

Abstract
The very impressive clinical results recently obtained in cancer patients treated with immune response checkpoint inhibitors boosted the interest in immunotherapy as a therapeutic choice in cancer treatment. However, these inhibitors require a pre-existing tumor specific immune response and the presence of tumor infiltrating T cells to be efficient. This immune response can be triggered by cancer vaccines. One of the main issues in tumor vaccination is the choice of the right antigen to target. All vaccines tested to date targeted tumor associated antigens (TAA) that are self-antigens and failed to show a clinical efficacy because of the immune self-tolerance to TAA. A new class of tumor antigens has recently been described, the neo-antigens that are created by point mutations of tumor expressing proteins and are recognized by the immune system as non-self. Neo-antigens exhibit two main properties: they are not involved in the immune self-tolerance process and are immunogenic. However, the majority of the neo-antigens are patient specific and their use as cancer vaccines requires their previous identification in each patient individualy that can be done only in highly specialized research centers. It is therefore evident that neo-antigens cannot be used for patient vaccination worldwide. This raises the question of whether we can find neo-antigen like vaccines, which would not be patient specific. In this review we show that optimized cryptic peptides from TAA are neo-antigen like peptides. Optimized cryptic peptides are recognized by the immune system as non-self because they target self-cryptic peptides that escape self-tolerance; in addition they are strongly immunogenic because their sequence is modified in order to enhance their affinity for the HLA molecule. The first vaccine based on the optimized cryptic peptide approach, Vx-001, which targets the widely expressed tumor antigen telomerase reverse transcriptase (TERT), has completed a large phase I clinical study and is currently being tested in a randomized phase II trial in non-small cell lung cancer (NSCLC) patients.
Keywords: Tumor associated antigens (TAA), tumor specific antigens (TSA), telomerase reverse transcriptase (TERT)
The recent advances in cancer treatment with immune response checkpoint inhibitors demonstrated that immunotherapy may play a key role in cancer therapy. Immune response checkpoint inhibitors are very efficient in patients with tumors highly infiltrated with tumor specific T cells. These tumor specific T cells can be triggered in non-immunogenic tumors or further amplified in immunogenic tumors by tumor vaccines.
The central problem in tumor vaccination is the choice of the optimal antigen to target.
Tumor antigens may be distinguished into two distinct groups:
The tumor associated antigens (TAA) that are normal proteins expressed by tumors but also by normal cells (self-antigens). Their very frequent expression by tumors made them very appealing candidates for broad-spectrum tumor vaccination. However, to date all the vaccines targeting TAA, which were tested in clinical studies, failed to show clinical efficacy. This is due to the fact that the immune system is tolerant to TAA because they are self-antigens (self-tolerance). Tolerance to TAA leads to the elimination or inactivation of all the TAA-specific high-affinity T cells that are the most efficient in inducing antitumor immunity;
The tumor specific antigens (TSA) that are abnormal proteins harboring somatic mutations. These mutations are either responsible for tumor transformation and may be shared by different tumors (driver mutations) or are the byproduct of the genomic instability of tumors and very often are patient specific (passenger mutations) (1-3). Passenger mutations are the most common cancer mutations. All these mutations can create T cell reactive epitopes (neo-epitopes or neo-antigens) that are not involved in the self-tolerance process are recognized by the immune system as non-self and could therefore be efficient targets for cancer vaccines. However, the absence of immune tolerance, although necessary, is not sufficient for a neo-antigen to be an efficient target for a vaccine. Neo-antigens must have two additional properties. First, they must have a high affinity for the HLA molecules in order to be immunogenic and second, they must be naturally processed by the proteasome/TAP machinery and presented at the surface of tumor cells. There are two functionally distinct locations of an immunogenic mutation in the sequence of a neo-antigen. Mutations may be located at the region that interacts with the TCR (TCR-contact mutation) creating a new non-self antigenic specificity. In this case the WT counterpart must exhibit high HLA affinity in order for the neo-antigen to be immunogenic. Alternatively they may be located at the region that interacts with the HLA and enhance the HLA affinity (HLA-contact mutation). In this latter case the mutation must result in an enhancement of the constitutively low affinity of the WT peptide for the HLA, that is the basis for this WT peptide to be ignored by the immune system thereby disallowing the self-tolerance process. There is now a body of evidence that neo-antigens may play an important role in tumor evolution and might be efficient cancer vaccines. Indeed, naturally occurring T cells infiltrating tumors are directed towards neo-antigens (4). The presence of these tumor infiltrating T cells (TILs) is associated with increased survival (5-8). Moreover, the number and the frequency of TILs are related to the number of mutations that in its turn defines the number of detectable neo-antigens (9). Microsatellite unstable colorectal cancers that accumulate mutations within DNA repeat sequences, because of defects of the DNA mismatch repair system, are known to have a better prognosis and a more dense intratumor T cell infiltration than microsatellite stable colorectal cancers (10). Finally, patients with at least one predictable immunogenic mutation show increased overall survival (9). The efficacy of neo-antigen based vaccines has been demonstrated in preclinical models in mice and in clinical studies in humans. Vaccination of mice with two Reps1 and Adpgk derived neo-antigens induced a protective tumor immunity, as strong as the immune checkpoint inhibitors (11). In humans adoptive transfer of TILs containing a high number of CD4 cells specific for a class II restricted neo-antigen led to partial regression of lung metastases in patients with cholangiocarcinoma (12). In another study transfer of TILs containing T cells specific for a melanoma neo-antigen led to a complete response (13). Finally, neo-antigen loaded dendritic cells induced a strong immune response in three melanoma patients (14). The presence of neo-antigens is also related to the efficacy of the immune response checkpoint inhibitors (15). Immune response checkpoint inhibitors are more efficient in tumors with high mutation load and presence of TILs that recognize neo-antigens. The higher the mutation load the higher the frequency of neo-antigen reactive TILs and the stronger the response to these inhibitors. In fact it has been suggested that predicted immunogenic neo-antigens correlate with clinical benefit from immune response checkpoint inhibitor treatment (16).
