

Abstract
Ph-like acute lymphoblastic leukemia (ALL) is a genetically defined high-risk ALL subtype with a generally poor prognosis. In this study, we evaluated the efficacy of birinapant, a small molecule mimetic of the apoptotic regulator SMAC, against a diverse set of ALL subtypes. Birinapant exhibited potent and selective cytotoxicity against B-cell precursor ALL (BCP-ALL) cells that were cultured ex vivo or in vivo as patient-derived tumor xenografts (PDX). Cytotoxicity was consistently most acute in Ph-like BCP-ALL. Unbiased gene expression analysis of BCP-ALL PDX specimens identified a 68-gene signature associated with birinapant sensitivity, including an enrichment for genes involved in inflammatory response, hematopoiesis and cell death pathways. All Ph-like PDXs analyzed clustered within this 68-gene classifier. Mechanistically, birinapant sensitivity was associated with expression of TNF receptor TNFR1 and was abrogated by interfering with the TNFα/TNFR1 interaction. In combination therapy, birinapant enhanced the in vivo efficacy of induction-type regimen of vincristine, dexamethasone and L-asparaginase against Ph-like ALL xenografts, offering a preclinical rationale to further evaluate this SMAC mimetic for BCP-ALL treatment.
Keywords: leukemia, SMAC mimetic, xenograft, preclinical, chemotherapy
Introduction
Apoptosis plays a critical role in the development and homeostasis of multicellular organisms, as well as normal and malignant hematopoiesis (1,2). The intrinsic (mitochondrial) and extrinsic (death receptor) apoptotic pathways are tightly regulated by pro- and anti-apoptotic proteins (3,4). The inhibitor of apoptosis (IAP) family are small anti-apoptotic proteins that promote cellular survival mediated by the tumor necrosis factor (TNF) superfamily of surface receptors and actively prevent the induction of apoptosis by inhibiting caspase activation. To date 8 IAPs have been identified in humans with X chromosome linked IAP (XIAP), cellular IAP1 (cIAP1) and cIAP2 playing the most prominent role in cell survival (5). SMAC (Second Mitochondria-derived Activator of Caspases), is an endogenous antagonist of IAPs and is released from the mitochondria in response to apoptotic stimuli, thus activating downstream caspases (6).
Several approaches have been utilized to target IAPs, and the development of various small molecule inhibitors, including SMAC mimetics, has recently been reported (5,7). Birinapant is a bivalent SMAC mimetic with high affinity for cIAP1/2 (8). Upon binding it promotes auto-ubiquitination of cIAP1/2 and proteasomal degradation leading to inactivation of NF-κB survival signaling and TNFα-dependent apoptosis (8). In response to TNFα receptor ligation (primarily via TNF receptor type 1, TNFR1, CD120a) birinapant also promotes caspase-8/RIPK1 (receptor interacting serine/threonine protein kinase-1) complex formation resulting in activation of caspase-8 and subsequent apoptosis. In some situations whereby caspase-8 function is impaired, an alternative cellular death pathway can be activated, whereby RIPK1 promotes RIPK3 phosphorylation and subsequent necroptosis (9,10). Birinapant is currently in clinical trials for both solid tumors and hematological malignancies and has so far been well tolerated and demonstrated excellent pharmacokinetic properties (8).
Various anti-apoptotic proteins including IAPs are overexpressed in many cancers (4,11,12) and are often associated with chemoresistance and treatment failure, making them an attractive target for therapeutic intervention (13). In pediatric acute myeloid leukemia cIAP1 was reported to be highly expressed in comparison to normal tissues (14), and aberrant expression of XIAP correlated with poor prognosis in acute lymphoblastic leukemia (ALL) (15). ALL accounts for approximately 80% of all pediatric leukemias (16) and, while the overall cure rate for pediatric ALL now exceeds 80%, certain high risk subtypes still experience shorter remissions and lower survival rates. A high risk subtype of B-cell precursor (BCP) ALL, termed Philadelphia chromosome–like (Ph-like) ALL, was identified based on a gene expression classifier similar to BCR-ABL1-positive cases despite the absence of the BCR-ABL1 translocation (17-21). Ph-like ALL represents approximately 10% of all pediatric BCP-ALL cases and increases in incidence with age (17-21). Activating mutations in the pseudokinase or kinase domains of Janus Kinases (JAKs) are observed in approximately 50% of Ph-like ALL cases, and are frequently accompanied by IKZF1 gene deletions. The presence of JAK mutations, IKZF1 deletions and the Ph-like gene expression signature are associated with poor outcome (17,20,22,23), highlighting the need for more targeted therapies.
Here we demonstrate that birinapant exerts potent anti-leukemic efficacy in vitro and in vivo against pediatric and adult BCP-ALL primary samples and patient-derived xenografts (PDXs), in particular those derived from Ph-like ALL, via a TNFα-dependent process associated with high expression of TNFR1. We also identified a gene expression signature in BCP-ALL associated with birinapant response that was enriched in genes involved in inflammatory response, hematopoiesis, and cell death pathways. Birinapant also significantly enhanced the anti-leukemic activity of an induction-type regimen of vincristine, dexamethasone and L-asparaginase (VXL) in vivo. These findings support the further clinical evaluation of birinapant in the treatment of BCP-ALL and other hematological malignancies.
Materials and Methods
Primary ALL cells and PDXs
Human leukemia cells used in this study were from peripheral blood or bone marrow biopsy specimens of ALL patients. Methods used to establish PDXs from pediatric ALL biopsies in NOD/SCID (NOD.CB17-Prkdcscid/J) or NOD/SCID/IL-2 receptor gamma-/- (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, NSG) mice have been previously described (24-29). Patient demographics of PDXs and primary patient samples are described in Supplementary Table S1. ALLs were classified as Ph-like using gene expression criteria previously described (18-21,30).
Apoptosis assays
PDX cells were resuspended in QBSF-60 medium (Quality Biological, Gaithersburg, MD) with 20 ng/ml Flt-3 ligand, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine. Patient samples were cultured in StemSpan media (Stem Cell Technologies, Vancouver, Canada) with 20 ng/ml IL-3, 20 ng/ml IL-6, 100 ng/ml FLT-3L, 100 ng/ml SCF. The NALM6 cell line (RIKEN BioResource Center, Ibaraki, Japan) was used within 3 months of culture following validation by short tandem repeat analysis, and maintained in RPMI with 10% heat inactivated fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine. For some experiments cells were pre-treated with 10 ng/ml TNFα (Lonza Australia, Gordon, NSW), 10 μg/ml Enbrel (etanercept, soluble TNFR2 fused to Fc; Clifford Hallam Healthcare, VIC, Australia), 10 μM Q-VD-Oph (QVD; Sigma-Aldrich, St Louis, MO) or 10 μM Necrostatin-1 (Nec-1; Selleck Chemicals, Houston, TX) for 2 h before the addition of birinapant (TetraLogic Pharmaceuticals, Malvern, PA). Apoptosis was determined using Annexin V/7-AAD staining 24 h later.
