

Background
Isoniazid (isonicotinic acid hydrazide, INH; PubChem ID 3767) [1] is a first-line anti-mycobacterial agent used to treat active or latent tuberculosis (TB) infections generated by Mycobacterium tuberculosis [2–4]. INH has been in clinical use for over 60 years [1] and standard regimens for active TB infections include two months treatment with INH, rifampicin, pyrazinamide and ethambutol or streptomycin, followed by an additional four months of INH and rifampicin treatment [2, 4, 5]. Management of latent TB infections typically involves administration of INH alone (for 6 or 9 months) or in combination with rifapentine (for 3 months) to individuals at high risk of developing active TB [6, 7]. Although effective, current therapeutic regimens are very lengthy and difficult to implement [8], and TB remains a major global health problem with more than 9 million new cases and 1.5 million deaths reported in 2013 [9].
INH formulations are available as tablets (50, 100, or 300 mg) or solution (50 mg/5 ml) for oral administration, or as injection solution (100 mg/ml) for intramuscular use. Two combination formulations have additionally been approved for anti-TB therapy: Rifamate® (capsules with 150 mg INH and 300 mg rifampin) and Rifater® (tablets with 50 mg INH, 120 mg rifampin and 300 mg pyrazinamide) [10]. All drug labels begin with a boxed warning regarding hepatotoxicity associated with INH therapy; peripheral neuropathy is another common adverse reaction that can be avoided by co-administration of pyridoxine supplements to susceptible individuals (e.g. malnourished, pregnant/breastfeeding, etc.) [2, 4, 5]. Treatment-induced hepatotoxicity and other serious adverse reactions cause discontinuation in up to 10% of patients treated with standard regimens of first-line anti-TB drugs, including INH [7, 11, 12]. Patients who develop INH-induced hepatotoxicity present with symptoms such as abdominal pain, jaundice, nausea and vomiting, whereas features of drug hypersensitivity (e.g. fever, rash, arthralgia, eosinophilia) are rare [4, 12, 13]. Although extensively studied, the underlying mechanisms for INH-induced hepatotoxicity remain unclear [14]. This is partly due to the complexity of these mechanisms, but also to the difficulty in distinguishing between drug-specific and patient-related factors that may determine susceptibility to INH toxicity [15]. This manuscript outlines the basic aspects of INH absorption, distribution, metabolism and excretion (ADME) in humans, with special emphasis on the influence of genetic polymorphisms in genes encoding xenobiotic-metabolizing enzymes that modulate INH pharmacokinetics and, consequently, their association with INH-induced hepatotoxicity.
ADME/Pharmacokinetics
Few studies have been conducted investigating the in situ intestinal permeability of INH alone, though studies have been performed showing that INH has low permeability in the stomach and high permeability in the three segments of the small intestine (duodenum, jejunum, ileum) of rats [16, 17]. While the apparent permeability of the intestines and intestinal absorption rate constant of INH appears to decrease upon simultaneous perfusion with pyridoxine, no significant effects were concomitantly observed on INH pharmacokinetics [18]. It is of note that the bioavailability of INH was not significantly affected in tuberculosis patients who had undergone surgical procedures involving resection of the stomach or parts of the intestinal tract [19, 20]. Absorption may be reduced by concomitant administration of sugar or following food intake. This is likely due to the conversion of INH to a hydrazone species, making it less available for absorption [21–23]. INH seems to be widely distributed to all fluids and tissues, according to the apparent value of distribution volume (0.6 L/kg on average), with the largest accumulation in the liver; the pharmacological model for INH seems to follow first-order kinetics [24].
In the liver and intestines, INH is predominantly metabolized (50–90%) via N-acetylation of its hydrazine functionality by arylamine N-acetyltransferase 2 (NAT2; E.C. 2.3.1.5) to N-acetylisoniazid (AcINH) (Figure 1) [14, 25–29]. INH can also be hydrolyzed to hydrazine (Hz) by amidase with concomitant formation of isonicotinic acid (INA); it can also be metabolized into oxoacid hydrazone species [14, 26–30]. In turn, AcINH may be enzymatically hydrolyzed by amidase to form acetylhydrazine (AcHz) and INA [14, 27–32]. Additionally, AcHz can be deacetylated to Hz via hydrolysis by amidase or be further acetylated by NAT2 to diacetylhydrazine [14, 27–30, 33]. Hz can be broken down to ammonia or acetylated to AcHz by NAT2 [27–29]. None of the metabolites have known antitubercular properties, apart from the hepatotoxic AcHz [23].
Figure 1.
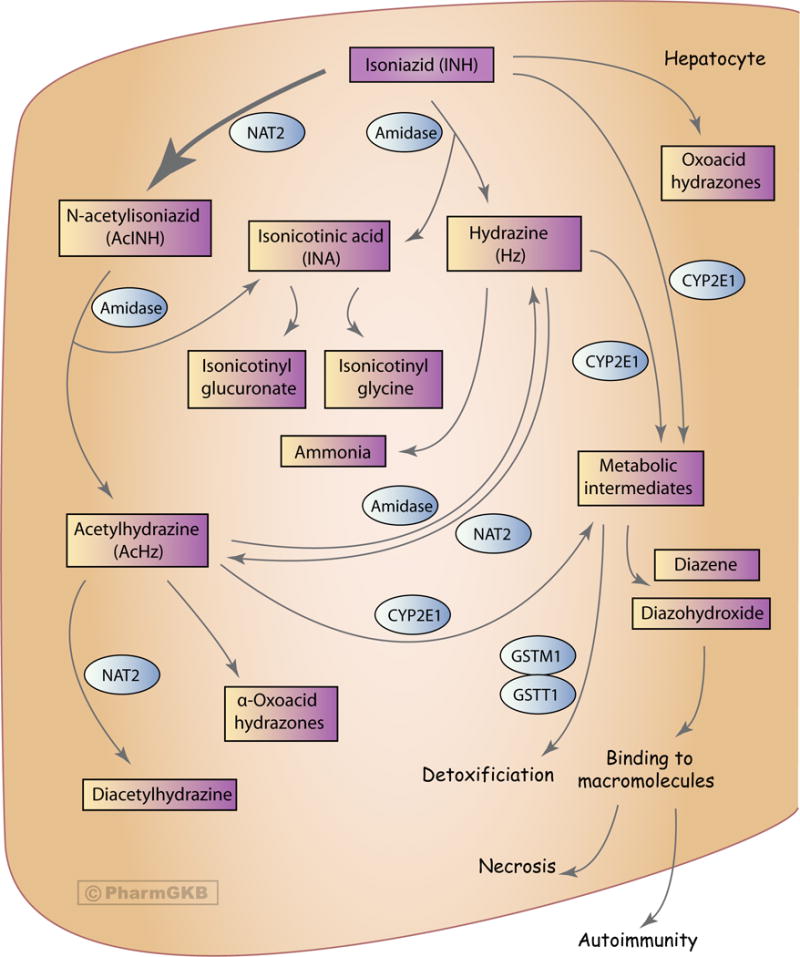
Graphic representation of the candidate genes involved in the pharmockinetics of isoniazid. A fully interactive version of this pathway is available online at PharmGKB at http://www.pharmgkb.org/pathway/PA166151813.
INH, AcHz, and Hz are likely oxidized, in part, by cytochrome P450 2E1 (CYP2E1) into potentially hepatotoxic intermediates, however explicit evidence of this has not yet been found [33–35]. These intermediates can then be dehydrated into compounds that covalently bind with macromolecules in hepatocytes causing necrosis and, possibly, autoimmunity [14, 28, 36]. The glutathione S-transferase (GST) enzyme family can conjugate these potentially harmful metabolites with glutathione, effectively removing these toxic metabolites [26, 35, 37]. Similar to CYP2E1, there have not been any studies explicitly detailing the metabolism of INH accomplished by the GST enzymes [37].
Urinary excretion is the primary elimination route (approximately 80%) of most INH metabolites (AcINH, AcHz, diacetylhydrazine) [38]; INA may be excreted as a free acid metabolite or a conjugated species with glycine (isonicotinyl glycine) [27–29]. Less than 10% of the oral INH dose is excreted in the feces [39].
INH and toxicity
Anti-tuberculosis treatment drug-induced liver injury (ATT-DILI) is an adverse reaction that may lead to poor compliance or interruption of treatment, and thus has implications for the control of TB infections [13, 40]. INH treatment is associated with increased activity of liver enzymes in 20% of patients and also with severe hepatotoxicity in 1–2% of patients [41–44]. INH thus constitutes the leading cause of hepatotoxicity in many countries [41–44]. A better understanding of the risk factors and mechanisms behind INH-induced hepatotoxicity may help in the prevention and mitigation of this complex drug reaction [42]. Reported possible risk factors include advanced age, female sex/pregnancy, low body weight/malnutrition, alcoholism, pre-existing abnormal liver function/liver transplantation, co-administration with other hepatotoxic agents, chronic hepatitis B and C infection, AIDS, and genetic factors [13, 42]. No consistent associations between race and INH-induced hepatotoxicity are evident [13]. Despite these postulated associations, the precise mechanism underlying INH-induced hepatotoxicity remains unclear; numerous different mechanisms are likely to be involved and influenced by multiple factors [13, 15, 42].
