

Abstract
The most common form of the childhood neurodegenerative disease late infantile neuronal ceroid lipofuscinosis (also called Batten disease) is caused by deficiency of the soluble lysosomal enzyme tripeptidyl peptidase 1 (TPP1) resulting from mutations in the TPP1 gene. We tested whether TPP1 gene transfer to the ependyma, the epithelial lining of the brain ventricular system, in TPP1-deficient dogs would be therapeutically beneficial. A one-time administration of recombinant adeno-associated virus (rAAV) expressing canine TPP1 (rAAV.caTPP1) resulted in high expression of TPP1 predominantly in ependymal cells and secretion of the enzyme into the cerebrospinal fluid leading to clinical benefit. Diseased dogs treated with rAAV.caTPP1 showed delays in onset of clinical signs and disease progression, protection from cognitive decline, and extension of life span. By immunostaining and enzyme assay, recombinant protein was evident throughout the brain and spinal cord, with correction of the neuropathology characteristic of the disease. This study in a naturally occurring canine model of TPP1 deficiency highlights the utility of AAV transduction of ventricular lining cells to accomplish stable secretion of recombinant protein for broad distribution in the central nervous system and therapeutic benefit.
INTRODUCTION
Late infantile neuronal ceroid lipofuscinosis (LINCL) is a childhood neurodegenerative disorder. Development is normal up to ages 2 to 4 years after which manifestations present as motor deterioration and mental decline, seizures, and visual deficits. Death generally occurs within the first decade of life (1). Most cases of LINCL are due to mutations in TPP1, which cause a deficiency of the soluble lysosomal enzyme tripeptidyl peptidase 1 (TPP1) (2). TPP1 is synthesized as a mannose-6-phosphate–decorated proenzyme, and similar to other soluble lysosomal hydrolases, the proenzyme is largely targeted to the lysosome but can also be released from the cell via the secretory pathway. Hence, cellular uptake by the same or neighboring cells, and subsequent lysosomal delivery and activation of the proenzyme to the active form, can occur (2, 3). These characteristics are important for cross-correction strategies including TPP1 protein replacement (4).
Helen Cserr and colleagues showed in earlier work that tracers, when delivered to the subarachnoid space of a living mammal, penetrated the brain parenchyma via the perivascular spaces (5). This provides a strong rationale for using the cerebrospinal fluid (CSF) to distribute recombinant protein throughout the brain. Indeed, enzyme replacement therapy has produced clinical benefit in TPP1 knockout mice (6) and TPP1-deficient dogs (7–9). In these studies, continuous infusion using implanted devices in rodents or repeated direct infusion of recombinant human pro-TPP1 into the CSF of dogs resulted in TPP1 biodistribution throughout the brain parenchyma. An associated decrease in the characteristic autofluorescence storage material, as well as decreases in astrocytic activation and neurodegeneration, was also found (7). The neuropathological changes were accompanied by attenuated progression of neurological signs (6, 9). Although promising, patients receiving TPP1 enzyme replacement therapy currently require biweekly infusion (ClinicalTrials.gov identifier: NCT01907087), which requires specialized lifelong care and geographical restriction to be close to major clinical centers. Additionally, problems have been reported associated with indwelling catheters required for CSF access in the TPP1-deficient dog model (8). Although patients treated for childhood brain cancers have indwelling catheters in place for up to 20 years, they are not accessed after cessation of cancer treatment in contrast to the repeated infusions required for TPP1 enzyme replacement therapy.
As an alternative, we tested the hypothesis that gene transfer predominantly to ependymal cells, which have direct access to the CSF, will provide long-term and widespread biodistribution of TPP1 in the LINCL dog model after a single unilateral infusion of recombinant adeno-associated virus (rAAV) expressing the canine form of TPP1 (caTPP1). The ependyma is composed of a single layer of epithelial cells lining the brain ventricular system and spinal cord central canal. Ependymal cells are multiciliated and postmitotic (10, 11), and they are essential for directional CSF flow and movement of paracrine signals, metabolites, and toxins through and out of the brain (10, 12–14). Ependymal cell transduction with rAAV expressing lysosomal hydrolases has been effective in reversing phenotypes in mouse models of lysosomal storage diseases (15, 16), but the utility of this approach in larger animal models is unknown. Here, we tested whether rAAV2 expressing canine TPP1 (rAAV.caTPP1) delivered into the cerebral ventricles for transduction of ependyma can provide enzyme replacement throughout the brain for therapeutic benefit.
RESULTS
Expression of TPP1 in canine CSF
The TPP1-null dachshund disease model has a frameshift mutation in TPP1 (17) with no detectable TPP1 protein or activity in blood or tissues and progressive neurodegenerative symptomatology that recapitulates human TPP1 deficiency (18, 19). First, we tested whether rAAV transduction of ependyma with TPP1 can provide widespread access of recombinant TPP1 to the central nervous system. In mice, rAAV4 is unique in that intrastriatal or intraventricular injection results in robust ependyma transduction (20). In contrast, we saw no ependyma transduction in canine brain after rAAV4 delivery. We additionally screened rAAV1, rAAV2, rAAV5, rAAV8, and rAAV9 serotypes expressing reporter genes and found rAAV2 to be optimal; intraventricular injection of 2 × 1012 vector genomes resulted in transduction of the ependyma lining the lateral third and fourth ventricles as evidenced by enhanced green fluorescent protein expression (fig. S1).
We next quantified TPP1 enzyme activity early after unilateral injection of rAAV. caTPP1 into the CSF in two presymptomatic TPP1−/− dogs (dogs DC013 and DC014; see table S1; all dogs were given the immunosuppressant cyclosporine before treatment). By as early as 5 days after injection, there were notable increases in TPP1 enzyme activity in the CSF, up to 30-fold greater than concentrations measured in the CSF of normal control dogs (Fig. 1A and fig. S2). However, recombinant TPP1 activity subsided to background levels 2 months later (Fig. 1A). As these dogs are TPP1-null, we tested whether this decline was coincident with an increase in neutralizing antibodies (NAbs). Canine TPP1 proenzyme was incubated with serial dilutions of CSF collected from rAAV.caTPP1-treated TPP1−/− dogs, and then the mixture was applied to Tpp1−/− mouse embryonic fibroblasts and the internalized TPP1 was quantified. We found that TPP1 activity in Tpp1−/− cell lysates was reduced when the recombinant protein was incubated with CSF from rAAV.caTPP1-treated dogs collected 1 month after injection (Fig. 1B).
Fig. 1. TPP1 and TPP1-NAbs in CSF after rAAV.caTPP1 infusion.