Protein expression analysis
PDX cells were treated with 1 μM birinapant for 0, 0.5 and 6 h. Analysis of cellular proteins by immunoblotting was performed as described previously (31). Membranes were probed with specific antibodies for the following proteins; cIAP1 (7065S), cIAP2 (3130S), xIAP (2045S), RIP1 (4926S), caspase 8 (9746S), cleaved caspase 8 (8592S), caspase 3 (9662S), cleaved caspase 3 (9664S), TNFR1 (3736S) (Cell Signaling, Beverly, MA) and actin (A2066; Sigma-Aldrich, St Louis, MO). The secondary antibody used was horseradish peroxidase (HRP) conjugates of anti-rabbit IgG (GE Healthcare, Little Chalfont, UK). Signals were detected by Immobilon Western Chemiluminescent HRP Substrate (Merck Millipore, Billerica, MA) and visualized using a VersaDoc 5000 Imaging System (Bio-Rad, Berkeley, CA).
RNA extraction and RT-PCR
Total RNA was isolated from PDX cells using the RNeasy mini plus kit (Qiagen, Germantown, MD). A high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) was used to synthesize cDNA from 1 μg total RNA. TNFR1 mRNA expression was measured using TaqMan Gene Expression Assays (Hs01042313_m1) (Applied Biosystems). EF1α was used as an endogenous control and data are expressed as fold change relative to normal peripheral blood mononuclear cells (PBMCs).
In vivo drug treatments
PDX cells were transplanted into 6-8 week old female NOD/SCID or NSG mice of 20-25 g weight by intravenous injection. Leukemia engraftment was monitored by flow cytometric quantification of the proportion of human CD45-positive (huCD45+) cells versus total CD45+ leukocytes (human + murine) in the peripheral blood, bone marrow or spleens as described previously (24,26-29). When the median %huCD45+ cells in the peripheral blood was above 1% for the entire cohort, mice were randomized and allocated to treatment groups (7-9 mice per group). For single agent efficacy studies birinapant was administered via intraperitoneal (IP) injection at a dose of 30 mg/kg, every 3 days x 5. Where indicated Enbrel was administered at 10 mg/kg IP, 4 h before birinapant treatment. For combination studies birinapant was given twice a week for two weeks at 10 mg/kg. VXL (Prince of Wales Hospital Pharmacy, NSW, Australia) was administered Mon-Fri for two weeks via IP injection, and consisted of vincristine (0.15 mg/kg) on Monday only followed by daily dexamethasone (5 mg/kg) and L-asparaginase (1000 U/kg) (32).
Determination of in vivo treatment response
Individual mouse event-free survival (EFS) was calculated as the time in days from treatment initiation until the %huCD45+ cells in the peripheral blood reached 25%, or until mice reached a humane end-point with evidence of leukemia-related morbidity. EFS time was represented graphically by Kaplan-Meier analysis and survival curves were compared by log-rank test. The response to drug treatment was evaluated by 2 methods: (1) progression delay (T-C), calculated as the difference between the median EFS of the drug-treated cohort (T) and the median EFS of the vehicle-treated cohort (C); and (2) using an objective response measure (ORM) modeled after stringent clinical criteria, which was assessed at Day 42 post treatment initiation as previously described (33) and detailed in the Supplemental Methods.
In vivo pharmacodynamic analysis
Mice were inoculated with ALL PDX cells and monitored until peripheral blood engraftment reached >80% huCD45+, at which stage a single dose of birinapant (30 mg/kg) or vehicle was administered. Groups of 3 mice were humanely killed and spleens harvested 6 h post-treatment. Mononuclear cells were isolated and samples confirmed to be >98% huCD45+ via flow cytometry. Protein expression was subsequently analyzed by immunoblotting.
Gene expression analysis
The raw expression data were pre-processed using Illumina BeadStudio (v2011.1) and Partek Genomics Suite (v6.6) including background subtractions, quantile normalization and log2 transformation. The normalized expression levels were further processed using the ComBat (v.3) algorithm, implementing a parametric estimator with one covariate (e.g. the leukemia subtype) to remove potential batch effects. The batch-removed expression levels were analyzed using the Analysis of Variance method (ANOVA) and 68 differentially expressed genes between Responders and Non-Responders (birinapant-signature) were identified using a P value <0.05 and a fold change >1.9 or <-1.9. The expression levels of the 68 genes within the birinapant-signature were scaled to a mean of zero and standard deviation of one and hierarchical clustering average linkage method and the Euclidean distance measure was performed. Similarly, the 68 genes within the birinapant-signature were analyzed using the Ingenuity IPA Core Analysis algorithm (v.24390178) and canonical, cellular, developmental and disease pathways, significantly associated with the molecular response to birinapant treatment, identified. Gene expression datasets can be accessed at www.ncbi.nlm.nih.gov/geo (accession no. GSE52991, GSE57795, GSE74460).
Statistical analysis
Quantitative data from in vitro assays and in vivo experiments were compared using the Mann-Whitney U test or Student's t-test. Correlations were analyzed using Pearsons correlation coefficient. P values <0.05 were considered significant. The exact log-rank test was used to compare EFS distributions between treatment and control groups (2-tailed), with P ≤ 0.05 considered significant. To evaluate interactions between drugs in vivo, therapeutic enhancement was considered if the EFS of mice treated with the combination treatment was significantly greater (P < 0.01) than those induced by both single arms of the study (34,35).
Study Approval
All studies had prior approval from the Human Research Ethics Committees of the University of New South Wales and the South Eastern Sydney Local Health District, and the Animal Care and Ethics Committee of the University of New South Wales. Written informed consent was received from participants prior to inclusion in the study.