One of the main issues of the use of neo-antigens is their identification. Whole exome sequencing coupled with either mass spectrometry or computational predictive models for HLA-I binding affinity and proteasome cleavage followed by in vitro validation are the two main approaches used to date. Using these approaches some potentially immunogenic neo-antigens have been identified both in mouse and human tumors. For instance, from 1,290 mutations in the MC-38 murine tumor, seven neo-antigens were identified by mass spectrometry but only two of them were immunogenic (17). Moreover, in the MCA murine tumor two potentially immunogenic neo-antigens were identified from 2,200 point mutations, using MHC binding affinity and proteasome cleavage predictive models (11). In humans potentially immunogenic mutations were detected in only 181 of the 515 tested patients (median of 3 mutations per patient) (9). Finally, two neo-antigens were identified by mass spectrometry among 1,019 mutations in melanoma patients (18). From these studies it would appear that immunogenic mutations are relatively rare and a small minority of total mutations (9). However, this conclusion was not confirmed by two studies showing that neo-antigens are present in 9 out of 10 gastrointestinal cancers and that in breast and colorectal cancer a new neo-antigen could be generated for every ten mutations (19,20). The relative rarity of potentially immunogenic neo-antigens is easily explained. For TCR-contact mutations the WT counterpart must already exhibit a high HLA affinity and the mutation must be a non-conservative amino acid substitution in order to create completely new non-self antigenic specificity. Scanning of an antigen, using HLA-I binding affinity predictive models, reveals a relatively low number of peptides with a predicted high HLA-I binding affinity. This is particularly true for peptides bound to HLA-I molecules that require the peptide to have quite rare residues at primary anchor positions in order to bind with high affinity (for example peptides bound to HLA-B7 that must have only a proline in the primary anchor position 2). For the HLA-contact mutations, these mutations must be located at the primary or sometimes at the secondary anchor positions and must introduce a favorable amino acid to replace a non-favorable one. Given the limited number of favorable amino acids, especially for some HLA-I molecules, the frequency of putative immunogenic neo-antigens must be very low. Moreover, these mutations, especially those that concern the secondary anchor motifs, must preserve the conformation of the peptide region that interacts with the TCR. This is particularly important in order to target not only mutation harboring tumor cells but also tumor cells that may miss the mutation that generated the neo-antigen.
Although neo-antigens seem to be extremely promising for developing efficient cancer vaccines several issues should be addressed.
Their use may be limited by the genetic heterogeneity of tumors. The analysis of the cancer genome has revealed that tumor mutation landscape is extremely variable among patients, among different lesions of the same tumor and even among different regions of the same lesion (3,21). This means that it is likely that neo-antigens are not expressed by all the lesions of the same tumor. Neo-antigen-free lesions could therefore escape neo-antigen induced immune responses. This is particularly true for neo-antigens generated by TCR-contact mutations. Neo-antigen specific T cells could not recognize the WT counterpart expressed in the neo-antigen-free lesions. This issue may not concern HLA-contact mutations, provided that the WT counterpart is naturally processed by tumor cells;
Their identification is time consuming and may be very expensive. Although significant progress was recently done in genome sequence, neo-antigen identification still requires several weeks when it is done in fully specialized research centers. Moreover, it requires adequate biopsy material that is not always available. This material can easily be obtained from accessible tumors, such as melanoma, but much less easily from other tumors, especially those where diagnosis in several clinical centers is based on cytology material containing few tumor cells. Moreover, neo-antigen identification requires fully qualified laboratories near to the bed of the patient. Vaccination with patient specific neo-antigens might therefore be a treatment that would be applied only in specialized centers, far from being a standard treatment given worldwide. The commercial failure of the prostate cancer vaccine Sipuleucel-T that uses the patient own dendritic cells shows that this highly personalized treatment approach may face some non-scientific barriers that are difficult to overcome. Another issue deals with their clinical development. Since neo-antigens are patient specific and can only be used for the treatment of the neo-antigen expressing patients, neo-antigen based vaccines can be considered to be an anticancer treatment approach rather than an anticancer drug. There will be as many different vaccines as the patients to be treated. It will be virtually impossible to evaluate potential toxicity and efficacy of the patient-specific vaccines before their approval as cancer treatments. And it will therefore be necessary to revise the classical drug clinical development and put in place new clinical development models adapted to these new cancer treatments.