INH-induced hepatotoxicity has traditionally been attributed to the cytotoxic effects of INH metabolites, particularly AcHz and Hz [14, 29]. However, several features of this hepatotoxicity, such as a delay in liver injury after drug onset or the activation of macrophages by INH, are indicative of an immune response (see recent review by Metushi and colleagues for a thorough account of possible immune-related components of INH DILI) [14]. A recent study also reported detection of covalent INH adducts with CYP2E1, CYP3A4 and CYP2C9 in the serum of INH-treated patients developing ATT-DILI, pointing to immunological response as the underlying mechanism of hepatotoxicity [45].
As previously discussed, INH, AcHz, and Hz can be oxidized, potentially by CYP2E1, to hydroxyl-hydrazine intermediates that are then dehydrated to more damaging metabolites. These metabolites have the capacity to covalently bind macromolecules, causing liver injury [14, 15, 28, 36]. Hz and ammonia may also contribute to toxicity [14, 26, 29]. AcINH is less toxic than INH, but is not a potential therapeutic alternative due to its 100-fold less anti-mycobacterial activity and its ability to be hydrolyzed to hepatotoxic AcHz [14, 26–29].
Mitochondrial abnormalities have been linked to the toxicity of an array of drugs [46]. With respect to INH-induced hepatotoxicity, mitochondrial dysfunction appears to be caused by Hz, support for which comes from the observation that rat liver cells develop megamitochondria upon exposure to this particular metabolite prior to apoptosis. This finding is consistent with adaptive responses to oxidant stress and/or reduced oxygen consumption rates [47]. Mechanistically, the finding that Hz inhibits the succinate dehydrogenase enzyme in a dose-dependent manner suggests that complex II and/or the tricarboxylic acid cycle may also be affected, but the nature of this effect is unclear [48]. The various postulated mechanisms underlying the toxicological hazard posed by INH on liver cells have been extensively discussed recently [12] and include oxidative stress and disruption of energy homeostasis attributable to mitochondrial damage.
It is also worth noting that INH is rarely administered alone and some of the toxic effects seen in patients treated with INH may be due to, or exacerbated by, drug-drug interactions [7, 12, 13, 49–54].
Pharmacogenetics
As ATT-DILI remains unpredictable, even when environmental factors and drug regimen are considered, polymorphisms within genes involved in the INH pharmacokinetic pathway have been investigated in order to identify possible biomarkers for hepatotoxicity risk. These associations and the possible reasons behind a lack of consensus between studies are discussed below and summarized in Table 1.
Table 1.
Genetic variants in genes within the isoniazid pharmacokinetic pathway associated with anti-TB treatment drug-induced liver injury (ATT-DILI).
| Gene | Genetic Variant | Pharmacogenetic Association | Referencesa |
|---|---|---|---|
| NAT2 | Slow acetylator genotype | NAT2 slow acetylator genotype is associated with an increased risk of ATT-DILI. | [35, 40, 62–81, 91]. |
| NAT2 slow acetylator genotype is not associated with an increased risk of ATT-DILI. | [82–86]. | ||
| GSTM1 | Nullb genotype | GSTM1 null genotype is associated with an increased risk of ATT-DILI. | [37, 102]. |
| GSTM1 null genotype is not associated with an increased risk of ATT-DILI. | [35, 63, 65, 74, 92, 93, 104–106]. | ||
| GSTT1 | Nullb genotype | GSTT1 null genotype is associated with an increased risk of ATT-DILI. | [106]. |
| GSTT1 null genotype is not associated with an increased risk of ATT-DILI. | [35, 37, 63, 65, 74, 93, 102, 104, 105]. | ||
| CYP2E1 | *1A, c1, D c1 is defined as the wildtype allele at rs2031920 (C) and rs3813867 (G), and D is defined as the presence of the DraI restriction site denoted by the wildtype allele at rs6413432 (T). |
CYP2E1*1A is associated with increased risk of ATT-DILI. | [85, 91, 92] |
| CYP2E1*1A is not associated with increased risk of ATT-DILI. | [35, 62, 63, 65–68, 71, 74, 75, 77, 78, 86, 93, 94]. | ||
| *6, C C is defined as the lack of the DraI restriction site denoted by the variant allele at rs6413432 (A). |
CYP2E1 *6 is associated with ATT-DILI. | [76, 84]. | |
| CYP2E1 *6 is not associated with ATT-DILI. | [35, 63, 66, 78, 93]. | ||
| rs2070672 NM_000773.3:c.-352A>G |
Presence of the G allele is not associated with ATT-DILI. | [75]. | |
| rs2070673 NM_000773.3:c.-333A>T |
Presence of the T allele is not associated with ATT-DILI. | [75]. | |
| HLA-DQA1 | *0102 | Lack of this allele is associated with ATT-DILI. | [109]. |
| HLA-DQB1 | *0201 | The presence of this allele is not associated with ATT-DILI. | [109] – before the Bonferroni correction, this allele was associated with ATT-DILI. |
| SOD2 | rs4880 NM_000636.2:c.47T>C |
Presence of the C allele is associated with increased risk of DILI when treated with an anti-TB drug regimen, as well as other drugs. | [102]. |
| NOS2A | rs11080344 NM_000625.4:c.1281+1205A>G |
Genotype CC (positive strand) is associated with increased risk of ATT-DILI. | [108]. |
| BACH1 | rs2070401 NM_001186.3:c.*331A>G |
Genotype GG is associated with increased risk of ATT-DILI. | [108]. |
| MAFK | rs4720833 NM_002360.3:c.-45+3869A>G |
Presence of the A allele is associated with increased risk of ATT-DILI. | [108]. |
See www.pharmgkb.org for detailed annotations for individual studies.
Null genotype refers to the absence of the gene.
NAT2
Studies unraveling the genetic basis of N-acetylation first appeared around 1990; an exhaustive review of INH acetylation pharmacogenetics in the NAT2 pre-genotyping era was published by Weber and Hein [55] and the subject has subsequently been reviewed by many authors, more recently by McDonagh and colleagues [56]. When examining plasma concentrations of INH over time after an oral dose, a bimodal pattern was originally observed; higher plasma levels and reduced clearance of the drug were seen in slow acetylators compared to rapid acetylators. More refined phenotypic analysis further demonstrated a trimodal population distribution pattern; subjects can be divided into rapid, intermediate or slow acetylators [55]. Following the development of genotyping techniques for the NAT2 gene, prediction of the acetylator phenotype has become possible through genetic testing. There are many different alleles described for the NAT2 gene (NAT Gene Nomenclature website) and an individual’s genotype can be predictive of rapid, slow or intermediate acetylator phenotype, depending on the presence of two “rapid” alleles, two “slow” alleles or one of each, respectively [57]. The reference NAT2*4 allele is the most common NAT2 allele conferring the rapid acetylator phenotype and is associated with increased metabolism and clearance of INH [56, 58, 59]. Conversely, the polymorphic NAT2 alleles of the main allelic groups *5, *6, *7 and *14 encode for slow acetylator enzyme variants that may compromise the drug-metabolizing ability of individuals [56].
An investigation of NAT2 genotype as a pharmacogenetic biomarker for personalization of INH therapeutic dosage demonstrated a linear relationship between clearance of the drug and the number (0 in slow, 1 in intermediate or 2 in rapid acetylators) of NAT2*4 alleles; this specific parameter accounted for 88% of the INH clearance variability observed in the Caucasian population studied [60]. Similar conclusions were drawn by another study with Japanese TB patients treated with INH and rifampicin. Genotyped NAT2 slow acetylators (no *4 allele) exhibited significantly decreased acetylation of INH and Hz, resulting in increased serum concentrations of INH compared to intermediate (one *4 allele) or rapid (two *4 alleles) acetylators [26]. Rapid acetylators display higher levels of AcINH and AcHz in serum and clear AcHz more quickly when compared to slow acetylators, whereas slow acetylators have higher exposure to AcHz and excrete more unchanged INH in urine [13, 26].
