

Abstract
The nucleosome remodeling and deacetylase (NuRD) complex remodels the genome in the context of both gene transcription and DNA damage repair. It is essential for normal development and is distributed across multiple tissues in organisms ranging from mammals to nematode worms. In common with other chromatin‐remodeling complexes, however, its molecular mechanism of action is not well understood and only limited structural information is available to show how the complex is assembled. As a step towards understanding the structure of the NuRD complex, we have characterized the interaction between two subunits: the metastasis associated protein MTA1 and the histone‐binding protein RBBP4. We show that MTA1 can bind to two molecules of RBBP4 and present negative stain electron microscopy and chemical crosslinking data that allow us to build a low‐resolution model of an MTA1‐(RBBP4)2 subcomplex. These data build on our understanding of NuRD complex structure and move us closer towards an understanding of the biochemical basis for the activity of this complex.
Keywords: NuRD complex, RBBP4, MTA1, transcription regulation, chromatin, protein structure
Introduction
Physical remodeling of the eukaryotic genome is an essential aspect of a wide range of processes, including transcription, replication, and DNA repair. ATP‐dependent chromatin remodeling complexes are instrumental in such remodeling events. Remodeling complexes all contain a DNA translocase enzyme that harnesses ATP‐derived energy to alter the positions, occupancy and composition of nucleosomes, thereby altering the accessibility of DNA to other DNA‐binding factors. The activity of the translocase is modulated by additional subunits that harbour, for example, domains that recognize specific chromatin modifications or other regulatory proteins.
Despite their central role in regulating the genome, a detailed mechanistic description of remodeling is still lacking and no high‐resolution structures of remodeling complexes are available. Four classes of remodeling complex have been defined, based on their central remodeling subunit: INO80, ISWI, SWI/SNF, and CHD type complexes. Low‐resolution (23–50 Å) models of several yeast remodeling complexes derived from single‐particle electron microscopy data have been reported (e.g.1, 2) and partial X‐ray crystal structures have been reported for several components or domains or other complexes3, 4, 5 but far less is known about CHD‐family remodeling complexes, the best‐described of which is the nucleosome remodeling and deacetylase (NuRD) complex.
The NuRD complex is conserved across all complex animals and is expressed in most, if not all, tissues. NuRD can repress or activate genes and its activity is required, for example, at all stages of haematopoiesis, regulating both haematopoietic stem cell (HSC) maintenance and differentiation of these cells into distinct lineages (e.g.6). NuRD is also emerging as a significant player in efforts to reprogram somatic cells into pluripotent stem cells.7, 8, 9
In mammalian cells, NuRD comprises the ATP‐dependent remodeling enzyme CHD4, the histone deacetylases HDAC1 and ‐2, the DNA‐binding proteins MBD2 and ‐3,10 the metastasis‐associated proteins MTA1, ‐2, and ‐3, the WD40‐repeat proteins RBBP4 and ‐7 and the poorly understood proteins GATAD2A and ‐B. The various isoforms of each protein are encoded by separate genes but little is known about their functional significance. The MTA‐family proteins [Fig. 1(A)] contain BAH and SANT domains, which in other proteins have been implicated in nucleosome recognition, whereas RBBP4 and ‐7 have been shown to bind the transcriptional coregulator FOG111 as well as both histone H312 and histone H4.13
Figure 1.
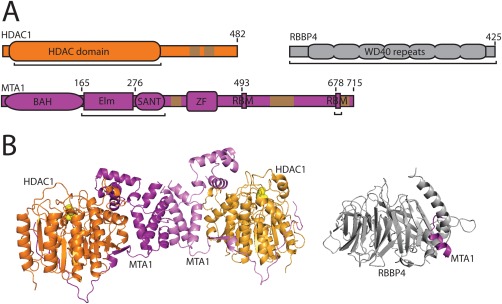
Schematics of HDAC1, RBBP4 and MTA1. A. Schematics of three components of the NuRD complex. Domains (shown as labelled ovals/boxes) have known structures or the structures of related domains are known. BAH, Elm, SANT, and ZF (zinc finger) are domains of MTA1, whereas RBM refers to RBBP Binding Motifs defined in this article. Brown regions are low complexity sequences. Black underlining refers to protein sections for which crystal structures are available. Colouring is preserved in B. B. Three‐dimensional structures of NuRD subcomplexes. Left: HDAC1‐MTA1ELM‐SANT dimer of dimers (PDB 4BKX). Right: RBBP4‐MTA1675‐686 (PBB 4PBY).