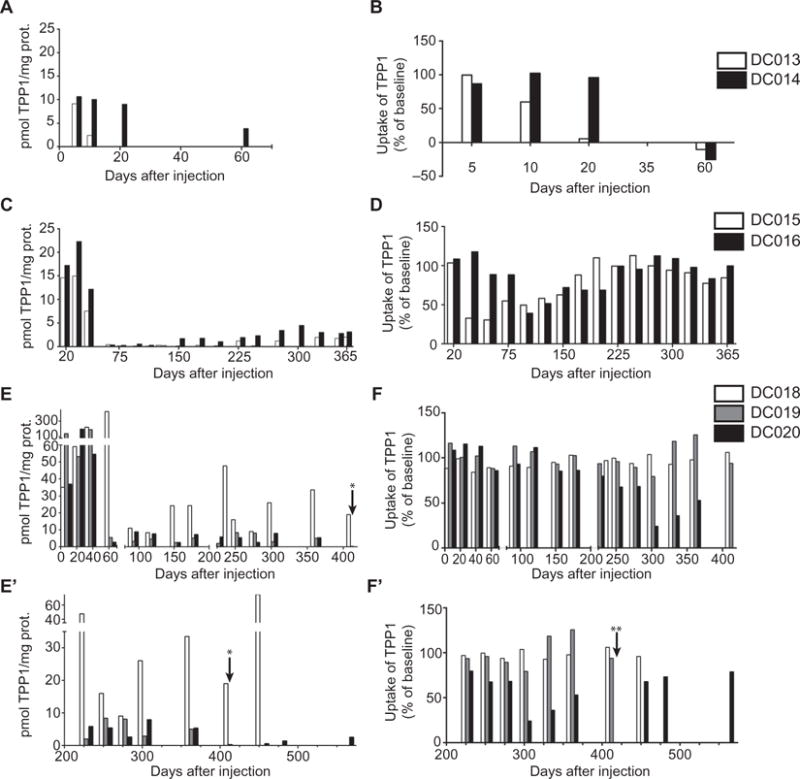
(A, C, E, and E′) An increase in TPP1 in CSF was measured with an enzyme activity assay (see also table S2). (B, D, F, and F′) NAbs in CSF were determined by a neutralization assay: TPP1-deficient mouse embryonic fibroblasts were exposed to a mixture of canine TPP1 plus dog CSF collected before and after treatment. Data were normalized to baseline (pretreatment) values. (A and B) TPP1 activity and NAbs in two dogs (DC013 and DC014) given mycophenolate mofetil treatment starting at day 44 relative to vector infusion (see table S1 for information on vector dose and drug treatment for each dog). (C and D) TPP1 activity and NAbs in two dogs (DC015 and DC016) given mycophenolate mofetil starting at day 33 relative to vector infusion. (E, E′, F, and F′) TPP1 activity and NAbs in three dogs (DC018, DC019, and DC020) given mycophenolate mofetil starting at day −5 relative to vector infusion. Vector infusions were at about 90 days of age in all cases. *TPP1 in CSF from DC019 was 0.3 pmol/mg protein or 100% of normal. **CSF from DC020 showed no detectable TPP1 uptake at this time point, indicating high levels of NAbs.
Mycophenolate mofetil plus cyclosporine is known to attenuate the immune response to dystrophin in a canine model of muscular dystrophy, extending the therapeutic window for rAAV gene therapy (21). On the basis of these findings, and the fact that some dogs were early post–rAAV.caTPP1 treatment during the time of the observed anti-TPP1 response, we instituted mycophenolate mofetil treatment in addition to ongoing cyclosporine treatment. Two dogs (DC013 and DC014) were administered mycophenolate mofetil starting at 44 days, and another two (DC015 and DC016) received mycophenolate mofetil at 33 days. The introduction of mycophenolate mofetil 44 days after vector administration did not result in a recovery of TPP1 activity (Fig. 1A). Initiating mycophenolate mofetil treatment by 33 days after vector administration resulted in lower NAb titers despite the additional dose of vector (Fig. 1, C and D, and table S1), and there was a coincident increase in TPP1 activity. Next, we treated a TPP1−/− dog with mycophenolate mofetil 5 days before rAAV.caTPP1 delivery (DC018). In this dog, TPP1 activity was robust and sustained, and anti-TPP1 antibodies did not appear (Fig. 1, E, E′, F, and F′). Given the stability of transgene expression and overall improvements in neurological symptoms, an additional two dogs (DC019 and DC020) were treated with rAAV.caTPP1 at day 5 after initiation of mycophenolate mofetil therapy.
Elevated TPP1 in CSF delays disease manifestations, extends life span, and improves in cognition
Untreated TPP1−/− dogs show onset of clinical signs as early as 5 months of age and generally require humane euthanasia at ~11 to 12 months (9). One of the earliest clinical signs in untreated TPP1−/− dogs is impaired proprioceptive responses in the hindlimbs (mean age at onset, 6 months; range, 4.6 to 8 months; n = 8). This clinical phenotype was delayed 4.5 months on average with rAAV.caTPP1 gene therapy (Fig. 2A; P < 0.05). In three dogs, abnormal nystagmus never appeared (untreated dogs: mean age at onset, 7.7 months; range, 4.6 to 10.9; n = 7; treated dogs: mean age at onset, 13.4 months; range, 12.6 to 14.3 months; n = 2) (Fig. 2B), nor did menace response deficits (untreated dogs: mean age at onset, 6.5 months; range, 4.8 to 8.3 months; n = 6; treated dogs: mean age at onset, 10 months; range, 9.9 to 10.2 months; n = 2) (Fig. 2C). Pupillary light reflex abnormalities were delayed in four treated dogs and never detected in one treated dog (untreated dogs: mean age at onset, 8.7 months; range, 6.7 to 10.1 months; n = 9; treated dogs: mean age at onset, 13.4 months; range, 10.6 to 18.7 months; n = 4) (Fig. 2D; P < 0.05). Cerebellar ataxia, which typically has an onset shortly after hindlimb proprioceptive deficits, was substantially delayed from a mean age of 7.3 months in untreated dogs (range, 5.9 to 9.4 months; n = 8) to 12.7 months in treated dogs (range, 12 to 13.8 months, n = 4; Fig. 2E; P < 0.05) (movies S1 and S2). Thoracic limb dysfunction was also delayed by 5.4 months and never appeared in one treated animal (Fig. 2F; P < 0.05).
Fig. 2. rAAV.caTPP1 delivery delays disease onset in TPP1-deficient dogs.
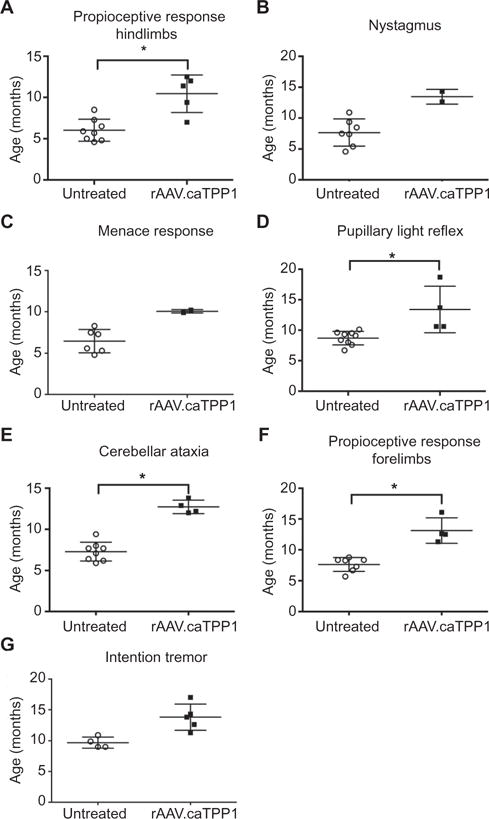
(A) Onset of proprioceptive response deficits in treated versus untreated TPP1-deficient dogs. (B and C) Onset of pathological nystagmus and menace response deficits in treated versus untreated TPP1-deficient dogs. (D) Onset of pupillary light reflex abnormalities in rAAV. caTPP1-treated TPP1-deficient dogs versus untreated dogs. (E) Onset of cerebellar ataxia in rAAV.caTPP1-treated TPP1-deficient dogs versus untreated dogs. (F) Onset of proprioceptive response deficits in the forelimbs of treated versus untreated TPP1-deficient dogs. (G) Intention tremor onset in rAAV.caTPP1-treated dogs compared to untreated TPP1-deficient dogs. *P < 0.05, nonparametric Mann-Whitney test (see also table S2).
We noted that the onset of neuroanatomical-localized abnormalities varied between treated and untreated TPP1-deficient dogs. The earliest abnormalities were general proprioceptive ataxia in untreated dogs and visual deficits in treated animals. This may reflect a lack of retinal penetration of recombinant protein secreted into the CSF. Nonetheless, there was notable sparing of the central pathways involved in the pupillary light reflex, which is initiated by the retina (Fig. 2, C and D). Overall, there were significant delays in the onset of movement abnormalities, the most striking example being the delay in onset of intention tremor (untreated dogs: mean age at onset, 9.1 months; range, 8.9 to 10.5 months; n = 6; treated dogs: mean age at onset, 13.8 months; range, 11.3 to 17 months; n = 5; Fig. 2G; P < 0.05), pathological nystagmus, and cerebellar ataxia, indicating improved function of the cerebellovestibular system.