Apart from INH acetylation, NAT2 also catalyzes the acetylation of AcHz to non-toxic diacetylhydrazine [55], and thus slow acetylator status causes accumulation of both Hz and AcHz to potentially hepatotoxic levels [15, 61]. Consequently, the NAT2 slow acetylator phenotype and genotype have been associated with an increased risk of INH-induced hepatotoxicity in the majority of published pharmacogenetic studies (Table 1); the patient cohorts genotyped are of various geographic origins, mainly from South America [62–66], East Asia [35, 67–75], South Asia [40, 76–78], Iran [79], Turkey [80] and Tunisia [81]. The labels of all INH-containing drug formulations currently approved by the FDA inform that slow acetylators may have increased blood levels of the compound, which results in an increased risk of hepatotoxicity and peripheral neuropathy [10]. The NAT2 gene is included in the pharmacogenomic biomarkers list of the FDA in relation with INH, but no specific actions are recommended on the basis of this information.
Contradictory results regarding the acetylator status and risk of INH hepatotoxicity have also been reported in the literature. An early study [36] attributed INH hepatotoxicity to the NAT2 rapid acetylator phenotype, presumably associated with increased plasma levels of AcHz. However, a series of ensuing pharmacological studies (reviewed by Weber and Hein [55]) showed the opposite or no association between the acetylator phenotype and INH hepatotoxicity. A small number of recent studies have also reported no association between the acetylator genotype and INH-induced hepatotoxicity [82–86] (Table 1).
A recent clinical trial reported a significantly lower relative risk of unfavorable events in patients treated with an INH dose based on NAT2 genotype, compared to those treated with the standard dose, supporting a clinically-relevant association between NAT2 variants and INH pharmacokinetics [87]. Importantly, in the genotype-based treatment group, efficacy of treatment in NAT2 slow acetylators was not reduced despite the lower doses administered. Also, incidence of INH-induced liver injury was not increased in rapid acetylators when INH was given in higher doses [87]. More independent studies are required to verify these results.
With regard to individual NAT2 single nucleotide polymorphisms (SNPs), rs1799930 (NM_000015.2:c.590G>A, signature SNP for the NAT2*6 allelic group) has been associated with decreased acetylation of INH and clearance of the drug, which correlated with an enhanced risk of drug-induced hepatitis [64, 75, 77]. The NAT2*6 signature polymorphism has been reported to confer an ultra-slow acetylator phenotype [88, 89], and this could explain the higher risk of INH-induced hepatotoxicity in slow acetylators carrying this particular SNP. A slow acetylator haplotype composed of rs4646244 (NM_00015.2:c.-1144T>A) allele A, rs4646267 (NM_000015.2:c.-949A>G) allele A, rs1799930 allele A, and rs1799931 (NM_000015.2:c.857G>A, signature SNP for the NAT2*7 allelic group) allele G, has been associated with an increased risk of hepatotoxicity. Patients with this haplotype had significantly decreased acetylation and clearance of INH, as compared to the other haplotypes examined, and this is likely attributed to the presence of the ultra-slow NAT2*6 signature SNP [75]. Another study has correlated genotype AA of rs1495741 (NC_000008.10:g.18272881G>A, a tag SNP located about 14 kb downstream of NAT2) with increased risk of INH hepatotoxicity [69]. This association is indirect, as the A allele of rs1495741 is likely to be in linkage disequilibrium with a slow NAT2 variant [90].
CYP2E1
CYP2E1 is involved in the oxidation of INH, AcHz, and Hz, resulting in likely hepatotoxic intermediates that undergo further dehydration to potentially harmful products [14, 26, 28, 33–35, 68]. Polymorphisms of the CYP2E1 gene have been examined in association with risk of INH-mediated ATT-DILI, mainly by investigators in South [40, 76–78, 84] and East [26, 35, 67, 68, 71, 74, 75, 91–93] Asia and South America [62, 63, 65, 66]. Although some studies have reported higher risk of ATT-DILI in INH-treated patients who bear high-activity alleles of CYP2E1, particularly *1A and *6 [76, 84, 85, 91, 92], other studies have found no association [35, 62, 63, 65–68, 71, 74, 75, 77, 78, 86, 93, 94] and, therefore, a direct role for CYP2E1 in INH-induced hepatotoxicity remains widely controversial (Table 1). Evidence suggests that CYP2E1 polymorphisms may be associated with increased severity of INH related ATT-DILI, rather than enhanced susceptibility to it [71]. Studies also report increased risk of ATT-DILI in INH-treated patients who combine high-activity CYP2E1 and slow NAT2 genotypes [66, 67, 76, 91, 95].
Since the generation of many INH metabolites does not depend exclusively on CYPs, attention has somewhat shifted away from CYP2E1 as a major predictor of hepatotoxicity [12, 34]. Nonetheless, the observation that CYP2E1 is expressed in mitochondria and is one of the CYP forms that generates relatively high levels of reactive oxygen species may point to a role for CYP2E1 in increasing the extent of oxidative stress [96, 97]. Moreover, anti-INH and anti-CYP2E1 antibodies have been found in the serum of patients with INH-induced liver failure; INH-CYP2E1 covalent adducts were also detected, as well as antibodies against them [45]. Such auto-antibodies are considered markers of CYP2E1-directed autoimmunity, which may lead to liver injury [98]. INH has been shown to induce CYP2E1 enzyme activity [99] and experiments with animal models support CYP2E1-mediated hepatotoxicity when INH is administered [100].
GSTM1 and GSTT1
The GSTs are involved in the detoxification of numerous drugs and oxidative stress products; due to this role, polymorphisms of the GSTM1 and GSTT1 genes have also been investigated in risk of ATT-DILI [101]. GSTs are known to be involved in the metabolism of anti-TB drugs via conjugation of potentially harmful metabolites; however, the specifics about this interaction are unknown [35, 37]. Deficiencies in GST activity due to homozygous null mutations in GSTM1 and GSTT1 may have the potential to modulate susceptibility to INH-induced hepatotoxicity [15]. Indeed, several published studies have reported an association between GSTM1 homozygous null genotype and increased risk of ATT-DILI [26, 37, 102, 103], although the majority of studies report no association found [35, 63, 65, 74, 92, 93, 104–106] (Table 1). A similar pattern is seen for the GSTT1 homozygous null genotype, with one study from Spain describing an association with increased risk of ATT-DILI [106], but the majority of studies (in Asia or Brazil) reporting no association [35, 37, 63, 65, 74, 93, 102–105] (Table 1). Studies have also shown an additive effect of combined GSTM1 and GSTT1 null genotypes on risk of INH hepatotoxicity [40, 103], as well as of combined NAT2, CYP2E1, GSTM1 and/or GSTT1 polymorphic genotypes [63]. There does not seem to be a consistent pattern of factors that indicate why these contradictory results have been observed.
Other genes
It is likely that multiple genetic factors and gene-gene interactions are involved in INH-induced hepatotoxicity risk, as is the case for other hepatotoxic drugs [107]. Indeed, significant associations with risk have been reported for genotypes of other genes [102, 108] (Table 1), which further complicates the assessment of possible risk factors. INH liver injury was associated with certain major histocompatibility complex (MHC) class II alleles, including human leukocyte antigen (HLA) haplotypes, which supports the role of the immune system in INH toxicity [14]. The absence of the HLA-DQA1*0102 allele and the presence of the HLA-DQB1*0201 allele were reported to be independently associated with increased risk of ATT-DILI in 331 TB patients [109]; however, the Bonferroni correction introduced to compare the distribution of both alleles in ATT-DILI and non-ATT-DILI patients could only confirm the negative association of DQA1*0102 with ATT-DILI, but was not clearly explained. Generally, only some cases of ATT-DILI associated with INH are suggested to be immune-mediated and HLA-associated, which may explain the discrepancy observed in some studies [14]. Overall, a direct association of ATT-DILI with HLA alleles may be difficult or not possible to establish at this stage [110].
One study in Japanese patients found associations between INH-induced hepatotoxicity and particular polymorphisms in the genes involved in one of the antioxidant pathways. Among these positive correlations, one polymorphism in NOS2A, which encodes inducible nitric oxide synthase, causes an increase in nitric oxide production. Polymorphisms in BACH1 (encoding basic leucine zipper transcription factor 1) and MAFK (encoding v-maf avian musculoaponeurotic fibrosarcoma oncogene homologue K) result in suppression of the nuclear factor erythroid 2-like 2 (Nrf2) pathway [108] (Table 1). However, it remains mechanistically unclear how these mutations may contribute to ATT-DILI.