Currently, we have little understanding of how all of these components come together to make the NuRD complex, although structures of a number of domains and several limited subcomplexes have been determined. These subcomplex structures [Fig. 1(B)] include an HDAC1‐MTA1 dimer of dimers involving the ELM and SANT domains of MTA114 and a complex formed between RBBP4 and a short motif at the C‐terminal end of MTA1.15
As part of efforts to delineate the architecture of the NuRD complex, we have examined the interaction between RBBP‐ and MTA‐family proteins and we show here that MTA1 and ‐2 carry two motifs that can each independently recruit a molecule of RBBP4. In vitro binding studies show that these two interactions can occur simultaneously and negative‐stain single particle electron microscopy combined with covalent crosslinking and mass spectrometry (XL‐MS) reveals the overall shape of an MTA1:(RBBP4)2 subcomplex. Taken together with the existing subcomplex structures of HDAC1:MTA1ELM‐SANT and RBBP4:MTA1, these data begin to provide an outline for the physical arrangement of subunits within the NuRD complex.
Results
The C‐terminal half of MTA1 and ‐2 can bind directly to two RBBP subunits
We previously demonstrated that a short sequence from the C‐terminal end of MTA1 is able to bind with micromolar affinity to either RBBP4 or RBBP7.15 This sequence encompasses a helical 678KRAARR motif (an RBBP‐binding motif, or RBM) that forms a number of electrostatic interactions with the RBBP partner. Examination of the MTA1 sequence revealed a second RBM with a closely related sequence that is highly conserved across complex eukaryotes [Figs. 1(A) and 2]. To assess whether this motif can also bind RBBP4, we coexpressed FLAG‐MTA1440–550 and HA‐RBBP4 and captured the HA‐RBBP4 with anti‐HA beads. As shown in Figure 3(A), this fragment was able to efficiently pull down FLAG‐MTA1440–550. We also carried out a triple coexpression, expressing FLAG‐MTA1440–550, HA‐RBBP4 and FLAG‐RBBP4. In this case, affinity captured HA‐RBBP4 still pulled down FLAG‐MTA1440–550 but FLAG‐RBBP4 did not copurify, indicating that FLAG‐MTA1440‐550 can bind only a single molecule of RBBP4 [Fig. 3(A)].
Figure 3.
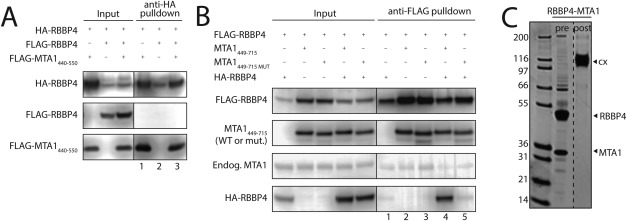
The C‐terminal half of MTA1 can bind two molecules of RBBP4. A. Immunoprecipitation analysis showing that MTA1440–550, which encompasses a single RBBP‐binding motif (RBM) at positions 493–498 (493WHAARH), is able to bind a single RBBP4. Cleared cell lysates (Input) of HEK293 cells expressing the proteins indicated on top, as well as anti‐HA immunoprecipitates (anti‐HA pulldown), were analysed by Western Blot using anti‐HA or anti‐FLAG antibodies, to detect the proteins indicated on the left. The absence of FLAG‐RBBP4 in the elution of the triple co‐transfection (lane 3) suggests that only one RBBP4 can bind to MTA1440–550. B. Immunoprecipitation analysis showing that MTA1449‐715, which contains the two RBMs at positions 493–498 and 678–683, is able to bind two molecules of RBBP4. Cleared cell lysates (Input) of HEK293 cells expressing the proteins indicated on top, as well as anti‐FLAG immunoprecipitates (anti‐FLAG pulldown), were analysed by Western Blot using anti‐FLAG, anti‐HA or anti‐MTA1 antibodies, to detect the proteins indicated on the left. The results show that FLAG‐RBBP4 can efficiently pull down HA‐RBBP4 in the presence of MTA1449–715, suggesting that this fragment can bind two molecules of RBBP4. However, this effect is not observed when using an MTA1449–715 version mutated at one of the RBMs (678KRAARR to AAAAAA). C. Sypro‐stained SDS‐PAGE showing purified RBBP4‐MTA1449–715 before and after treatment with glutaraldehyde. The crosslinked complex runs at a molecular weight consistent with the formation of a 2:1 (RBBP4:MTA1449–715) complex.
