

Abstract
Age-related macular degeneration (AMD) afflicts one out of every 40 individuals worldwide, causing irreversible central blindness in millions. The transformation of various tissue layers within the macula in the retina has led to competing conceptual models of the molecular pathways, cell types, and tissues responsible for the onset and progression of AMD. A model that has persisted for over 6 decades is the hemodynamic, or vascular theory of AMD progression, which states that vascular dysfunction of the choroid underlies AMD pathogenesis. Here, we re-evaluate this hypothesis in light of recent advances on molecular, anatomic, and hemodynamic changes underlying choroidal dysfunction in AMD. We propose an updated, detailed model of hemodynamic dysfunction as a mechanism of AMD development and progression.
Age-related macular degeneration (AMD) (see Glossary) is the leading cause of blindness in industrialized nations, affecting an estimated 2.5% of the world’s population [1], and the main risk factors include age and smoking. AMD is progressive, with the first clinical manifestation being the development of drusen or subretinal drusenoid deposits (SDD), depending on their location relative to the layers of the eye. These extracellular deposits of lipoproteins and inflammatory constituents in isolation, are considered subclinical. Over time, the deposits expand and coalesce, at which stage the disease is termed ‘early’ or ‘intermediate’. The stage depends on the size, number and location of deposits in the eye and the presence or absence of pigmentary changes, indicative of RPE disease [2]. In natural history studies, drusen reabsorption is closely succeeded by progression to ‘advanced’, blinding stages of AMD [3]. Most typically, advanced or ‘late’ AMD manifests as geographic atrophy (GA), a neovascular, or ‘wet’ form. GA is considered the ‘default’ end stage of AMD pathogenesis, typified by progressive deterioration of essentially all retinal layers, as well as the choroid and the retinal pigmented epithelium (RPE) (Figure 1, Box 1). GA causes irreversible blindness in over 1 million Americans and no approved therapies exist yet to prevent, halt or reverse its effects.
Figure 1. The Retina-Choroid Interface in AMD: Schematic Diagram of the Choroid and Retina in the Eye.
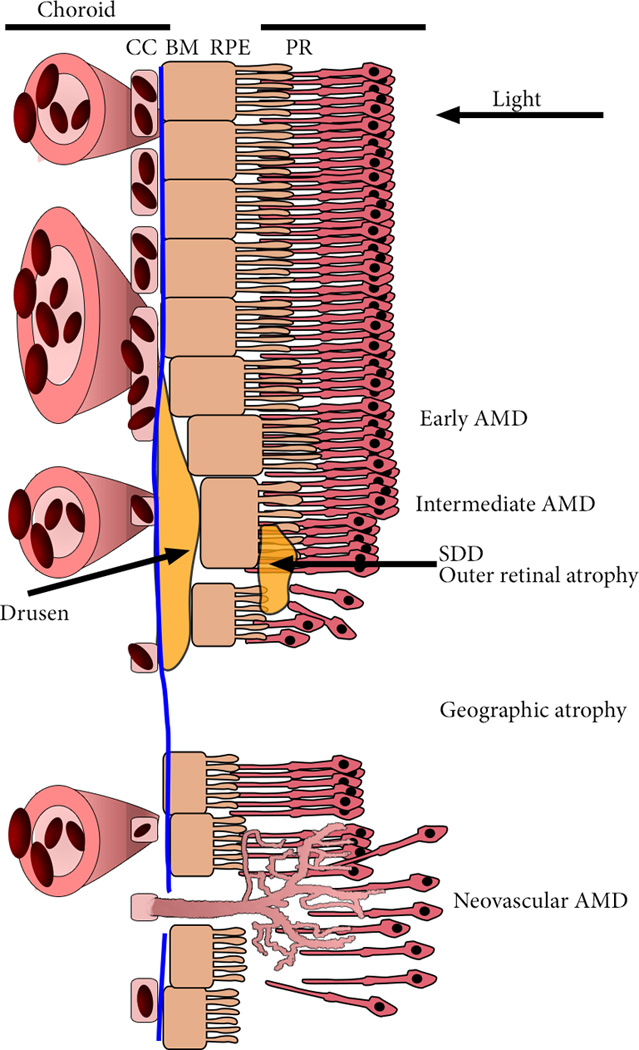
Light entering from the right is focused on the outer retinal photoreceptors (PR). PR are supported by the retinal pigmented epithelium (RPE) and the choroidal vasculature. The innermost layer of the choroid is the choriocapillaris (CC), a thin, dense, highly anastomotic microvascular structure. Between the RPE and the CC is Bruch’s membrane (BM), a specialized multi-layered extracellular matrix that divides the ‘inside’ and ‘outside’ of the eye. The first clinical presentation of AMD is the presence of drusen, extracellular debris that most often develop basal to the RPE, in between CC lumens, and subretinal drusenoid deposits (SDD) that develop between the RPE and photoreceptor layer, and cause outer retinal degeneration. AMD progresses to either geographic atrophy (GA), the gradual atrophy of the choroid, RPE and photoreceptors, and neovascular AMD, aberrant angiogenesis of the choroid through ruptures in BM and the RPE to the PR. Leakage of blood constituents into the retina causes disorganization and swelling of the retina, frequently incompatible with vision.
Box 1. The Retinal Pigmented Epithelium.
The RPE performs multiple functions including recycling components of the visual cycle, phagocytosis and lysosomal degradation of expired photoreceptor outer segments, light absorption, maintenance of the outer blood-retinal barrier by tight intracellular junctions, maintenance of the photoreceptor and choroidal layers by secretion of trophic factors such as VEGF A, and innate immune cell activation. Currently, and for the past dozen years, the RPE has been considered the most crucial cell layer to understand AMD. Drusen, the predominant clinical characteristic of early AMD, are derived from RPE and interrupt the RPE monolayer. Drusen constituents are implicated in AMD pathogenesis such as complement factors, amyloid β, and double-stranded RNA, inducing inflammatory and degenerative effects in RPE [107]. Early onset hereditary macular degenerations such as Stargardt disease are caused by genetic mutations in genes such as ABCA4 leading to RPE atrophy [108]. In addition to atrophy emanating from RPE-dependent signals, overexpression of VEGFA by the RPE is sufficient to induce aberrant subclinical choroidal neovascularization (CNV) [109], which may represent an early preclinical stage of neovascular AMD.
Thus, the choroid and photoreceptor layers have often been viewed as bystanders to the pathological process, passively following cues from the degenerating RPE. The importance of this view is manifested by recent efforts to develop cell-based therapies for advanced AMD. Endeavors thus far have exclusively utilized differentiated RPE cells derived from human embryonic stem cells [110, 111]. However, AMD is not in all cases an RPE cell-based disease, but rather, a disease where the hemodynamic microenvironment plays a decisive role, so replacing degenerating RPE with “healthy” “RPE” may be a fruitless effort, like sowing seeds in barren ground.
In neovascular AMD, the other form of advanced AMD, immature blood vessels, most commonly emanating from the choroid, but occasionally from the retinal vasculature, invade the outer retina. Exudation from these aberrant vessels causes anatomic disruption of the normally highly organized retinal architecture, leading to vision loss. Incidence of neovascular AMD is approximately twice that of GA [4, 5]. Clinical management of neovascular AMD was transformed by the approval and broad adoption of therapeutics that target the vascular endothelial growth factor-A (VEGFA) signaling pathway, but significant challenges remain for treatment and maintaining vision gains in these patients (Box 2).
Box 2. Anti-VEGFA Therapies for Neovascular AMD.
These therapies include the monoclonal antibody (Fab antibody portion) ranibizumab (Lucentis®, Genentech), its related full-length humanized monoclonal antibody bevacizumab (Avastin®, Genentech), and the VEGF-trap (recombinant fusion protein of VEGFR1 and VEGFR2) aflibercept (Eylea®, Regeneron), and pegatinib (Macugen®, Bausch and Lomb), a VEGFA binding aptamer. Although intraocular anti-VEGFA therapies have had clinical success, many patients do not attain significant visual improvement [112, 113]. For example, in neovascular AMD, over half of anti-VEGF recipients do not achieve 20/40 (driving) vision [114]. The long term prognosis for these individuals is sobering, where despite treatment, nearly 25% (representing millions of people) [1] have 20/200 vision (legal blindness) or worse [115], a majority of eyes develop untreatable central retinal atrophy [115, 116], and most short-term gains in visual acuity are lost within 4 or 5 years [116, 117]. Although a causal relationship between prolonged anti-VEGF therapy treatment and vision loss in humans has not been established, adverse effects of VEGFA neutralization on multiple retinal cell types including retinal neurons have been observed in animal models [118–122], and in patients treated with anti-VEGFA drugs for several years [115, 123, 124]. Thus, delineating the underlying causes of advanced AMD represents a major global health need.