Meta-analyses of NAT2, CYP2E1, GSTM1 and GSTT1 genotypes
Several meta-analyses have been published to examine the association between ATT-DILI and genetic variants of drug metabolizing enzymes. A large meta-analysis of 38 studies found NAT2 slow acetylator genotype to be significantly associated with risk of ATT-DILI [111]. A meta-analysis of 4 studies found an association between increased risk of ATT-DILI and NAT2 slow acetylator status (defined as non-carrier of the *4 allele, as opposed to rapid acetylator status defined as heterozygous or homozygous for *4) in TB patients of Asian ethnicity (OR=2.52, CI=1.49–4.26, p-value not provided) [112]. However, in another larger meta-analysis with some overlapping studies, a significant association between ATT-DILI and NAT2 slow acetylator status was observed in both Asian and non-Asian patients [113]. The same meta-analysis, which also analyzed different treatment combinations, found a significantly increased risk of ATT-DILI with NAT2 slow acetylator status compared to the rapid status in patients treated with INH, rifampicin, pyrazinamide and ethambutol (9 studies, OR=4.09, 95% CI=2.78–6.03, p<0.001), or INH and rifampicin (3 studies, OR=34.30, 95% CI=10.41–113.00, p<0.001), but not with INH only (2 studies, OR=2.36, 95% CI=0.52–10.73, p<0.266) [113].
One meta-analysis showed that the CYP2E1*1A/*1A high-activity genotype is significantly associated with risk of ATT-DILI, but only in East Asian patients [111]. CYP2E1*1A/*1A genotype was significantly associated with increased risk of ATT-DILI compared to all other genotypes in a meta-analysis of 4 studies (OR=2.22, 95% CI=1.06–4.66, p=0.03) [112]. Finally, the CYP2E1*1A/*1A genotype was determined to be a risk factor for ATT-DILI, particularly when combined with the slow acetylator NAT2 genotype (OR=3.10, 95% CI=1.83–5.26. p<0.0001) [95].
While two meta-analyses have shown significant associations of the GSTM1 null genotype with risk of ATT-DILI, neither meta-analysis found such an association for the GSTT1 null genotype [111, 112]. A recent large-scale meta-analysis of GST variants, which included 13 and 12 case-control studies for the GSTM1 and GSTT1 null genotypes, respectively (approximately 900 cases and 1900 controls for each gene), found evidence that the null genotype of GSTM1, but not GSTT1, was associated with marginally increased susceptibility to ATT-DILI [114], which was consistent with the previous meta-analyses [111, 112].
Conclusion
The current consensus among the literature is that one mechanism is unlikely to explain INH-induced hepatotoxicity and that numerous pathways are probably involved. Different drug-specific mechanisms have been suggested, but most supporting data have been generated from cellular and animal models and thus do not account for the multitude of factors that may contribute to susceptibility to INH-induced hepatotoxicity in clinical settings. Variants of enzymes involved in the INH metabolic pathway have been associated with ATT-DILI, particularly the slow acetylator variant of NAT2. However, upon comparing studies in Table 1, there does not seem to be an obvious underlying factor that explains why some studies found an association and others did not; it is likely that many factors, such as inconsistent genotyping and phenotyping methods, study design, anti-TB drug regimen and the overall condition of patients, may contribute to risk of INH-induced hepatotoxicity. Future investigations that utilize DNA sequencing may lead to further identification of variants contributing to ATT-DILI. Large-scale, robust analyses of these underlying genetic and environmental risk factors in clinical settings will help uncover the full picture of these important and complex adverse reactions.
Acknowledgments
This work is supported by the NIH/NIGMS (R24 GM61374).
Abbreviations
- INH
Isoniazid
- AcINH
N-acetylisoniazid
- INA
isonicotinic acid
- AcHz
acetylhydrazine
- Hz
hydrazine
- ATT-DILI
anti-tuberculosis treatment drug-induced liver injury
Footnotes
Conflicts of Interest
RBA is a stockholder in Personalis, Inc. TEK is a paid scientific advisor to Rxight Pharmacogenetics. All other authors declare no conflicts of interest.
References
- 1.Isoniazid. Tuberculosis (Edinb) 2008;88:112–116. doi: 10.1016/S1472-9792(08)70011-8. [DOI] [PubMed] [Google Scholar]
- 2.Blumberg HM, Burman WJ, Chaisson RE, Daley CL, Etkind SC, Friedman LN, Fujiwara P, Grzemska M, Hopewell PC, Iseman MD, et al. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: Treatment of Tuberculosis. Am J Respir Crit Care Med. 2003;167:603–662. doi: 10.1164/rccm.167.4.603. [DOI] [PubMed] [Google Scholar]
- 3.Migliori GB, Zellweger JP, Abubakar I, Ibraim E, Caminero JA, De Vries G, D’Ambrosio L, Centis R, Sotgiu G, Menegale O, et al. European Union Standards for Tuberculosis Care. Eur Respir J. 2012;39:807–819. doi: 10.1183/09031936.00203811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization. Treatment of Tuberculosis: Guidelines. 4th. Geneva: World Health Organization; 2010. Stop TB Initiative (World Health Organization) [Google Scholar]
- 5.Migliori GB, Raviglione MC, Schaberg T, Davies PD, Zellweger JP, Grzemska M, Mihaescu T, Clancy L, Casali L. Tuberculosis Management in Europe. Task Force of the European Respiratory Society (Ers), the World Health Organisation (Who) and the International Union against Tuberculosis and Lung Disease (Iuatld) Europe Region. Eur Respir J. 1999;14:978–992. doi: 10.1183/09031936.99.14497899. [DOI] [PubMed] [Google Scholar]
- 6.Chapman HJ, Lauzardo M. Advances in Diagnosis and Treatment of Latent Tuberculosis Infection. J Am Board Fam Med. 2014;27:704–712. doi: 10.3122/jabfm.2014.05.140062. [DOI] [PubMed] [Google Scholar]
- 7.Forget EJ, Menzies D. Adverse Reactions to First-Line Antituberculosis Drugs. Expert Opin Drug Saf. 2006;5:231–249. doi: 10.1517/14740338.5.2.231. [DOI] [PubMed] [Google Scholar]
- 8.Handbook of Anti-Tuberculosis Agents. Introduction. Tuberculosis (Edinb) 2008;88:85–86. doi: 10.1016/S1472-9792(08)70002-7. [DOI] [PubMed] [Google Scholar]
- 9.World Health Organization. Global Tuberculosis Report 2014. Geneva: World Health Organization; 2014. [Google Scholar]
- 10.U.S. FDA Drugs@Fda Database. https://www.accessdata.fda.gov/scripts/cder/drugsatfda/
- 11.Schaberg T, Rebhan K, Lode H. Risk Factors for Side-Effects of Isoniazid, Rifampin and Pyrazinamide in Patients Hospitalized for Pulmonary Tuberculosis. Eur Respir J. 1996;9:2026–2030. doi: 10.1183/09031936.96.09102026. [DOI] [PubMed] [Google Scholar]
- 12.Boelsterli UA, Lee KK. Mechanisms of Isoniazid-Induced Idiosyncratic Liver Injury: Emerging Role of Mitochondrial Stress. J Gastroenterol Hepatol. 2014;29:678–687. doi: 10.1111/jgh.12516. [DOI] [PubMed] [Google Scholar]
- 13.Saukkonen JJ, Cohn DL, Jasmer RM, Schenker S, Jereb JA, Nolan CM, Peloquin CA, Gordin FM, Nunes D, Strader DB, et al. An Official Ats Statement: Hepatotoxicity of Antituberculosis Therapy. Am J Respir Crit Care Med. 2006;174:935–952. doi: 10.1164/rccm.200510-1666ST. [DOI] [PubMed] [Google Scholar]
