 6 in the range reported for molecules with well-behaved kinetics, as well as no direct FcRn interactions at neutral pH (Table 1). 32,33 In addition, the BsAbs showed reasonable Tm and poor interaction with heparin sulfate at salt concentrations that were at or below those expected to be present in biological fluids, indicating that neither of these properties could sufficiently account for the observed rapid clearance (Table 1 and data not shown). Interestingly, we did find that, when IgG was fused to the ECD, the overall hydrophobicity of the BsAb increased, as indicated by increased retention times on the HIC column (Table 1). The increased overall hydrophobicity was also noted when the ECD was fused to an IgG4-derived Fc, suggesting the ECD imparts these properties upon fusion to the Fc and IgGs. Increased overall hydrophobicity did not occur with the IgG-scFv construct (GB-sc2) we evaluated (Table 1). However, across the panel of molecules evaluated, the absolute retention time was found to be a poor predictor for the increased clearance preference of the BsAbs (Tables 1, 2). For instance, a large change in retention time was observed for Fc-ECD relative to Fc, but a marginal PK difference was observed between the 2 constructs; in contrast, in the case of G2-ECD, a smaller difference in retention time was measured relative to G2, but there was a marked effect on BsAb clearance compared to the G2 mAb. Although increased hydrophobicity was clearly not causative in our observations, such properties may in part contribute as a potential limited mechanism for increased clearance, and further studies into its overall impact are warranted. In addition, since all the BsAbs examined use IgG4 parental mAbs that have been engineered with additional residue changes in the Fc region to eliminate interactions with Fcγ receptors, direct binding with blood cells is not a viable clearance mechanism. Given the lack of clearly identified differences between the BsAbs and their parental mAbs with regards to their overall biophysical parameters that are known to affect pharmacokinetics, the data suggested that a unique mechanism was involved in the clearance of these BsAb molecules.
Another plausible mechanism that we and others have suggested can negatively affect the pharmacokinetics of mAbs is the slowing of the rate of the dissociation of the IgG:FcRn as the pH increases, which would impede mAb recycling within the endosomal compartment. 25,27,34 Limited in vitro analyses of some of our BsAbs using previously published approaches showed that our fusion constructs dissociated from FcRn similarly compared to their respective wild-type mAb (data not shown).25,27 These preliminary findings suggest factor(s) other than altered dissociation from FcRn contribute to the faster clearance of our BsAb molecules. More recent studies with briakinumab and ustekinumab suggest unintended charge-related interactions of the variable fragment (Fv) region of briakinumab with FcRn may in part be an underlying mechanism eliciting the altered FcRn interactions, which facilitate degradation of mAbs in the lysosomes instead of release back into the blood circulation.25,27,34 Interestingly, these studies showed the Fv interactions with FcRn affected the elimination phase (β phase) of the pharmacokinetic profile, which explained the shorter half-life of briakinumab in humans relative to ustekinumab.34 In contrast, our BsAbs have a pronounced difference in the distribution phase (α phase) of the kinetic time course relative to their parental mAbs. Thus, our data do not appear to be consistent with altered Fv interactions with FcRn as the mechanism for the rapid BsAb clearance. However, additional interrogation of our BsAb constructs using other approaches including FcRn column chromatography and cell-based trafficking approaches may provide further insight for future studies.
As mentioned above, the aberrant clearance of the BsAb molecules described by Rossi and coworkers, Yazaki et al. and in this report was not readily explained by the typical elements of mAb clearance.9,10,18 Previous investigations did find empirically that the fusion position of the protein domain to the mAb can influence the bispecific molecule's peripheral clearance. However, these groups did not explore or tie these findings to biochemical or in vivo factors to mechanistically explain the observation.9,18 Similar to these published findings on positional effects of fusion on clearance, we observed that the pharmacokinetics of a Fc-ECD C-terminal protein fusion was not negatively affected relative to the Fc alone. This observation suggests some specific conformational aspects of the IgG-ECD constructs play a role in the clearance, and minimizes the possibility that the ECD, through cellular or other interaction, accounts for the phenomenon. The rapid clearance of the IgG-scFv constructs also points to some conformational influence related to C-terminal fusion to the IgG, which may be modulated by altering the fusion position as reported previously.9 Interestingly, the negative kinetic impact of C-terminal fusion of a scFv to IgG is not universal, as we have constructed a variety of mAb-scFv combinations that have very IgG-like pharmacokinetic properties (data not shown). This observation again seems to support some conformational change or misfolding in certain combinations of BsAbs. Importantly, it must be restated that such alterations in conformational structure must occur in vivo, or perhaps are transient in nature, because the DSC data collected did not indicate any gross perturbation of the antibody domain stability in vitro (Table 1). Although outside of the current scope of these studies, additional structural analyses using X-ray crystallography under different crystallization conditions may provide additional insight into these issues. Thus, while we and others indicate the orientation/nature of the fusion partner can affect the pharmacokinetics of a BsAb, the role is unpredictable, and at best requires empirical and case-by-case assessments.