A third, recently described type of atrophy in AMD consists of outer retinal atrophy associated with SDD deposition [6]. However, the relationship between the choroid and this type of AMD is unclear, and therefore restrict our discussion to GA and neovascular AMD.
Here, we describe the anatomic and hemodynamic properties of the choroid and, in light of recent advances in the field, revisit the hemodynamic theory of AMD, which posits that blood flow disturbances are the root cause of AMD. Re-evaluating this theory, we propose that hemodynamic changes in the choroid represent a key determinant of local manifestations of AMD pathogenesis.
The Retina in Age-Related Macular Degeneration
Deterioration of macular photoreceptors in AMD is devastating for vision and quality of life. Juxtaposed to the photoreceptor layer is the RPE, whose apical extensions intercalate with the dense layer of photoreceptor outer segments, and whose basal layer resides on Bruch’s membrane, separating the outer retina from the choroid (Figure 1).
With the focus on the RPE as the principal cell layer in determining the fate of the retina in AMD, the hemodynamic theory of AMD pathogenesis has received far less attention from cell and molecular biologists. However, emerging data collectively suggest that local choroidal hemodynamics may play a crucial role in the manifestation of AMD pathologies.
Friedman’s Hemodynamic Theory - Support and Need for Re-Evaluation
The concept that aberrant choroidal blood flow is a pathogenic stimulus for AMD dates back to at least 1905, when Possek proposed atherosclerotic changes to the ocular blood supply as a cause of what is now termed neovascular AMD [7]. The modern hemodynamic theory of AMD proposed by Friedman in 1997 [8], and updated over the next decade [9–11], hypothesizes that AMD arises due to alterations in choroidal blood flow resulting from increased stiffening of Bruch’s membrane and sclera. Stiffening of these structures is thought to be due to the accumulation of lipoproteins in a manner similar to lipid deposition in arterial walls during atherosclerosis [8]. Such stiffening increases choroidal vascular resistance, which results in either an increase in choriocapillaris hydrostatic pressure, or a decrease in choroidal perfusion, depending on the relative resistances of the ophthalmic artery (which supplies the choroid) and the cerebral artery (Figure 2).
Figure 2. Friedman’s Hemodynamic Theory of AMD.

In 1998 Friedman formally postulated that increased stiffness of the sclera, (supportive fibrous structure that lies posterior to the choroid), and BM, promotes AMD pathogenesis. By this model, the progression towards ‘wet’ or ‘dry’ AMD depends on whether the ophthalmic artery (OA), which provides blood the retinal and choroidal circulations, or the middle cerebral artery (MCA) provides greater vascular resistance. In the case where the OA provides less resistance due to intimal constriction of the the MCA, results in elevated blood pressure in the choroidal vasculature. Because of increased scleral stiffness to the posterior, and intraocular pressure to the anterior, elevated pressure within the choroidal vasculature results in hydrostatic pressure. Friedman postulated that this elevated hydrostatic pressure prevents the clearance of debris from the RPE layer to the choroid, and that this was the cause of neovascular AMD. If, on the other hand, the OA provides more resistance due to intimal constriction of the MCA, the choroid is hypoperfused. The choroid thins or collapses between intraocular pressure and a rigid sclera. Friedman postulated that choroidal hypoperfusion caused hypoxia and was responsible for geographic atrophy.
The concept that choroidal hemodynamics contribute to AMD progression is supported by recent clinical and experimental evidence. Altered choroidal hemodynamic parameters, such as reduced choroidal blood flow [12–16] and focal hypoperfusion [17] have been observed in human AMD. Analyses of postmortem human donor eyes have found that areas of decreased choriocapillaris density extends beyond the margin of RPE loss [18–21], which implies that choriocapillaris hemodynamic changes precede manifestation of advanced AMD. Studies in eyes of human AMD patients using optical coherence tomography angiography (OCTA) have also found that perfusion deficits in choriocapillaris extend beyond the margin of RPE atrophy [22, 23]. Protein expression of endothelial nitric oxide synthase, a major blood flow regulator, is significantly reduced in AMD-stricken choroid relative to age-matched healthy eyes [24]. Collectively, these studies support that choroidal hemodynamics alterations are associated with, and may be an early cause of AMD.
However, Friedman’s theory does not account for the finding that neither elevated intraocular pressure, nor Bruch’s membrane stiffening, while associated with normal aging, are associated with AMD pathology [25]. Still, no studies have ruled out the possibility that local variation in stiffness might contribute to local flow disturbances. Friedman’s hypothesis that AMD manifests due to vascular resistance imbalances in larger arteries does not itself explain the exquisitely focal nature of drusen deposits, or AMD pathologies overall. Both GA and CNV have been observed concomitantly in the same eye in humans [26], which is difficult to reconcile with Friedman’s model of ophthalmic artery disturbances as being the discriminating factor between these pathologies. Similarly, choroidal hypoperfusion and atrophy have been reported in both neovascular and atrophic AMD [21], suggesting that at a macro-scale, hemodynamic changes might be insufficient to discriminate between AMD outcomes.
These limitations in Friedman’s theory could be reconciled by considering choroidal hemodynamics in a more discrete manner. Others have suggested AMD to be a hypoxic response to hypoperfusion of the choroidal watershed zones which localize to the macula [27, 28], and CNV frequently localizes to watershed zones [29]. Postmortem analysis of human donor eyes also suggests AMD pathologic features spatially correlate with hemodynamic properties of choriocapillaris on a much finer length scale. For example, reduced density of capillaries and accumulation of ghost capillaries within the choriocapillaris are associated with drusen formation and disease severity [30]. Moreover, AMD pathologic features such as deposition of complement-related proteins, C-reactive protein and advanced glycation end products demonstrate punctate distribution in the RPE and Bruch’s membrane that spatially correlates to the spaces between vessels of the choriocapillaris, even in unaffected eyes [31–34].
One explanation for these observations is that spatial distribution of biochemical factors and drusen implicated in AMD are selectively deposited due to local heterogeneity in hemodynamic parameters. Taking this concept to its logical conclusion, we hypothesize that local variation in hemodynamic parameters, such as wall shear stress and residence time within the choriocapillaris govern the exquisitely focal nature of AMD pathogenesis, which is a centrally defining, yet unexplained, feature of the disease correlating strongly with retinal function [35].
An important caveat to this hypothesis is that local hemodynamic variabilities that exist within choriocapillaris alone are not sufficient to cause AMD. Choroidal ischemia in hypertensive choroidopathy, where blood flow within the choroid is diminished or absent, and Alzheimer’s disease where choroidal thinning is observed [36], are distinct entities from AMD. Moreover, computational modeling predicts that within the healthy choriocapillaris, highly heterogeneous flow rates and pressures exist which can vary by orders of magnitude within tens of microns [37, 38]. In analogy to the effects of local hemodynamics of large arteries on atherosclerotic plaque development, we predict that local hemodynamics within choriocapillaris are critical determinants of the location, severity and progression of disease, but in isolation, are not sufficient to explain all manifestations of the complex disease that is AMD.
Description of Choroidal Hemodynamics
The choroid performs multiple functions necessary for vision, among which are nutrient transport, waste removal and immune cell trafficking [39]. Macular photoreceptors, whose oxygen demand is among the largest in the human body [40], derive the majority of oxygen via diffusion from the choroidal circulation. Transport between the choroid and RPE/retina occurs at the level of the choriocapillaris, a highly anastomotic and extremely dense vascular bed. In the submacular choriocapillaris, the vascular lumen accounts for 80% of the volume of the tissue. The choriocapillaris has be computationally modeled as a thin (7–10 µm) sheet of blood interrupted by intercapillary pillars [37, 38]. Blood is supplied and drained from the choriocapillaris by small and mid-sized arterioles and venules that comprise Sattler’s layer (Figure 3). Despite high blood velocities in the choroidal arterioles and venules, empirical measurements of blood flow within the choriocapillaris suggest an average blood velocity far slower, with significant variability observed within the plane of the layer [41]. This may be due to the unique anatomic arrangement of the arteriorole, capillary, and venule network within the posterior pole and macula of the eye. Here, the arterioles and venules join the choriocapillaris nearly orthogonal to the capillary plane. This causes blood entering from the arteriole to rapidly dissipate centrifugally towards collecting venules. This dissipation is observed in computational modeling [37, 38] and empirical observation in non-human primates [42, 43], which together suggest that the choriocapillaris exhibits substantial gradients in hemodynamic parameters as a function of distance to arteriolar and venule junctions. Although pending further validation, these findings suggest that shear stresses also vary significantly within the choriocapillaris, based on the relative distance from a feeding arteriole and draining venule. Similar to arterial branch points and curvatures where atherosclerotic plaques are focally deposited, the hemodynamic environment of the choriocapillaris is extremely anatomically heterogeneous. We hypothesize that this heterogeneity renders the choroid, and adjacent RPE and retina that rely upon it, protected from or prone to AMD pathogenesis.