In addition to neurological signs, TPP1−/− dogs exhibit cognitive decline that can be quantified by a T-maze (18). We enrolled four dogs, two treated and two untreated, in the T-maze test beginning at 4 months of age. Similar to earlier work (18), untreated TPP1−/− dogs made more mistakes on the T-maze and were unable to complete the task by 7 to 9 months of age (Fig. 3A). However, dogs receiving rAAV.caTPP1 navigated the maze in a similar manner to healthy dogs [for data on healthy dogs, see (9)] and continued to perform well months after untreated TPP1−/− dogs reached the end stage of disease (Fig. 3A). One treated dog retained cognitive abilities until the end stage of disease (~20 months) and was highly interactive with its environment, caregivers, and other dogs in the colony (movie S3). There was also a significant extension of life span (Fig. 3B; mean age to humane euthanasia was 10.4 months for untreated TPP1−/− dogs versus 17.5 months for rAAV.caTPP1-treated TPP1−/− dogs). The delay in disease manifestations (Fig. 2), retention of cognitive abilities, and life-span extension (Fig. 3) were greater in rAAV.caTPP1-treated TPP1−/− dogs compared to TPP1−/− dogs from the same colony receiving biweekly infusion of recombinant TPP1 into the intrathecal or ventricular space (9).
Fig. 3. rAAV.caTPP1 treatment delays onset of cognitive deficits and increases life span of TPP1-deficient dogs.
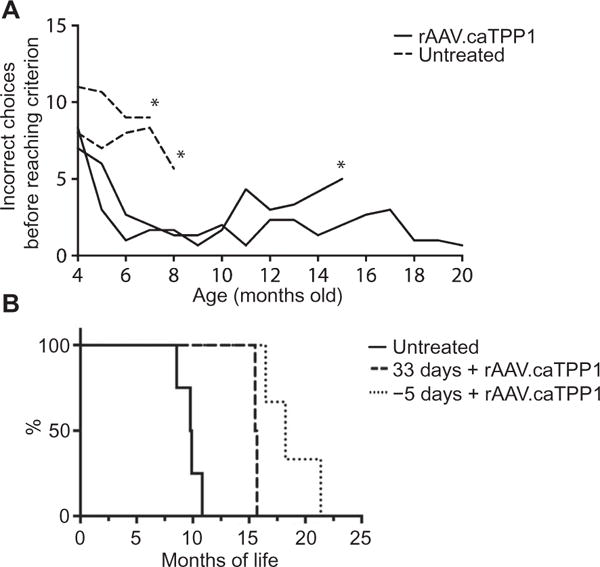
(A) Cognitive deficits were assessed by the T-maze (18) in rAAV.caTPP1-treated dogs (DC019 and DC020) and untreated dogs (DC024 and DC025) from 4 months (1 month after injection) onward (see also table S2). The asterisk denotes the last time point that dogs were capable of completing the T-maze because of motor impairment or behavioral problems. (B) Survival plot of untreated TPP1-deficient dogs (solid line) and rAAV.caTPP1-treated TPP1-deficient dogs (dashed and dotted lines). Mycophenolate mofetil treatment was initiated 33 days after vector delivery (dashed line) or 5 days before vector delivery (dotted line).
Recombinant TPP1 is widely distributed in brain parenchyma
Thus far, our results support the hypothesis that rAAV.caTPP1 gene transfer predominantly to the ependyma results in TPP1 activity in the brain that is sufficient for clinical benefit. It is expected that recombinant TPP1 in the CSF flows from the ventricles to the subarachnoid space and ultimately diffuses along perivascular spaces to the underlying parenchyma. In transgenic mice, this results in improvement in disease-associated readouts (6, 15), but we did not know whether there would be widespread delivery and regional variability in TPP1 penetration in a larger brain or whether there would be preferential uptake by certain cell types in the brain.
Using an enzyme assay for TPP1, we found significant increases in TPP1 activity from rostral to caudal brain regions in tissues harvested 60 days (2 months; DC013 and DC014), 300 days (10 months; DC015 and DC016), 444 days (~15 months; DC019), 545 days (~18 months; DC018), and 641 days (~21 months; DC020) after injection of rAAV. caTPP1. TPP1 activity was increased two- to sevenfold over normal in areas close to the lateral ventricle (Fig. 4A). Enzyme concentrations increased 7- to 10-fold in samples harvested from the caudate nucleus, thalamus, and medulla relative to tissues from TPP+/+ dogs (Fig. 4B). TPP1 activity was also detected in the occipital cortex, suggesting that the proenzyme can reach the subarachnoid space and penetrate the cerebral cortex (Fig. 4B).
Fig. 4. TPP1 activity in brain parenchyma of TPP1-deficient dogs.
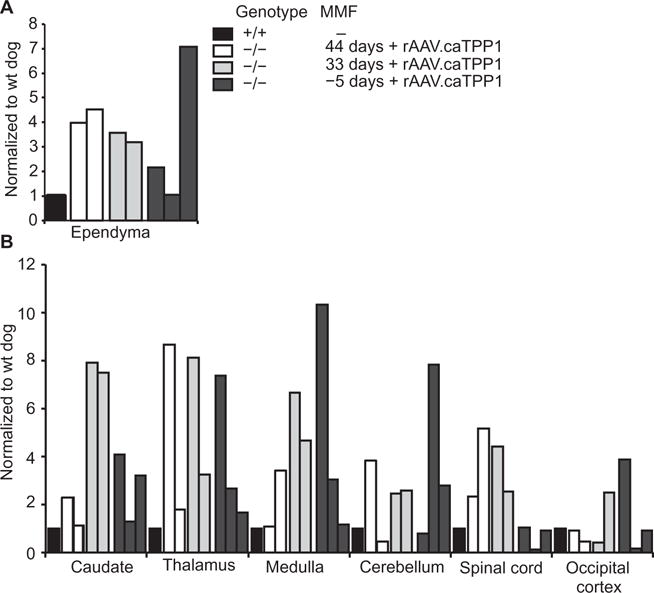
TPP1 activity at the experimental end points taken from punches from the indicated regions (see also table S2). (A) TPP1 activity in ependyma. wt, wild type. (B) TPP1 activity in periventricular or meningeal regions. TPP1 activity is relative to that found in normal dogs (see fig. S2). Open bars, dogs DC013 and DC014, which received mycophenolate mofetil treatment starting at day 44 relative to vector infusion; gray bars, dogs DC015 and DC016, which received mycophenolate mofetil treatment starting at day 33 relative to vector infusion; dark gray bars, dogs DC018, DC019, and DC020, which received mycophenolate mofetil treatment starting at day −5 relative to vector infusion; black bars, normal dogs.
To further examine TPP1 distribution in the brain, we performed immunohistochemistry for canine TPP1. We found robust enzyme staining throughout the brain, in contrast to TPP1−/− dogs (Fig. 5). TPP1 was evident in ependymal cells lining the entire ventricular system. TPP1 penetrated lateral ventricle subependymal regions (for example, septum and caudate; Fig. 5A and fig. S3A), the third ventricle (for example, hypothalamus; Fig. 5A), the fourth ventricle (for example, vestibular area; Fig. 5A), and the central canal of the spinal cord (Fig. 5B). Although unilateral injection of rAAV.caTPP1 was performed, TPP1-positive ependymal cells were evident bilaterally, resulting in a symmetrical or near-symmetrical distribution of TPP1 between the hemispheres (fig. S3A). TPP1-positive cells were also found along the cerebral and cerebellar cortices of rAAV.caTPP1-treated dogs (Fig. 5C). As expected, a caudal-to-rostral gradient of immunopositive cells was found. Most TPP1-positive cells were morphologically equivalent to pyramidal neurons, with punctate immune reactivity within the soma and the major dendrites. In the prefrontal cortex, most TPP1-positive cells were in layer V (Fig. 5C and fig. S3B), with similar regions showing positivity among all dogs, albeit with varying intensity (fig. S3B). More caudally, TPP1-immunopositive cells were found in all cortical layers (Fig. 5C). In the piriform cortex, the intracellular immunoreactivity was present in the soma and multiple cellular processes (Fig. 5C). In the cerebellar cortex, the Purkinje neurons and other cells within the molecular layers showed the most robust TPP1 staining (Fig. 5C). Overall, TPP1-positive immunoreactivity was most evident in neurons, with little evidence of uptake in astrocytes or glia. Whether this reflects a sensitivity limitation of the antibody or a true lack of uptake is unclear at this time.