Results
Birinapant exerts potent anti-leukemic efficacy against BCP-ALL PDXs and primary cells in vitro, and in particular Ph-like ALL
We have previously described the development and characterization of panels of PDXs representative of diverse pediatric ALL subtypes, including BCP-ALL, Ph-like ALL, infant MLL-rearranged ALL (MLL-ALL), T-ALL and early T-cell precursor ALL (ETP-ALL) (24-29), details of which can be found in Supplementary Table S1. Since another small molecule SMAC mimetic (BV6) had previously shown in vitro efficacy against pediatric ALL cell lines and primary samples (36) we wished to determine whether birinapant exhibited any subtype-specific efficacy. Therefore, we assessed the in vitro sensitivity of 41 PDXs after 24 h exposure to 100 nM birinapant (Figure 1A). Using an arbitrary cutoff of 50% of control cell viability, 9/9 Ph-like ALL PDXs were sensitive to birinapant, compared with 7/13 BCP-ALL, 3/7 MLL-ALL, 1/9 T-ALL and 1/3 ETP-ALL. When stratified according to B-lineage (BCP-ALL, Ph-like ALL, infant MLL-ALL) or T-lineage (T-ALL, ETP-ALL), the B subtype was significantly more sensitive than T-lineage (P < 0.001, Figure 1B). Within the B-lineage ALL PDXs the Ph-like subtype was significantly more sensitive than other high risk subtypes such as infant MLL-ALL (Figure 1C). To verify that the sensitivity of Ph-like PDX cells was not an artifact of xenografting we next tested the birinapant sensitivity of primary ALL cells derived from 6 pediatric and 3 adult patients with Ph-like ALL, alongside 3 BCP-ALL primary samples. Using the same cutoff as for the PDX cells 9/9 primary Ph-like biopsies were sensitive to birinapant, and 2/3 non Ph-like samples were resistant (Figure 1D, Supplementary Table S1). The predisposition of Ph-like ALL PDXs and primary samples to birinapant sensitivity did not appear to be due to the presence of specific kinase lesions per se, since in the BCP-ALL cohort of PDXs and primary biopsies kinase lesions were detected in 4/8 sensitive and 3/8 resistant samples (Figure 1A and D, and Supplementary Table S1). Moreover of the 4 BCR-ABL1-positive samples, 2 were sensitive (ALL-56 and #110) while 2 were resistant (ALL-4 and ALL-55).
Figure 1. Preferential subtype sensitivity of ALL cells to birinapant in vitro.
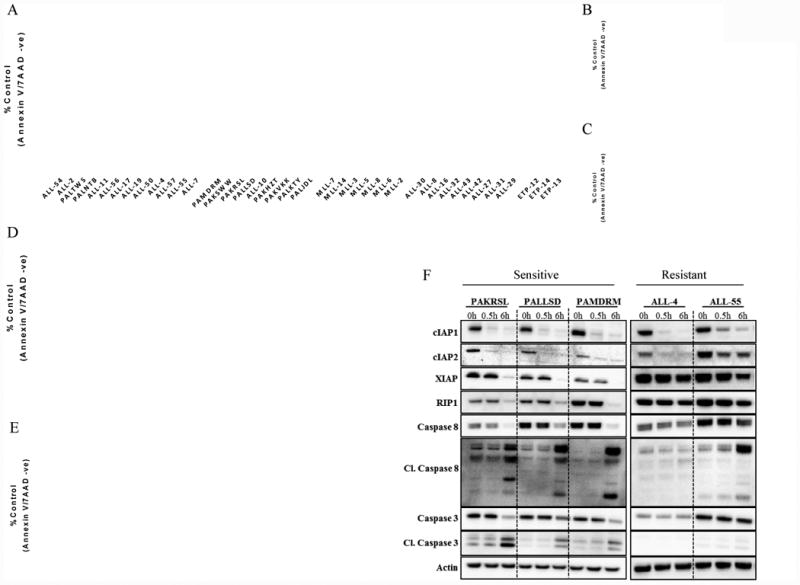
(A) viability of 41 PDX lines relative to controls 24 h after exposure to 100 nM birinapant. (B) Birinapant sensitivity of 41 PDX lines (% control after treatment with 100 nM) stratified by B and T lineage ALL. (C) Birinapant sensitivity in BCP-ALL xenografts stratified by subtype. (D) Percentage of viable primary ALL cells (annexin V-/7AAD–) cells after 24 h treatment with 100 nM birinapant. (E) sensitive and resistant BCP-ALL xenografts were incubated with birinapant (0.1 and 1 μM respectively), QVD (10 μM), Nec1 (10 μM), or birinapant + QVD/Nec1 for 24 h. (F) protein levels of cIAP1, cIAP2, XIAP, RIP1, total/cleaved caspase 8 (Cl.caspase 8) and total/cleaved caspase 3 were analyzed by immunoblotting in three Ph-like BCP-ALL xenografts (PAKRSL, PALLSD and PAMDRM) and 2 Ph+ BCP-ALL xenografts (ALL-4 and ALL-55) after treatment with 1 μM birinapant. Data in A and E represent the mean ± SEM from ≥ 3 biological replicates, while data in D are from 3 technical replicates.
To further investigate the mechanism by which birinapant kills Ph-like ALL cells, PDX cells were pre-incubated with the pan-caspase inhibitor QVD or the RIPK1 inhibitor Nec-1 prior to birinapant treatment. QVD significantly attenuated birinapant-induced apoptosis in 3/4 sensitive PDXs, while Nec-1 caused significant inhibition in 2/4 PDXs (Figure 1E). These results indicate that birinapant kills Ph-like ALL cells via caspase- and RIPK-mediated pathways.
Both cIAP1 and cIAP2 were rapidly degraded in sensitive (n=3) and resistant (n=2) PDXs within 30 min of birinapant treatment (Figure 1F). The cleaved (activated) forms of both caspase-3 and caspase-8 were markedly upregulated in sensitive PDXs within 6 h of birinapant treatment in contrast to the resistant PDXs, indicating that resistance occurred downstream of cIAP1/2 degradation. Consistent with the decreased ability of birinapant to inhibit XIAP-dependent signaling pathways compared with cIAP1/2 (8), XIAP protein levels only markedly decreased in the 3 sensitive PDXs after 6 h of birinapant treatment concomitant with caspase activation, suggesting that XIAP degradation is a consequence, rather than a cause, of cell death.
The sensitivity of BCP-ALL to birinapant is replicated in vivo
When administered at a dose of 30 mg/kg birinapant induced significant progression delays in 17/19 PDXs relative to control vehicle -treated mice, with progression delays (T-C) ranging from 2-76.9 days (Supplementary Table S2). Representative plots of leukemia progression over time and corresponding Kaplan-Meier survival plots illustrating Ph-like PDX responses to birinapant are shown in Figure 2A, which indicates that prolonged extension of mouse event free survival (EFS) could be induced despite a brief treatment window (12 days). Using stringent objective response criteria modeled after the clinical setting (33), 11/19 PDXs achieved objective responses (1 partial response, PR; 7 complete responses, CRs; 3 maintained CRs, MCRs; Supplementary Table S2, Figure 2B and C, Supplementary Figures S1 and S2), including 6/6 Ph-like ALL PDXs. The birinapant treatments were well tolerated and caused no significant decrease in body weight or perturbations in hematological parameters (Supplementary Figure S3). A complete summary of results is provided in Supplementary Table S3. Of note, only 1/165 (<1%) of birinapant treated mice experienced toxicity-related events.
Figure 2. Birinapant exhibits significant single-agent efficacy against Ph-like BCP-ALL PDXs in vivo.