The complexity of these challenges raises the following question: can we find neo-antigen like vaccines that share all the neo-antigen properties (escaping self-tolerance and immunogenicity) but are not confounded by the issues discussed above?
Optimized cryptic peptides
The various peptides derived from the processing of a given antigen are not equally presented by antigen presenting cells. Two classes of peptides can be defined: the immunodominant and the cryptic peptides. For peptides presented by the HLA-I molecules, this distinction depends mainly on their affinity for the HLA-I (22). Immunodominant peptides exhibit a high affinity and form stable peptide/HLA-I complexes, while cryptic peptides exhibit a low affinity and form unstable peptide/HLA-I complexes. Given the strong correlation between HLA-I affinity and immunogenicity, immunodominant peptides are immunogenic, while cryptic peptides are not (23). We and others have previously demonstrated that dominant and cryptic peptides from self-proteins are not equally involved in the self-tolerance process. Tolerance to self antigens, such as TAA, concerns immunodominant rather than cryptic peptides (24-34). As a consequence the T cell repertoire that is available to be stimulated by TAA-derived immunodominant peptides is limited and composed mainly by T cells with low affinity for the antigen (Figure 1). Immunodominant TAA-derived peptides, although potentially immunogenic because of their high HLA-I affinities, are not highly immunogenic because the specific T cells repertoire has been rendered tolerant.
Figure 1.

Immune self-tolerance (clonal T cell deletion in the thymus) to immunodominant and cryptic self-peptides.
The inability of immunodominant TAA-derived peptides to induce an antitumor immunity has been demonstrated in several preclinical models. We have previously shown that vaccination of HLA-A*0201 transgenic mice with two immunodominant HLA-A2 restricted peptides derived from the TERT TAA failed to induce an antitumor protective immunity (Figure 2). Similar results were obtained with two immunodominant peptides derived from the HER-2/neu TAA (Figure 2) (28). These studies came as a confirmation of the claim made by others that an efficient antitumor immunity cannot be induced by immunodominant peptides when using different mouse models (35-41). This also explains why all cancer vaccines tested in clinical studies to date failed to show clinical benefit. All of these vaccines targeted TAA immunodominant peptides.
Figure 2.

HLA-A*0201 transgenic HHD mice were vaccinated with two immunodominant HER-2/neu-derived and two immunodominant TERT-derived peptides. Seven days later TERT vaccinated mice were grafter with HLA-A*0201 expressing EL-4HHD tumor cells and HER-2/neu vaccinated mice were grafted with HLA-A*0201 and HER-2/neu expressing EL-4HHD/HER-2 tumor cells.
In contrast to immunodominant peptides, cryptic peptides escape, completely or partially, self-tolerance. This was first shown by Cibotti et al. (24). Using a transgenic mouse expressing hen-egg lysozyme as self-antigen we showed that there is an hierarchy in the tolerance to self-peptides, and that tolerance to subdominant/cryptic peptides is partial or completely absent. More recently we showed that HLA-A*0201 transgenic mice are not tolerant to HLA-A*0201 restricted cryptic peptides from the TERT (28). These observations were confirmed by others in different mouse models (31-34,42). This means that cryptic peptides from self-antigens are considered by the immune system as non-self. The absence of tolerance to cryptic self-peptides does not create any risk of autoimmunity, since cryptic peptides because of their non-immunogenicity are ignored by the immune system. This also means that the T cell repertoire specific for the TAA-derived cryptic peptides is fully available to be mobilized by cryptic peptide targeting vaccines and contains high affinity T cells (Figure 1). This is extremely important because an efficient antitumor immunity requires the stimulation of high affinity T cells.