Figure 3. Hemodynamics of the Choriocapillaris.

In this diagram, light is depicted as entering the eye upwards. Blood enters the choriocapillaris through terminal arterioles that form junctions orthogonal to the capillary plane, and exit through venules in much the same manner. As blood exits the arteriole, it spreads centrifugally. This unique anatomic scenario causes a substantial drop in hemodynamic parameters from relatively high pressures, shear stresses, and radiative heat capacity at the arterial inlet, to the relatively low pressures, shear stresses and radiative capacity at the venous exist sites. These parameters therefore are predicted to vary substantially over a relatively short distance. Residence time follows an inverse pattern. The local variability in hemodynamics is reminiscent of arterial branches, where local hemodynamics exhibit strong anatomic dependence.
How Choroidal Hemodynamics Affect Function
Given the apparent heterogeneity of blood velocity in the choriocapillaris measured empirically and as predicted by computational modeling, it is important to understand the influence of local blood flow on choroidal endothelial cell (EC) function. For example, one would expect transport of oxygen, glucose and macromolecules from the choroid to the RPE layer to depend greatly on local blood flow. Computational modeling of choroidal geometry also predicts a strong interrelationship between anatomical parameters and blood residence time [37], a measure of the time which a substance remains within a unit system and a critical parameter in heat dissipation [44]. Another parameter affected by local blood flow is the magnitude of shear stress imparted on the vascular endothelium. Shear stress is a mechanical drag that is linearly related to both the shear rate (the spatial gradient of fluid velocity orthogonal to the vessel wall) and the viscosity of the fluid. As described below, shear stress dramatically influences EC behavior.
Computational modeling of choriocapillary blood flow predicts that local pressure and velocity are inextricably linked to anatomic architecture of the choroidal functional unit [37, 38]. The anatomy of the choroid is fundamentally transformed in aging and in AMD as described below. Therefore, it is critical to understand how anatomic changes in the choroid, as observed in healthy aging or in diseases such as AMD, affect local hemodynamics. For example, choriocapillaris dropout is exacerbated in AMD [18–21]. One would predict that focal choriocapillaris atrophy would increase local vascular resistance. Holding flow rate constant, this implies that capillary atrophy would cause a local increase in both blood velocity (by conservation of mass) and consequently shear stress in the residual capillaries. This assumption is complicated by the observation that bulk measurements of choroidal blood flow have shown significant reductions in severe AMD relative to healthy control human subjects [13]. Thus, precisely how local hemodynamic parameters are affected by choriocapillaris anatomic changes occurring in AMD is currently unknown. Nevertheless, descriptions of the anatomic changes in the choroid during AMD progression have advanced considerably. As described above, recent studies have reported that in GA, the atrophy of the choriocapillaris extends beyond the margin of RPE loss, suggesting that it precedes RPE atrophy in AMD [18–21]. The implication of these findings is that the extent of choriocapillaris atrophy affects hemodynamic parameters such as shear stress and residence time, and consequently, the pathophysiologic impact of these parameters could contribute to AMD progression in other layers such as the RPE and retina. We hypothesize that initial hemodynamic gradients within the choriocapillaris render areas susceptible to atrophy or proliferation. Once the disease begins, anatomic transformation of the choriocapillaris as a consequence of disease causes further hemodynamic alterations which in turn exacerbate disease progression. This concept is analogous to the notion that local hemodynamic changes due to atherosclerotic plaque development are responsible for distal expansion of the plaque.
Molecular and Cellular Consequences of Choroidal Hemodynamics
Although studies directly assessing the role of mechanical forces on choroidal endothelium are lacking, data generated using other vascular beds, or cultured ECs derived from other vascular beds have provided potentially meaningful insights into the molecular mechanisms implicated in AMD pathogenesis. An important caveat is that vascular beds exhibit substantial regional heterogeneity. For example, transcriptional, proteomic, and functional profiling of primary human choroidal and retinal EC have displayed substantial divergence [45–48]. Consequently, hemodynamic responses of commonly utilized ECs (such as human umbilical vein (HUVEC)) need not be conserved in the unique hemodynamic environment and cellular millieu of the choroid. To our knowledge, no published study exists that directly interrogates the role of hemodynamics on choroidal EC biology. Nonetheless, several responses of the EC shear response are relevant for AMD pathogenesis. Broadly, the endothelial shear stress response can be divided into three groups, based on properties of shear stress stimulation: i) early responses to flow initiation, ii) responses to prolonged high-magnitude, unidirectional shear stress, often referred to as atheroprotective shear stress, (with signals imparted to the endothelium lining the large ‘straight portions’ of arteries resistant to atherosclerosis) and iii) responses to prolonged low-magnitude, oscillatory shear stress, or, atheroprone shear stress, (with signals imparted to the endothelium lining ‘branched or curved portions’ of arteries susceptible to atherosclerosis). Another important aspect to the theory is that choroidal anatomy is fundamentally different in the macula compared to the periphery. Whereas the junction of arterioles and venules are almost orthogonal in the macula, venules and arterioles intersect with choriocapillaris at more gradual angles in the periphery. This suggests that the hemodynamic environment of the periphery is more likely to be uniform compared to the highly heterogeneous macula, as observed in non-human primates [42]. Consequently, we predict that the extreme heterogeneity of shear stress in the macula is more uniform in the periphery. It follows that peripheral areas may be less ‘primed’ by lack of exposure to ‘atheroprone’ shear stress, and thus, less susceptible to disease progression.
VEGF Signaling and Angiogenesis
Anti-VEGF therapies have revolutionized the management of neovascular AMD (Box 2). VEGFA is also a trophic factor for both the healthy choroid and the neuroretina, and prolonged use of these therapies can eventually induce macular atrophy. VEGF signaling is an indispensable component of endothelial shear stress responses. VEGFR2 and VEGFR3 are components of a mechanosensory complex which mediates a number of shear stress responses [49, 50]. Acute application of arterial levels of shear stress (12 dyne/cm2) in vitro using a cone and plate viscometer induces rapid VEGFR2 phosphorylation in primary bovine ECs, similar to stimulation with VEGFA [51]. Whether VEGFR2 activation by shear stress depends on ligand binding is controversial. For instance, pretreatment of primary bovine EC with a VEGFA neutralizing antibody does not inhibit flow-induced VEGR2 phosphorylation [51]. Conversely, other studies argue that VEGR2 activation by flow relies instead on a pool of VEGFA normally inaccessible, either bound within the extracellular matrix, or released either by flow stimulation by matrix metalloproteinases [52], or between EC in a juxtacrine manner [53]. In either case, shear stress-dependent VEGF signaling may be recalcitrant to anti-VEGFA therapies utilized clinically. Prolonged application of atheroprotective shear stress increases VEGFA protein expression in HUVEC and primary bovine distal pulmonary microvascular ECs [53–55], enhances EC survival under serum starvation challenge in vitro [53]. Atheroprotective shear stress also significantly blunts primary bovine EC proliferation as measured by cell cycle analysis [56, 57], suggesting that EC residing in the area exposed to this shear stress regime exhibit robust autocrine-juxtacrine VEGFA-dependent cytoprotection against starvation conditions, and proliferative quiescence. By comparison, areas of the vasculature exposed to atheroprone shear stresses that do not confer proliferative quiescence may be particularly susceptible to mitogenic signaling in EC, a process that contributes to angiogenic expansion of the choroidal vascular network [58].
In addition to the effects on resident ECs, circulating angiogenic cells (CACs), formerly referred to as endothelial progenitor cells (EPC), have been thought to contribute to the development and elaboration of the neovascular lesion in wet AMD by providing paracrine support of the aberrant angiogenic resident endothelium. In human CNV tissues and in laser injury-induced CNV in mice, CAC-derived cells account for a significant fraction of cellular content of the neovascular membrane [59–61]. Thus, inhibiting CAC trafficking is a proposed therapeutic strategy [60, 61]. Because, atheroprone shear stress is an important stimulus for CAC trafficking into the vascular endothelium [62], reduced hemodynamic forces could potentially promote CAC recruitment. Shear stress promotes CAC differentiation [63, 64], although it is not known whether CAC subtypes implicated in neovascular AMD are sensitive to shear stress. Collectively, we propose that atheroprone shear stress in the choriocapillaris is a critical determinant of angiogenesis through the activation and recruitment of multiple cell types that collaborate to promote aberrant neovascularization.
Immune Signaling, Cell Trafficking and Inflammation
Shear stress strongly influences diverse endothelial inflammatory responses, including those associated with AMD. In vitro application of atheroprotective shear stress induces expression of the transcription factors Kruppel like factor 2 (KLF2) and nuclear factor, erythroid 2 like 2 (NFE2L2) whose transcriptional targets suppress NF-κB activation in a variety of cell culture model systems [65]. Further, expression of these transcription factors localizes to regions of the arterial tree exposed to atheroprotective shear stress in vivo, in mouse and human studies. Conversely, atheroprone shear stress in vitro in animal models, and in vivo in primary human aortic ECs, induces a variety of signals including induction of inflammatory transcription factor NF-κB [66], and the danger sensor Receptor of Advanced Glycation Endproducts (RAGE) [67]. These signaling pathways, among others, sensitize ECs exposed to atheroprone shear stress to endothelial barrier dysfunction and inflammation.