The similarity in observations across the variety of BsAb constructs examined in this study also raises the questions of whether more universal/common clearance mechanisms could be responsible or if differential clearance mechanisms that manifest similarly in terms of LSEC mediated clearance across our BsAbs are involved. The inability to slow or saturate the peripheral clearance with a higher dose with one of the BsAbs was consistent with a high capacity clearance mechanism(s) in a highly vascularized organ such as the liver. It is generally accepted that extracellular macromolecules, as well as entire cells, are turned over by uptake and lysosomal degradation in specialized scavenger cells, namely, macrophages and scavenger endothelial cells (SECs).35 Functionally, macrophages represent the professional phagocyte responsible for the elimination of larger particles.35 While clearance of the BsAbs via resident liver macrophages was plausible, the lack of effect of macrophage depletion on clearance (Fig. 4), along with cellular staining of the tissue, ruled this out as a primary site/mechanism (Figs. 6–7). The numerous sinusoids of the liver are lined by an endocytically highly active endothelium, known as LSECs, which have been demonstrated to be the major elimination site for an array of physiological and pathophysiological soluble waste macromolecules.36,37 These cells carry high-affinity endocytic receptors (scavenger receptors, SR) that efficiently mediate the uptake and subsequent degradation of a great number of soluble macromolecules. Although we demonstrated here that LSECs appear responsible for the unusually rapid clearance of several of our IgG-scFv and IgG-ECD fusion constructs, we acknowledge that additional studies are needed to generalize this as a mechanism with common threads across multiple BsAbs and formats. It is, however, now appreciated that these SRs recognize an extremely diverse range of ligands that includes unmodified endogenous proteins and lipoproteins, as well as a number of conserved microbial structures, such as bacterial lipopolysaccharide and lipoteichoic acid.36,37 Owing to the expanding number of cognate ligands, these scavenger receptors are considered to be a subclass of the membrane-bound pattern recognition receptors.37,38 The wide range of scavenger receptor ligands suggests that epitopes generated by oxidation, charge or conformation may be involved in recognition of LSECs by SRs.37,38 To that, it has been proposed that peroxidation of endogenous proteins or lipoproteins resemble microbial structures facilitating recognition.37,38 In the context of our BsAb observations, since these structures are dynamic, it is plausible that they can assume conformations that appear ‘foreign’ in the absence or presence of a soluble antigen interaction, facilitating binding to SRs. Alternately, the BsAb structure could be mistaken as ‘antigen loaded’, imparting some ‘specific’ unintended non-specific binding to FcγRIIb, or other Fcγ receptors, that are expressed on LSECs and play a role in clearing soluble immune complexes.39-41 While the exact, and perhaps case-by-case, biochemical interaction(s) has not been defined, this mechanism of clearance is consistent with the physiological role of LSECs in the liver.
In summary, our data show that the mechanisms involved in the in vivo clearance of mAbs and various mAb structures can be complicated. Recent disclosures have revealed many biophysical and biochemical nuances that can be related to antibody pharmacokinetics in vivo.13,15,16,25,26,29,34 Clearance mechanisms related to FcRn binding, antigen interaction and charge have been characterized and are becoming better understood.13,15,16,25,26,29 However, directed modulation of these for improved molecular properties is still in its early stages, and the broad translatability of these mechanisms is still being evaluated. With the advent of more complicated mAb-like structures, such as multi-specifics, the elucidation of additional in vivo clearance mechanisms warrants further consideration and individual study.
Materials and methods
Expression and purification of recombinant monoclonal and bispecific antibody proteins
The mAbs and BsAbs described here were expressed in either transient HEK-293 or stably transfected Chinese hamster ovary cells that were generated at Eli Lilly and Company (Indianapolis, IN, USA). Purification of these compounds from concentrated cell culture supernatants proceeded via a 2-step purification method. In a first step, crude conditioned cell culture media containing antibody was captured on Protein A column, and, following column washing, bound antibody was eluted using a single step gradient of 10 mM citrate, pH 3.0, containing 150 mM sodium chloride. Molecules were further purified using a second step that utilized preparative gel filtration column chromatography with an isocratic gradient of 1X phosphate-buffered saline (PBS), pH 7.4. Protein was pooled based on size properties and purity was assessed by SDS-PAGE gel analysis and was generally found to be >95% pure.