(A) individual %huCD45+ cells in the peripheral blood over time (top panels) and Kaplan–Meier curves for EFS (bottom panels) for mice engrafted with 3 Ph-like BCP-ALL PDXs. Vehicle controls, dashed lines; birinapant treated (30 mg/kg), red solid lines. Black boxes on the x-axis indicate treatment days. (B) distributions of objective response measures (ORMs) of individual mice from 19 PDX lines treated with birinapant. (C) Midpoint difference representation of the median objective response scores in mice engrafted with ALL PDXs and treated with birinapant. A score of -5 to 0 indicates that an objective response was not achieved for a given xenograft; whereas a score of > 0 to 5 indicates an objective response. Red bars indicate treatments for which the EFS is significantly different between control and treated mice, blue bars indicate no statistical difference in EFS. (D) In vivo birinapant efficacy stratified according to xenograft ALL subtype and quantified by median EFS T-C values. (E) In vivo birinapant efficacy stratified according to PDX ALL subtype and quantified by median EFS T/C values. (F) Correlation of in vitro and in vivo response as determined by T-C. Regression line and 95% confidence intervals are shown. (G) Groups of 3 mice engrafted with PDXs ALL-2 or ALL-7 were treated with birinapant (30 mg/kg) or vehicle control and euthanized 6 h later. Lysates were prepared from spleen-derived cells (>95% huCD45+) and immunoblotted for cIAP1 and downstream signaling proteins. Each lane represents one mouse.
Similar to the in vitro data described above, the Ph-like PDXs were significantly more sensitive to birinapant in vivo compared to the BCP-ALL and MLL-ALL PDX panels, when assessed by EFS T-C (Figure 2D) or T/C values (Figure 2E). Moreover, the in vitro birinapant sensitivity (% of control at 100 nM) of the 19 PDXs tested significantly correlated with their in vivo responses (T-C) (R2 = 0.41, P < 0.005) (Figure 2F). Consistent with the in vitro results, birinapant also caused rapid (within 6 h) degradation of cIAP1 in vivo regardless of the PDX response (Figure 2G), despite clear differences in caspase-3, -7, -8 and -9 activation between a sensitive (ALL-2) and resistant (ALL-7) PDX.
Birinapant induces significant regressions in Ph-like ALL PDXs over a broad dose range and demonstrates synergy with established drugs
A desirable characteristic for an anti-cancer drug is that it is effective over a broad dose range and not only at its maximum tolerated dose (MTD). To test this quality for birinapant we treated mice engrafted with 2 Ph-like PDXs with doses as low as 1/8th of its MTD (30 mg/kg). Birinapant significantly delayed the progression of both PAMDRM and PALLSD at all doses tested, resulting in significantly increased EFS of mice across all treatment groups (Figure 3A and Supplementary Table S2). The efficacy of birinapant was dose-dependent, with T-C values of 24.6-64.7 days for PAMDRM and 5.5-24.9 days for PALLSD. Objective responses were achieved at all 4 dose levels for PAMDRM and at the 2 highest doses for PALLSD (Supplementary Table S2).
Figure 3. Birinapant exhibits in vivo efficacy over a broad dose range.
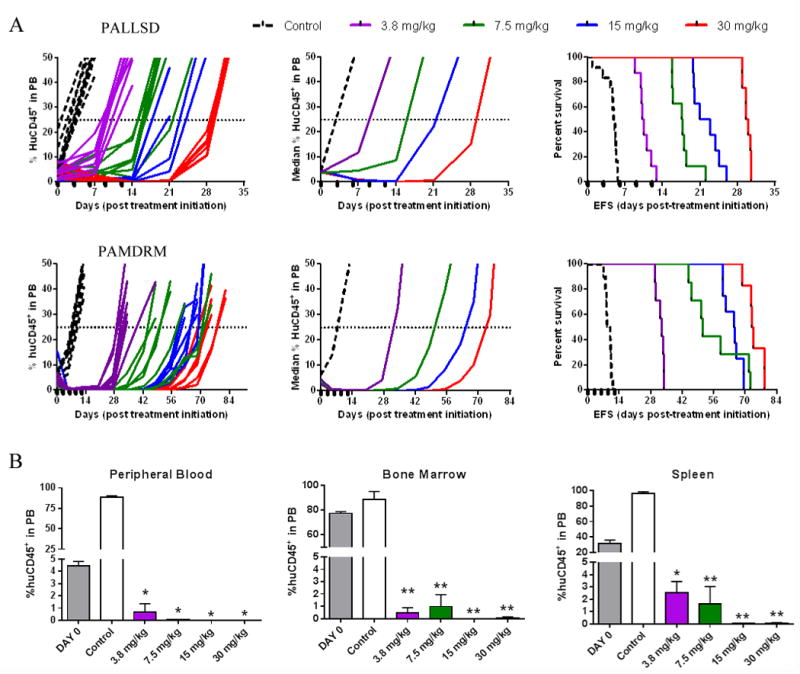
(A) individual and median (left and middle panels) %huCD45+ cells in the peripheral blood of mice and Kaplan–Meier curves for EFS (right panels) for 2 Ph-like BCP-ALL PDXs, PALLSD and PAMDRM. Black boxes on the x-axis indicate treatment days. (B) Assessment of disease dissemination in the peripheral blood, bone marrow and spleen of the Ph-like xenograft PAMDRM, 14 days post treatment initiation. Data represent the mean ± SEM, n ≥ 3 mice/ group. *P<0.05, **P<0.001, Student's t-test.
Assessment of PAMDRM at Days 0 and 14 revealed a profound and significant reduction in leukemia infiltration in all organs at all 4 dose levels tested (Figure 3B), with >97% clearance of human leukemia cells. These results demonstrate consistency in measuring anti-leukemic drug responses by assessing the peripheral blood, bone marrow or spleen, and that birinapant can induce significant leukemia regressions over a broad dose range.
An additional desirable quality for an anti-cancer drug is that it enhances the efficacy of established therapy, or in the least is not antagonistic. This is particularly relevant in pediatric ALL, in which effective combination chemotherapy treatments are available for the vast majority of patients. Mice engrafted with PAKHZT and PALLSD were subjected to a two week treatment schedule combining birinapant with an induction type regimen of VXL (32). Both VXL and birinapant (10 mg/kg, 1/3rd its MTD) significantly delayed the progression of PAKHZT and PALLSD compared to vehicle controls (Figure 4A, Supplementary Table S4). Birinapant achieved progressive disease 2 (PD2) in PALLSD (P < 0.0001) and a CR in PAKHZT (P = 0.001), while VXL achieved CRs in both PDXs. The combination of VXL plus birinapant significantly extended disease remission to 35.3 and 40.9 days for PALLSD and PAKHZT, respectively, elicited CRs and resulted in therapeutic enhancement for both PDXs.
Figure 4. Birinapant is effective in vivo in combination with VXL and against relapsed disease.