Atheroprone shear stress in HUVEC also induces NLRP3 inflammasome activation [68], an inflammatory pathway implicated in AMD pathogenesis in RPE cell cultures and rodent models, as well as analyses of postmortem human eyes [69–73]. Of note, NLRP3 inflammasome activation in the choroid has been detected in human AMD donor eyes [72].
Human eyes with intermediate and advanced AMD also exhibit robust increases in macrophage content [74, 75], and macrophage depletion significantly reduces CNV in a mouse model of laser injury [76]. Atheroprone shear stress also increases endothelial-monocyte adhesion to HUVEC [77] and mouse aortic EC [78]. Furtermore, shear stress is a strong predictor of macrophage extravasation in the aorta of atherosclerosis prone mice [79], suggesting the hemodynamic environment is an important regulator of immune cell trafficking.
The most thoroughly studied immune pathway in AMD is the complement cascade, first observed in blood vessels of surgically excised neovascular AMD membranes [80] and subsequently popularized by genome-wide association studies, which report that polymorphisms in several complement related genes confer statistical risk for AMD development (reviewed in [81]). Activation of the complement membrane attack complex (MAC) is strongest in the choriocapillaris of AMD donor eyes relative to age-matched healthy controls [18]. A cell culture study of a rhesus chorioretinal EC line suggests that MAC deposition may contribute to AMD pathogenesis via induction of characteristic phenotypes of choroidal endothelium in AMD, namely, a concomitant atrophy and proliferation [82]. CD59 is an important negative regulator of complement activation, which in mice protects the RPE/choroid from inflammatory damage [83] and may represent a putative therapeutic target for AMD [84, 85]. In vitro atheroprotective, (but not atheroprone) shear stress induces CD59 expression in HUVEC, and CD59 is upregulated in atheroprotected areas of mouse aorta [86]. Collectively, these findings indicate that areas of the choriocapillaris exposed to atheroprone shear stress may be more susceptible to inflammatory activation. However, further experimental validation will be required to fully establish these associations.
Vascular Barrier Function
Fluid leakage into the retina from aberrant neovessels is a clinically relevant outcome of neovascular AMD [87]. Barrier function within the healthy choriocapillaris is unique. The apical surface of the choriocapillaris is fenestrated, allowing the free transport of large macromolecules through passive diffusion. Little is known about the effects of shear stress on EC fenestrations. However, whether neovascular lesions are indolent or active is thought to depend on stability of EC-cell-cell junctions. Shear stress causes dynamic remodeling of the intercellular junctions, including reorganization of the adherens junction [88–90], and eventually the formation of stable tight barriers to facilitate transport [91, 92]. Prolonged exposure to atheroprone shear stress induces activation of p21-activated kinase which controls endothelial cell-cell junction integrity [93, 94], leading to impaired trans-endothelial resistance [94]. These findings imply that atheroprone shear stress in the developing neovascular lesion may contribute to impaired barrier function.
Overall, a better understanding of the hemodynamic environment within the degenerating choroid and the neovascular lesion, and its influence on choroidal EC behavior could shed light on endothelial transport function in AMD.
Heat Dissipation
One role of the choroid is to act as a cooling radiator to remove excess heat which accumulates due to absorption of focused light. Reduced choroidal perfusion decreases its heat radiating capacity in an experimental model of intraocular pressure induced choroidal flow impairment [95] and in a computational model [96]. Moreover, production of VEGFA by the RPE is temperature dependent [97–99], suggesting that an indirect effect of reduced choroidal perfusion in AMD contributes to heat-induced VEGFA secretion. Another potential deleterious consequence of decreased heat dissipation by the choroid is induction of Alu RNAs, endogenous activators of the NLPR3 inflammasome which have been implicated in GA in human AMD specimens, mouse models and in primary cell cultures [73, 100, 101]. Alu RNAs and analogous rodent transcripts are upregulated by heat stress in HEK293, HeLa, K562, and 3T3 cells [102, 103]. Conversely, in a mouse laser injury model of CNV, heat preconditioning promotes anti-angiogenic matrix deposition in RPE cells, thus reducing neovascularization [104]. Heat pre-treatment of RPE cells confers protection against oxidative damage, blunts oxidative stress-induced VEGFA secretion [105], and enhances heat shock protein 70 expression [106]. Therefore at the onset of AMD pathology, the loss of the choroid’s capacity to dissipate retinal heat could modulate the immunologic activation and angiogenic capacity as the disease progresses.
Concluding Remarks - A revised Model of Choroidal Hemodynamic Dysfunction in AMD
We propose two major revisions to Friedman’s hemodynamic model (Figure 4, Key Figure). The first is that choroidal hemodynamics contribute to AMD pathogenesis in a substantially more localized manner than has been appreciated. Given emerging empirical and computational evidence of significant heterogeneity of hemodynamics in the choriocapillaris, we hypothesize that hemodynamic parameters within individual capillaries are pivotal factors which promote or prevent AMD pathogenesis. For example, based on observations from other vascular beds, we envisage that local reductions in hemodynamic shear stress promotes (or licenses) choroidal proliferation. By comparison, local elevations in shear stress are predicted to prevent choroidal EC remodeling, immune cell trafficking and transcellular transport. Our theory suggests that local hemodynamics, which likely vary on the scale of a single capillary, determine whether the choroidal endothelium undergoes apoptosis or proliferation under conditions of environmental, genetic or systemic stress. In this way, the present theory may combine with other proposed disease mechanisms by providing an explanation for the focal manifestation of systemic AMD risk contributors such as genetics, smoking, and comorbid cardiovascular risk factors. The second major revision we propose is that the consequences of altered choroidal perfusion extend beyond hypoxia and passive transport of waste from the RPE. Instead, we posit that hemodynamic parameters themselves (e.g. endothelial shear stress, residence time, heat dissipation) comprise fundamental micro-environmental cues that transform the molecular trajectory of the choroid, and indirectly, the RPE and outer retina. Clinicians have long recognized the value of quantifying choroidal blood flow in the diagnosis and prognosis of this disease (Box 3). New technologies and detailed mechanistic studies have provided a more resolved picture of the choroidal landscape of AMD pathogenesis, although many questions and further validation of the proposed model still remain (see Oustanding Questions). Now is the time to bring the power of molecular biology to confirm or refute decades of old hypotheses of choroidal hemodynamics as an underlying cause of AMD.
Key Figure, Figure 4. A Revised Hemodynamic Model of AMD.

The model posits that the normal, healthy choriocapillaris consists of resistant (red) and susceptible (brown) areas, based on the local hemodynamic environment. Relevant factors in addition to those mentioned by Friedman include distance from the terminal arteriole, density of arterioles and venules, as well as choriocapillaris thickness and density. With aging, susceptible areas may eventually adopt an atrophic phenotype (beige), which alters local hemodynamics. By virtue of decreased CC density, atrophied areas lose heat-radiating capacity, thereby increasing inflammatory mediators such as complement and Alu RNA. In areas of the CC, atrophy induces elevated shear stress in adjacent capillaries, rendering the choroid ill-suited to remove retinal waste and incapable of vascular repair. This ultimately leads to GA by choroidal RPE and retinal atrophy. Other areas of the CC experience reduced shear stress, thereby promoting an ‘activated’ endothelial phenotype. The angiogenic and inflammatory influences of low shear stress and hyperthermia of the RPE conspire to produce a choroidal neovascular membrane.
Box 3. Clinician’s Corner.
The development of age-related macular degeneration (AMD) is strongly associated with hemodynamic abnormalities in the choroidal vasculature. However, measurements of the hemodynamic changes preceding AMD are poorly resolved with respect to the discrete appearance of disease manifestations (e.g. drusen, choriocapillaris dropout).
Hemodynamic forces such as endothelial wall shear stress are important pathologic parameters predicting the development of vascular diseases such as atherosclerosis as well as aneurysms. Shear stress is an important mediator of endothelial cell health.
A newer model of AMD pathogenesis emerges, positing that local hemodynamic aberrations within the choriocapillaris could be causative factors contributing to the pathogenesis of AMD.
In the future, delineating the relationship between choriocapillaris hemodynamics might provide new tools to predict areas that may be susceptible to AMD development, as well as new insights into altered pathways stemming from aberrant hemodynamics and leading to putative molecular targets to treat this disease.
Trends.
The choroid is an anatomically unique vascular bed that provides essential trophic support for the retina.