Recombinant, soluble cynomolgus monkey FcRn was expressed in 293EBNA cells transfected with the plasmids encoding for the soluble portion αFcRn and β2m, and the protein was purified as previously described.27,42
Evaluation of the FcRn binding affinity
The interaction of the parental and bispecific mAbs with recombinant cynomolgus monkey FcRn was monitored by SPR detection using a BIAcore T-200 instrument (Biacore Inc.). Briefly, cynomolgus monkey FcRn was immobilized directly to a CM5 chip at ∼120 RUs (resonance units) using the standard amine-coupling kit methodology.
Antibodies and BsAbs samples were prepared in running buffer (PBS, pH 6.0, 0.0005% Tween 20) at 5 μM, followed by 2-fold dilutions in running buffer to 19 nM for a total of 9 concentrations evaluated in duplicate. Samples were injected for 30 seconds at a flow rate of 100 μL/min and a dissociation time of 300 seconds at 25°C. BsAbs and mAbs were dissociated from FcRn using 1x PBS (pH 7.4) dissociation buffer. Kinetic binding constants were determined through global fits of the average of 2 data sets collected on separate days using Biacore T200 Evaluation, version 1.0. The kinetics (association and dissociation rates) were simultaneously fit to a heterogeneous ligand binding model to determine the equilibrium dissociation constant (KD) value for each FcRn interaction. The fitted curves to the sensorgrams had low residuals and χ2 values. Data from both the high affinity and low affinity sites from the fit are presented. BsAb and mAb binding to FcRn was also evaluated at pH 7.4 (PBS, pH 7.4, 0.0005% Tween 20) at single concentration (5 μM diluted PBS, pH pH 7.4, 0.0005% Tween 20). Data collected at pH 7.4 was not fit since there was no observable signal.
Heparin sepharose binding chromatographic assay
The interaction of the antibodies with heparin was measured using a chromatographic method in which ∼100 μg of each antibody variant in 1X PBS, pH 7.4 was injected onto a heparin sepharose fastflow column (GE Healthcare, Waukesha, WI) that had been equilibrated in 20 mM potassium phosphate, pH 7.0. Bound protein was eluted using a linear gradient of 0–100% 20 mM potassium phosphate, 1 M sodium chloride, pH 7.0 and extent of heparin sulfate binding activity was assessed based on the observed column retention (measured in minutes) and the corresponding required concentration of sodium chloride to elute the bound protein from the column.
Differential scanning calorimetry analysis
The thermal melting properties of the mAbs and BsAbs were assessed using DSC (TA instruments, New Castle, DE). Samples were dialyzed into 1X PBS, pH 7.4 and diluted to a final concentration of 1 mg/ml. The samples were then subjected to increased temperatures gradients with a temperature ramp of 1°C/min under a constant cell pressure of 45 psi. Resultant data obtained from sample scans was buffer scan subtracted, converted to molar heat capacity and fit to a 2-state scaled model to obtain Tm.
Evaluation of mAb and BsAb isoelectric points (pIs)
The pIs of the mAbs and BsAbs evaluated in this report were determined by capillary isoelectric focusing (iCEF), Protein Simple iCE3 system (Protein Simple, San Jose CA). Samples were prepared by mixing 30 µL (5–10 μg) of protein sample with 170 µL of master mix (700 µL of 1% v/v methylcellulose, 750 µL of 8 M urea, 10 µL of pI 4.22 marker stock, 10 ul of pI 9.46 marker stock, 100 ul of Pharmalyte 3–10 (GE Healthcare), 130 ul of water), and briefly vortexed to mix. The electrophoresis and data collection were performed using a focusing step voltage of 15 kgvolts for 1 minute, followed by a second focusing phase with a step voltage of 3 kgvolts for 8 minutes. During this second focusing period, image “snapshots” of 280nm signal were acquired every 30 seconds to monitor focusing progress to equilibrium. Resultant data were exported and plotted as a function of A280 signal versus pH and main peak, and the corresponding pI were calculated using chemstation software (Agilent).
Hydrophobic interaction chromatography analysis
The gross hydrophobicity of the molecules evaluated in this study was assessed using HIC methods. Proteins were diluted to 0.5 mg/ml with 100 mM potassium phosphate, pH 6.7 containing 2 M ammonium sulfate. These “salted” samples were then injected onto a NPR butyl HIC column (Tosho). Bound protein was then eluted using a linear gradient of 0–100% 50 mM potassium phosphate, pH 6.7 and hydrophobicity of individual compounds was inferred based on the observed column retention (measured in minutes) and the corresponding minimal concentration of ammonium sulfate required for column retention.