Fig. 5. Biodistribution of TPP1 in rAAV.caTPP1-treated TPP1-deficient dogs.
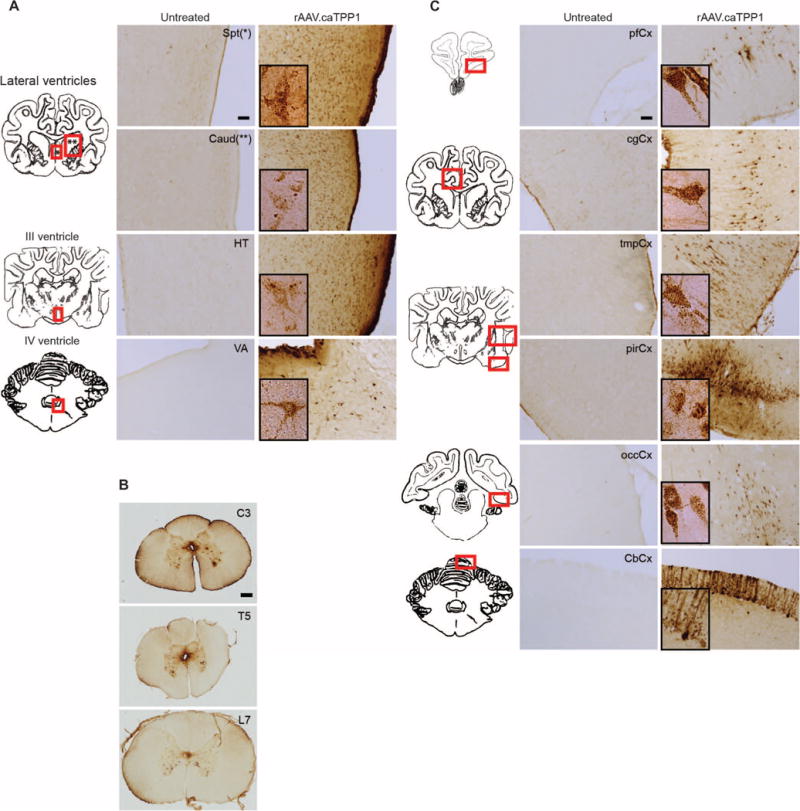
Representative immunohistochemical staining for canine TPP1 in treated versus untreated TPP1-deficient dogs. (A) TPP1-immunopositive cells in the septum (Spt) and caudate nucleus (Caud) in the forebrain, hypothalamus (HT) at the level of the third ventricle, and the vestibular area (VA) near the fourth ventricle. Scale bar, 100 μm. (B) TPP1-immunopositive cells near the central canal and neurons along the spinal cord: cervical 3 (C3), thoracic 5 (T5), and lumbar 7 (L7). Scale bar, 400 μm. (C) TPP1-immunopositive cells spanning caudal to rostral regions. pfCx, prefrontal cortex; cgCX, cingulate cortex; tmpCx, temporal cortex; pirCx, piriform cortex; occCx, occipital cortex; CbCx, cerebellar cortex. Scale bar, 100 μm. (Insets) High-magnification images of TPP1-positive cells demonstrate cellular morphology and the punctate staining typical of lysosomal localization.
We also quantified viral vector genomes in tissue punches harvested from ependyma, thalamus, cerebellar cortex, meninges, heart, liver, spleen, and kidney. rAAV.caTPP1 viral genomes were predominantly localized to the ependyma (fig. S4), and none were detected in the meninges or peripheral tissues. Together, these results corroborate the notion that enzyme secreted from ependyma can reach the subarachnoid space and be internalized along perivascular spaces throughout multiple brain regions.
Decreased astrocyte activation and storage burden after rAAV.caTPP1 transduction
Progressive astrocyte activation has been described in the brains of rodent models and patients with TPP1 deficiency (6). This activation is characterized by increased intensity of immunostaining for glial fibrillary acidic protein (GFAP) in astrocytic processes. We found intense GFAP staining in untreated TPP1-null dogs (Fig. 6). Intraventricular injection of rAAV.caTPP1 resulted in a notable decrease in GFAP immunoreactivity even in tissues from the end stage of disease (Fig. 6). The septum, caudate nucleus, motor cortex, and cingulate cortex in rAAV.caTPP1-injected dogs showed weak GFAP staining by immunohistochemistry and fewer immunoreactive processes relative to those areas in untreated TPP1-deficient dogs (Fig. 6). A similar decrease in GFAP immunoreactivity was noted in the amygdala (fig. S5A). Cumulatively, these data indicate that the presence of TPP1 in these dogs reduced the progressive astrocytosis. Because of the observed anti-TPP1 responses, we also assessed whether microglial activation was enhanced in dogs treated with rAAV.caTPP1. Similar to earlier work in this dog model treated with enzyme replacement therapy (7, 22), we did not detect microglia activation as assessed by immunohistochemistry for Iba1 (fig. S5B).
Fig. 6. rAAV.caTPP1 treatment reduces astrocytic activation.
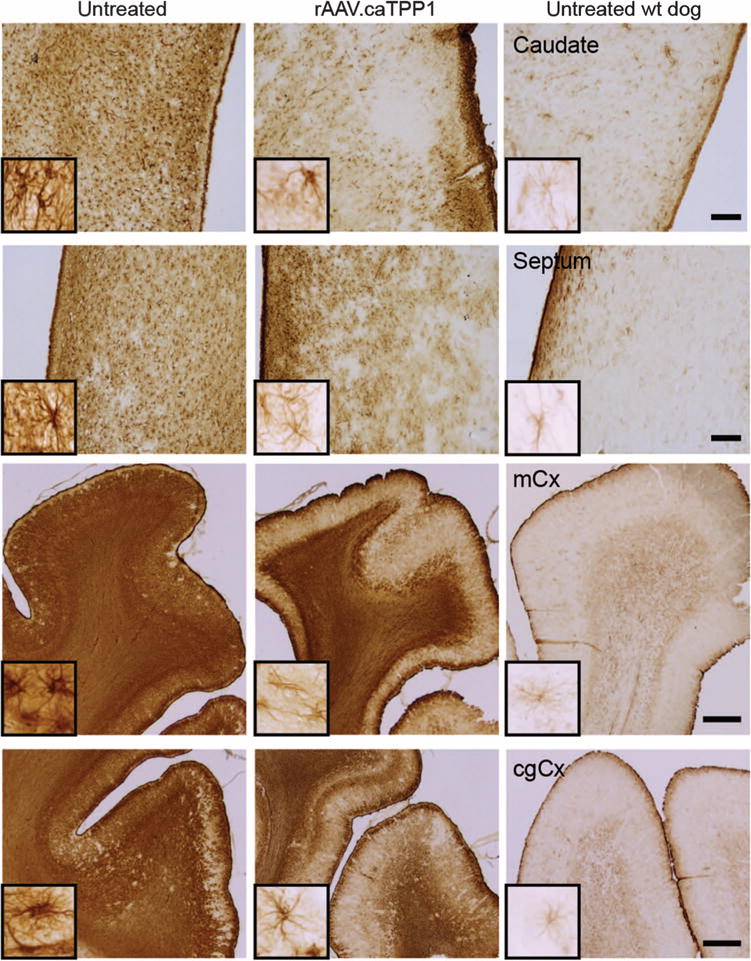
Glial astrocytosis measured by immunoreactivity for GFAP in treated versus untreated TPP1-deficient dogs. Representative photomicrographs from the caudate, septum, motor cortex (mCx), and cingulate cortex (cgCx) of untreated (n = 3) and rAAV.caTPP1-treated dogs (n = 5). GFAP staining of an untreated wt dog is shown for reference (n = 1). Scale bars, 500 μm for caudate and septum and 1 mm for mCx and cgCx photomicrographs.