Although TAA-derived cryptic peptides can be considered by the immune system as non-self, their use as cancer vaccines must overcome their non immunogenicity (23,43). Cryptic peptides are non immunogenic because they exhibit a low HLA-I affinity. Enhancement of their immunogenicity is a prerequisite for cryptic peptides to be used as cancer vaccines and such enhancement can be achieved by the improvement of their HLA-I affinity. It is well known that the affinity of the HLA-I/peptide interaction depends on the presence of favorable residues in well-defined positions called primary and secondary anchor positions. The simplest way to increase HLA-I affinity would be to introduce HLA-I binding favorable amino acids in the primary and secondary anchor positions of the cryptic peptide sequence. Primary anchor positions that play a major role in defining the strength of the HLA-I/peptide interaction are located in the two extremities of the peptide (position 2 and position 9/10). This is true for peptides bound to almost all HLA molecules. In contrast the favorable residues in these positions are HLA-I specific. As an example, high affinity peptides bound to HLA-A*0201 have a hydrophobic residue (M, L, I, V) in position 2 and L or V in position 9, while high affinity peptides bound to HLA-B*0702 have a proline in position 2 and a L in position 9. Secondary anchor motifs are more randomly located in the sequence of peptides bound to different HLA molecules. Increasing affinity for HLA-I by introducing sequence modifications is necessary in order to render cryptic peptides immunogenic (optimized cryptic peptides) but these sequence modifications must preserve the conformation of the peptide segment that interacts with the TCR. Only under this condition will T cells that are stimulated by the immunogenic optimized cryptic peptides recognize the WT counterparts that are presented by tumor cells. Our previous work with HLA-A*0201 restricted cryptic peptides showed that substitution of the first residue, whatever this residue might be, by a tyrosine enhances HLA-A*0201 binding affinity of almost all the HLA-A*0201 restricted cryptic peptides (43). This is illustrated in Figure 3. Forty-seven WT cryptic peptides from different TAA and their optimized variants were tested for their binding affinity for HLA-A*0201 and their immunogenicity. As expected all WT cryptic peptides exhibited a low binding affinity and were non-immunogenic. In contrast, 42 of the 47 optimized variants had a high affinity and were immunogenic. Importantly, T cells stimulated by the optimized cryptic variants cross-recognized the WT counterpart. Similar results were obtained with cryptic peptides presented by two other common HLA molecules, the HLA-B*0702 and the HLA-A*2402. In both cases, affinity enhancement required modifications of the peptide sequence in position 9 and/or in position 1 (data not shown).
Figure 3.
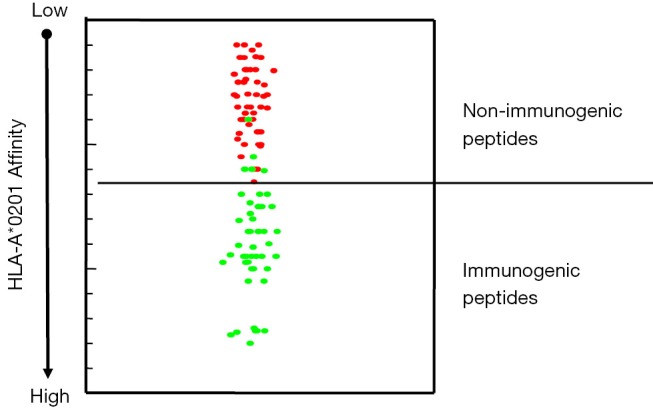
HLA-A*0201 binding affinity and immunogenicity of HLA-A*0201 restricted WT cryptic peptides and their optimized variants. Red spots: WT cryptic peptides. Green spots: optimized cryptic peptides.
Using an HLA-A*0201 expressing mouse model, we showed that vaccination with optimized cryptic peptides from TAA induces tumor immunity. In fact, mice vaccinated with four optimized cryptic peptides from two different TAA (HER-2/neu and TERT) developed protective tumor immunity (Figure 4) (28).
Figure 4.

HLA-A*0201 transgenic HHD mice were vaccinated with two optimized cryptic HER-2/neu-derived and two optimized cryptic TERT-derived peptides. Seven days later TERT vaccinated mice were grafter with HLA-A*0201 expressing EL-4HHD tumor cells and HER-2/neu vaccinated mice were grafted with HLA-A*0201 and HER-2/neu expressing EL-4HHD/HER-2 tumor cells.
Targeting cryptic tumor peptides by using heteroclitic or xenogeneic vaccines has been described by others (37-41,44-50). It is noteworthy that in the case of xenogeneic vaccines, the immune response was directed towards cryptic tumor peptides and was induced by naturally occurring optimized variants of the tumor cryptic peptides that are present in the sequence of the xenogeneic vaccines. Moreover, one of the two Db-restricted immunogenic neo-antigens identified from MC-38 tumors although resulting from an TCR-contact mutation (mutation in position 4 that has two anchor residues in its neighborhood, a primary anchor residue in position 5 and a secondary anchor residue in the position 3) has a significantly higher Db affinity than the WT counterpart that exhibits an intermediate Db affinity (17). Finally, three out of the 17 immunogenic neo-antigens identified in cancer patients appear to be natural optimized cryptic peptides. They exhibit a strong HLA-I affinity (two peptides for HLA-A*0301 and one for HLA-A*2402), while their WT counterpart had a very low affinity. In all three cases neo-antigens were created by an HLA-contact mutation (51).
In conclusion, optimized cryptic peptides from TAA had the two main properties of the neo-antigens: they escape self-immune tolerance and are immunogenic. They can therefore be considered as universal neo-antigen like peptides (Table 1).
Table 1. Properties of neo-antigens and TAA-derived optimized cryptic peptides.
| Properties | Neo-antigens | Optimized cryptic peptides from TAA |
|---|---|---|
| Escaping self-tolerance | Yes | Yes |
| Immunogenicity | Yes | Yes |
| Broad spectrum application | No | Yes |
TAA, tumor associated antigens.