Cynomolgus monkey pharmacokinetic studies
A number of independent cynomolgus monkey pharmacokinetic studies were performed at a contract research organization (Covance Laboratories, Madison, WI). The animal studies were conducted within approved IACUC guidelines. In six studies, 2 to 3 male cynomolgus monkeys (2.3–3.2 kg) were assigned to each study group, and all animals received a single IV bolus dose via the saphenous vein. In the first 4 studies, there were 2 animals per group. Each animal in the first study received either: G1, G1-ECD, G3 or G3-ECD dissolved in PBS (pH ∼7.4) at 2.0 mg/kg. In the second study, each animal received G2 dissolved in PBS (pH ∼7.4) at 10.0 mg/kg, while each animal in the third study received G2-ECD dissolved in PBS (pH ∼7.4) at 15.0 mg/kg. Animals in the fourth study received either Fc or Fc-ECD dissolved in PBS (pH ∼7.4) at 2 mg/kg. In the fifth study, 3 animals per group received GB dissolved in 10 mM citrate/150 mM NaCl (pH ∼6.5) at 5 mg/kg, and in the sixth study, 2 animals per group received GB-sc2 dissolved in 10 mM citrate/150 mM NaCl (pH ∼6.0) at 5 mg/kg. In all 6 studies, each animal had blood samples collected via a femoral vein. Data is reported from blood samples collected at predose through 672 hours post dose. Blood samples were collected into tubes containing no anticoagulant (serum separator tubes) or sodium citrate or potassium EDTA anticoagulant. To obtain serum, blood samples were allowed to clot at ambient temperature prior to centrifugation. To obtain plasma, blood samples were maintained in chilled cyroracks and centrifuged.
Cynomolgus monkey pharmacokinetic studies with clodronate liposome treatment
The kinetics of G1-ECD in cynomolgus monkeys pretreated with liposomes containing clodronate to deplete macrophage cells in vivo was also evaluated (Covance Laboratories, Madison, WI). In this study, 4 male cynomolgus monkeys (2.2–3.0 kg) were assigned to one of 2 study groups. The pretreatment materials, liposomal clodronate (Clodrosome®) and control liposomes (Encapsome®) were received from Encapsula NanoSciences as pre-formulated suspensions in PBS (pH ∼7.4). Approximately 24 hours prior to G1-ECD administration, each animal received a single 13 mL IV infusion of either liposomal clodronate at ∼20–23 mg/kg or control liposomes given at 1 mL/minute via a saphenous vein. Each animal received an IV bolus dose of G1-ECD dissolved in PBS (pH ∼7.4) at 2.0 mg/kg. Blood samples were collected from each animal via a femoral vein prior to dosing and at scheduled intervals from 1 to 168 hours after administration of the dose into tubes containing sodium citrate anticoagulant. The blood samples were maintained in chilled cryoracks and centrifuged to obtain plasma.
Bioanalytical assays and pharmacokinetic data analysis
Concentrations of the IgG, IgG-ECD, Fc, Fc-ECD and IgG-scFv molecules in cynomolgus monkey serum or plasma were determined using anti-human IgG or anti-human kappa ELISAs for each of the molecules. In brief, each well of a microtiter plate was coated with either goat anti-human IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) or goat anti-human kappa antibody (Southern Biotech, Birmingham, AL). After sample pretreatment, washing and blocking, all the standards, control samples, and study samples were added to the plates, then incubated for one hour at room temperature. After washing, the bound molecules were detected with a horseradish peroxidase-conjugated mouse anti-human IgG (Fc) antibody (Southern Biotech, Birmingham, AL) via TMB Microwell Peroxidase Substrate System (KPL, Gaithersburg, MD) for a colorimetric response. Plates were read at 450–493 nm with a reference of 630 nm. Concentrations from plasma or serum samples were determined from a standard curve prepared with known amounts of G1, G1-ECD, G2, G2-ECD, G3, G3-ECD, Fc, Fc-ECD, GB or GB-sc2 in the appropriate cynomolgus monkey matrix using a 4/5-parameter algorithm. The standard curve range for G1, G3 and G3-ECD was from 3.91 to 500 ng/mL, and the lower limit of quantitation (LLOQ) was defined as 25 ng/mL. For G1-ECD, the standard curve range was from 0.98 to 125 ng/mL, and the LLOQ was defined as 6 ng/mL. G2 had a standard curve range of 0.78 to 50 ng/mL with an LLOQ defined as 0.78 mg/mL, while G2-ECD had a standard curve range from 4.88 to 156.25 ng/mL and an LLOQ defined as 20 ng/mL. Both Fc and Fc-ECD had a standard curve range from 7.81 to 1000 ng/mL with a defined LLOQ as 25 ng/mL. The standard curve range for GB was from 25 to 300 ng/mL with a defined LLOQ as 25 ng/mL and GB-sc2 standard curve range was from 1.95 to 250 ng/mL with a defined LLOQ as 6 ng/mL.
Plasma or serum concentration-time data following IV administration was described using a model-independent method according to the statistical moment theory using either the WinNonlin® Professional 5.0 or Phoenix® WinNonlin® software package (Pharsight, A Certara™ Company, St. Louis, MO). The parameters calculated included the maximum plasma or serum concentration (Cmax), area under the curve (AUC0-∞), clearance (CL), and elimination half-life (T1/2).