To assess whether the entire C‐terminal half of MTA1 can bind two RBBP4 subunits simultaneously, we coexpressed combinations of FLAG‐RBBP4, HA‐RBBP4 and a longer (untagged) MTA1449–715 construct in HEK293 cells and purified the FLAG‐RBBP4 using anti‐FLAG Sepharose beads. Western blot analysis [Fig. 3(B), lane 4] showed that HA‐RBBP4 was robustly pulled down when MTA1449–715 was coexpressed, indicating that this portion of MTA1 is able to bind to two RBBP4 molecules simultaneously. Mutation of the 678KRAARR motif to AAAAAA, however, abrogates the ability of MTA1449–715 to bind the second RBBP4 [Fig. 3(B), lane 5]. For corroboration, we purified a coexpressed RBBP4‐MTA1449–715 complex from HEK293 cells and subjected it to covalent crosslinking using glutaraldehyde. Figure 3(C) shows that the crosslinked complex runs on denaturing SDS‐PAGE with an apparent molecular mass of ∼130 kDa, which is consistent with the expected mass of a 2:1 RBBP4‐MTA1449–715 complex (128 kDa).
Covalent crosslinking combined with mass spectrometry (XL‐MS)
We next sought to map the RBBP4‐MTA1449–715 interface using XL‐MS. Purified RBBP4‐MTA1449–715 complex was crosslinked with either disuccinimidyl suberate (DSS) or adipic acid dihydrazide (ADH) as previously described.16, 17 The use of two crosslinkers provides complementary data. Briefly, DSS specifically crosslinks primary amines (i.e., lysine residues and the protein N‐terminus) while ADH crosslinks carboxylic acids (i.e., glutamate, aspartate residues and the protein C‐terminus). In addition, in the ADH reaction, a separate side‐reaction arising from the 4‐(4,6‐dimethoxy‐1,3,5‐triazin‐2‐yl)‐4‐methylmorpholinium chloride (DMTMM) used can also lead to “zero‐length” crosslinks to form between carboxylic acids and primary amines; a direct condensation reaction between the primary amine and carboxylic acid. These crosslinks will be referred to as “ZLXL” crosslinks from here on forth. Post‐crosslinking, the samples were digested with trypsin, fractionated via size exclusion to enrich for crosslinked peptides and then analysed by LC‐MS/MS to identify crosslinked peptides from RBBP4 and MTA1. We identified 58 high‐confidence crosslinks (Supporting Information Table I). Amongst these 58 crosslinks, 13 could be mapped to our previously determined crystal structure of RBBP4 bound to MTA1670–695 (15), including RBBP4K22‐MTA1K686, RBBP4K317‐MTA1K686, and RBBP4‐RBBP4 intra‐protein crosslinks. Importantly, all 13 of these crosslinks were within the maximum distance limits for the crosslinker used, providing confidence in the XL‐MS data. We note that a small number of crosslinks were also detected that involved residues from other NuRD components, including MBD3, HDAC1, GATA2DA, and CHD4. These most likely indicate that a fraction of our expressed FLAG‐RBBP4, which was used as the purification “handle” in our experiments, has docked with endogenous NuRD subunits.