Accumulation of autofluorescent material is a hallmark of the neuronal ceroid lipofuscinoses. rAAV.caTPP1 reduced storage of autofluorescent material in the brain and spinal cord (Fig. 7A). This was accompanied by a measurable decrease in adenosine triphosphate (ATP) synthase subunit C accumulation evaluated by immunohistochemistry (Fig. 7B) and Western blot (Fig. 7C and fig. S6). We also evaluated p62 expression, which was recently shown to be elevated in TPP1-null mice (23). rAAV.caTPP1 reduced p62 storage burden relative to that found in untreated dogs (Fig. 7C and fig. S6).
Fig. 7. rAAV.caTPP1 treatment reduces storage burden.
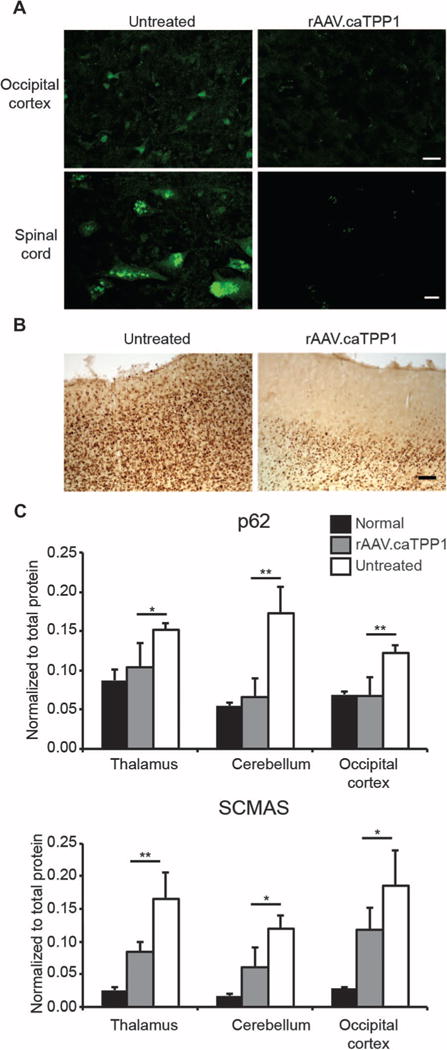
(A) Representative images of autofluorescent material in layer II/III of the occipital cortex (top panels) or spinal cord (layer C3, bottom panels) observed by confocal microscopy. Scale bar, 20 μm. (B) Immunolabeling for subunit C of mitochondrial ATP synthase (SCMAS) accumulation in rAAV.caTPP1-treated versus untreated TPP1-deficient dogs. Scale bar, 100 μm. (C) p62 and SCMAS quantified by Western blot in tissue lysates harvested from thalamus, cerebellum, or occipital cortex. *P < 0.05; **P < 0.01 by nonparametric Mann-Whitney test of treated (n = 6) versus untreated (n = 6) TPP1-deficient dogs.
Recombinant TPP1 in peripheral organs
Although the most relevant symptoms in children with mutations in TPP1 are neurological in origin, TPP1 is a ubiquitously expressed enzyme (24). TPP1 expressed in CSF after ependymal cell transduction could provide a source of enzyme systemically because protein in CSF can pass through the arachnoid villi to reach the peripheral vasculature. To test if this occurs in treated dogs, recombinant TPP1 was quantified by an assay for enzymatic activity in liver, spleen, kidney, and heart in untreated and rAAV.caTPP1-treated dogs. We found significantly elevated TPP1 activity in spleen and heart in rAAV.caTPP1-treated dogs relative to untreated dogs (Fig. 8; P < 0.05). Although total activity was elevated, immunohistochemistry of available heart samples showed patchy, robust staining in some myoblasts, with more uniform staining in spleen (fig. S7). TPP1 activity was not different between TPP1-null and healthy dogs in liver or kidney possibly due to the presence of other hydrolases capable of cleaving the artificial substrate (Fig. 8). The normalization of activity in the heart is relevant to the clinical manifestation because cardiomyopathies have been reported and may contribute to disease progression in other forms of the neuronal ceroid lipofuscinoses (25). Indeed, there has been one case report of cardiac involvement in a 23-year-old patient with TPP1 deficiency (26). In TPP1-null dogs treated with rAAV.caTPP1, enzymatic activity was one order of magnitude greater than that in untreated TPP1-null animals in heart and twofold higher in spleen (Fig. 8). Quantitative polymerase chain reaction (qPCR) analysis of vector genomes (fig. S4) suggests that the enzyme activity detected is from recombinant TPP1 expressed in brain, rather than rAAV2/2 vectors leaking into the periphery during or after infusion. These results support the notion that CSF-resident enzymes may provide a source of TPP1 for peripheral organs.
Fig. 8. TPP1 activity in peripheral organs.
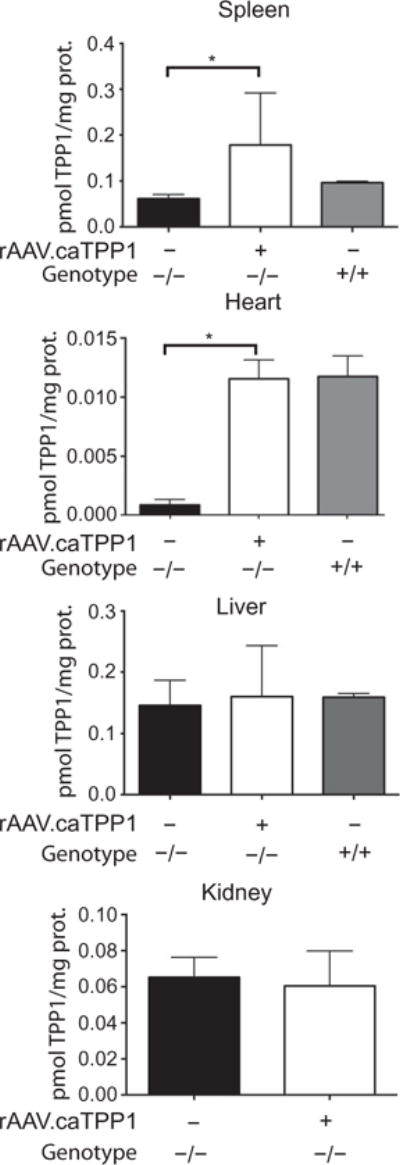
TPP1 activity in homogenized samples of peripheral tissues (spleen, heart, liver, and kidney) of untreated or rAAV.caTPP1-treated TPP1-deficient dogs or normal healthy dogs (see table S2). Nonparametric Kruskal-Wallis (P < 0.01) test with Dunn’s post hoc test (*P < 0.05).
DISCUSSION
Safety studies of gene therapy for treatment of LINCLs due to TPP1 deficiency have been performed in patients using 12 injections of recombinant AAV2 into the brain parenchyma (27). A second clinical study using a similar delivery method has been initiated with AAV serotype rh10, a serotype that shows broader distribution after intraparenchymal injection (ClinicalTrials.gov identifier: NCT01161576). In both trials, expression of virus was restricted to the cerebral parenchyma. We reasoned that global delivery of recombinant protein to the brain of patients would be better achieved via the CSF, which could be accomplished after a single intraventricular injection or a combined intraventricular and cisternal injection. Moreover, these methods do not require major neurosurgery needing extended sedation, an important consideration for children with a neurodegenerative disease. In dogs, this approach resulted in broad distribution of recombinant TPP1, with delays in disease onset, disease progression, and protection from cognitive decline. This therapy also produced neurological improvements, increased life span, and, as evidenced from the videos, improved quality of life.