Very importantly, optimized cryptic peptides from TAA not only share the properties of the neo-antigens but they are not affected by the two issues that can be barriers to the use of neo-antigens as cancer vaccines:
Their use is not limited by the genetic heterogeneity of tumors, especially if the TAA targeted by an optimized peptide based vaccine is a universal antigen involved in the tumorigenesis and/or the survival of tumor cells, such as TERT and survivin. All tumor cells express these universal antigens and the probability of antigen loss is relatively low;
They are not patient-specific and they do not need to be identified in each patient individually. Optimized cryptic peptides look more like classical anticancer drugs, which can undergo a classical clinical development leading to a broad-spectrum patient treatment.
Vx-001 vaccine
Vx-001 is the first tumor vaccine based on optimized cryptic peptides. It targets the universal tumor antigen TERT (TElomerase Reverse Transcriptase) that is expressed by more than 85% of tumors of different histology and origin (52-54) and can be used for the treatment of cancer patients expressing HLA-A*0201 (43–45% of the population). Vx-001 is composed of two peptides of nine amino acids: the WT cryptic TERT572 and its optimized variant TERT572Y. These two peptides are administered separately, along with the adjuvant Montanide ISA51®VG. The optimized immunogenic TERT572Y is given in the first two vaccinations, in order to trigger a large immune response. The WT TERT572 is given in the following vaccinations, in order to select among all the TERT572Y stimulated T cells those with the highest specificity for the WT TERT572 that is presented in the surface of tumor cells associated with the HLA-A*0201. This particular protocol was validated in mouse models and in a clinical study in cancer patients (55). Patients were vaccinated two times with the optimized TERT572Y and then separated into two groups. The first group received four additional vaccinations with the same optimized TERT572Y, while the second received four vaccinations with the WT TERT572. Comparison of the affinity of WT TERT572 specific T cells generated in these two groups revealed that affinity was higher in the patients that received two vaccinations with the optimized TERT572Y followed by four vaccinations with the WT TERT572. These results may explain previous results obtained with the gp100 209M peptide that showed that multiple vaccinations with the optimized gp100 209M peptide stimulates T cells that in their majority are unable to efficiently recognize the WT gp100 209 and gp100 expressing tumor cells (56-58).
Vx-001 has been tested in a large phase I clinical study with 116 patients with different types of tumors, mainly NSCLC, prostate, breast and colorectal cancer (59-61). Seventy percent of the patients had metastatic disease and 70% had progressive disease when entered the study. All patients but one (a hepatocellular carcinoma) had received at least one previous treatment. The results of this study showed that Vx-001 is safe, since only grade I/II vaccine related toxicities were observed, mainly local skin reactions at the site of injection. This reaction was due to the adjuvant Montanide ISA51®VG. They also showed that Vx-001 is highly immunogenic. An IFNg ELISpot assay was used for all vaccinated patients to detect WT TERT572 specific T cells. In some patients immune response was confirmed using TERT572Y specific tetramers and a perforin ELISpot assay. WT TERT572 specific T cells were detected after the second vaccination in half of the evaluable patients (62), while 68% of the evaluable vaccinated patients developed an immune response after the sixth vaccination. In some patients the response was maintained by boost vaccinations as long as five years after the first vaccination.
Vx-001 showed signs of clinical activity. Four objective responses (one complete and three partial) were observed in breast cancer, NSCLC and hepatocarcinoma patients. Very interestingly, the partial response observed in the metastatic hepatocarcinoma patient who received Vx-001 as first line treatment lasted five years and the patient progressed when the WT TERT572 specific T cells were no more detectable. Moreover, long lasting (more than six months) disease stabilization was observed in more than 40% of the NSCLC patients. Survival of the NSCLC patients who entered the study with disease control was 18 months, much longer than expected from the historical controls (Figure 5). Finally, we observed a correlation between immune and clinical response. NSCLC patients with a WT TERT572 specific immune response survived longer than patients who failed to mount such an immune response (Figure 6).
Figure 5.
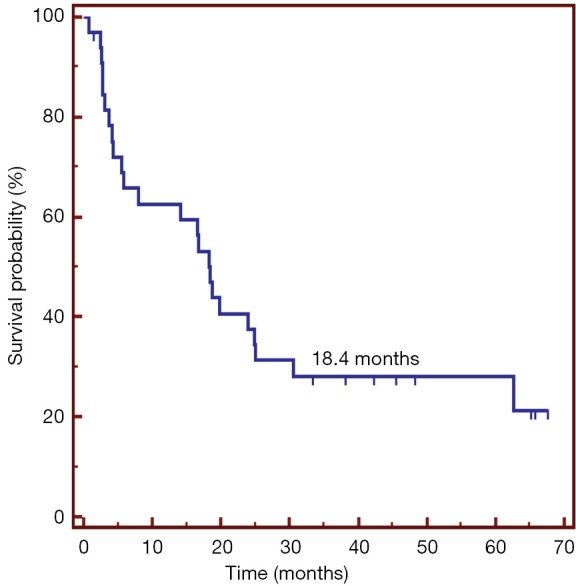
Survival of locally advanced and metastatic NSCLC patients who entered the Vx-001 phase I study with disease control. NSCLC, non-small cell lung cancer.