Negative‐stain EM of an (RBBP4)2‐MTA1449–715 complex
To gain structural insight into the RBBP4‐MTA1 interaction, we first purified the RBBP4‐MTA1449–715 complex in two steps, using FLAG‐affinity chromatography and sucrose gradient centrifugation combined with mild glutaraldehyde crosslinking [using the GraFix protocol18; Fig. 4(A)]. We then subjected the complex to negative‐stain electron microscopy (EM). Figure 4(B) shows examples of individual particles and 2D class averages obtained using RELION.19 Approximately 1,700 particles were manually picked and used to generate templates for automatic particle picking; this process yielded a total of 12,000 particles, which was reduced to 9,000 following manual inspection of the dataset and further reduced to 4,000 particles following 2D classification and 3D classification. Two cycles of unsupervised 3D classification in RELION were used to generate first ten, then four 3D classes. The best class from the first cycle was used to generate a new starting model for the next cycle, which resulted in models that had a similar overall shape [Fig. 4(C)]. The most heavily populated 3D class, which contained 1,700 particles, was further refined to obtain a final model with an estimated resolution of 29.8 Å according to gold standard Fourier shell correlation (0.143 cut‐off). The 2D projections were calculated from the 3D model and match well to independently generated 2D class averages [Fig. 4(B)].
Figure 4.
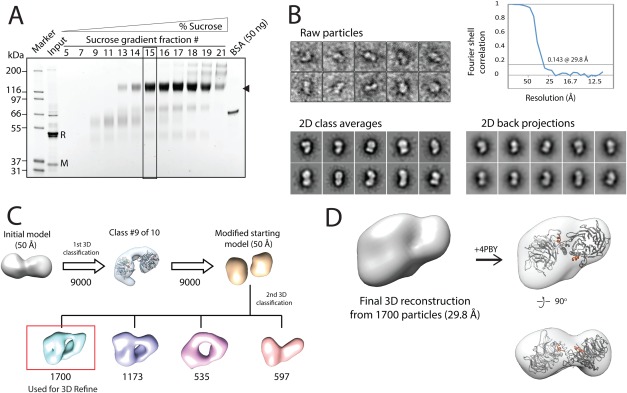
Negative stain EM of the RBBP4:MTA1449–715 complex. A. Sypro‐Ruby stained SDS‐PAGE showing selected fractions of a 2–25% sucrose plus 0–0.15% glutaraldehyde “GraFix” gradient after anti‐FLAG affinity purification of the complex. The position of the species of interest is indicated on the right by a triangle, and fraction #15, which was selected for the EM analysis, is boxed. The lane corresponding to the non‐crosslinked sample loaded onto the sucrose gradient is labelled as “Input”. “R” indicates the position of RBBP4, and “M” the position of MTA1449–715. B. Examples of typical individual particles (top left) and reference‐free 2D class averages observed in negative‐stain EM analysis of RBBP4‐MTA1449–715 (bottom left). An FSC plot (top right) and sample 2D projections of the final reconstruction (bottom right) are shown. C. The 3D reconstruction workflow for RBBP4‐MTA1449–715 obtained from RELION. Numbers shown under models refer to the number of particles associated with each stage of model development. D. Refined 3D reconstruction of negatively stained RBBP4‐MTA1449–715. A copy of the RBBP4 crystallographic dimer (PDB 4PBY) is fitted into the envelope as a rigid body (right). The RBM helices from MTA1 are shown in orange and the RBBP4 subunits are shown in grey.
Simple correlation‐based fitting of the crystallographic dimer observed for the RBBP4‐MTA1 structure (PDB: 4PBY) into the EM‐derived envelope (using the “fit in map” tool in UCSF Chimera) revealed a very good overall fit for the two RBBP subunits [Fig. 4(D)]. Placement of the MTA1449–715 into the envelope was not, however, as straightforward. Bioinformatic analysis does not reveal a clear prediction for the structure of this region of MTA1 (or MTA2 or ‐3). Disorder predictions suggest that the region encompassing residues 545–650 has a high probability of being disordered, although a number of Molecular Recognition Features (MoRFs, short sequences that undergo a disorder‐to‐order transition upon binding their partner20) are also predicted within this region. Secondary structure and domain predictions predict several helical segments within MTA1449–715 [Fig. 5(A)] but no identifiable domains, suggesting that MTA1 might undergo a significant conformational change upon binding RBBP4 and form an extended interface with the two RBBP4 subunits, similar to the structure observed for the N‐terminal part of the MTA1 ELM domain that binds to HDAC1.14
Figure 5.