Liposome-treated cynomolgus monkey clinical chemistry and hematology test methods
In the cynomolgus monkey pharmacokinetic studies with clodronate liposome treatment, blood samples were collected from each animal via a femoral vein into tubes containing no anticoagulant (serum separator tubes) (clinical chemistry) or tubes containing K2EDTA anticoagulant (hematology). Routine clinical chemistry and hematology tests were conducted and the parameters are listed in Supplemental Table 1.
Liposome-treated cynomolgus monkey liver biopsy and terminal liver collections
Animals were fasted overnight prior to liver biopsy and terminal liver sample collections according to approved IACUC guidelines. Briefly, for non-terminal collection time points, liver biopsy samples were collected laparoscopically from each animal treated with clodronate or empty liposomes only. Animals were placed under general anesthesia using ketamine and sevoflurane (an analog of isoflurane). The abdomen was insufflated with CO2 and each biopsy sample was taken from the liver margin. For terminal liver collections, animals were sacrificed via exsanguination under sodium pentobarbital anesthesia and the left lateral lobe was excised from each animal.
Liposome-treated cynomolgus monkey liver immunohistochemistry
Each liver tissue (including biopsy tissues) was fixed in 10% (neutral buffered formalin solution) for preparation of Formalin-Fixed Paraffin Embedding (FFPE). Liver sections of ∼5 µm were stained with hematoxylin and eosin (H&E) and CD68 immunohistochemistry (IHC) (described in section Cynomolgus monkey liver distribution studies below) for macrophage evaluation. For scoring macrophage numbers, an independent board certified pathologist used a nonparametric scale as described in the results section (Table 3 footnote).
Cynomolgus monkey liver distribution studies
Male cynomolgus monkeys (∼2- to 3 kg) were IV administered compounds G1, G1-ECD or GB-sc2 (n = 1 animal/molecule) at doses of 2 mg/kg, 2 mg/kg, and 5 mg/kg, respectively, as a single bolus injection via a saphenous vein. Liver biopsy samples were collected from each animal at 1 or 6 hours post-dose. Liver tissues were processed to FFPE and sectioned as previously described. FFPE sections were deparaffinized and rehydrated prior to immunofluorescent staining to detect G1, G1-ECD or GB-sc2, as well as endothelial cells (CD31 marker) and resident macrophages (CD68 marker).43,44 Antigen retrieval was performed using 1x heat-induced epitope retrieval solution, Diva Decloaker (BioCare Medical, Concord, CA) for 30 seconds at 125°C under pressure. After rinsing with 1x TBS-T (Dako Wash Buffer, Dako Carpinteria, CA), liver sections were incubated with a polyclonal anti-human IgG (Bethyl Laboratories, Montgomery, TX) at 10 µg/mL to detect antibodies; a monoclonal anti-human CD31 (Dako, Carpinteria, CA) at 10 µg/mL and a monoclonal anti-human CD68 (Thermo Scientific, Waltham, MA) at a dilution of 1:50. Species-specific control IgG obtained from Jackson ImmunoResearch, (West Grove, PA) or R&D Systems, (Minneapolis, MN), respectively, were used at concentrations equivalent to the primary antibodies as a control to determine the specificity of BsAb detection. Following a 1 hour incubation with the primary antibody or control IgGs, slides were rinsed a minimum of 4 times with 1x wash buffer followed by detection with donkey anti-species Alexa dye-conjugated reagents each at 10 µg/mL (Life Technologies, (Grand Island, NY) or Jackson ImmunoResearch (West Grove, PA)) in combination with DAPI at a 1:500 dilution for 30 minutes prior to being water rinsed. Stained slides were coverslipped using Dako fluorescent mounting media. Reagent volumes ranged between 150–200 uL/slide, dependent upon tissue area to be covered. All steps were performed at ambient temperature and with slides and reagents protected from light.
Images of the stained slides were collected on a 3-D HISTECH (Budapest, Hungary) scanner having Plan-Apochromat 40x objective lenses at ambient temperature. The fluorochromes used were Alexa-488, Alexa-555, Alexa-647 and DAPI. A PCO.edge camera was used to capture images in the JPEG medium and the Pannoramic Viewer software (3DHISTECH) was used for image acquisition. Brightness and contrast parameters were applied consistently to all images.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Jirong Lu, Ling Liu, Victor Obungu, Derrick Witcher and Jeffrey Boyles for helpful discussions.