Figure 6.
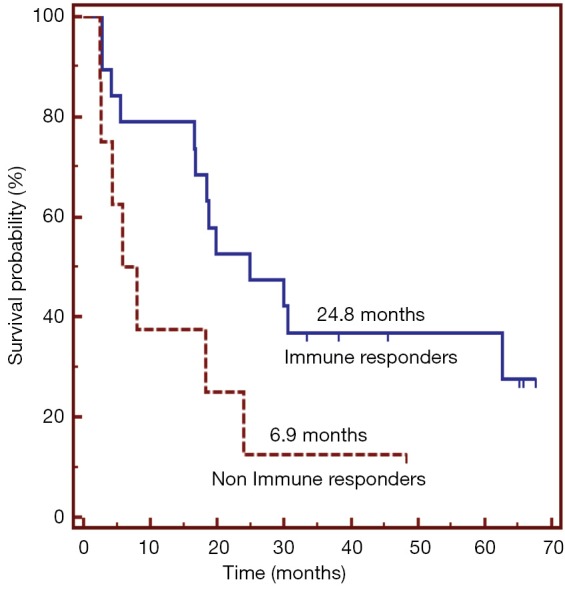
Correlation between immune and clinical response in locally advanced and metatstatic NSCLC patients treated with the Vx-001 vaccine. NSCLC, non-small cell lung cancer.
Vx-001 is currently tested in a randomized phase IIb clinical trial in metastatic or recurrent NSCLC patients who experienced disease control after four cycles of platinum based chemotherapy. Patients need to be HLA-A*0201 positive and their tumor must express TERT. The primary objective of the study is overall survival. Two hundred twenty one patients are randomized (1,400 patients are screened) in a 1:1 ratio in 70 sites located in eight European countries (63). The study is ongoing and is expected to be completed in September 2016.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflict of interest to declare.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57-70. 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science 2013;339:1546-58. 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lennerz V, Fatho M, Gentilini C, et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci U S A 2005;102:16013-8. 10.1073/pnas.0500090102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang WT, Adams SF, Tahirovic E, et al. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol 2012;124:192-8. 10.1016/j.ygyno.2011.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moudgil KD, Sercarz EE. The T cell repertoire against cryptic self determinants and its involvement in autoimmunity and cancer. Clin Immunol Immunopathol 1994;73:283-9. 10.1006/clin.1994.1200 [DOI] [PubMed] [Google Scholar]
- 7.Yamada N, Oizumi S, Kikuchi E, et al. CD8+ tumor-infiltrating lymphocytes predict favorable prognosis in malignant pleural mesothelioma after resection. Cancer Immunol Immunother 2010;59:1543-9. 10.1007/s00262-010-0881-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oble DA, Loewe R, Yu P, et al. Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in human melanoma. Cancer Immun 2009;9:3. [PMC free article] [PubMed] [Google Scholar]
- 9.Brown SD, Warren RL, Gibb EA, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014;24:743-50. 10.1101/gr.165985.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maby P, Tougeron D, Hamieh M, et al. Correlation between Density of CD8+ T-cell Infiltrate in Microsatellite Unstable Colorectal Cancers and Frameshift Mutations: A Rationale for Personalized Immunotherapy. Cancer Res 2015;75:3446-55. 10.1158/0008-5472.CAN-14-3051 [DOI] [PubMed] [Google Scholar]
- 11.Gubin MM, Artyomov MN, Mardis ER, et al. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 2015;125:3413-21. 10.1172/JCI80008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014;344:641-5. 10.1126/science.1251102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robbins PF, Lu YC, El-Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013;19:747-52. 10.1038/nm.3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carreno BM, Magrini V, Becker-Hapak M, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015;348:803-8. 10.1126/science.aaa3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Rooij N, van Buuren MM, Philips D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 2013;31:e439-42. 10.1200/JCO.2012.47.7521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189-99. 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yadav M, Jhunjhunwala S, Phung QT, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014;515:572-6. 10.1038/nature14001 [DOI] [PubMed] [Google Scholar]
- 18.Kalaora S, Barnea E, Merhavi-Shoham E, et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget 2016;7:5110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran E, Ahmadzadeh M, Lu YC, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015;350:1387-90. 10.1126/science.aad1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Segal NH, Parsons DW, Peggs KS, et al. Epitope landscape in breast and colorectal cancer. Cancer Res 2008;68:889-92. 10.1158/0008-5472.CAN-07-3095 [DOI] [PubMed] [Google Scholar]
- 21.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883-92. 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gammon G, Shastri N, Cogswell J, et al. The choice of T-cell epitopes utilized on a protein antigen depends on multiple factors distant from, as well as at the determinant site. Immunol Rev 1987;98:53-73. 10.1111/j.1600-065X.1987.tb00519.x [DOI] [PubMed] [Google Scholar]
- 23.Sette A, Vitiello A, Reherman B, et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J Immunol 1994;153:5586-92. [PubMed] [Google Scholar]