(A) Individual (left panels) and median (middle panels) %huCD45+ cells in the peripheral blood of mice and Kaplan–Meier curves for EFS (right panels), for PAKHZT (top) and PALLSD (bottom). Vehicle controls, black dotted lines; VXL, green lines; birinapant, blue lines; birinapant/VXL combination, red lines. Black boxes on the x-axis indicate birinapant treatments, grey bars indicate VXL treatment. (B) Schematic of experimental plan for birinapant treatment of relapsed disease. (C) Percentage of huCD45+ cells in the PB of mice treated with one round of VXL followed by one round of birinapant post relapse. (D) Infiltration of leukemic cells in the PB, BM and SP of mice at relapse or 14 days post birinapant treatment (Group 1). (E) Percentage of huCD45+ cells in the PB of mice treated with birinapant followed by a second round of birinapant post relapse. (F) Infiltration of leukemic cells in the PB, BM and SP of mice at relapse or 14 days post birinapant treatment (Group 2). *P<0.001, **P<0.05, Student's t-test.
Due to the efficacy of conventional treatment regimens in pediatric ALL, any Phase 1/2 clinical trial of birinapant is likely to focus on relapsed/refractory patients. Therefore, we have emphasized the preclinical testing of birinapant against PDXs derived from high-risk pediatric ALL subgroups, or from patients who have relapsed (Supplementary Table S1). To directly test the in vivo efficacy of birinapant on previously treated ALL PDXs, mice engrafted with PAKHZT were treated with birinapant after they had relapsed with >25%huCD45+ cells in the peripheral blood following prior VXL or birinapant treatment (Figure 4B-F). Birinapant (10 mg/kg) was highly effective in reducing high leukemic burden in the peripheral blood, bone marrow and spleens of mice that had been pre-treated with either regimen, indicating that birinapant is likely to be effective against relapsed disease.
A unique gene expression signature distinguishes birinapant sensitive and resistant PDXs
To evaluate a potential molecular predisposition for birinapant response in BCP-ALL we interrogated gene expression profiles of 12 BCP-ALL PDXs. ANOVA identified that a signature of 68 genes was differentially expressed between Responders and Non-Responders in BCP-ALL (birinapant-signature; Figure 5A, Supplementary Table S5). Hierarchical clustering of the birinapant-signature across the 12 BCP-ALL PDXs confirmed that these samples were optimally separated into two major classes corresponding with response to birinapant treatment (Figure 5A). Importantly, hierarchical clustering of the same BCP-ALL PDXs but with an additional 6 Ph-like PDXs (PAKHZT, PAKSWW, PAKRSL, ALL-10, PAMDRM and PALLSD) confirmed that the Ph-like PDXs, consistent with their functional response, all grouped with other birinapant Responders (Figure 5B and C). Ingenuity Pathway Analysis (IPA) further revealed that the birinapant-signature had the highest enrichment for genes associated with Inflammatory Response (P = 3.48E-06-6; Disease Pathways), Hematological System Development and Function (P = 6.03E-07; Developmental Pathways) and Cell Death and Survival (P = 3.22E-06, Cellular Pathways) (Figure 5D). When visualized using the “organic” layout algorithm the birinapant-signature was enriched for components of the TNFα signaling pathway including up-regulation of TNFR1 and its direct targets NOS2 and IL12A in Responders (Figure 5E and Supplementary Table S5). Strikingly, when visualized using the “hierarchical” layout algorithm TNFR1 was identified as the founder node and major regulator of the underlying network (Supplementary Figure S4).
Figure 5. Generation of a birinapant sensitivity gene expression signature.
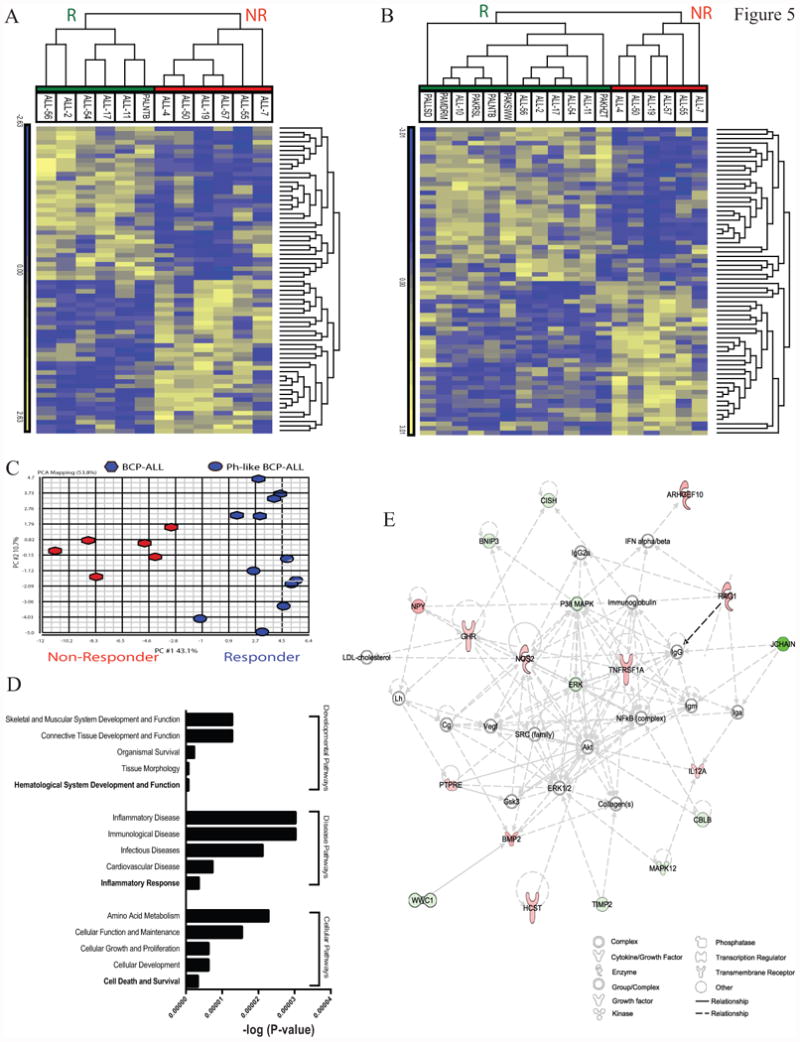
(A) hierarchical clustering of 12 BCP-ALL PDX samples (columns), based on the pre-treatment expression levels of the Birinapant-signature genes (rows), grouped as Responders (green) or Non-Responders (red). (B) hierarchical clustering of the same 12 PDXs plus an additional 6 Ph-like PDXs showing that the Birinapant-signature was maintained in Ph-like ALL Responders. (C) Principal Component Analysis of the same samples identified that the first principal component was sufficient to separate Responders (blue) from Non-Responders (red). (D) IPA of the birinapant-signature identified the most enriched pathways associated with Developmental, Diseases and Cellular expressed as –log(P-value). (E) Network analysis (IPA) of the birinapant-signature using the organic layout algorithm identified a network centered on TNFR1.