- 14.Metushi IG, Cai P, Zhu X, Nakagawa T, Uetrecht JP. A Fresh Look at the Mechanism of Isoniazid-Induced Hepatotoxicity. Clin Pharmacol Ther. 2011;89:911–914. doi: 10.1038/clpt.2010.355. [DOI] [PubMed] [Google Scholar]
- 15.Ramappa V, Aithal GP. Hepatotoxicity Related to Anti-Tuberculosis Drugs: Mechanisms and Management. J Clin Exp Hepatol. 2013;3:37–49. doi: 10.1016/j.jceh.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennernas H, Hussain AS, Junginger HE, Stavchansky SA, Midha KK, Shah VP, Amidon GL. Molecular Properties of Who Essential Drugs and Provisional Biopharmaceutical Classification. Molecular pharmaceutics. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 17.Mariappan TT, Singh S. Regional Gastrointestinal Permeability of Rifampicin and Isoniazid (Alone and Their Combination) in the Rat. The international journal of tuberculosis and lung disease: the official journal of the International Union against Tuberculosis and Lung Disease. 2003;7:797–803. [PubMed] [Google Scholar]
- 18.Zhou Y, Jiao Y, Wei YH, Zhang GR, Zhang JP, Ren JX, Zhang F, Zhang GQ, Duan HG, Wu XA. Effects of Pyridoxine on the Intestinal Absorption and Pharmacokinetics of Isoniazid in Rats. European journal of drug metabolism and pharmacokinetics. 2013;38:5–13. doi: 10.1007/s13318-012-0106-9. [DOI] [PubMed] [Google Scholar]
- 19.Kleber FX. Absorption of Anti-Tuberculous Drugs after Gastric Surgery (Author’s Transl) Praxis und Klinik der Pneumologie. 1979;33:38–44. [PubMed] [Google Scholar]
- 20.Polk RE, Tenenbaum M, Kline B. Isoniazid and Ethambutol Absorption with Jejunoileal Bypass. Annals of internal medicine. 1978;89:430–431. doi: 10.7326/0003-4819-89-3-430_2. [DOI] [PubMed] [Google Scholar]
- 21.Lin MY, Lin SJ, Chan LC, Lu YC. Impact of Food and Antacids on the Pharmacokinetics of Anti-Tuberculosis Drugs: Systematic Review and Meta-Analysis. The international journal of tuberculosis and lung disease: the official journal of the International Union against Tuberculosis and Lung Disease. 2010;14:806–818. [PubMed] [Google Scholar]
- 22.Rao KV, Kailasam S, Menon NK, Radhakrishna S. Inactivation of Isoniazid by Condensation in a Syrup Preparation. Indian J Med Res. 1971;59:1343–1353. [PubMed] [Google Scholar]
- 23.Becker C, Dressman JB, Amidon GL, Junginger HE, Kopp S, Midha KK, Shah VP, Stavchansky S, Barends DM. Biowaiver Monographs for Immediate Release Solid Oral Dosage Forms: Isoniazid. Journal of pharmaceutical sciences. 2007;96:522–531. doi: 10.1002/jps.20765. [DOI] [PubMed] [Google Scholar]
- 24.O’Neil MJ, Merck Research Laboratories . The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals. 14th. Whitehouse Station, N.J.: Merck; 2006. [Google Scholar]
- 25.Windmill KF, Gaedigk A, Hall PM, Samaratunga H, Grant DM, McManus ME. Localization of N-Acetyltransferases Nat1 and Nat2 in Human Tissues. Toxicological sciences: an official journal of the Society of Toxicology. 2000;54:19–29. doi: 10.1093/toxsci/54.1.19. [DOI] [PubMed] [Google Scholar]
- 26.Fukino K, Sasaki Y, Hirai S, Nakamura T, Hashimoto M, Yamagishi F, Ueno K. Effects of N-Acetyltransferase 2 (Nat2), Cyp2e1 and Glutathione-S-Transferase (Gst) Genotypes on the Serum Concentrations of Isoniazid and Metabolites in Tuberculosis Patients. J Toxicol Sci. 2008;33:187–195. doi: 10.2131/jts.33.187. [DOI] [PubMed] [Google Scholar]
- 27.Mahapatra S, Woolhiser LK, Lenaerts AJ, Johnson JL, Eisenach KD, Joloba ML, Boom WH, Belisle JT. A Novel Metabolite of Antituberculosis Therapy Demonstrates Host Activation of Isoniazid and Formation of the Isoniazid-Nad+ Adduct. Antimicrob Agents Chemother. 2012;56:28–35. doi: 10.1128/AAC.05486-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Preziosi P. Isoniazid: Metabolic Aspects and Toxicological Correlates. Curr Drug Metab. 2007;8:839–851. doi: 10.2174/138920007782798216. [DOI] [PubMed] [Google Scholar]
- 29.Tostmann A, Boeree MJ, Aarnoutse RE, de Lange WC, van der Ven AJ, Dekhuijzen R. Antituberculosis Drug-Induced Hepatotoxicity: Concise up-to-Date Review. J Gastroenterol Hepatol. 2008;23:192–202. doi: 10.1111/j.1440-1746.2007.05207.x. [DOI] [PubMed] [Google Scholar]
- 30.Sarich TC, Adams SP, Petricca G, Wright JM. Inhibition of Isoniazid-Induced Hepatotoxicity in Rabbits by Pretreatment with an Amidase Inhibitor. J Pharmacol Exp Ther. 1999;289:695–702. [PubMed] [Google Scholar]
- 31.Ellard GA, Gammon PT. Pharmacokinetics of Isoniazid Metabolism in Man. J Pharmacokinet Biopharm. 1976;4:83–113. doi: 10.1007/BF01086149. [DOI] [PubMed] [Google Scholar]
- 32.Ellard GA, Gammon PT, Wallace SM. The Determination of Isoniazid and Its Metabolites Acetylisoniazid, Monoacetylhydrazine, Diacetylhydrazine, Isonicotinic Acid and Isonicotinylglycine in Serum and Urine. Biochem J. 1972;126:449–458. doi: 10.1042/bj1260449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daly AK, Day CP. Genetic Association Studies in Drug-Induced Liver Injury. Drug Metab Rev. 2012;44:116–126. doi: 10.3109/03602532.2011.605790. [DOI] [PubMed] [Google Scholar]
- 34.Cheng J, Krausz KW, Li F, Ma X, Gonzalez FJ. Cyp2e1-Dependent Elevation of Serum Cholesterol, Triglycerides, and Hepatic Bile Acids by Isoniazid. Toxicol Appl Pharmacol. 2013;266:245–253. doi: 10.1016/j.taap.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sotsuka T, Sasaki Y, Hirai S, Yamagishi F, Ueno K. Association of Isoniazid-Metabolizing Enzyme Genotypes and Isoniazid-Induced Hepatotoxicity in Tuberculosis Patients. In Vivo. 2011;25:803–812. [PubMed] [Google Scholar]
- 36.Mitchell JR, Thorgeirsson UP, Black M, Timbrell JA, Snodgrass WR, Potter WZ, Jollow HR, Keiser HR. Increased Incidence of Isoniazid Hepatitis in Rapid Acetylators: Possible Relation to Hydranize Metabolites. Clin Pharmacol Ther. 1975;18:70–79. doi: 10.1002/cpt197518170. [DOI] [PubMed] [Google Scholar]
- 37.Roy B, Chowdhury A, Kundu S, Santra A, Dey B, Chakraborty M, Majumder PP. Increased Risk of Antituberculosis Drug-Induced Hepatotoxicity in Individuals with Glutathione S-Transferase M1 ‘Null’ Mutation. J Gastroenterol Hepatol. 2001;16:1033–1037. doi: 10.1046/j.1440-1746.2001.02585.x. [DOI] [PubMed] [Google Scholar]
- 38.Baselt RC. Disposition of Toxic Drugs and Chemicals in Man. 6th. Foster City: Biomedical Publications; 2002. [Google Scholar]
- 39.Holdiness MR. Clinical Pharmacokinetics of the Antituberculosis Drugs. Clinical pharmacokinetics. 1984;9:511–544. doi: 10.2165/00003088-198409060-00003. [DOI] [PubMed] [Google Scholar]
- 40.Singla N, Gupta D, Birbian N, Singh J. Association of Nat2, Gst and Cyp2e1 Polymorphisms and Anti-Tuberculosis Drug-Induced Hepatotoxicity. Tuberculosis (Edinb) 2014;94:293–298. doi: 10.1016/j.tube.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 41.Garibaldi RA, Drusin RE, Ferebee SH, Gregg MB. Isoniazid-Associated Hepatitis. Report of an Outbreak. Am Rev Respir Dis. 1972;106:357–365. doi: 10.1164/arrd.1972.106.3.357. [DOI] [PubMed] [Google Scholar]