- 24.Cibotti R, Kanellopoulos JM, Cabaniols JP, et al. Tolerance to a self-protein involves its immunodominant but does not involve its subdominant determinants. Proc Natl Acad Sci U S A 1992;89:416-20. 10.1073/pnas.89.1.416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cabaniols JP, Cibotti R, Kourilsky P, et al. Dose-dependent T cell tolerance to an immunodominant self peptide. Eur J Immunol 1994;24:1743-9. 10.1002/eji.1830240804 [DOI] [PubMed] [Google Scholar]
- 26.Theobald M, Biggs J, Hernández J, et al. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J Exp Med 1997;185:833-41. 10.1084/jem.185.5.833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hernández J, Lee PP, Davis MM, et al. The use of HLA A2.1/p53 peptide tetramers to visualize the impact of self tolerance on the TCR repertoire. J Immunol 2000;164:596-602. 10.4049/jimmunol.164.2.596 [DOI] [PubMed] [Google Scholar]
- 28.Gross DA, Graff-Dubois S, Opolon P, et al. High vaccination efficiency of low-affinity epitopes in antitumor immunotherapy. J Clin Invest 2004;113:425-33. 10.1172/JCI200419418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moudgil KD, Sercarz EE. The T cell repertoire against cryptic self determinants and its involvement in autoimmunity and cancer. Clin Immunol Immunopathol 1994;73:283-9. 10.1006/clin.1994.1200 [DOI] [PubMed] [Google Scholar]
- 30.Moudgil KD, Southwood S, Ametani A, et al. The self-directed T cell repertoire against mouse lysozyme reflects the influence of the hierarchy of its own determinants and can be engaged by a foreign lysozyme. J Immunol 1999;163:4232-7. [PubMed] [Google Scholar]
- 31.Boisgérault F, Anosova NG, Tam RC, et al. Induction of T-cell response to cryptic MHC determinants during allograft rejection. Hum Immunol 2000;61:1352-62. 10.1016/S0198-8859(00)00209-3 [DOI] [PubMed] [Google Scholar]
- 32.Sinha P, Chi HH, Kim HR, et al. Mouse lysozyme-M knockout mice reveal how the self-determinant hierarchy shapes the T cell repertoire against this circulating self antigen in wild-type mice. J Immunol 2004;173:1763-71. 10.4049/jimmunol.173.3.1763 [DOI] [PubMed] [Google Scholar]
- 33.Friedman RS, Spies AG, Kalos M. Identification of naturally processed CD8 T cell epitopes from prostein, a prostate tissue-specific vaccine candidate. Eur J Immunol 2004;34:1091-101. 10.1002/eji.200324768 [DOI] [PubMed] [Google Scholar]
- 34.Anderton SM, Viner NJ, Matharu P, et al. Influence of a dominant cryptic epitope on autoimmune T cell tolerance. Nat Immunol 2002;3:175-81. 10.1038/ni756 [DOI] [PubMed] [Google Scholar]
- 35.Naftzger C, Takechi Y, Kohda H, et al. Immune response to a differentiation antigen induced by altered antigen: a study of tumor rejection and autoimmunity. Proc Natl Acad Sci U S A 1996;93:14809-14. 10.1073/pnas.93.25.14809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vierboom MP, Nijman HW, Offringa R, et al. Tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. J Exp Med 1997;186:695-704. 10.1084/jem.186.5.695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weber LW, Bowne WB, Wolchok JD, et al. Tumor immunity and autoimmunity induced by immunization with homologous DNA. J Clin Invest 1998;102:1258-64. 10.1172/JCI4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Overwijk WW, Tsung A, Irvine KR, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of "self"-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med 1998;188:277-86. 10.1084/jem.188.2.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowne WB, Srinivasan R, Wolchok JD, et al. Coupling and uncoupling of tumor immunity and autoimmunity. J Exp Med 1999;190:1717-22. 10.1084/jem.190.11.1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hawkins WG, Gold JS, Dyall R, et al. Immunization with DNA coding for gp100 results in CD4 T-cell independent antitumor immunity. Surgery 2000;128:273-80. 10.1067/msy.2000.107421 [DOI] [PubMed] [Google Scholar]
- 41.Palomba ML, Roberts WK, Dao T, et al. CD8+ T-cell-dependent immunity following xenogeneic DNA immunization against CD20 in a tumor challenge model of B-cell lymphoma. Clin Cancer Res 2005;11:370-9. [PubMed] [Google Scholar]
- 42.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med 2003;198:569-80. 10.1084/jem.20030590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tourdot S, Scardino A, Saloustrou E, et al. A general strategy to enhance immunogenicity of low-affinity HLA-A2. 1-associated peptides: implication in the identification of cryptic tumor epitopes. Eur J Immunol 2000;30:3411-21. [DOI] [PubMed] [Google Scholar]