Validation of an essential role for the TNFα pathway in birinapant-induced death of ALL PDXs and primary cells
We next sought to validate the role of the TNFα pathway in the mode of action of birinapant in ALL PDXs and primary samples. Both TNFα and TNFR1 gene expression significantly correlated with in vivo sensitivity to birinapant (Figure 6A and B). Moreover, TNFR1 mRNA and protein were expressed at moderate to high levels in 5/5 birinapant sensitive PDXs (Figure 6C and D), while 2/3 resistant PDXs expressed low to undetectable levels. Consistent with the differential expression of TNFR1 between the 3 birinapant resistant PDXs (ALL-55, ALL-4 and -7) exogenous TNFα was unable to sensitize resistant xenografts to birinapant, but was cytotoxic by itself to ALL-55 (Figure 6E).
Figure 6. Expression levels of TNFα and related genes in BCP-ALL PDXs and their correlation with in vivo birinapant response.

(A) and (B) correlation between TNFα (A) and TNFR1 (B) gene expression and in vivo sensitivity (T-C) for 19 BCP-ALL PDXs. Regression line and 95% confidence intervals are shown. (C) mRNA expression of TNFR1 by RT-PCR for 8 xenografts. Results are expressed as fold change relative to normal PBMNCs. (D) Western blot analysis of TNFR1 protein levels. (E) Resistant xenografts were treated with birinapant ± 10 ng/ml TNFα. Viability was assessed after 24 h using annexin V 7AAD staining. (F) TNFα protein levels in the plasma of control and birinapant treated mice as quantified by ELISA, ND (not detectable). (G) NALM6 and PAMDRM cells treated in vitro with birinapant ± exogenous TNFα (10 ng/ml) in either RMPI1640 or QBSF-60 based media. Viability is expressed as a percent of untreated controls. Data represent the mean ± SEM from ≥ 3 independent experiments.
Plasma from mice engrafted with ALL was next evaluated for TNFα levels. Birinapant (10 mg/kg for 6 hr) significantly increased TNFα production in 4/4 PDX models, including 2 resistant xenografts, with no detectable levels present in control bearing animals (Figure 6F). Like many cell lines NALM6 cells only responded to birinapant in the presence of exogenous TNFα (Figure 6G), which is clearly not the case for primary ALL cells and PDXs (Figure 1A and D). To ensure that the culture media was not contributing to birinapant cytotoxicity, both NALM6 and a Ph-like PDX PAMDRM (Figure 6G) were cultured in RPMI and QBSF media prior to treatment. No significant differences in response were observed for either cell type regardless of the media used. These data suggest that any potential trace amounts of TNFα present in the media are insufficient to induce birinapant mediated cell death in ALL PDXs.
We next showed that Enbrel, a clinical grade soluble TNFR2 fused to Fc, offered almost complete protection against birinapant-induced cell death of ALL PDXs and primary cells in vitro (Figure 7A and B). More importantly, Enbrel inhibited the anti-leukemic effects of birinapant against the Ph-like PAKHZT PDX in vivo. Birinapant alone profoundly reduced leukemic infiltration in the murine peripheral blood (<0.2%), bone marrow (<5%) and spleens (<1%) at Day 14 following treatment initiation (Figure 7C), and substantially delayed leukemia progression (Figure 7D and E) and induced a CR (Supplementary Table S4). In contrast, Enbrel co-treatment resulted in leukemia persistence in the bone marrow, spleens and peripheral blood (Figure 7C), and significantly accelerated disease relapse (Figure 7D and E, Supplementary Table S4).
Figure 7. Birinapant efficacy in vitro and in vivo is TNFα dependent.

(A) Sensitive PDXs were treated with birinapant ± 10 ng/ml TNFα or 10 μg/ml Enbrel (Enb). Annexin V/7AAD negative cells are shown as a percent of untreated controls (n=4 ± SEM). *P<0.0001, **P<0.0005, Student's t-test. (B) Primary patient cells were treated with birinapant ± 10 μg/ml Enbrel. Data represent the mean ± SEM from ≥ 3 technical replicates, *P<0.0001, **P<0.01, Student's t-test. (C) Assessment of disease dissemination in the peripheral blood, bone marrow and spleen of mice engrafted with the Ph-like xenograft PAKHZT 14 days post treatment initiation. Insert shows %huCD45+ cells in the peripheral blood for the birinapant and combination groups. n = 4 mice/group. (D) Individual %huCD45+ cells in the peripheral blood over time for PAKHZT. E, Kaplan–Meier curves for EFS for PAKHZT.
Discussion
The IAP family of proteins represents a promising therapeutic target in pediatric ALL due to the fact that overexpression can contribute to evasion of apoptosis. In 2011 The Pediatric Preclinical Testing Program (PPTP) reported that LCL-161, a monovalent IAP inhibitor, was ineffective against a panel of pediatric ALL PDXs (37). Birinapant, however, is reported to be superior to other IAP inhibitors, primarily due to its bivalent structure and ability to target multiple IAP proteins simultaneously (8). Here we report that the majority of a large panel of BCP-ALL PDXs are sensitive to birinapant in vitro at sub-micromolar concentrations, including 9/9 Ph-like BCP-ALL PDXs. Furthermore 100% of primary Ph-like ALL samples were also sensitive to birinapant, confirming that the sensitivity of Ph-like subtype was not an artifact of xenografting.
Overall, these findings are of particular interest given that many previous studies have reported that SMAC mimetics are ineffective against primary tumor cells when administered as a single agent, with the majority of cells being resistant or only being effected at high micromolar concentrations (38-41). In addition, we have demonstrated significant therapeutic efficacy of birinapant against models of high-risk, aggressive or chemoresistant disease in vivo, a property that lends itself to the use of birinapant in the relapsed/refractory disease setting.
The clinical development of any potential new agent may be impaired if the drug is only effective at its MTD. Here we have shown that birinapant was still able to induce significant leukemia regressions in vivo when administered at 1/8th of its MTD, not only clearing blasts from the peripheral blood but also from the major hematopoietic organs including the spleen and bone marrow. Moreover, the significant anti-leukemic efficacy observed in this study was achieved using doses of birinapant that are not only well tolerated in mice but are also reflective of drug levels attainable in humans (42).
Despite the high overall survival rate for pediatric ALL, treatment outcomes remain poor for the approximate 20% of patients who experience relapse. Attempts to improve outcomes of primary high-risk disease by intensification of existing treatment, while largely successful, carry the burden of high toxicity (43,44), thus highlighting the need for targeted therapies that could be incorporated to strengthen current combination chemotherapy regimens. Similar to a recent study using BV6 (45) combining birinapant with an induction-type regimen (VXL) significantly enhanced objective responses and prolonged leukemia regressions in two Ph-like PDXs in vivo. Importantly, no additional side effects were observed when birinapant was combined with VXL combination therapy in vivo. Furthermore, we have clearly demonstrated that birinapant remains effective against leukemia cells previously exposed to either standard chemotherapy (VXL) or birinapant in vivo, highlighting a potential use for birinapant in the relapsed/refractory ALL clinical setting.