Alterations in choroidal anatomy and hemodynamics are strongly associated with development of age-related macular degeneration (AMD), the leading cause of blindness in the industrialized world.
Rather than passive conduits to flow, the vascular endothelium is highly active, and strongly influenced by local blood flow.
A variety of AMD-associated signaling pathways are hemodynamically regulated in the vascular endothelium.
A revised model of choroidal hemodynamics as an underlying cause of AMD pathogenesis is emerging, wherein local hemodynamic parameters influence choroidal and retinal pigmented epithelial cell health.
OUTSTANDING QUESTIONS BOX.
-
How does the choriocapillaris endothelium shear stress response (SSR) affect its surrounding microenvironment such as Bruch’s membrane and the RPE layer?
Studies of endothelial cells (EC) in other vascular beds provide insight into the SSR conserved between diverse tissue areas. How the choroidal EC SSR influences surrounding cell types such as the RPE and resident non-endothelial choroidal cells, and structures such as Bruch’s membrane is unknown.
-
How does the RPE and surrounding choroidal microenvironment modulate the choroidal EC SSR?
How the choroidal EC SSR is modulated by unique cellular and molecular constituents comprising the choroid and RPE is unknown.
-
In what ways does anatomic transformation of the choroid influence local hemodynamics forces and parameters?
Simplified anatomic models of healthy choriocapillaris demonstrate substantial regional heterogeneity of hemodynamic parameters. Understanding how these parameters are affected by choroidal neovascularization and atrophy is critical in determining how the hemodynamic response may aggravate or mitigate disease progression.
-
Can choriocapillaris hemodynamics be utilized to predict localized AMD risk?
The focal development of atherosclerosis is predictable if given a thorough hemodynamic description. Can ever-improving ocular imaging modalities and computational modeling be utilized to provide a spatially resolved risk profile for the development of AMD pathologies?
-
Can correcting choroidal hemodynamics represent a therapeutic strategy for AMD?
Given the association of hemodynamic abnormalities with AMD pathogenesis, are there surgical or pharmacologic strategies that can be deployed to prevent, delay or reverse the progression of AMD? Alternatively, can the deleterious effects of the abnormal hemodynamic environment be circumvented by molecular targeting of the choroidal endothelium to treat AMD?
Glossary of terms
- Macula
specialized area of the retina responsible for color vision and fine visual acuity. The vascular supply and retinal cells are unique in this area, reflective of its extreme metabolic demand.
- Choroid
One of two blood supplies nourishing the retina. Whereas the retinal vasculature supplies the inner retinal layers, the choroid provides the majority of nutrients required by the outer retina (photoreceptors), and RPE. The choroid is divided into three layers: Outermost, includes Haller’s layer of large arterioles and venules; medial or Sattler’s layer; and innermost, the choriocapillaris.
- Sattler’s layer
layer of the choroid which is comprised of medium-sized arterioles and venules. These vessels supply blood to the choriocapillaris.
- Choriocapillaris
innermost layer of the choroid, comprised of a thin single layer of fenestrated capillaries. The capillary bed is dense and highly interconnected (anastomosed).
- Bruch’s membrane
thick acellular membrane lying between the choriocapillaris and the RPE. Ruptures in Bruch’s membrane are a critical step in choroidal neovascularization.
- Drusen
Extracellular deposits mostly accumulating within (or adjacent to) Bruch’s membrane. Subretinal drusenoid deposits are a subtype forming between the RPE and photoreceptor layers.; Drusen are the first clinical sign of AMD, and their biogenesis and reabsorption are thought to be both a sign and symptom of age-related pathology.
- Geographic atrophy (GA)
The advanced and irreversible binding form of ‘dry’ AMD demarked by confluent areas of macular RPE atrophy.
- Choroidal neovascularization (CNV)
pathological growth of aberrant blood vessels from the choroid, across Bruch’s membrane and into the sub-RPE or subretinal space. Leakage of CNV vessels is the most common cause of vision loss in neovascular AMD, and occurs in a number of visual diseases such as myopia.
- Neovascular age-related macular degeneration
the most prevalent advanced form of AMD characterized by leakage blood constituents from CNV lesions (or occasionally aberrant growth of retinal vessels). Neovascular AMD is managed by anti-VEGF therapies, but significant healthcare challenges remain.
- Stargardt disease
inherited juvenile macular degeneration, whose clinical manifestation bear similarities to atrophic AMD. Most commonly, Stargardt disease is caused by a mutation in the ABCA4 gene, which causes degeneration of the RPE.
- Optical coherence tomography angiography (OCTA)
non-invasive imaging technique to visualize the retinal and choroidal circulation. Recent advances in OCTA have made observations of choroidal vascular changes in AMD progression easier, more quantitative, and better resolved than previously possible.
- Exudate
Fluid leakage from aberrant neovessels disrupting the retinal architecture. Exudate is strongly and negatively correlated with visual acuity.
- Hemodynamics
fluid mechanics of blood flow. Parameters include pressure, velocity, flow rate, viscosity, shear rate and shear stress. A hemodynamic description of blood flow also provides information about how the fluid interacts with the tissue environment, such as in terms of nutrient, waste and heat transport.
- Shear stress
frictional drag that a moving fluid exerts on a solid interface. In the context of the circulatory system, as blood flows across a vessel wall it exerts shear stress on the endothelium. Endothelial cells are sensitive to their shear stress environment.
- Atheroprotective and atheroprone shear stress
Types of shear stress patterns observed in regions resistant to or predisposed to atherosclerosis development respectively. Atheroprotective shear stress is high magnitude, unidirectional, and pulsatile. Atheroprone flow is low magnitude and changes directions.
- Vascular endothelial growth factor A (VEGFA)
promotes the survival and development of new blood vessels. Extracellular VEGFA also reduces cell-cell integrity which increases the exudation of choroidal neovessels.
- Anti-VEGF therapies
compounds that prevent extracellular VEGFA from signaling. Three such therapies are approved for the treatment of neovascular AMD.
- NLRP3 inflammasome
innate immune signaling complex whose activation is implicated in the pathogenesis of AMD. It is activated by a variety of AMD-associated stressors including Alu RNA, amyloid β, and cigarette smoke. NLRP3 inflammasome activation causes secretion of inflammatory cytokines IL-1β and IL-18, both thought to be involved in AMD pathogenesis.
- Retinal pigmented epithelium (RPE)
single cell layer situated between the photoreceptors and Bruch’s membrane. It provides trophic support for the choroid and photoreceptor layer. Degeneration of the RPE layer is a hallmark of GA.
- Photoreceptors
Specialized neurons that convert light into electrochemical signals necessary for vision. The two major types are rods, sensitive to low levels of light, and cones, capable of distinguishing wavelengths (colors) of light.
- Outer segments
Processes of the photoreceptors in which light is absorbed. Constituents of outer segments are periodically shed and recycled by the RPE to support continuous rejuvenation of the phototransduction machinery.
- Sclera
white fibrous, protective coating of the eye which is situated posterior to the choroid. Scleral rigidity is implicated in AMD pathogenesis.
- Hypoxic response
coordinated cellular response to conditions of poor or no oxygenation. One such adaptive response is the development of new blood vessels to restore transport of oxygen and metabolic nutrients.
- Watershed zones
Anatomic areas whose vascular supply is at the extreme distal end of two or more feeding arteries. These areas are thought to be susceptible to hypoperperfusion; tissue metabolism requires full perfusion of both feeding arteries as the vascular networks that branch from the arteries are non-redundant.
- Posterior pole
posterior hemisphere of the eye surrounding the macula, encircled by the periphery. The choroidal vasculature in the posterior pole is distinct from both the macula and the periphery by presenting a greater density of feeding arterioles and draining venules.
- Neuroretina
refers to the neuronal layers that comprise the retina, excluding the RPE and blood vessels.
- Membrane attack complex (MAC)
multiprotein complex that is a terminal effector of the complement cascade activation. Deposition of MAC on cells surfaces causes cell lysis, and in submaximal conditions (sublytic) conditions, induces cell inflammation and dysfunction. MAC deposition in choroidal endothelial cells is one proposed mechanism of choroidal atrophy in AMD.
- Adherens junction
type of cell-cell junction thought to promote monolayer maturation and integrity, as well as serve as a signaling hub for the endothelial shear stress response.