A striking observation from this work was the difference in the order in which clinical signs first appeared. Untreated TPP1-deficient dogs first displayed deficits in proprioceptive placement and ataxia with weakness of the hindlimbs. They also showed very early cognitive impairment reflected by poor performance in the T-maze. The earliest symptoms in children with TPP1 deficiency are similar. Affected children generally display early language deficits and either a delay in developmental milestones or a loss of milestones already acquired, as well as ataxia and proprioceptive deficits (1). In rAAV.caTPP1-treated dogs, the features of general proprioceptive deficits and cerebellar ataxia, postural reaction deficits, and cognitive deficits were markedly delayed regarding onset, and in some treated animals, they never appeared. Instead, the earliest clinical signs in rAAV.caTPP1-treated animals were visual abnormalities. Nonetheless, treated dogs showed delay of clinical signs that would be caused by defects in central pathways receiving retinal input. Additionally, the histological and enzyme assay results showed TPP1 activity in these and other regions necessary for normal vision, but we did not detect TPP1 in photoreceptor cells or in the retinal pigmented epithelium. Thus, directed correction of the retina via intraocular injection together with intraventricular delivery of rAAV.caTPP1 could likely preserve vision in these animals.
Histologically, we detected a reduction in ATP synthase subunit C accumulation and glial activation in many areas of the treated dog brain, but the results were variable regarding the extent of that reduction. TPP1-deficient dogs with the most profound clinical benefit had lower residual autofluorescent material and ATP synthase subunit C accumulation. Untreated TPP1-deficient dogs, euthanized from 9 to 11 months of age, showed extensive autofluorescence and glial activation throughout all brain regions, similar to what has been reported for TPP1-null mice (28). Tissues from treated animals harvested at 13 to 22 months of age (10 to 19 months after gene transfer) showed a reduction in ATP synthase subunit C storage, autofluorescent inclusions, p62 protein levels, and glial activation. These data are interesting in light of the fact that there were anti-TPP1 responses to the recombinant protein in some dogs.
When vector is infused through the circulation, NAb titers to AAV are used as inclusion/exclusion criteria because NAbs effectively preclude transduction of the target tissue. In nonhuman primates with variable but detectable expression of preexisting cross-reactive antibodies to AAV, there is no impact on the efficiency of transduction after parenchymal infusion into the brain (29). Here, we did not test dog blood for cross-reactive antibodies to rAAV2/2 before vector infusion. The impact of modest to high titers of anti-AAV NAbs on transduction efficiency in the ependyma, and secretion of the transgene product into CSF, would be interesting to determine. In addition to NAb responses, T cell responses to both AAV capsid and transgene product are possible (30). In the case of T cell responses, immune suppressants can be used to protect against CD8-positive T cell–mediated destruction of transduced cells, resulting in prolonged transgene expression and efficacy (31). Here, most rAAV genomes were in the ependyma, yet this cell layer remained structurally intact. Thus, in the setting of a TPP1-null dog, rAAV2/2 transduction with expression and secretion of TPP1 into the CSF does not appear to elicit a destructive T cell response.
Here, early anti-TPP1 responses were most robust in dogs not pretreated with mycophenolate mofetil. Introduction of the drug at the time of or before rAAV delivery reduced, inhibited, or significantly delayed anti-TPP1 immune responses. In earlier work in this same dog model, immune responses to recombinant human TPP1 infused into the lumbar space or brain ventricles also induced robust anti-TPP1 responses (22). Anaphylactic reactions were noted with a bolus infusion that was better managed or did not develop with slower, continuous infusion protocols. To date, there are no reports of anti-TPP1 responses affecting children being treated biweekly with recombinant protein. Whether this reflects differences between immune responses to recombinant protein in CSF between children and dogs deficient in TPP1 is unknown at this time. Immune responses to neoantigens in children receiving enzyme therapy for life-threatening lysosomal storage diseases can be managed, however, as shown by the noteworthy successes seen in the treatment of patients with Pompe’s disease (32). Similar medical interventions could be applied to TPP1-deficient children after rAAV. TPP1 delivery of NAbs limits efficacy.
The mycophenolate mofetil used in this work may or may not be used in children because other treatment options are available (32). Here, mycophenolate mofetil was variably tolerated in TPP1-deficient dogs and was therefore tapered at different times (table S1), with no correlation between the timing of that tapering and clinical efficacy. Additionally, in TPP1-deficient children with residual protein, it would be expected that their responses to the recombinant protein would be diminished. Although the dose of vector applied here resulted in clinical efficacy, we do not know if this is the minimal effective dose; lower doses would presumably result in less protein expression and less robust immune responses. As carrier dogs are normal, expressing 50% wild-type protein continuously throughout life may be sufficient.
Similar to enzyme replacement therapy in TPP1-deficient dogs, even those animals that responded well to gene therapy eventually succumbed to disease. However, several neurological signs including cognitive decline were delayed or prevented with rAAV.caTPP1 gene therapy, resulting in an improved quality of life throughout their extended life span. Although we found evidence for enzyme reaching the periphery from the CSF (elevated expression in heart and spleen), this expression may be insufficient to rescue systemic manifestations that may appear as the dogs live longer owing to improved neurological function. Nonetheless, the approach presented here holds promise for children with TPP1 deficiency and may be preferable to current clinical treatments including multiple intraparenchymal injections for gene therapy or implant of a device used to deliver repeated enzyme replacement infusions into the brain. Our gene therapy approach would not require extensive time under sedation for vector or device implant or recurring intensive care for infusions. Finally, this approach could reduce risk to patients, as it would obviate the need for lifelong enzyme infusions that would require repeated access to the brain.
MATERIALS AND METHODS
Study design
The overall objective of this work was to test the utility of rAAV delivery to ependymal cells for subsequent secretion into CSF and distribution to the brain via CSF flow pathways. The model tested is a naturally occurring dog model of the late infantile form of Batten disease due to TPP1 deficiency. To test this approach, heterozygous dogs were bred and homozygous TPP1-deficient dogs were enrolled in the study over the course of 4 years. Most of the dogs were treated with rAAV.caTPP1 or were left untreated or were treated with an anti-inflammatory regimen only (table S1). All dogs were monitored daily and neurological examinations were performed weekly. Clinical findings were documented until humane euthanasia was warranted. The neurologists, neurosurgeons, and most caregivers were not blinded to the treatment regimen.
rAAV vector production
rAAV2 vectors expressing canine TPP1 under control of the cytomegalovirus early enhancer/chicken β-actin promoter were generated using standard triple transfection methods and purification by CsCl gradient centrifugation (33). Titers were quantified by silver stain after gel electrophoresis (SDS–polyacrylamide gel electrophoresis) and qPCR.
Animals
An outbred strain of long-haired miniature dachshunds was bred and housed at the University of Missouri in facilities overseen by the Office of Animal Resources (OAR). TPP1-null dogs were generated from heterozygous breeding (17). All puppies were genotyped by PCR using an allelic discrimination assay. In addition to the standard care provided by OAR, dogs were socialized on a daily basis throughout the study and neurological exams were performed weekly as previously described (9). All procedures involving dogs were reviewed and approved by the University of Missouri Animal Care and Use Committee and conformed to the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. Decision for humane euthanasia was when end-stage disease was reached on the basis of defined criteria including loss of cognition, severe mentation abnormalities, treatment-refractory myoclonic jerks, and inability to eat without significant assistance. As dogs were able to live longer with attenuation of their neurologic signs, other disease-related effects were evident in other organ systems, especially the cardiac muscle.