We next carried out an unbiased analysis of global gene expression profiles and identified a set of 68 genes associated with birinapant sensitivity in BCP-ALL. This gene set was distinct from that identified by others to classify Ph-like ALL (18,20,21,30), and was enriched in genes associated with the TNFR1 pathway. Further evaluation of gene expression data revealed a significant correlation between TNFα expression and in vivo response to birinapant. There appears to be an absolute requirement for TNFα (or other inflammatory cytokines i.e TRAIL) for birinapant to exert its effect in both oncology and infectious disease settings (46,47). Birinapant efficacy against PDXs and primary ALL patient samples was TNFα dependent, as demonstrated by reversing drug-induced cell death using Enbrel. Nevertheless, the source of TNFα in ALL PDXs and primary cells remains unclear and may be due to autocrine production; perhaps stimulated by birinapant treatment itself (40,41,48). Not surprisingly, expression of TNFR1 appeared necessary, but not sufficient, for birinapant sensitivity in pediatric ALL cells, with high level expression detected cross the Ph-like panel.
Similar to bortezomib and topotecan (49,50), birinapant showed significant single-agent efficacy against the PPTP ALL PDX models. Since bortezomib and topotecan also both showed significant clinical efficacy against pediatric ALL when combined with an induction-type regimen (51,52), it can be predicted that birinapant would be highly efficacious in the management of relapsed/refractory ALL patients in combination with established drugs.
In summary, we have demonstrated that birinapant at clinically relevant doses displays profound anti-leukemic activity as a single agent and in combination with chemotherapy in PDX models of high-risk pediatric ALL, including Ph-like ALL. Birinapant exhibited superior efficacy against BCP-ALL compared to T-ALL in a large panel of patient derived xenografts, with Ph-like ALL the most sensitive BCP-ALL subtype. The identification of a gene expression signature associated with birinapant sensitivity may lead to biomarker-driven clinical trials to identify ALL patients who might benefit from birinapant treatment. Overall, birinapant appears to be a promising new drug for the treatment of high-risk pediatric BCP-ALL, and further clinical investigations into its potential applications appear warranted.
Supplementary Material
Acknowledgments
The authors thank Elvira van Straten for technical assistance and TetraLogic Pharmaceuticals Corporation for providing birinapant. Children's Cancer Institute Australia is affiliated with UNSW Australia and the Sydney Children's Hospitals Network.
Financial Support: This research was funded by grants from the National Cancer Institute (NOI-CM-42216 and NOI-CM-91001-03). RBL is supported by a Senior Research Fellowship (1059804) from the Australian National Health and Medical Research Council.
Footnotes
Conflict of Interest Disclosure: The authors declare no conflict of interest.
Authorship Contributions: Contributions: JR, HC, PJH, MAS and RBL designed the study; JR, AR, KE and SH acquired the data; JR, CAB, RTK, DB and JEP analyzed the data; RS, GMM and DW provided patient samples and data; JR, MAS and RBL interpreted the data and wrote the manuscript.
References
- 1.Schimmer AD. Apoptosis in leukemia: from molecular pathways to targeted therapies. Best Pract Res Clin Haematol. 2008;21(1):5–11. doi: 10.1016/j.beha.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Fulda S. Cell death in hematological tumors. Apoptosis. 2009;14(4):409–23. doi: 10.1007/s10495-008-0306-6. [DOI] [PubMed] [Google Scholar]
- 3.Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25(34):4798–811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 4.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 5.Fulda S. Inhibitor of apoptosis (IAP) proteins: novel insights into the cancer-relevant targets for cell death induction. ACS Chem Biol. 2009;4(7):499–501. doi: 10.1021/cb900150g. [DOI] [PubMed] [Google Scholar]
- 6.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 7.Fulda S. Smac mimetics as IAP antagonists. Semin Cell Dev Biol. 2015;39:132–8. doi: 10.1016/j.semcdb.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Condon SM, Mitsuuchi Y, Deng Y, LaPorte MG, Rippin SR, Haimowitz T, et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J Med Chem. 2014;57(9):3666–77. doi: 10.1021/jm500176w. [DOI] [PubMed] [Google Scholar]
- 9.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39(3):443–53. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 10.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 11.Gordon GJ, Appasani K, Parcells JP, Mukhopadhyay NK, Jaklitsch MT, Richards WG, et al. Inhibitor of apoptosis protein-1 promotes tumor cell survival in mesothelioma. Carcinogenesis. 2002;23(6):1017–24. doi: 10.1093/carcin/23.6.1017. [DOI] [PubMed] [Google Scholar]
- 12.Kirkin V, Joos S, Zornig M. The role of Bcl-2 family members in tumorigenesis. Biochim Biophys Acta. 2004;1644(2-3):229–49. doi: 10.1016/j.bbamcr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11(2):109–24. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 14.Wuchter C, Richter S, Oltersdorf D, Karawajew L, Ludwig WD, Tamm I. Differences in the expression pattern of apoptosis-related molecules between childhood and adult de novo acute myeloid leukemia. Haematologica. 2004;89(3):363–4. [PubMed] [Google Scholar]
- 15.Hundsdoerfer P, Dietrich I, Schmelz K, Eckert C, Henze G. XIAP expression is post-transcriptionally upregulated in childhood ALL and is associated with glucocorticoid response in T-cell ALL. Pediatr Blood Cancer. 2010;55(2):260–6. doi: 10.1002/pbc.22541. [DOI] [PubMed] [Google Scholar]
- 16.Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood. 2012;119(1):34–43. doi: 10.1182/blood-2011-04-347872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2):125–34. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116(23):4874–84. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2009;106(23):9414–8. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–15. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):153–66. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–80. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Veer A, Waanders E, Pieters R, Willemse ME, Van Reijmersdal SV, Russell LJ, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013;122(15):2622–9. doi: 10.1182/blood-2012-10-462358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liem NL, Papa RA, Milross CG, Schmid MA, Tajbakhsh M, Choi S, et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood. 2004;103(10):3905–14. doi: 10.1182/blood-2003-08-2911. [DOI] [PubMed] [Google Scholar]