- Alu RNAs
Long non-coding RNAs derived from Alu repetitive genetic elements, ubiquitous in the human genome. Accumulation of Alu RNA is implicated in inflammatory-mediated cell death in AMD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wong WL, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106–e116. doi: 10.1016/S2214-109X(13)70145-1. [DOI] [PubMed] [Google Scholar]
- 2.Ferris FL, 3rd, et al. Clinical classification of age-related macular degeneration. Ophthalmology. 2013;120(4):844–851. doi: 10.1016/j.ophtha.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlanitz FG, et al. Drusen volume development over time and its relevance to the course of age-related macular degeneration. Br J Ophthalmol. 2016 doi: 10.1136/bjophthalmol-2016-308422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chew EY, et al. Ten-year follow-up of age-related macular degeneration in the age-related eye disease study: AREDS report no. 36. JAMA Ophthalmol. 2014;132(3):272–277. doi: 10.1001/jamaophthalmol.2013.6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonasson F, et al. Prevalence of age-related macular degeneration in old persons: Age, Gene/environment Susceptibility Reykjavik Study. Ophthalmology. 2011;118(5):825–830. doi: 10.1016/j.ophtha.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spaide RF. Outer retinal atrophy after regression of subretinal drusenoid deposits as a newly recognized form of late age-related macular degeneration. Retina. 2013;33(9):1800–1808. doi: 10.1097/IAE.0b013e31829c3765. [DOI] [PubMed] [Google Scholar]
- 7.Possek E. Ueber senile Maculaveränderung bei Arteriosklerose. Zeitschrift für Augenheilkunde. 1905;13:771–779. [Google Scholar]
- 8.Friedman E. A hemodynamic model of the pathogenesis of age-related macular degeneration. Am J Ophthalmol. 1997;124(5):677–682. doi: 10.1016/s0002-9394(14)70906-7. [DOI] [PubMed] [Google Scholar]
- 9.Friedman E. The pathogenesis of age-related macular degeneration. Am J Ophthalmol. 2008;146(3):348–349. doi: 10.1016/j.ajo.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 10.Friedman E. Update of the vascular model of AMD. Br J Ophthalmol. 2004;88(2):161–163. doi: 10.1136/bjo.2003.036277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman E. The role of the atherosclerotic process in the pathogenesis of age-related macular degeneration. Am J Ophthalmol. 2000;130(5):658–663. doi: 10.1016/s0002-9394(00)00643-7. [DOI] [PubMed] [Google Scholar]
- 12.Metelitsina TI, et al. Foveolar choroidal circulation and choroidal neovascularization in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49(1):358–363. doi: 10.1167/iovs.07-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grunwald JE, et al. Reduced foveolar choroidal blood flow in eyes with increasing AMD severity. Invest Ophthalmol Vis Sci. 2005;46(3):1033–1038. doi: 10.1167/iovs.04-1050. [DOI] [PubMed] [Google Scholar]
- 14.Grunwald JE, et al. Foveolar choroidal blood flow in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1998;39(2):385–390. [PubMed] [Google Scholar]
- 15.Berenberg TL, et al. The association between drusen extent and foveolar choroidal blood flow in age-related macular degeneration. Retina. 2012;32(1):25–31. doi: 10.1097/IAE.0b013e3182150483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boltz A, et al. Choroidal blood flow and progression of age-related macular degeneration in the fellow eye in patients with unilateral choroidal neovascularization. Invest Ophthalmol Vis Sci. 2010;51(8):4220–4225. doi: 10.1167/iovs.09-4968. [DOI] [PubMed] [Google Scholar]
- 17.Gewaily DY, et al. Delayed patchy choroidal filling in the Comparison of Age-Related Macular Degeneration Treatments Trials (CATT) Am J Ophthalmol. 2014;158(3):525–531. doi: 10.1016/j.ajo.2014.06.004. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mullins RF, et al. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol. 2014;184(11):3142–3153. doi: 10.1016/j.ajpath.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biesemeier A, et al. Choriocapillaris breakdown precedes retinal degeneration in age-related macular degeneration. Neurobiol Aging. 2014;35(11):2562–2573. doi: 10.1016/j.neurobiolaging.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med. 2012;33(4):295–317. doi: 10.1016/j.mam.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLeod DS, et al. Relationship between RPE and choriocapillaris in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50(10):4982–4991. doi: 10.1167/iovs.09-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waheed NK, et al. Optical Coherence Tomography Angiography of Dry Age-Related Macular Degeneration. Dev Ophthalmol. 2016;56:91–100. doi: 10.1159/000442784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi W, et al. Ultrahigh-Speed, Swept-Source Optical Coherence Tomography Angiography in Nonexudative Age-Related Macular Degeneration with Geographic Atrophy. Ophthalmology. 2015;122(12):2532–2544. doi: 10.1016/j.ophtha.2015.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhutto IA, et al. Low nitric oxide synthases (NOSs) in eyes with age-related macular degeneration (AMD) Exp Eye Res. 2010;90(1):155–167. doi: 10.1016/j.exer.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ugarte M, Hussain AA, Marshall J. An experimental study of the elastic properties of the human Bruch’s membrane-choroid complex: relevance to ageing. Br J Ophthalmol. 2006;90(5):621–626. doi: 10.1136/bjo.2005.086579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaszubski P, et al. Geographic Atrophy and Choroidal Neovascularization in the Same Eye: A Review. Ophthalmic Res. 2016;55(4):185–193. doi: 10.1159/000443209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayreh SS. Submacular choroidal vascular bed watershed zones and their clinical importance. Am J Ophthalmol. 2010;150(6):940–941. doi: 10.1016/j.ajo.2010.08.011. author reply 941-2. [DOI] [PubMed] [Google Scholar]
- 28.Ross RD, et al. Presumed macular choroidal watershed vascular filling, choroidal neovascularization, and systemic vascular disease in patients with age-related macular degeneration. Am J Ophthalmol. 1998;125(1):71–80. doi: 10.1016/s0002-9394(99)80237-2. [DOI] [PubMed] [Google Scholar]
- 29.Mendrinos E, Pournaras CJ. Topographic variation of the choroidal watershed zone and its relationship to neovascularization in patients with age-related macular degeneration. Acta Ophthalmol. 2009;87(3):290–296. doi: 10.1111/j.1755-3768.2008.01247.x. [DOI] [PubMed] [Google Scholar]
- 30.Mullins RF, et al. Choriocapillaris vascular dropout related to density of drusen in human eyes with early age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52(3):1606–1612. doi: 10.1167/iovs.10-6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fett AL, et al. Immunohistochemical localization of complement regulatory proteins in the human retina. Histol Histopathol. 2012;27(3):357–364. doi: 10.14670/HH-27.357. [DOI] [PubMed] [Google Scholar]
- 32.Bhutto IA, et al. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br J Ophthalmol. 2011;95(9):1323–1330. doi: 10.1136/bjo.2010.199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kochounian H, Johnson LV, Fong HK. Accumulation of extracellular RGR-d in Bruch’s membrane and close association with drusen at intercapillary regions. Exp Eye Res. 2009;88(6):1129–1136. doi: 10.1016/j.exer.2009.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Handa JT, et al. Increase in the advanced glycation end product pentosidine in Bruch’s membrane with age. Invest Ophthalmol Vis Sci. 1999;40(3):775–779. [PubMed] [Google Scholar]
- 35.Pilotto E, et al. Microperimetry Features of Geographic Atrophy Identified With En Face Optical Coherence Tomography. JAMA Ophthalmol. 2016 doi: 10.1001/jamaophthalmol.2016.1535. [DOI] [PubMed] [Google Scholar]
- 36.Gharbiya M, et al. Choroidal thinning as a new finding in Alzheimer’s disease: evidence from enhanced depth imaging spectral domain optical coherence tomography. J Alzheimers Dis. 2014;40(4):907–917. doi: 10.3233/JAD-132039. [DOI] [PubMed] [Google Scholar]
- 37.Zouache MA, Eames I, Luthert PJ. Blood flow in the choriocapillaris. Journal of Fluid Mechanics. 2015 Jul;774:37–66. [Google Scholar]
- 38.Flower RW, et al. Theoretical investigation of the role of choriocapillaris blood flow in treatment of subfoveal choroidal neovascularization associated with age-related macular degeneration. Am J Ophthalmol. 2001;132(1):85–93. doi: 10.1016/s0002-9394(01)00872-8. [DOI] [PubMed] [Google Scholar]
- 39.Nickla DL, Wallman J. The multifunctional choroid. Prog Retin Eye Res. 2010;29(2):144–168. doi: 10.1016/j.preteyeres.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alm A, Bill A. Ocular and optic nerve blood flow at normal and increased intraocular pressures in monkeys (Macaca irus): a study with radioactively labelled microspheres including flow determinations in brain and some other tissues. Exp Eye Res. 1973;15(1):15–29. doi: 10.1016/0014-4835(73)90185-1. [DOI] [PubMed] [Google Scholar]