Intraventricular infusions were performed over 25 min using the Brainsight neuronavigation system equipped with a 26-gauge 12-cm needle. Cyclosporine (10 mg/kg twice daily by mouth) was administered to all of the dogs in this study starting 1 to 2 weeks before the gene therapy vector administration. The dose was gradually tapered after 2 months to once daily, which was continued for the duration of the study. We instituted a daily dose (10 to 20 mg/kg by mouth) of mycophenolate mofetil as indicated in Results, which was tapered after two to several months depending on tolerability.
Statistics
GraphPad Prism 6 software (GraphPad Software) was used for statistical analyses. For clinical assessments, data were analyzed by nonparametric tests as the groups did not pass the D’Agostino normality test. Mann-Whitney tests were performed when only two groups were compared; Kruskal-Wallis tests were used in the case of three or more groups.
Supplementary Material
Fig. S1. Representative image of rAAV serotype screening for transduction of canine ependymal cells.
Fig. S2. TPP1 activity in the CSF from normal dogs.
Fig. S3. Unilateral injection of rAAV.caTPP1 results in bilateral distribution of TPP1 activity and TPP1-immunopositive cortical pyramidal cells.
Fig. S4. rAAV.caTPP1 biodistribution.
Fig. S5. rAAV.caTPP1 improves astroglial activation and does not induce microglial activation.
Fig. S6. Representative transfer membranes used for Western blot of SCMAS and p62 in three brain regions (thalamus, occipital cortex, and cerebellar cortex).
Fig. S7. Recombinant TPP1 in peripheral organs of rAAV.caTPP1-treated dogs.
Table S1. TPP1-null dachshund dogs used for vector and/or drug infusions.
Table S2. Tissue and clinical data.
Movie S1. Representative video of the gait of a 9.7-month-old untreated TPP1-null dog (DC024).
Movie S2. Representative video of the gait of a 9.8-month-old TPP1-null dog 6.3 months after rAAV.caTPP1 injection (DC020).
Movie S3. Life quality and social interactions of a 17.5-month-old TPP1-null dog (DC020), 14.5 months after rAAV.caTPP1 injection.
Acknowledgments
We thank those who contributed to the care and health of the dogs and colony management at the University of Missouri, and P. Lobel for recombinant human TPP1 for assay setup.
Funding: This work was funded in part by the NIH (NS068099 to B.L.D. and M.L.K., NS077516 to B.L.D., and EY023968 to M.L.K.); the Batten Disease Support and Research Association, Jasper Against Batten Fund at Partnership for Cures, and Blake’s Purpose Foundation (B.L.D. and M.L.K.); and the Roy J. Carver Trust and the Children’s Hospital of Philadelphia Research Institute (B.L.D.).
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/7/313/313ra180/DC1
Materials and Methods
Reference (34)
Author contributions: L.T., Y.C., and B.L.D. designed the study, developed the vector, and analyzed the tissues; M.L.K., L.T., Y.C., J.R.C., and B.L.D. analyzed the experimental and clinical data; M.L.K. assisted with study design and managed all aspects of the dog colony and surgical interventions and sample collections; J.R.C. supervised brain sampling and oversaw all neurologic care and clinical ratings, as well as monitoring of the colony with assistance from B.G.W. and W.M.Y.; R.E.H.W. designed and performed the visual system assessments; E.L. performed experiments; G.C.J. assisted with necropsies and analyzed clinical specimens; F.A.W. developed the surgical approach, performed neurosurgery on all dogs, and provided perioperative care.
Competing interests: B.L.D. is a cofounder of Spark Therapeutics Inc., a gene therapy company. B.L.D. is on the scientific advisory board of Sarepta Therapeutics and Marina Biotech and consults for Wave Life Biosciences.
Data and materials availability: The vectors used in this work are available according to the guidelines of The Children’s Hospital of Philadelphia Research Institute and the University of Iowa. The dog colony is available according to the guidelines of the University of Missouri (Columbia, MO) and the NIH.
REFERENCES AND NOTES
- 1.Chang M, Cooper JD, Davidson BL, van Diggelen OP, Elleder M, Goebel HH, Golabek AA, Kida E, Kohlschütter A, Lobel P, Mole SE, Schulz A, Sleat DE, Warburton M, Wisniewski KE. The Neuronal Ceroid Lipofuscinoses (Batten Disease) Oxford Univ Press; New York: Contemporary Neurology Series. 2011. pp. 80–109. [Google Scholar]
- 2.Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- 3.Sohar I, Sleat DE, Jadot M, Lobel P. Biochemical characterization of a lysosomal protease deficient in classical late infantile neuronal ceroid lipofuscinosis (LINCL) and development of an enzyme-based assay for diagnosis and exclusion of LINCL in human specimens and animal models. J Neurochem. 1999;73:700–711. doi: 10.1046/j.1471-4159.1999.0730700.x. [DOI] [PubMed] [Google Scholar]
- 4.Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases. Mol Ther. 2006;13:839–849. doi: 10.1016/j.ymthe.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Ichimura T, Fraser PA, Cserr HF. Distribution of extracellular tracers in perivascular spaces of the rat brain. Brain Res. 1991;545:103–113. doi: 10.1016/0006-8993(91)91275-6. [DOI] [PubMed] [Google Scholar]
- 6.Chang M, Cooper JD, Sleat DE, Cheng SH, Dodge JC, Passini MA, Lobel P, Davidson BL. Intraventricular enzyme replacement improves disease phenotypes in a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol Ther. 2008;16:649–656. doi: 10.1038/mt.2008.9. [DOI] [PubMed] [Google Scholar]
- 7.Vuillemenot BR, Katz ML, Coates JR, Kennedy D, Tiger P, Kanazono S, Lobel P, Sohar I, Xu S, Cahayag R, Keve S, Koren E, Bunting S, Tsuruda LS, O’Neill CA. Intrathecal tripeptidyl-peptidase 1 reduces lysosomal storage in a canine model of late infantile neuronal ceroid lipofuscinosis. Mol Genet Metab. 2011;104:325–337. doi: 10.1016/j.ymgme.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 8.Vuillemenot BR, Kennedy D, Cooper JD, Wong AMS, Sri S, Doeleman T, Katz ML, Coates JR, Johnson GC, Reed RP, Adams EL, Butt MT, Musson DG, Henshaw J, Keve S, Cahayag R, Tsuruda LS, O’Neill CA. Nonclinical evaluation of CNS-administered TPP1 enzyme replacement in canine CLN2 neuronal ceroid lipofuscinosis. Mol Genet Metab. 2014;114:281–293. doi: 10.1016/j.ymgme.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Katz ML, Coates JR, Sibigtroth CM, Taylor JD, Carpentier M, Young WM, Wininger FA, Kennedy D, Vuillemenot BR, O’Neill CA. Enzyme replacement therapy attenuates disease progression in a canine model of late-infantile neuronal ceroid lipofuscinosis (CLN2 disease) J Neurosci Res. 2014;92:1591–1598. doi: 10.1002/jnr.23423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Del Bigio MR. The ependyma: A protective barrier between brain and cerebrospinal fluid. Glia. 1995;14:1–13. doi: 10.1002/glia.440140102. [DOI] [PubMed] [Google Scholar]
- 11.Spassky N, Merkle FT, Flames N, Tramontin AD, García-Verdugo JM, Alvarez-Buylla A. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci. 2005;25:10–18. doi: 10.1523/JNEUROSCI.1108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roth Y, Kimhi Y, Edery H, Aharonson E, Priel Z. Ciliary motility in brain ventricular system and trachea of hamsters. Brain Res. 1985;330:291–297. doi: 10.1016/0006-8993(85)90688-2. [DOI] [PubMed] [Google Scholar]