- 25.Maude SL, Dolai S, Delgado-Martin C, Vincent T, Robbins A, Selvanathan A, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125(11):1759–67. doi: 10.1182/blood-2014-06-580480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moradi Manesh D, El-Hoss J, Evans K, Richmond J, Toscan CE, Bracken LS, et al. AKR1C3 is a biomarker of sensitivity to PR-104 in preclinical models of T-cell acute lymphoblastic leukemia. Blood. 2015;126(10):1193–202. doi: 10.1182/blood-2014-12-618900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richmond J, Carol H, Evans K, High L, Mendomo A, Robbins A, et al. Effective targeting of the P53-MDM2 axis in preclinical models of infant MLL-rearranged acute lymphoblastic leukemia. Clin Cancer Res. 2015;21(6):1395–405. doi: 10.1158/1078-0432.CCR-14-2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suryani S, Bracken LS, Harvey RC, Sia KC, Carol H, Chen IM, et al. Evaluation of the in vitro and in vivo efficacy of the JAK inhibitor AZD1480 against JAK-mutated acute lymphoblastic leukemia. Mol Cancer Ther. 2015;14(2):364–74. doi: 10.1158/1535-7163.MCT-14-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suryani S, Carol H, Chonghaile TN, Frismantas V, Sarmah C, High L, et al. Cell and molecular determinants of in vivo efficacy of the BH3 mimetic ABT-263 against pediatric acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2014;20(17):4520–31. doi: 10.1158/1078-0432.CCR-14-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yeung DT, Moulton DJ, Heatley SL, Nievergall E, Dang P, Braley J, et al. Relapse of BCR-ABL1-like ALL mediated by the ABL1 kinase domain mutation T315I following initial response to dasatinib treatment. Leukemia. 2015;29(1):230–2. doi: 10.1038/leu.2014.256. [DOI] [PubMed] [Google Scholar]
- 31.Bachmann PS, Gorman R, Papa RA, Bardell JE, Ford J, Kees UR, et al. Divergent mechanisms of glucocorticoid resistance in experimental models of pediatric acute lymphoblastic leukemia. Cancer Res. 2007;67(9):4482–90. doi: 10.1158/0008-5472.CAN-06-4244. [DOI] [PubMed] [Google Scholar]
- 32.Szymanska B, Wilczynska-Kalak U, Kang MH, Liem NL, Carol H, Boehm I, et al. Pharmacokinetic modeling of an induction regimen for in vivo combined testing of novel drugs against pediatric acute lymphoblastic leukemia xenografts. PLoS ONE. 2012;7(3):e33894. doi: 10.1371/journal.pone.0033894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49(7):928–40. doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
- 34.Houghton PJ, Morton CL, Gorlick R, Lock RB, Carol H, Reynolds CP, et al. Stage 2 combination testing of rapamycin with cytotoxic agents by the Pediatric Preclinical Testing Program. Mol Cancer Ther. 2010;9(1):101–12. doi: 10.1158/1535-7163.MCT-09-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rose WC, Wild R. Therapeutic synergy of oral taxane BMS-275183 and cetuximab versus human tumor xenografts. Clin Cancer Res. 2004;10(21):7413–7. doi: 10.1158/1078-0432.CCR-04-1045. [DOI] [PubMed] [Google Scholar]
- 36.Belz K, Schoeneberger H, Wehner S, Weigert A, Bonig H, Klingebiel T, et al. Smac mimetic and glucocorticoids synergize to induce apoptosis in childhood ALL by promoting ripoptosome assembly. Blood. 2014;124(2):240–50. doi: 10.1182/blood-2013-05-500918. [DOI] [PubMed] [Google Scholar]
- 37.Houghton PJ, Kang MH, Reynolds CP, Morton CL, Kolb EA, Gorlick R, et al. Initial testing (stage 1) of LCL161, a SMAC mimetic, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2012;58(4):636–9. doi: 10.1002/pbc.23167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allensworth JL, Sauer SJ, Lyerly HK, Morse MA, Devi GR. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-alpha-independent mechanism. Breast Cancer Res Treat. 2013;137(2):359–71. doi: 10.1007/s10549-012-2352-6. [DOI] [PubMed] [Google Scholar]
- 39.Krepler C, Chunduru SK, Halloran MB, He X, Xiao M, Vultur A, et al. The novel SMAC mimetic birinapant exhibits potent activity against human melanoma cells. Clin Cancer Res. 2013;19(7):1784–94. doi: 10.1158/1078-0432.CCR-12-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131(4):669–81. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 41.Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131(4):682–93. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 42.Amaravadi RK, Schilder RJ, Martin LP, Levin M, Graham MA, Weng DE, et al. A Phase I study of the SMAC-mimetic birinapant in adults with refractory solid tumors or lymphoma. Mol Cancer Ther. 2015;14(11):2569–75. doi: 10.1158/1535-7163.MCT-15-0475. [DOI] [PubMed] [Google Scholar]
- 43.Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol. 2012;30(14):1663–9. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marshall GM, Dalla Pozza L, Sutton R, Ng A, de Groot-Kruseman HA, van der Velden VH, et al. High-risk childhood acute lymphoblastic leukemia in first remission treated with novel intensive chemotherapy and allogeneic transplantation. Leukemia. 2013;27(7):1497–503. doi: 10.1038/leu.2013.44. [DOI] [PubMed] [Google Scholar]
- 45.Schirmer M, Trentin L, Queudeville M, Seyfried F, Demir S, Tausch E, et al. Intrinsic and chemo-sensitizing activity of SMAC-mimetics on high-risk childhood acute lymphoblastic leukemia. Cell Death Dis. 2016;7:e2052. doi: 10.1038/cddis.2015.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Probst BL, Liu L, Ramesh V, Li L, Sun H, Minna JD, et al. Smac mimetics increase cancer cell response to chemotherapeutics in a TNF-alpha-dependent manner. Cell Death Differ. 2010;17(10):1645–54. doi: 10.1038/cdd.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ebert G, Allison C, Preston S, Cooney J, Toe JG, Stutz MD, et al. Eliminating hepatitis B by antagonizing cellular inhibitors of apoptosis. Proc Natl Acad Sci USA. 2015;112(18):5803–8. doi: 10.1073/pnas.1502400112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell. 2007;12(5):445–56. doi: 10.1016/j.ccr.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carol H, Houghton PJ, Morton CL, Kolb EA, Gorlick R, Reynolds CP, et al. Initial testing of topotecan by the pediatric preclinical testing program. Pediatr Blood Cancer. 2010;54(5):707–15. doi: 10.1002/pbc.22352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Houghton PJ, Morton CL, Kolb EA, Lock R, Carol H, Reynolds CP, et al. Initial testing (stage 1) of the proteasome inhibitor bortezomib by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50(1):37–45. doi: 10.1002/pbc.21214. [DOI] [PubMed] [Google Scholar]
- 51.Hijiya N, Stewart CF, Zhou Y, Campana D, Coustan-Smith E, Rivera GK, et al. Phase II study of topotecan in combination with dexamethasone, asparaginase, and vincristine in pediatric patients with acute lymphoblastic leukemia in first relapse. Cancer. 2008;112(9):1983–91. doi: 10.1002/cncr.23395. [DOI] [PubMed] [Google Scholar]
- 52.Messinger YH, Gaynon PS, Sposto R, van der Giessen J, Eckroth E, Malvar J, et al. Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood. 2012;120(2):285–90. doi: 10.1182/blood-2012-04-418640. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.