- 42.Huang YS. Recent Progress in Genetic Variation and Risk of Antituberculosis Drug-Induced Liver Injury. J Chin Med Assoc. 2014;77:169–173. doi: 10.1016/j.jcma.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 43.Scharer L, Smith JP. Serum Transaminase Elevations and Other Hepatic Abnormalities in Patients Receiving Isoniazid. Ann Intern Med. 1969;71:1113–1120. doi: 10.7326/0003-4819-71-6-1113. [DOI] [PubMed] [Google Scholar]
- 44.Tafazoli S, Mashregi M, O’Brien PJ. Role of Hydrazine in Isoniazid-Induced Hepatotoxicity in a Hepatocyte Inflammation Model. Toxicol Appl Pharmacol. 2008;229:94–101. doi: 10.1016/j.taap.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Metushi IG, Sanders C, Lee WM, Uetrecht J. Detection of Anti-Isoniazid and Anti-Cytochrome P450 Antibodies in Patients with Isoniazid-Induced Liver Failure. Hepatology. 2014;59:1084–1093. doi: 10.1002/hep.26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Labbe G, Pessayre D, Fromenty B. Drug-Induced Liver Injury through Mitochondrial Dysfunction: Mechanisms and Detection During Preclinical Safety Studies. Fundam Clin Pharmacol. 2008;22:335–353. doi: 10.1111/j.1472-8206.2008.00608.x. [DOI] [PubMed] [Google Scholar]
- 47.Karbowski M, Kurono C, Wozniak M, Ostrowski M, Teranishi M, Nishizawa Y, Usukura J, Soji T, Wakabayashi T. Free Radical-Induced Megamitochondria Formation and Apoptosis. Free Radic Biol Med. 1999;26:396–409. doi: 10.1016/s0891-5849(98)00209-3. [DOI] [PubMed] [Google Scholar]
- 48.Lee KK, Fujimoto K, Zhang C, Schwall CT, Alder NN, Pinkert CA, Krueger W, Rasmussen T, Boelsterli UA. Isoniazid-Induced Cell Death Is Precipitated by Underlying Mitochondrial Complex I Dysfunction in Mouse Hepatocytes. Free Radic Biol Med. 2013;65:584–594. doi: 10.1016/j.freeradbiomed.2013.07.038. [DOI] [PubMed] [Google Scholar]
- 49.Apostolova N, Gomez-Sucerquia LJ, Moran A, Alvarez A, Blas-Garcia A, Esplugues JV. Enhanced Oxidative Stress and Increased Mitochondrial Mass During Efavirenz-Induced Apoptosis in Human Hepatic Cells. Br J Pharmacol. 2010;160:2069–2084. doi: 10.1111/j.1476-5381.2010.00866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blas-Garcia A, Apostolova N, Ballesteros D, Monleon D, Morales JM, Rocha M, Victor VM, Esplugues JV. Inhibition of Mitochondrial Function by Efavirenz Increases Lipid Content in Hepatic Cells. Hepatology. 2010;52:115–125. doi: 10.1002/hep.23647. [DOI] [PubMed] [Google Scholar]
- 51.Guo YX, Xu XF, Zhang QZ, Li C, Deng Y, Jiang P, He LY, Peng WX. The Inhibition of Hepatic Bile Acids Transporters Ntcp and Bsep Is Involved in the Pathogenesis of Isoniazid/Rifampicin-Induced Hepatotoxicity. Toxicol Mech Methods. 2015:1–6. doi: 10.3109/15376516.2015.1033074. [DOI] [PubMed] [Google Scholar]
- 52.Hoffmann CJ, Charalambous S, Thio CL, Martin DJ, Pemba L, Fielding KL, Churchyard GJ, Chaisson RE, Grant AD. Hepatotoxicity in an African Antiretroviral Therapy Cohort: The Effect of Tuberculosis and Hepatitis B. AIDS. 2007;21:1301–1308. doi: 10.1097/QAD.0b013e32814e6b08. [DOI] [PubMed] [Google Scholar]
- 53.Li F, Lu J, Cheng J, Wang L, Matsubara T, Csanaky IL, Klaassen CD, Gonzalez FJ, Ma X. Human Pxr Modulates Hepatotoxicity Associated with Rifampicin and Isoniazid Co-Therapy. Nat Med. 2013;19:418–420. doi: 10.1038/nm.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki A, Yuen NA, Ilic K, Miller RT, Reese MJ, Brown HR, Ambroso JI, Falls JG, Hunt CM. Comedications Alter Drug-Induced Liver Injury Reporting Frequency: Data Mining in the Who Vigibase. Regul Toxicol Pharmacol. 2015;72:481–490. doi: 10.1016/j.yrtph.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weber WW, Hein DW. N-Acetylation Pharmacogenetics. Pharmacol Rev. 1985;37:25–79. [PubMed] [Google Scholar]
- 56.McDonagh EM, Boukouvala S, Aklillu E, Hein DW, Altman RB, Klein TE. Pharmgkb Summary: Very Important Pharmacogene Information for N-Acetyltransferase 2. Pharmacogenet Genomics. 2014;24:409–425. doi: 10.1097/FPC.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stanley LA, Sim E. Update on the Pharmacogenetics of Nats: Structural Considerations. Pharmacogenomics. 2008;9:1673–1693. doi: 10.2217/14622416.9.11.1673. [DOI] [PubMed] [Google Scholar]
- 58.Vatsis KP, Weber WW, Bell DA, Dupret JM, Evans DA, Grant DM, Hein DW, Lin HJ, Meyer UA, Relling MV, et al. Nomenclature for N-Acetyltransferases. Pharmacogenetics. 1995;5:1–17. doi: 10.1097/00008571-199502000-00001. [DOI] [PubMed] [Google Scholar]
- 59.Sabbagh A, Darlu P, Crouau-Roy B, Poloni ES. Arylamine N-Acetyltransferase 2 (Nat2) Genetic Diversity and Traditional Subsistence: A Worldwide Population Survey. PLoS One. 2011;6:e18507. doi: 10.1371/journal.pone.0018507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kinzig-Schippers M, Tomalik-Scharte D, Jetter A, Scheidel B, Jakob V, Rodamer M, Cascorbi I, Doroshyenko O, Sorgel F, Fuhr U. Should We Use N-Acetyltransferase Type 2 Genotyping to Personalize Isoniazid Doses? Antimicrob Agents Chemother. 2005;49:1733–1738. doi: 10.1128/AAC.49.5.1733-1738.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Timbrell JA, Scales MD, Streeter AJ. Studies on Hydrazine Hepatotoxicity. 2. Biochemical Findings. J Toxicol Environ Health. 1982;10:955–968. doi: 10.1080/15287398209530309. [DOI] [PubMed] [Google Scholar]
- 62.Chamorro JG, Castagnino JP, Musella RM, Nogueras M, Aranda FM, Frias A, Visca M, Aidar O, Peres S, de Larranaga GF. Sex, Ethnicity, and Slow Acetylator Profile Are the Major Causes of Hepatotoxicity Induced by Antituberculosis Drugs. J Gastroenterol Hepatol. 2013;28:323–328. doi: 10.1111/jgh.12069. [DOI] [PubMed] [Google Scholar]
- 63.Costa GN, Magno LA, Santana CV, Konstantinovas C, Saito ST, Machado M, Di Pietro G, Bastos-Rodrigues L, Miranda DM, De Marco LA, et al. Genetic Interaction between Gstm1 Gstt1 Cyp2e1, and Environmental Factors Is Associated with Adverse Reactions to Anti-Tuberculosis Drugs. Mol Diagn Ther. 2012;16:241–250. doi: 10.1007/BF03262213. [DOI] [PubMed] [Google Scholar]
- 64.Possuelo LG, Castelan JA, de Brito TC, Ribeiro AW, Cafrune PI, Picon PD, Santos AR, Teixeira RL, Gregianini TS, Hutz MH, et al. Association of Slow N-Acetyltransferase 2 Profile and Anti-Tb Drug-Induced Hepatotoxicity in Patients from Southern Brazil. Eur J Clin Pharmacol. 2008;64:673–681. doi: 10.1007/s00228-008-0484-8. [DOI] [PubMed] [Google Scholar]
- 65.Teixeira RL, Morato RG, Cabello PH, Muniz LM, Moreira Ada S, Kritski AL, Mello FC, Suffys PN, Miranda AB, Santos AR. Genetic Polymorphisms of Nat2, Cyp2e1 and Gst Enzymes and the Occurrence of Antituberculosis Drug-Induced Hepatitis in Brazilian Tb Patients. Mem Inst Oswaldo Cruz. 2011;106:716–724. doi: 10.1590/s0074-02762011000600011. [DOI] [PubMed] [Google Scholar]