References
- 1.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol 2010; 10:301-16; PMID:20414204; http://dx.doi.org/ 10.1038/nri2761 [DOI] [PubMed] [Google Scholar]
- 2.Grandjenette C, Dicato M, Diederich M. Bispecific antibodies: an innovative arsenal to hunt, grab and destroy cancer cells. Curr Pharm Biotechnol 2015; 16:670-83; PMID:25941884; http://dx.doi.org/ 10.2174/1389201016666150505124037 [DOI] [PubMed] [Google Scholar]
- 3.Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today 2015; 20(7):838-47; PMID:25728220; http://dx.doi.org/ 10.1016/j.drudis.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 4.Nunez-Prado N, Compte M, Harwood S, Alvarez-Mendez A, Lykkemark S, Sanz L, Alvarez-Vallina L. The coming of age of engineered multivalent antibodies. Drug Discov today 2015; 20:588-94; PMID:25757598; http://dx.doi.org/ 10.1016/j.drudis.2015.02.013 [DOI] [PubMed] [Google Scholar]
- 5.Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67(2 Pt A):95-106; PMID:25637431; http://dx.doi.org/ 10.1016/j.molimm.2015.01.003 [DOI] [PubMed] [Google Scholar]
- 6.Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, Hattori K. Engineering the variable region of therapeutic IgG antibodies. mAbs 2011; 3:243-52; PMID:21406966; http://dx.doi.org/ 10.4161/mabs.3.3.15234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dela Cruz JS, Trinh KR, Morrison SL, Penichet ML. Recombinant anti-human HER2/neu IgG3-(GM-CSF) fusion protein retains antigen specificity and cytokine function and demonstrates antitumor activity. J Immunol 2000; 165:5112-21; http://dx.doi.org/ 10.4049/jimmunol.165.9.5112 [DOI] [PubMed] [Google Scholar]
- 8.Dong J, Sereno A, Aivazian D, Langley E, Miller BR, Snyder WB, Chan E, Cantele M, Morena R, Joseph IB, et al.. A stable IgG-like bispecific antibody targeting the epidermal growth factor receptor and the type I insulin-like growth factor receptor demonstrates superior anti-tumor activity. mAbs 2011; 3:273-88; PMID:21393993; http://dx.doi.org/ 10.4161/mabs.3.3.15188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossi EA, Chang CH, Cardillo TM, Goldenberg DM. Optimization of multivalent bispecific antibodies and immunocytokines with improved in vivo properties. Bioconjugate Chem 2013; 24:63-71; PMID:23116517; http://dx.doi.org/ 10.1021/bc300488f [DOI] [PubMed] [Google Scholar]
- 10.Rossi EA, Goldenberg DM, Cardillo TM, Stein R, Chang CH. Hexavalent bispecific antibodies represent a new class of anticancer therapeutics: 1. Properties of anti-CD20/CD22 antibodies in lymphoma. Blood 2009; 113:6161-71; PMID:19372261; http://dx.doi.org/ 10.1182/blood-2008-10-187138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croasdale R, Wartha K, Schanzer JM, Kuenkele KP, Ries C, Mayer K, Gassner C, Wagner M, Dimoudis N, Herter S, et al.. Development of tetravalent IgG1 dual targeting IGF-1R-EGFR antibodies with potent tumor inhibition. Arch Biochem Biophys 2012; 526:206-18; PMID:22464987; http://dx.doi.org/ 10.1016/j.abb.2012.03.016 [DOI] [PubMed] [Google Scholar]
- 12.Dall'Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol 2002; 169:5171-80; http://dx.doi.org/ 10.4049/jimmunol.169.9.5171 [DOI] [PubMed] [Google Scholar]
- 13.Datta-Mannan A, Thangaraju A, Leung D, Tang Y, Witcher DR, Lu J, Wroblewski VJ. Balancing charge in the complementarity-determining regions of humanized mAbs without affecting pI reduces non-specific binding and improves the pharmacokinetics. mAbs 2015; 7:483-93; PMID:25695748; http://dx.doi.org/ 10.1080/19420862.2015.1016696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Datta-Mannan A, Wroblewski VJ. Application of FcRn binding assays to guide mab development. Drug Metab Dispos 2014; 42:1867-72; PMID:25024401; http://dx.doi.org/ 10.1124/dmd.114.059089 [DOI] [PubMed] [Google Scholar]
- 15.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, et al.. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel 2010; 23:385-92; PMID:20159773; http://dx.doi.org/ 10.1093/protein/gzq009 [DOI] [PubMed] [Google Scholar]