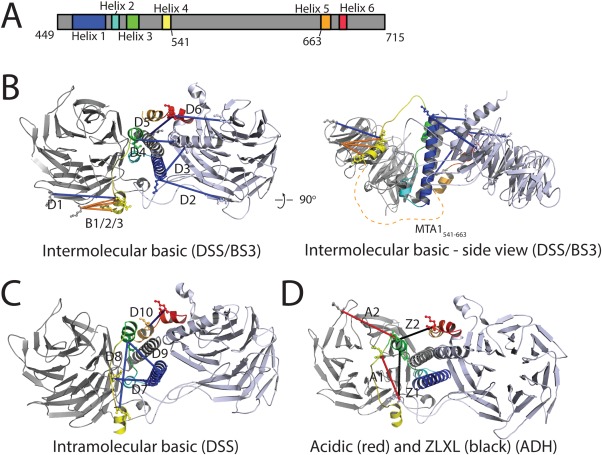
Proposed model of RBBP4‐MTA1449–715 with associated crosslinking data. A. Schematic of MTA1449–715 showing the predicted helices. Colour coding is used in the rest of the Figure. B–D. Model of the complex. The RBBP4 crystallographic dimer taken from PDB 4PBY is shown in two shades of grey. Modeled MTA1449–715 helices are: Helix 1 (461–488), Helix 2 (also RBM1, 491–499), Helix 3 (505–515), Helix 4 (534–541), Helix 5 (663–671), and Helix 6 (also RBM2, 677–684). A large putative disordered region (541–663) is not included in the model; the position of this region is indicated by a dashed orange line in B (right). Crosslinking data are shown as blue (DSS—basic), red (ADH—acidic), black (ZLXL), and orange (BS3—basic; from (23)) and are labelled accordingly.
To model the likely overall structure and placement of MTA1449–715 within the complex, we made use of the PORTER secondary structure prediction server21 and the PRALINE multiple alignment server22 to define six putative helical regions for MTA1449–715 [Fig. 4(D)]. Helix 6 is the RBM taken directly from the RBBP4‐MTA1 crystal structure (PDBi: 4PBY), and Helix 2 (MTA1491‐499)—corresponding to the RBM identified above (Fig. 2 )—was modeled on Helix 6 (MTA1677–684) and placed in an equivalent binding location in the second RBBP4 monomer. Helices 1 (461–488), 3 (505–515), 4 (534–541), and 5 (663–671) were modeled based on helical predictions made by the PRALINE sequence alignments (Supporting Information Figs. 1 and 2). We also made use of distance restraints derived from 19 crosslinks detected in the XL‐MS data (Table 1). This subset comprised the crosslinks between RBBP4 and MTA1 (or between two residues in MTA1 that are >10 residues apart), excluding those that involved MTA1 residues that were not represented in the model (e.g., the MTA1541–663 region that is predicted to be disordered). We used the XL‐MS data to guide manual placement of the six predicted MTA1 helices into the EM‐derived molecular envelope into which had been fitted two copies of RBBP4. Care was taken to keep charged residues relatively exposed and, where possible, to juxtapose hydrophobic surfaces. The lengths of the sequences linking the six MTA1 helices were also considered in the course of the modelling.
Figure 2.
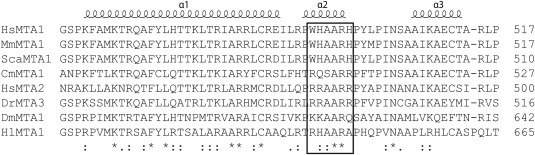
Sequence alignment of a portion of MTA1/2 from a range of eukaryotes. The alignment highlights the high conservation of the first of the RBBP‐binding motifs across a wide range of species, including human (HsMTA1—UniprotID: Q13330, HsMTA2—O94776), Mus musculus (MmMTA1—Q8K4B0), Struthio camelus australis (ScaMTA1—A0A093HEK0), Callorhinchus milii (CmMTA1—V9KH55), Danio rerio (DrMTA3—E7EY65), Drosophila melanogaster (DmMTA1—Q9VNF6), and Habropoda laboriosa (HlMTA1—A0A0L7R1K5).
Table 1.