- 41.Zhu L, et al. Feasibility of extracting velocity distribution in choriocapillaris in human eyes from ICG dye angiograms. J Biomech Eng. 2006;128(2):203–209. doi: 10.1115/1.2165692. [DOI] [PubMed] [Google Scholar]
- 42.Hirata Y, et al. Analysis of choriocapillaris flow patterns by continuous laser-targeted angiography in monkeys. Invest Ophthalmol Vis Sci. 2004;45(6):1954–1962. doi: 10.1167/iovs.03-0759. [DOI] [PubMed] [Google Scholar]
- 43.Kiryu J, et al. Noninvasive visualization of the choriocapillaris and its dynamic filling. Invest Ophthalmol Vis Sci. 1994;35(10):3724–3731. [PubMed] [Google Scholar]
- 44.Diller KR. Therapeutic Recruitment of Thermoregulation in Humans by Selective Thermal Stimulation along the Spine. In: Sparrow EM, Abraham JP, Gorman JM, editors. Advances in Heat Transfer. Academic Press; 2015. pp. 341–396. [Google Scholar]
- 45.Saker S, et al. The effect of hyperglycaemia on permeability and the expression of junctional complex molecules in human retinal and choroidal endothelial cells. Exp Eye Res. 2014;121:161–167. doi: 10.1016/j.exer.2014.02.016. [DOI] [PubMed] [Google Scholar]
- 46.Browning AC, et al. Comparative gene expression profiling of human umbilical vein endothelial cells and ocular vascular endothelial cells. Br J Ophthalmol. 2012;96(1):128–132. doi: 10.1136/bjophthalmol-2011-300572. [DOI] [PubMed] [Google Scholar]
- 47.Zamora DO, et al. Proteomic profiling of human retinal and choroidal endothelial cells reveals molecular heterogeneity related to tissue of origin. Mol Vis. 2007;13:2058–2065. [PubMed] [Google Scholar]
- 48.Smith JR, et al. Unique gene expression profiles of donor-matched human retinal and choroidal vascular endothelial cells. Invest Ophthalmol Vis Sci. 2007;48(6):2676–2684. doi: 10.1167/iovs.06-0598. [DOI] [PubMed] [Google Scholar]
- 49.Coon BG, et al. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J Cell Biol. 2015;208(7):975–986. doi: 10.1083/jcb.201408103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tzima E, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437(7057):426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 51.Jin ZG, et al. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res. 2003;93(4):354–363. doi: 10.1161/01.RES.0000089257.94002.96. [DOI] [PubMed] [Google Scholar]
- 52.dela Paz NG, Melchior B, Frangos JA. Early VEGFR2 activation in response to flow is VEGF-dependent and mediated by MMP activity. Biochem Biophys Res Commun. 2013;434(3):641–646. doi: 10.1016/j.bbrc.2013.03.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.dela Paz NG, et al. Role of shear-stress-induced VEGF expression in endothelial cell survival. J Cell Sci. 2012;125(Pt 4):831–843. doi: 10.1242/jcs.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goettsch W, et al. Flow-dependent regulation of angiopoietin-2. J Cell Physiol. 2008;214(2):491–503. doi: 10.1002/jcp.21229. [DOI] [PubMed] [Google Scholar]
- 55.Li M, et al. High pulsatility flow induces adhesion molecule and cytokine mRNA expression in distal pulmonary artery endothelial cells. Ann Biomed Eng. 2009;37(6):1082–1092. doi: 10.1007/s10439-009-9684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin K, et al. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc Natl Acad Sci U S A. 2000;97(17):9385–9389. doi: 10.1073/pnas.170282597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levesque MJ, Nerem RM, Sprague EA. Vascular endothelial cell proliferation in culture and the influence of flow. Biomaterials. 1990;11(9):702–707. doi: 10.1016/0142-9612(90)90031-k. [DOI] [PubMed] [Google Scholar]
- 58.Giani A, et al. In vivo evaluation of laser-induced choroidal neovascularization using spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2011;52(6):3880–3887. doi: 10.1167/iovs.10-6266. [DOI] [PubMed] [Google Scholar]
- 59.Sheridan CM, et al. The presence of AC133-positive cells suggests a possible role of endothelial progenitor cells in the formation of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2006;47(4):1642–1645. doi: 10.1167/iovs.05-0779. [DOI] [PubMed] [Google Scholar]
- 60.Sengupta N, et al. Preventing stem cell incorporation into choroidal neovascularization by targeting homing and attachment factors. Invest Ophthalmol Vis Sci. 2005;46(1):343–348. doi: 10.1167/iovs.04-0153. [DOI] [PubMed] [Google Scholar]
- 61.Sengupta N, et al. The role of adult bone marrow-derived stem cells in choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44(11):4908–4913. doi: 10.1167/iovs.03-0342. [DOI] [PubMed] [Google Scholar]
- 62.Lewis DM, et al. Endothelial progenitor cell recruitment in a microfluidic vascular model. Biofabrication. 2015;7(4):045010. doi: 10.1088/1758-5090/7/4/045010. [DOI] [PubMed] [Google Scholar]
- 63.Obi S, et al. Fluid shear stress induces differentiation of circulating phenotype endothelial progenitor cells. Am J Physiol Cell Physiol. 2012;303(6):C595–C606. doi: 10.1152/ajpcell.00133.2012. [DOI] [PubMed] [Google Scholar]
- 64.Xia WH, et al. Age-related decline in reendothelialization capacity of human endothelial progenitor cells is restored by shear stress. Hypertension. 2012;59(6):1225–1231. doi: 10.1161/HYPERTENSIONAHA.111.179820. [DOI] [PubMed] [Google Scholar]
- 65.Boon RA, Horrevoets AJ. Key transcriptional regulators of the vasoprotective effects of shear stress. Hamostaseologie. 2009;29(1):39–40. 41-3. [PubMed] [Google Scholar]
- 66.Harry BL, et al. Endothelial cell PECAM-1 promotes atherosclerotic lesions in areas of disturbed flow in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28(11):2003–2008. doi: 10.1161/ATVBAHA.108.164707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeVerse JS, et al. Shear stress modulates RAGE-mediated inflammation in a model of diabetes-induced metabolic stress. Am J Physiol Heart Circ Physiol. 2012;302(12):H2498–H2508. doi: 10.1152/ajpheart.00869.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xiao H, et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 2013;128(6):632–642. doi: 10.1161/CIRCULATIONAHA.113.002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, et al. NLRP3 Upregulation in Retinal Pigment Epithelium in Age-Related Macular Degeneration. Int J Mol Sci. 2016;17(1) doi: 10.3390/ijms17010073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ardeljan CP, et al. Inflammation and Cell Death in Age-Related Macular Degeneration: An Immunopathological and Ultrastructural Model. J Clin Med. 2014;3(4):1542–1560. doi: 10.3390/jcm3041542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kerur N, et al. TLR-independent and P2×7-dependent signaling mediate Alu RNA-induced NLRP3 inflammasome activation in geographic atrophy. Invest Ophthalmol Vis Sci. 2013;54(12):7395–7401. doi: 10.1167/iovs.13-12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tseng WA, et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2013;54(1):110–120. doi: 10.1167/iovs.12-10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tarallo V, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149(4):847–859. doi: 10.1016/j.cell.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lad EM, et al. Abundance of infiltrating CD163+ cells in the retina of postmortem eyes with dry and neovascular age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2015;253(11):1941–1945. doi: 10.1007/s00417-015-3094-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cherepanoff S, et al. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br J Ophthalmol. 2010;94(7):918–925. doi: 10.1136/bjo.2009.165563. [DOI] [PubMed] [Google Scholar]
- 76.Sakurai E, et al. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003;44(8):3578–3585. doi: 10.1167/iovs.03-0097. [DOI] [PubMed] [Google Scholar]
- 77.Fan W, et al. Shear-sensitive microRNA-34a modulates flow-dependent regulation of endothelial inflammation. J Cell Sci. 2015;128(1):70–80. doi: 10.1242/jcs.154252. [DOI] [PubMed] [Google Scholar]
- 78.Hwang J, et al. Oscillatory shear stress stimulates endothelial production of O2- from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J Biol Chem. 2003;278(47):47291–47298. doi: 10.1074/jbc.M305150200. [DOI] [PubMed] [Google Scholar]
- 79.De Wilde D, et al. Shear Stress Metrics and Their Relation to Atherosclerosis: An In Vivo Follow-up Study in Atherosclerotic Mice. Ann Biomed Eng. 2015 doi: 10.1007/s10439-015-1540-z. [DOI] [PubMed] [Google Scholar]
- 80.Baudouin C, et al. Immunohistological study of subretinal membranes in age-related macular degeneration. Jpn J Ophthalmol. 1992;36(4):443–451. [PubMed] [Google Scholar]