- 16.Li B, Tesar D, Boswell CA, Cahaya HS, Wong A, Zhang J, Meng YG, Eigenbrot C, Pantua H, Diao J, et al.. Framework selection can influence pharmacokinetics of a humanized therapeutic antibody through differences in molecule charge. mAbs 2014; 6:1255-64; PMID:25517310; http://dx.doi.org/ 10.4161/mabs.29809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vugmeyster Y, Szklut P, Wensel D, Ross J, Xu X, Awwad M, Gill D, Tchistiakov L, Warner G. Complex pharmacokinetics of a humanized antibody against human amyloid beta peptide, anti-abeta Ab2, in nonclinical species. Pharm Res 2011; 28:1696-706; PMID:21424161; http://dx.doi.org/ 10.1007/s11095-011-0405-x [DOI] [PubMed] [Google Scholar]
- 18.Yazaki PJ, Lee B, Channappa D, Cheung CW, Crow D, Chea J, Poku E, Li L, Andersen JT, Sandlie I, et al.. A series of anti-CEA/anti-DOTA bispecific antibody formats evaluated for pre-targeting: comparison of tumor uptake and blood clearance. Protein Eng Des Sel 2013; 26:187-93; PMID:23175797; http://dx.doi.org/ 10.1093/protein/gzs096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen Y, Zeng L, Novosyadlyy R, Forest A, Zhu A, Korytko A, Zhang H, Eastman SW, Topper M, Hindi S, et al.. A bi-functional antibody-receptor domain fusion protein simultaneously targeting IGF-IR and VEGF for degradation. mAbs 2015; 7:931-45; PMID:26073904; http://dx.doi.org/ 10.1080/19420862.2015.1055442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molskness TA, Stouffer RL, Burry KA, Gorrill MJ, Lee DM, Patton PE. Circulating levels of free and total vascular endothelial growth factor (VEGF)-A, soluble VEGF receptors-1 and -2, and angiogenin during ovarian stimulation in non-human primates and women. Hum Reprod 2004; 19:822-30; PMID:15033950; http://dx.doi.org/ 10.1093/humrep/deh132 [DOI] [PubMed] [Google Scholar]
- 21.Xin Y, Li J, Wu J, Kinard R, Weekes CD, Patnaik A, Lorusso P, Brachmann R, Tong RK, Yan Y, et al.. Pharmacokinetic and pharmacodynamic analysis of circulating biomarkers of anti-NRP1, a novel antiangiogenesis agent, in two phase I trials in patients with advanced solid tumors. Clin Cancer Res 2012; 18:6040-8; PMID:22962439; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1652 [DOI] [PubMed] [Google Scholar]
- 22.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al.. High Resolution Mapping of the Binding Site on Human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and Design of IgG1 Variants with Improved Binding to the FcγR. J Biol Chem 2001; 276:6591-604; PMID:11096108; http://dx.doi.org/ 10.1074/jbc.M009483200 [DOI] [PubMed] [Google Scholar]
- 23.Stubenrauch K, Wessels U, Regula JT, Kettenberger H, Schleypen J, Kohnert U. Impact of molecular processing in the hinge region of therapeutic IgG4 antibodies on disposition profiles in cynomolgus monkeys. Drug Metab Dispos 2010; 38:84-91; PMID:19850673; http://dx.doi.org/ 10.1124/dmd.109.029751 [DOI] [PubMed] [Google Scholar]
- 24.Boswell CA, Tesar DB, Mukhyala K, Theil F-P, Fielder PJ, Khawli LA. Effects of Charge on Antibody Tissue Distribution and Pharmacokinetics. Bioconjugate Chem 2010; 21:2153-63; PMID:21053952; http://dx.doi.org/ 10.1021/bc100261d [DOI] [PubMed] [Google Scholar]
- 25.Datta-Mannan A, Chow CK, Dickinson C, Driver D, Lu J, Witcher DR, Wroblewski VJ. FcRn affinity-pharmacokinetic relationship of five human IgG4 antibodies engineered for improved in vitro FcRn binding properties in cynomolgus monkeys. Drug Metab Dispos 2012; 40:1545-55; PMID:22584253; http://dx.doi.org/ 10.1124/dmd.112.045864 [DOI] [PubMed] [Google Scholar]
- 26.Hotzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al.. A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012; 4:753-60; PMID:23778268; http://dx.doi.org/ 10.4161/mabs.22189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Jiang W, Wroblewski VJ. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: relationship to pharmacokinetics in mice and primates. Drug Metab Dispos 2007; 35:86-94; PMID:17050651; http://dx.doi.org/ 10.1124/dmd.106.011734 [DOI] [PubMed] [Google Scholar]