Crosslinks Used as EM Modelling Restraints Obtained From XL–MS Analyses
| Id | Crosslinker | Protein 1 | Residue | Protein 2 | Residue | Length in model (Å) |
|---|---|---|---|---|---|---|
| Crosslinks from this study | ||||||
| D1 | DSS | RBBP7a | 263 | MTA1 | 532 | 28.1 |
| D2 | DSS | RBBP4 | 120 | MTA1 | 462 | 33.2 |
| D3 | DSS | RBBP4/7 | 22/21 | MTA1 | 462 | 17.5 |
| D4 | DSS | RBBP4 | 26 | MTA1 | 509 | 17.1 |
| D5 | DSS | RBBP4/7 | 22/21 | MTA1 | 686 | 25.6 |
| D6 | DSS | RBBP4 | 317 | MTA1 | 686 | 21.7 |
| D7 | DSS | MTA1 | 477 | MTA1 | 527 | 25.9 |
| D8 | DSS | MTA1 | 509 | MTA1 | 532 | 27.0 |
| D9 | DSS | MTA1 | 462 | MTA1 | 509 | 19.7 |
| D10 | DSS | MTA1 | 670 | MTA1 | 686 | 22.0 |
| D11 | DSS | RBBP7 | 119 | RBBP4 | 156 | b |
| D12 | DSS | RBBP7 | 211 | RBBP4 | 156 | b |
| D13 | DSS | RBBP7 | 119 | RBBP4 | 160 | b |
| A1 | ADH | RBBP4 | 104 | MTA1 | 518 | 22.9 |
| A2 | ADH | RBBP4 | 166 | MTA1 | 511 | 30.7 |
| A3 | ADH | MTA1 | 488 | MTA1 | 518 | b |
| A4 | ADH | MTA1 | 511 | MTA1 | 686 | b |
| Z1 | ZLXL | RBBP4 | 104 | MTA1 | 509 | 19.5c |
| Z2 | ZLXL | MTA1 | 511 | MTA1 | 686 | 16.4 |
| Crosslinks from in Kloet et al., 2015 | ||||||
| B1 | BS3d | RBBP4/7 | 306/307 | MTA2a | 531 | 17.7 |
| B2 | BS3d | RBBP4/7 | 306/307 | MTA2a | 533 | 18.9 |
| B3 | BS3d | RBBP4/7 | 306/307 | MTA2a | 539 | 14.3 |
Note:
Where possible, RBBP7 and MTA2 residues are mapped onto corresponding RBBP4 and MTA1 residues, respectively, in the EM model.
These crosslinks were not compatible with the model.
While this linker distance exceeds the theoretical limit of ∼14 Å, residue RBBP4104 is located on a flexible loop.
BS3 refers to the use of bis‐sulfosuccinimidyl suberate as the crosslinker.
Using this strategy, we were able to generate a model that satisfied 14 of 19 crosslinks (Fig. 5). Given that our XL‐MS dataset included crosslinks that involved endogenous NuRD components, it is possible that some or all of the unsatisfied crosslinks correspond to connections made in larger NuRD subcomplexes. In addition, a recent XL‐MS study of the full NuRD complex23 yielded three RBBP4‐MTA2 crosslinks that correspond to pairs of residues found in our model [B1, B2, and B3 in Fig. 5(B)]. These crosslinks are entirely consistent with our model. Spectra for crosslinks that satisfied the modeling are shown in Supporting Information Figure 3. Helix 1 was placed in a position such that it formed a coiled coil with the N‐terminal helix (RBBP43–31) of one of the RBBP4 monomers, consistent with coiled coil predictions for this portion of MTA1 from PAIRCOIL2.24 It is notable that this arrangement juxtaposes a cluster of arginines (R484, R484 and R487) at the C‐terminal end of Helix 1 with a cluster of acidic residues on the RBBP4 monomer (E357, D358, and E360). Helix 3 was also placed to interact with RBBP43–31 on one side of the model, linking across to Helix 4 on the other side of the model through an 18‐residue linker. This arrangement was important for satisfying crosslinks A2 and Z2 for helix 3, and D1 for Helix 4 (Table 1). The disordered region (MTA1541–663) is assumed to pass under the model as indicated in Figure 5(B), connecting with Helix 5 on the opposite side of the model. Helix 5 leads in turn to the crystallographically defined Helix 6. Notably, a loop region of the RBBP4 crystal structure (4PBY—residues 86–113) had to be remodeled to accommodate MTA1 helices 1 and 2. This loop is absent from other RBBP4‐MTA1 structures (PDBs 4PBZ, 4PC0), suggesting it is relatively mobile. Collectively, the model posits that a large portion of MTA1449–715 forms a helical core that lies at the interface between two RBBP4 subunits.