- 66.Santos NP, Callegari-Jacques SM, Ribeiro Dos, Santos AK, Silva CA, Vallinoto AC, Fernandes DC, de Carvalho DC, Santos SE, Hutz MH. N-Acetyl Transferase 2 and Cytochrome P450 2e1 Genes and Isoniazid-Induced Hepatotoxicity in Brazilian Patients. Int J Tuberc Lung Dis. 2013;17:499–504. doi: 10.5588/ijtld.12.0645. [DOI] [PubMed] [Google Scholar]
- 67.An HR, Wu XQ, Wang ZY, Zhang JX, Liang Y. Nat2 and Cyp2e1 Polymorphisms Associated with Antituberculosis Drug-Induced Hepatotoxicity in Chinese Patients. Clin Exp Pharmacol Physiol. 2012;39:535–543. doi: 10.1111/j.1440-1681.2012.05713.x. [DOI] [PubMed] [Google Scholar]
- 68.Cho HJ, Koh WJ, Ryu YJ, Ki CS, Nam MH, Kim JW, Lee SY. Genetic Polymorphisms of Nat2 and Cyp2e1 Associated with Antituberculosis Drug-Induced Hepatotoxicity in Korean Patients with Pulmonary Tuberculosis. Tuberculosis (Edinb) 2007;87:551–556. doi: 10.1016/j.tube.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 69.Ho HT, Wang TH, Hsiong CH, Perng WC, Wang NC, Huang TY, Jong YJ, Lu PL, Hu OY. The Nat2 Tag Snp Rs1495741 Correlates with the Susceptibility of Antituberculosis Drug-Induced Hepatotoxicity. Pharmacogenet Genomics. 2013;23:200–207. doi: 10.1097/FPC.0b013e32835e95e1. [DOI] [PubMed] [Google Scholar]
- 70.Huang YS, Chern HD, Su WJ, Wu JC, Lai SL, Yang SY, Chang FY, Lee SD. Polymorphism of the N-Acetyltransferase 2 Gene as a Susceptibility Risk Factor for Antituberculosis Drug-Induced Hepatitis. Hepatology. 2002;35:883–889. doi: 10.1053/jhep.2002.32102. [DOI] [PubMed] [Google Scholar]
- 71.Lee SW, Chung LS, Huang HH, Chuang TY, Liou YH, Wu LS. Nat2 and Cyp2e1 Polymorphisms and Susceptibility to First-Line Anti-Tuberculosis Drug-Induced Hepatitis. Int J Tuberc Lung Dis. 2010;14:622–626. [PubMed] [Google Scholar]
- 72.Ohno M, Yamaguchi I, Yamamoto I, Fukuda T, Yokota S, Maekura R, Ito M, Yamamoto Y, Ogura T, Maeda K, et al. Slow N-Acetyltransferase 2 Genotype Affects the Incidence of Isoniazid and Rifampicin-Induced Hepatotoxicity. Int J Tuberc Lung Dis. 2000;4:256–261. [PubMed] [Google Scholar]
- 73.Shimizu Y, Dobashi K, Mita Y, Endou K, Moriya S, Osano K, Koike Y, Higuchi S, Yabe S, Utsugi M, et al. DNA Microarray Genotyping of N-Acetyltransferase 2 Polymorphism Using Carbodiimide as the Linker for Assessment of Isoniazid Hepatotoxicity. Tuberculosis (Edinb) 2006;86:374–381. doi: 10.1016/j.tube.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 74.Xiang Y, Ma L, Wu W, Liu W, Li Y, Zhu X, Wang Q, Ma J, Cao M, Wang Q, et al. The Incidence of Liver Injury in Uyghur Patients Treated for Tb in Xinjiang Uyghur Autonomous Region, China, and Its Association with Hepatic Enzyme Polymorphisms Nat2, Cyp2e1, Gstm1 and Gstt1. PLoS One. 2014;9:e85905. doi: 10.1371/journal.pone.0085905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim SH, Kim SH, Bahn JW, Kim YK, Chang YS, Shin ES, Kim YS, Park JS, Kim BH, Jang IJ, et al. Genetic Polymorphisms of Drug-Metabolizing Enzymes and Anti-Tb Drug-Induced Hepatitis. Pharmacogenomics. 2009;10:1767–1779. doi: 10.2217/pgs.09.100. [DOI] [PubMed] [Google Scholar]
- 76.Bose PD, Sarma MP, Medhi S, Das BC, Husain SA, Kar P. Role of Polymorphic N-Acetyl Transferase2 and Cytochrome P4502e1 Gene in Antituberculosis Treatment-Induced Hepatitis. J Gastroenterol Hepatol. 2011;26:312–318. doi: 10.1111/j.1440-1746.2010.06355.x. [DOI] [PubMed] [Google Scholar]
- 77.Mishra S, Daschakraborty S, Shukla P, Kapoor P, Aggarwal R. N-Acetyltransferase and Cytochrome P450 2e1 Gene Polymorphisms and Susceptibility to Antituberculosis Drug Hepatotoxicity in an Indian Population. Natl Med J India. 2013;26:260–265. [PubMed] [Google Scholar]
- 78.Gupta VH, Amarapurkar DN, Singh M, Sasi P, Joshi JM, Baijal R, Ramegowda PH, Amarapurkar AD, Joshi K, Wangikar PP. Association of N-Acetyltransferase 2 and Cytochrome P450 2e1 Gene Polymorphisms with Antituberculosis Drug-Induced Hepatotoxicity in Western India. J Gastroenterol Hepatol. 2013;28:1368–1374. doi: 10.1111/jgh.12194. [DOI] [PubMed] [Google Scholar]
- 79.Khalili H, Fouladdel S, Sistanizad M, Hajiabdolbaghi M, Azizi E. Association of N-Acetyltransferase-2 Genotypes and Anti-Tuberculosis Induced Liver Injury; First Case-Controlled Study from Iran. Curr Drug Saf. 2011;6:17–22. doi: 10.2174/157488611794479946. [DOI] [PubMed] [Google Scholar]
- 80.Bozok Cetintas V, Erer OF, Kosova B, Ozdemir I, Topcuoglu N, Aktogu S, Eroglu Z. Determining the Relation between N-Acetyltransferase-2 Acetylator Phenotype and Antituberculosis Drug Induced Hepatitis by Molecular Biologic Tests. Tuberk Toraks. 2008;56:81–86. [PubMed] [Google Scholar]
- 81.Ben Mahmoud L, Ghozzi H, Kamoun A, Hakim A, Hachicha H, Hammami S, Sahnoun Z, Zalila N, Makni H, Zeghal K. Polymorphism of the N-Acetyltransferase 2 Gene as a Susceptibility Risk Factor for Antituberculosis Drug-Induced Hepatotoxicity in Tunisian Patients with Tuberculosis. Pathol Biol (Paris) 2012;60:324–330. doi: 10.1016/j.patbio.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 82.Leiro-Fernandez V, Valverde D, Vazquez-Gallardo R, Botana-Rial M, Constenla L, Agundez JA, Fernandez-Villar A. N-Acetyltransferase 2 Polymorphisms and Risk of Anti-Tuberculosis Drug-Induced Hepatotoxicity in Caucasians. Int J Tuberc Lung Dis. 2011;15:1403–1408. doi: 10.5588/ijtld.10.0648. [DOI] [PubMed] [Google Scholar]
- 83.Lv X, Tang S, Xia Y, Zhang Y, Wu S, Yang Z, Li X, Tu D, Chen Y, Deng P, et al. Nat2 Genetic Polymorphisms and Anti-Tuberculosis Drug-Induced Hepatotoxicity in Chinese Community Population. Ann Hepatol. 2012;11:700–707. [PubMed] [Google Scholar]
- 84.Roy B, Ghosh SK, Sutradhar D, Sikdar N, Mazumder S, Barman S. Predisposition of Antituberculosis Drug Induced Hepatotoxicity by Cytochrome P450 2e1 Genotype and Haplotype in Pediatric Patients. J Gastroenterol Hepatol. 2006;21:784–786. doi: 10.1111/j.1440-1746.2006.04197.x. [DOI] [PubMed] [Google Scholar]
- 85.Vuilleumier N, Rossier MF, Chiappe A, Degoumois F, Dayer P, Mermillod B, Nicod L, Desmeules J, Hochstrasser D. Cyp2e1 Genotype and Isoniazid-Induced Hepatotoxicity in Patients Treated for Latent Tuberculosis. Eur J Clin Pharmacol. 2006;62:423–429. doi: 10.1007/s00228-006-0111-5. [DOI] [PubMed] [Google Scholar]
- 86.Yamada S, Tang M, Richardson K, Halaschek-Wiener J, Chan M, Cook VJ, Fitzgerald JM, Elwood RK, Brooks-Wilson A, Marra F. Genetic Variations of Nat2 and Cyp2e1 and Isoniazid Hepatotoxicity in a Diverse Population. Pharmacogenomics. 2009;10:1433–1445. doi: 10.2217/pgs.09.66. [DOI] [PubMed] [Google Scholar]
- 87.Azuma J, Ohno M, Kubota R, Yokota S, Nagai T, Tsuyuguchi K, Okuda Y, Takashima T, Kamimura S, Fujio Y, et al. Nat2 Genotype Guided Regimen Reduces Isoniazid-Induced Liver Injury and Early Treatment Failure in the 6-Month Four-Drug Standard Treatment of Tuberculosis: A Randomized Controlled Trial for Pharmacogenetics-Based Therapy. Eur J Clin Pharmacol. 2013;69:1091–1101. doi: 10.1007/s00228-012-1429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patillon B, Luisi P, Poloni ES, Boukouvala S, Darlu P, Genin E, Sabbagh A. A Homogenizing Process of Selection Has Maintained an “Ultra-Slow” Acetylation Nat2 Variant in Humans. Hum Biol. 2014;86:185–214. doi: 10.13110/humanbiology.86.3.0185. [DOI] [PubMed] [Google Scholar]