- 28.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Investig 2005; 115:56-65; PMID:15630444; http://dx.doi.org/ 10.1172/JCI200522675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khawli LA, Goswami S, Hutchinson R, Kwong ZW, Yang J, Wang X, Yao Z, Sreedhara A, Cano T, Tesar D, et al.. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. mAbs 2010; 2:613-24; PMID:20818176; http://dx.doi.org/ 10.4161/mabs.2.6.13333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, Moriyama C, Watanabe T, Takubo R, Doi Y, et al.. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol 2010; 28:1203-7; PMID:20953198; http://dx.doi.org/ 10.1038/nbt.1691 [DOI] [PubMed] [Google Scholar]
- 31.Zheng Y, Scheerens H, Davis JC, Deng R, Fischer SK, Woods C, Fielder PJ, Stefanich EG. Translational Pharmacokinetics and Pharmacodynamics of an FcRn-Variant Anti-CD4 Monoclonal Antibody From Preclinical Model to Phase I Study. Clin Pharmacol Ther 2011; 89:283-90; PMID:21191378; http://dx.doi.org/ 10.1038/clpt.2010.311 [DOI] [PubMed] [Google Scholar]
- 32.Gurbaxani B, Dela Cruz LL, Chintalacharuvu K, Morrison SL. Analysis of a family of antibodies with different half-lives in mice fails to find a correlation between affinity for FcRn and serum half-life. Mol Immunol 2006; 43:1462-73; PMID:16139891; http://dx.doi.org/ 10.1016/j.molimm.2005.07.032 [DOI] [PubMed] [Google Scholar]
- 33.Gurbaxani B, Dostalek M, Gardner I. Are endosomal trafficking parameters better targets for improving mAb pharmacokinetics than FcRn binding affinity? Mol Immunol 2013; 56:660-74; PMID:23917469; http://dx.doi.org/ 10.1016/j.molimm.2013.05.008 [DOI] [PubMed] [Google Scholar]
- 34.Schoch A, Kettenberger H, Mundigl O, Winter G, Engert J, Heinrich J, Emrich T. Charge-mediated influence of the antibody variable domain on FcRn-dependent pharmacokinetics. Proc Natl Acad Sci U S A 2015; 112(19):5997-6002; http://dx.doi.org/ 10.1073/pnas.1408766112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sorensen KK, McCourt P, Berg T, Crossley C, Le Couteur D, Wake K, Smedsrod B. The scavenger endothelial cell: a new player in homeostasis and immunity. Am J Physiol Regul Integr Comp Physiol 2012; 303:R1217-30; PMID:23076875; http://dx.doi.org/ 10.1152/ajpregu.00686.2011 [DOI] [PubMed] [Google Scholar]
- 36.Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Ann Rev Immunol 2015; 33:643-75; PMID:25861979; http://dx.doi.org/ 10.1146/annurev-immunol-032414-112220 [DOI] [PubMed] [Google Scholar]
- 37.Zani IA, Stephen SL, Mughal NA, Russell D, Homer-Vanniasinkam S, Wheatcroft SB, Ponnambalam S. Scavenger receptor structure and function in health and disease. Cells 2015; 4:178-201; PMID:26010753; http://dx.doi.org/ 10.3390/cells4020178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ashraf MZ, Sahu A. Scavenger receptors: a key player in cardiovascular diseases. Biomolecular Concepts 2012; 3:371-80; PMID:25436543; http://dx.doi.org/ 10.1515/bmc-2012-0003 [DOI] [PubMed] [Google Scholar]
- 39.Jodo S, Kung JT, Xiao S, Chan DV, Kobayashi S, Tateno M, Lafyatis R, Ju ST. Anti-CD95-induced lethality requires radioresistant Fcgamma RII+ cells. A novel mechanism for fulminant hepatic failure. J Biol Chem 2003; 278:7553-7; PMID:12477718; http://dx.doi.org/ 10.1074/jbc.M211229200 [DOI] [PubMed] [Google Scholar]
- 40.Roux KH, Strelets L, Brekke OH, Sandlie I, Michaelsen TE. Comparisons of the ability of human IgG3 hinge mutants, IgM, IgE, and IgA2, to form small immune complexes: a role for flexibility and geometry. J Immunol 1998; 161:4083-90. [PubMed] [Google Scholar]
- 41.Taylor RP, Lindorfer MA. Fcgamma-receptor-mediated trogocytosis impacts mAb-based therapies: historical precedence and recent developments. Blood 2015; 125:762-6; PMID:25498911; http://dx.doi.org/ 10.1182/blood-2014-10-569244 [DOI] [PubMed] [Google Scholar]
- 42.Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Wroblewski VJ. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J Biol Chem 2007; 282:1709-17; PMID:17135257; http://dx.doi.org/ 10.1074/jbc.M607161200 [DOI] [PubMed] [Google Scholar]
- 43.Holness CL, Simmons DL. Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 1993; 81:1607-13; PMID:7680921 [PubMed] [Google Scholar]
- 44.Kishimoto T, Goyert S, Kikutani H, Mason D, Miyasaka M, Moretta L, Ohno T, Okumura K, Shaw S, Springer TA, et al.. Update: new CD antigens, 1996. Tissue Antigens 1997; 49:287-8; PMID:9098941; http://dx.doi.org/ 10.1111/j.1399-0039.1997.tb02755.x [DOI] [PubMed] [Google Scholar]