- 13.Mirzadeh Z, Han YG, Soriano-Navarro M, García-Verdugo JM, Alvarez-Buylla A. Cilia organize ependymal planar polarity. J Neurosci. 2010;30:2600–2610. doi: 10.1523/JNEUROSCI.3744-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohata S, Nakatani J, Herranz-Pérez V, Jr, Cheng G, Belinson H, Inubushi T, Snider WD, García-Verdugo JM, Wynshaw-Boris A, Álvarez-Buylla A. Loss of Dishevelleds disrupts planar polarity in ependymal motile cilia and results in hydrocephalus. Neuron. 2014;83:558–571. doi: 10.1016/j.neuron.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu G, Martins I, Wemmie JA, Chiorini JA, Davidson BL. Functional correction of CNS phenotypes in a lysosomal storage disease model using adeno-associated virus type 4 vectors. J Neurosci. 2005;25:9321–9327. doi: 10.1523/JNEUROSCI.2936-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamazaki Y, Hirai Y, Miyake K, Shimada T. Targeted gene transfer into ependymal cells through intraventricular injection of AAV1 vector and long-term enzyme replacement via the CSF. Sci Rep. 2014;4:5506. doi: 10.1038/srep05506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Awano T, Katz ML, O’Brien DP, Sohar I, Lobel P, Coates JR, Khan S, Johnson GC, Giger U, Johnson GS. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006;89:254–260. doi: 10.1016/j.ymgme.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 18.Sanders DN, Kanazono S, Wininger FA, Whiting REH, Flournoy CA, Coates JR, Castaner LJ, O’Brien DP, Katz ML. A reversal learning task detects cognitive deficits in a Dachshund model of late-infantile neuronal ceroid lipofuscinosis. Genes Brain Behav. 2011;10:798–804. doi: 10.1111/j.1601-183X.2011.00718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katz ML, Coates JR, Cooper JJ, O’Brien DP, Jeong M, Narfström K. Retinal pathology in a canine model of late infantile neuronal ceroid lipofuscinosis. Invest Ophthalmol Vis Sci. 2008;49:2686–2695. doi: 10.1167/iovs.08-1712. [DOI] [PubMed] [Google Scholar]
- 20.Davidson BL, Stein CS, Heth JA, Martins I, Kotin RM, Derksen TA, Zabner J, Ghodsi A, Chiorini JA. Recombinant adeno-associated virus type 2, 4, and 5 vectors: Transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci USA. 2000;97:3428–3432. doi: 10.1073/pnas.050581197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin JH, Yue Y, Srivastava A, Smith B, Lai Y, Duan D. A simplified immune suppression scheme leads to persistent micro-dystrophin expression in Duchenne muscular dystrophy dogs. Hum Gene Ther. 2012;23:202–209. doi: 10.1089/hum.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vuillemenot BR, Kennedy D, Cooper JD, Wong AMS, Sri S, Doeleman T, Katz ML, Coates JR, Johnson GC, Reed RP, Adams EL, Butt MT, Musson DG, Henshaw J, Keve S, Cahayag R, Tsuruda LS, O’Neill CA. Nonclinical evaluation of CNS-administered TPP1 enzyme replacement in canine CLN2 neuronal ceroid lipofuscinosis. Mol Genet Metab. 2015;114:281–293. doi: 10.1016/j.ymgme.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Micsenyi MC, Sikora J, Stephney G, Dobrenis K, Walkley SU. Lysosomal membrane permeability stimulates protein aggregate formation in neurons of a lysosomal disease. J Neurosci. 2013;33:10815–10827. doi: 10.1523/JNEUROSCI.0987-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kida E, Golabek AA, Walus M, Wujek P, Kaczmarski W, Wisniewski KE. Distribution of tripeptidyl peptidase I in human tissues under normal and pathological conditions. J Neuropathol Exp Neurol. 2001;60:280–292. doi: 10.1093/jnen/60.3.280. [DOI] [PubMed] [Google Scholar]
- 25.Østergaard JR, Rasmussen TB, Mølgaard H. Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease) Neurology. 2011;76:1245–1251. doi: 10.1212/WNL.0b013e31821435bd. [DOI] [PubMed] [Google Scholar]
- 26.Fukumura S, Saito Y, Saito T, Komaki H, Nakagawa E, Sugai K, Sasaki M, Oka A, Takamisawa I. Progressive conduction defects and cardiac death in late infantile neuronal ceroid lipofuscinosis. Dev Med Child Neurol. 2012;54:663–666. doi: 10.1111/j.1469-8749.2011.04170.x. [DOI] [PubMed] [Google Scholar]
- 27.Worgall S, Sondhi D, Hackett NR, Kosofsky B, Kekatpure MV, Neyzi N, Dyke JP, Ballon D, Heier L, Greenwald BM, Christos P, Mazumdar M, Souweidane MM, Kaplitt MG, Crystal RG. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. 2008;19:463–474. doi: 10.1089/hum.2008.022. [DOI] [PubMed] [Google Scholar]
- 28.Sleat DE, Wiseman JA, El-Banna M, Kim KH, Mao Q, Price S, Macauley SL, Sidman RL, Shen MM, Zhao Q, Passini MA, Davidson BL, Stewart GR, Lobel P. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24:9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, Davidson BL. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol Ther. 2011;19:2152–2162. doi: 10.1038/mt.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. 2011;11:321–330. doi: 10.2174/156652311796150354. [DOI] [PubMed] [Google Scholar]
- 31.Nathwani AC, Reiss UM, Tuddenham EGD, Rosales C, Chowdary P, McIntosh J, Della Peruta M, Lheriteau E, Patel N, Raj D, Riddell A, Pie J, Rangarajan S, Bevan D, Recht M, Shen YM, Halka KG, Basner-Tschakarjan E, Mingozzi F, High KA, Allay J, Kay MA, Ng CYC, Zhou J, Cancio M, Morton CL, Gray JT, Srivastava D, Nienhuis AW, Davidoff AM. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Messinger YH, Mendelsohn NJ, Rhead W, Dimmock D, Hershkovitz E, Champion M, Jones SA, Olson R, White A, Wells C, Bali D, Case LE, Young SP, Rosenberg AS, Kishnani PS. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. 2012;14:135–142. doi: 10.1038/gim.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayuso E, Mingozzi F, Montane J, Leon X, Anguela XM, Haurigot V, Edmonson SA, Africa L, Zhou S, High KA, Bosch F, Wright JF. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17:503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- 34.Tian Y, Sohar I, Taylor JW, Lobel P. Determination of the substrate specificity of tripeptidylpeptidase I using combinatorial peptide libraries and development of improved fluorogenic substrates. J Biol Chem. 2006;281:6559–6572. doi: 10.1074/jbc.M507336200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Representative image of rAAV serotype screening for transduction of canine ependymal cells.
Fig. S2. TPP1 activity in the CSF from normal dogs.
Fig. S3. Unilateral injection of rAAV.caTPP1 results in bilateral distribution of TPP1 activity and TPP1-immunopositive cortical pyramidal cells.
Fig. S4. rAAV.caTPP1 biodistribution.
Fig. S5. rAAV.caTPP1 improves astroglial activation and does not induce microglial activation.
Fig. S6. Representative transfer membranes used for Western blot of SCMAS and p62 in three brain regions (thalamus, occipital cortex, and cerebellar cortex).
Fig. S7. Recombinant TPP1 in peripheral organs of rAAV.caTPP1-treated dogs.
Table S1. TPP1-null dachshund dogs used for vector and/or drug infusions.
Table S2. Tissue and clinical data.
Movie S1. Representative video of the gait of a 9.7-month-old untreated TPP1-null dog (DC024).
Movie S2. Representative video of the gait of a 9.8-month-old TPP1-null dog 6.3 months after rAAV.caTPP1 injection (DC020).
Movie S3. Life quality and social interactions of a 17.5-month-old TPP1-null dog (DC020), 14.5 months after rAAV.caTPP1 injection.