- 44.Dyall R, Bowne WB, Weber LW, et al. Heteroclitic immunization induces tumor immunity. J Exp Med 1998;188:1553-61. 10.1084/jem.188.9.1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moudgil KD, Sercarz EE. Dominant determinants in hen eggwhite lysozyme correspond to the cryptic determinants within its self-homologue, mouse lysozyme: implications in shaping of the T cell repertoire and autoimmunity. J Exp Med 1993;178:2131-8. 10.1084/jem.178.6.2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Disis ML, Shiota FM, Cheever MA. Human HER-2/neu protein immunization circumvents tolerance to rat neu: a vaccine strategy for 'self' tumour antigens. Immunology 1998;93:192-9. 10.1046/j.1365-2567.1998.00424.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fong L, Brockstedt D, Benike C, et al. Dendritic cell-based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol 2001;167:7150-6. 10.4049/jimmunol.167.12.7150 [DOI] [PubMed] [Google Scholar]
- 48.Srinivasan R, Houghton AN, Wolchok JD. Induction of autoantibodies against tyrosinase-related proteins following DNA vaccination: unexpected reactivity to a protein paralogue. Cancer Immun 2002;2:8. [PubMed] [Google Scholar]
- 49.Hawkins WG, Gold JS, Blachere NE, et al. Xenogeneic DNA immunization in melanoma models for minimal residual disease. J Surg Res 2002;102:137-43. 10.1006/jsre.2001.6302 [DOI] [PubMed] [Google Scholar]
- 50.Bergman PJ, McKnight J, Novosad A, et al. Long-term survival of dogs with advanced malignant melanoma after DNA vaccination with xenogeneic human tyrosinase: a phase I trial. Clin Cancer Res 2003;9:1284-90. [PubMed] [Google Scholar]
- 51.van Buuren MM, Calis JJ, Schumacher TN. High sensitivity of cancer exome-based CD8 T cell neo-antigen identification. Oncoimmunology 2014;3:e28836. 10.4161/onci.28836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994;266:2011-5. 10.1126/science.7605428 [DOI] [PubMed] [Google Scholar]
- 53.Meyerson M, Counter CM, Eaton EN, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 1997;90:785-95. 10.1016/S0092-8674(00)80538-3 [DOI] [PubMed] [Google Scholar]
- 54.Kumaki F, Kawai T, Hiroi S, et al. Telomerase activity and expression of human telomerase RNA component and human telomerase reverse transcriptase in lung carcinomas. Hum Pathol 2001;32:188-95. 10.1053/hupa.2001.21567 [DOI] [PubMed] [Google Scholar]
- 55.Vetsika EK, Papadimitraki E, Aggouraki D, et al. Sequential administration of the native TERT572 cryptic peptide enhances the immune response initiated by its optimized variant TERT(572Y) in cancer patients. J Immunother 2011;34:641-50. 10.1097/CJI.0b013e31823284a6 [DOI] [PubMed] [Google Scholar]
- 56.Dudley ME, Nishimura MI, Holt AK, et al. Antitumor immunization with a minimal peptide epitope (G9-209-2M) leads to a functionally heterogeneous CTL response. J Immunother 1999;22:288-98. 10.1097/00002371-199907000-00002 [DOI] [PubMed] [Google Scholar]
- 57.Parkhurst MR, Salgaller ML, Southwood S, et al. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol 1996;157:2539-48. [PubMed] [Google Scholar]
- 58.Speiser DE, Baumgaertner P, Voelter V, et al. Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen. Proc Natl Acad Sci U S A 2008;105:3849-54. 10.1073/pnas.0800080105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kotsakis A, Vetsika EK, Christou S, et al. Clinical outcome of patients with various advanced cancer types vaccinated with an optimized cryptic human telomerase reverse transcriptase (TERT) peptide: results of an expanded phase II study. Ann Oncol 2012;23:442-9. 10.1093/annonc/mdr396 [DOI] [PubMed] [Google Scholar]
- 60.Bolonaki I, Kotsakis A, Papadimitraki E, et al. Vaccination of patients with advanced non-small-cell lung cancer with an optimized cryptic human telomerase reverse transcriptase peptide. J Clin Oncol 2007;25:2727-34. 10.1200/JCO.2006.10.3465 [DOI] [PubMed] [Google Scholar]
- 61.Mavroudis D, Bolonakis I, Cornet S, et al. A phase I study of the optimized cryptic peptide TERT(572y) in patients with advanced malignancies. Oncology 2006;70:306-14. 10.1159/000096252 [DOI] [PubMed] [Google Scholar]
- 62.Vetsika EK, Konsolakis G, Aggouraki D, et al. Immunological responses in cancer patients after vaccination with the therapeutic telomerase-specific vaccine Vx-001. Cancer Immunol Immunother 2012;61:157-68. 10.1007/s00262-011-1093-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Georgoulias V, Douillard JY, Khayat D, et al. A multicenter randomized phase IIb efficacy study of Vx-001, a peptide-based cancer vaccine as maintenance treatment in advanced non-small-cell lung cancer: treatment rationale and protocol dynamics. Clin Lung Cancer 2013;14:461-5. 10.1016/j.cllc.2013.02.001 [DOI] [PubMed] [Google Scholar]