- 89.Selinski S, Blaszkewicz M, Ickstadt K, Hengstler JG, Golka K. Refinement of the Prediction of N-Acetyltransferase 2 (Nat2) Phenotypes with Respect to Enzyme Activity and Urinary Bladder Cancer Risk. Arch Toxicol. 2013;87:2129–2139. doi: 10.1007/s00204-013-1157-7. [DOI] [PubMed] [Google Scholar]
- 90.Garcia-Closas M, Hein DW, Silverman D, Malats N, Yeager M, Jacobs K, Doll MA, Figueroa JD, Baris D, Schwenn M, et al. A Single Nucleotide Polymorphism Tags Variation in the Arylamine N-Acetyltransferase 2 Phenotype in Populations of European Background. Pharmacogenet Genomics. 2011;21:231–236. doi: 10.1097/FPC.0b013e32833e1b54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang YS, Chern HD, Su WJ, Wu JC, Chang SC, Chiang CH, Chang FY, Lee SD. Cytochrome P450 2e1 Genotype and the Susceptibility to Antituberculosis Drug-Induced Hepatitis. Hepatology. 2003;37:924–930. doi: 10.1053/jhep.2003.50144. [DOI] [PubMed] [Google Scholar]
- 92.Wang T, Yu HT, Wang W, Pan YY, He LX, Wang ZY. Genetic Polymorphisms of Cytochrome P450 and Glutathione S-Transferase Associated with Antituberculosis Drug-Induced Hepatotoxicity in Chinese Tuberculosis Patients. J Int Med Res. 2010;38:977–986. doi: 10.1177/147323001003800324. [DOI] [PubMed] [Google Scholar]
- 93.Tang SW, Lv XZ, Zhang Y, Wu SS, Yang ZR, Xia YY, Tu DH, Deng PY, Ma Y, Chen DF, Zhan SY. Cyp2e1, Gstm1 and Gstt1 Genetic Polymorphisms and Susceptibility to Antituberculosis Drug-Induced Hepatotoxicity: A Nested Case-Control Study. J Clin Pharm Ther. 2012;37:588–593. doi: 10.1111/j.1365-2710.2012.01334.x. [DOI] [PubMed] [Google Scholar]
- 94.Leiro-Fernandez V, Valverde D, Vazquez-Gallardo R, Constenla L, Fernandez-Villar A. Genetic Variations of Nat2 and Cyp2e1 and Isoniazid Hepatotoxicity in a Diverse Population. Pharmacogenomics. 2010;11:1205–1206. doi: 10.2217/pgs.10.109. author reply 1207–1208. [DOI] [PubMed] [Google Scholar]
- 95.Sheng YJ, Wu G, He HY, Chen W, Zou YS, Li Q, Zhong L, Huang YM, Deng CL. The Association between Cyp2e1 Polymorphisms and Hepatotoxicity Due to Anti-Tuberculosis Drugs: A Meta-Analysis. Infect Genet Evol. 2014;24:34–40. doi: 10.1016/j.meegid.2014.01.034. [DOI] [PubMed] [Google Scholar]
- 96.Knockaert L, Fromenty B, Robin MA. Mechanisms of Mitochondrial Targeting of Cytochrome P450 2e1: Physiopathological Role in Liver Injury and Obesity. FEBS J. 2011;278:4252–4260. doi: 10.1111/j.1742-4658.2011.08357.x. [DOI] [PubMed] [Google Scholar]
- 97.Lieber CS. Cytochrome P-4502e1: Its Physiological and Pathological Role. Physiol Rev. 1997;77:517–544. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- 98.Sutti S, Rigamonti C, Vidali M, Albano E. Cyp2e1 Autoantibodies in Liver Diseases. Redox Biol. 2014;3:72–78. doi: 10.1016/j.redox.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Park KS, Sohn DH, Veech RL, Song BJ. Translational Activation of Ethanol-Inducible Cytochrome P450 (Cyp2e1) by Isoniazid. Eur J Pharmacol. 1993;248:7–14. doi: 10.1016/0926-6917(93)90019-m. [DOI] [PubMed] [Google Scholar]
- 100.Yue J, Peng RX, Yang J, Kong R, Liu J. Cyp2e1 Mediated Isoniazid-Induced Hepatotoxicity in Rats. Acta Pharmacol Sin. 2004;25:699–704. [PubMed] [Google Scholar]
- 101.Daly AK. Drug-Induced Liver Injury: Past, Present and Future. Pharmacogenomics. 2010;11:607–611. doi: 10.2217/pgs.10.24. [DOI] [PubMed] [Google Scholar]
- 102.Huang YS, Su WJ, Huang YH, Chen CY, Chang FY, Lin HC, Lee SD. Genetic Polymorphisms of Manganese Superoxide Dismutase, Nad(P)H:Quinone Oxidoreductase, Glutathione S-Transferase M1 and T1, and the Susceptibility to Drug-Induced Liver Injury. J Hepatol. 2007;47:128–134. doi: 10.1016/j.jhep.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 103.Gupta VH, Singh M, Amarapurkar DN, Sasi P, Joshi JM, Baijal R, H RP, Amarapurkar AD, Joshi K, Wangikar PP. Association of Gst Null Genotypes with Anti-Tuberculosis Drug Induced Hepatotoxicity in Western Indian Population. Ann Hepatol. 2013;12:959–965. [PubMed] [Google Scholar]
- 104.Chatterjee S, Lyle N, Mandal A, Kundu S. Gstt1 and Gstm1 Gene Deletions Are Not Associated with Hepatotoxicity Caused by Antitubercular Drugs. J Clin Pharm Ther. 2010;35:465–470. doi: 10.1111/j.1365-2710.2009.01101.x. [DOI] [PubMed] [Google Scholar]
- 105.Kim SH, Kim SH, Yoon HJ, Shin DH, Park SS, Kim YS, Park JS, Jee YK. Gstt1 and Gstm1 Null Mutations and Adverse Reactions Induced by Antituberculosis Drugs in Koreans. Tuberculosis (Edinb) 2010;90:39–43. doi: 10.1016/j.tube.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 106.Leiro V, Fernandez-Villar A, Valverde D, Constenla L, Vazquez R, Pineiro L, Gonzalez-Quintela A. Influence of Glutathione S-Transferase M1 and T1 Homozygous Null Mutations on the Risk of Antituberculosis Drug-Induced Hepatotoxicity in a Caucasian Population. Liver Int. 2008;28:835–839. doi: 10.1111/j.1478-3231.2008.01700.x. [DOI] [PubMed] [Google Scholar]
- 107.Daly AK. Pharmacogenomics of Adverse Drug Reactions. Genome medicine. 2013;5:5. doi: 10.1186/gm409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nanashima K, Mawatari T, Tahara N, Higuchi N, Nakaura A, Inamine T, Kondo S, Yanagihara K, Fukushima K, Suyama N, et al. Genetic Variants in Antioxidant Pathway: Risk Factors for Hepatotoxicity in Tuberculosis Patients. Tuberculosis (Edinb) 2012;92:253–259. doi: 10.1016/j.tube.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 109.Sharma SK, Balamurugan A, Saha PK, Pandey RM, Mehra NK. Evaluation of Clinical and Immunogenetic Risk Factors for the Development of Hepatotoxicity During Antituberculosis Treatment. Am J Respir Crit Care Med. 2002;166:916–919. doi: 10.1164/rccm.2108091. [DOI] [PubMed] [Google Scholar]
- 110.Bothamley GH. Treatment Tuberculosis Human Leukocyte Antigen. Am J Respir Crit Care Med. 2002;166:907–908. doi: 10.1164/rccm.2207001. [DOI] [PubMed] [Google Scholar]
- 111.Cai Y, Yi J, Zhou C, Shen X. Pharmacogenetic Study of Drug-Metabolising Enzyme Polymorphisms on the Risk of Anti-Tuberculosis Drug-Induced Liver Injury: A Meta-Analysis. PLoS One. 2012;7:e47769. doi: 10.1371/journal.pone.0047769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sun F, Chen Y, Xiang Y, Zhan S. Drug-Metabolising Enzyme Polymorphisms and Predisposition to Anti-Tuberculosis Drug-Induced Liver Injury: A Meta-Analysis. Int J Tuberc Lung Dis. 2008;12:994–1002. [PubMed] [Google Scholar]
- 113.Wang PY, Xie SY, Hao Q, Zhang C, Jiang BF. Nat2 Polymorphisms and Susceptibility to Anti-Tuberculosis Drug-Induced Liver Injury: A Meta-Analysis. Int J Tuberc Lung Dis. 2012;16:589–595. doi: 10.5588/ijtld.11.0377. [DOI] [PubMed] [Google Scholar]
- 114.Li C, Long J, Hu X, Zhou Y. Gstm1 and Gstt1 Genetic Polymorphisms and Risk of Anti-Tuberculosis Drug-Induced Hepatotoxicity: An Updated Meta-Analysis. Eur J Clin Microbiol Infect Dis. 2013;32:859–868. doi: 10.1007/s10096-013-1831-y. [DOI] [PubMed] [Google Scholar]