- 81.van Lookeren Campagne M, Strauss EC, Yaspan BL. Age-related macular degeneration: Complement in action. Immunobiology. 2016;221(6):733–739. doi: 10.1016/j.imbio.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 82.Zeng S, et al. Molecular response of chorioretinal endothelial cells to complement injury: implications for macular degeneration. J Pathol. 2016;238(3):446–456. doi: 10.1002/path.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Herrmann P, et al. Cd59a deficiency in mice leads to preferential innate immune activation in the retinal pigment epithelium-choroid with age. Neurobiol Aging. 2015;36(9):2637–2648. doi: 10.1016/j.neurobiolaging.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 84.Bora NS, et al. Recombinant membrane-targeted form of CD59 inhibits the growth of choroidal neovascular complex in mice. J Biol Chem. 2010;285(44):33826–33833. doi: 10.1074/jbc.M110.153130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ramo K, Cashman SM, Kumar-Singh R. Evaluation of adenovirus-delivered human CD59 as a potential therapy for AMD in a model of human membrane attack complex formation on murine RPE. Invest Ophthalmol Vis Sci. 2008;49(9):4126–4136. doi: 10.1167/iovs.08-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kinderlerer AR, et al. KLF2-dependent, shear stress-induced expression of CD59: a novel cytoprotective mechanism against complement-mediated injury in the vasculature. J Biol Chem. 2008;283(21):14636–14644. doi: 10.1074/jbc.M800362200. [DOI] [PubMed] [Google Scholar]
- 87.Roberts P, et al. A quantitative approach to identify morphological features relevant for visual function in ranibizumab therapy of neovascular AMD. Invest Ophthalmol Vis Sci. 2014;55(10):6623–6630. doi: 10.1167/iovs.14-14293. [DOI] [PubMed] [Google Scholar]
- 88.Shay-Salit A, et al. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci U S A. 2002;99(14):9462–9467. doi: 10.1073/pnas.142224299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ukropec JA, Hollinger MK, Woolkalis MJ. Regulation of VE-cadherin linkage to the cytoskeleton in endothelial cells exposed to fluid shear stress. Exp Cell Res. 2002;273(2):240–247. doi: 10.1006/excr.2001.5453. [DOI] [PubMed] [Google Scholar]
- 90.Noria S, et al. Transient and steady-state effects of shear stress on endothelial cell adherens junctions. Circ Res. 1999;85(6):504–514. doi: 10.1161/01.res.85.6.504. [DOI] [PubMed] [Google Scholar]
- 91.Seebach J, et al. Endothelial barrier function under laminar fluid shear stress. Lab Invest. 2000;80(12):1819–1831. doi: 10.1038/labinvest.3780193. [DOI] [PubMed] [Google Scholar]
- 92.Phelps JE, DePaola N. Spatial variations in endothelial barrier function in disturbed flows in vitro. Am J Physiol Heart Circ Physiol. 2000;278(2):H469–H476. doi: 10.1152/ajpheart.2000.278.2.H469. [DOI] [PubMed] [Google Scholar]
- 93.Orr AW, et al. p21-activated kinase signaling regulates oxidant-dependent NF-kappa B activation by flow. Circ Res. 2008;103(6):671–679. doi: 10.1161/CIRCRESAHA.108.182097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Orr AW, et al. Matrix-specific p21-activated kinase activation regulates vascular permeability in atherogenesis. J Cell Biol. 2007;176(5):719–727. doi: 10.1083/jcb.200609008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Parver LM, Auker C, Carpenter DO. Choroidal blood flow as a heat dissipating mechanism in the macula. Am J Ophthalmol. 1980;89(5):641–646. doi: 10.1016/0002-9394(80)90280-9. [DOI] [PubMed] [Google Scholar]
- 96.Wainwright PR. Computational modelling of temperature rises in the eye in the near field of radiofrequency sources at 380, 900 and 1800 MHz. Phys Med Biol. 2007;52(12):3335–3350. doi: 10.1088/0031-9155/52/12/002. [DOI] [PubMed] [Google Scholar]
- 97.Faby H, et al. Hyperthermia-induced upregulation of vascular endothelial growth factor in retinal pigment epithelial cells is regulated by mitogen-activated protein kinases. Graefes Arch Clin Exp Ophthalmol. 2014;252(11):1737–1745. doi: 10.1007/s00417-014-2750-z. [DOI] [PubMed] [Google Scholar]
- 98.Coassin M, et al. Hypothermia reduces secretion of vascular endothelial growth factor by cultured retinal pigment epithelial cells. Br J Ophthalmol. 2010;94(12):1678–1683. doi: 10.1136/bjo.2009.168864. [DOI] [PubMed] [Google Scholar]
- 99.Cordeiro S, et al. Heat-sensitive TRPV channels in retinal pigment epithelial cells: regulation of VEGF-A secretion. Invest Ophthalmol Vis Sci. 2010;51(11):6001–6008. doi: 10.1167/iovs.09-4720. [DOI] [PubMed] [Google Scholar]
- 100.Kaneko H, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471(7338):325–330. doi: 10.1038/nature09830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gelfand BD, et al. Iron Toxicity in the Retina Requires Alu RNA and the NLRP3 Inflammasome. Cell Rep. 2015;11(11):1686–1693. doi: 10.1016/j.celrep.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li TH, Schmid CW. Differential stress induction of individual Alu loci: implications for transcription and retrotransposition. Gene. 2001;276(1–2):135–141. doi: 10.1016/s0378-1119(01)00637-0. [DOI] [PubMed] [Google Scholar]
- 103.Liu WM, et al. Cell stress and translational inhibitors transiently increase the abundance of mammalian SINE transcripts. Nucleic Acids Res. 1995;23(10):1758–1765. doi: 10.1093/nar/23.10.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sekiyama E, et al. Heat treatment of retinal pigment epithelium induces production of elastic lamina components and antiangiogenic activity. FASEB J. 2012;26(2):567–575. doi: 10.1096/fj.11-184127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Iwami H, et al. Protective effect of a laser-induced sub-lethal temperature rise on RPE cells from oxidative stress. Exp Eye Res. 2014;124:37–47. doi: 10.1016/j.exer.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 106.Inagaki K, et al. Sublethal Photothermal Stimulation with a Micropulse Laser Induces Heat Shock Protein Expression in ARPE-19 Cells. J Ophthalmol. 2015;2015:729792. doi: 10.1155/2015/729792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. 2013;13(6):438–451. doi: 10.1038/nri3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Allikmets R, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 109.Schwesinger C, et al. Intrachoroidal neovascularization in transgenic mice overexpressing vascular endothelial growth factor in the retinal pigment epithelium. Am J Pathol. 2001;158(3):1161–1172. doi: 10.1016/S0002-9440(10)64063-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Song WK, et al. Treatment of macular degeneration using embryonic stem cell-derived retinal pigment epithelium: preliminary results in Asian patients. Stem Cell Reports. 2015;4(5):860–872. doi: 10.1016/j.stemcr.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schwartz SD, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet. 2015;385(9967):509–516. doi: 10.1016/S0140-6736(14)61376-3. [DOI] [PubMed] [Google Scholar]
- 112.Krebs I, et al. Non-responders to treatment with antagonists of vascular endothelial growth factor in age-related macular degeneration. Br J Ophthalmol. 2013;97(11):1443–1446. doi: 10.1136/bjophthalmol-2013-303513. [DOI] [PubMed] [Google Scholar]
- 113.Lux A, et al. Non-responders to bevacizumab (Avastin) therapy of choroidal neovascular lesions. Br J Ophthalmol. 2007;91(10):1318–1322. doi: 10.1136/bjo.2006.113902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rosenfeld PJ, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- 115.Rofagha S, et al. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP) Ophthalmology. 2013;120(11):2292–2299. doi: 10.1016/j.ophtha.2013.03.046. [DOI] [PubMed] [Google Scholar]
- 116.Comparison of Age-related Macular Degeneration Treatments Trials Research, G. et al. Five-Year Outcomes with Anti-Vascular Endothelial Growth Factor Treatment of Neovascular Age-Related Macular Degeneration: The Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology. 2016 doi: 10.1016/j.ophtha.2016.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Singer MA, et al. HORIZON: an open-label extension trial of ranibizumab for choroidal neovascularization secondary to age-related macular degeneration. Ophthalmology. 2012;119(6):1175–1183. doi: 10.1016/j.ophtha.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 118.Kurihara T, et al. Targeted deletion of Vegfa in adult mice induces vision loss. J Clin Invest. 2012;122(11):4213–4217. doi: 10.1172/JCI65157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nishijima K, et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171(1):53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Takeda A, et al. CCR3 is a target for age-related macular degeneration diagnosis and therapy. Nature. 2009;460(7252):225–230. doi: 10.1038/nature08151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Saint-Geniez M, et al. Endogenous VEGF is required for visual function: evidence for a survival role on muller cells and photoreceptors. PLoS One. 2008;3(11):e3554. doi: 10.1371/journal.pone.0003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Marneros AG, et al. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am J Pathol. 2005;167(5):1451–1459. doi: 10.1016/S0002-9440(10)61231-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Comparison of Age-related Macular Degeneration Treatments Trials Research, G. et al. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology. 2012;119(7):1388–1398. doi: 10.1016/j.ophtha.2012.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Grunwald JE, et al. Risk of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2014;121(1):150–161. doi: 10.1016/j.ophtha.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]