Discussion
Our data demonstrate that MTA1 can coordinate the binding of two RBBP subunits through two closely related motifs that are highly conserved throughout phyla that assemble a NuRD complex, suggesting that this arrangement is a characteristic aspect of NuRD structure. The structure of HDAC1 bound to an N‐terminal fragment of MTA114 displays a 2:2 stoichiometry, indicating that the MTA1‐RBBP4 interactions observed in our work can rationalize the presence of four RBBP4 subunits in the NuRD complex. Given the presence of a number of regions of MTA1 that have strong predictions of disorder (e.g., MTA1350–380 and MTA1541–663), it is tempting to speculate that a certain degree of flexibility will be observed in the intact NuRD structure—both in the region bordered by the ELM‐SANT region and our MTA1449–715 (MTA1350–380) and even perhaps between the two RBBP subunits.
What will be the function of these four RBBP subunits? Published work suggests that although RBBP4/7 interacts with histone H4, the interaction makes use of (and interacts similarly with) the same surface on RBBP4 that is contacted by the C‐terminal RBM.13 It therefore seems unlikely that any of the four RBBPs will make direct contact with histone H4 in the context of an assembled NuRD complex. In contrast, the surface that contacts both histone H3 and FOG1 is more likely to be available. Figure 6 shows a simple model of a 2:2:4 HDAC1‐MTA1‐RBBP4 complex. The arrangements shown makes it clear that the N‐terminal tail of a histone H3 tail (shown as a blue dashed line) in the context of a nucleosome can readily contact both the histone H3 binding site on an RBBP4 subunit and the HDAC1 active site, which might act on the tail (or on other acetylated lysines in the nucleosome). Similar interactions can take place with the other copy of histone H3 via the second (right‐hand side) RBBP4‐MTA1 unit. It is tempting to speculate that these RBBP4‐containing “arms” might simultaneously grasp the nucleosome from opposite sides, allowing the chromatin remodeling component of the NuRD complex, CHD4 (not shown), to operate on the nucleosomal DNA, moving it relative to the histone octamer. Alternatively, it is possible that the substrate of NuRD is a dinucleosome in which each MTA‐RBBP42 entity might contact a separate nucleosome, allowing CHD4 to act on a two‐nucleosome unit.
Figure 6.
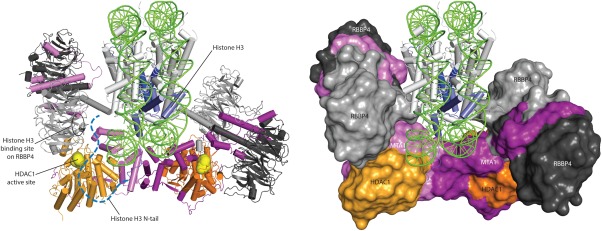
Model of the HDAC‐MTA1‐RBBP4 portion of the NuRD complex. The model has been made by simply juxtaposing the HDAC‐MTA1 crystal structure with two copies of our model of the RBBP4‐MTA1449–715 complex and adding the crystal structure of the nucleosome (1AOI). The HDAC1 active sites are shown as yellow spheres and the histone H3 binding site on RBBP4 is circled. The N‐terminal 40‐residue tail of histone H3 is indicated schematically to show that it is within reach of both the histone‐H3‐binding site on RBBP4 and the HDAC1 active site. Similar interactions are possible for the right‐hand RBBP4 subunits, which could interact with the second histone H3 tail on the back of the model as drawn.
It is worth noting that the first of two papers that quantitatively analysed the stoichiometry of the NuRD complex suggested that NuRD harbours six RBBP subunits.25 The second paper suggested that some RBBP subunits might be less tightly associated with NuRD. If these additional subunits are present, they might well be attached to NuRD via a different interaction—which would then leave the MTA1/histone H4 binding site on these RBBP4 subunits available for interactions with nucleosome substrates. Finally, given that FOG1 and several other coregulators make use of the histone H3 binding surface of RBBP4, there is clearly still much to learn about how the different RBBP subunits in the NuRD complex coordinate their activity to recognize both histone substrates and coregulatory binding partners such as FOG1.
Materials and Methods
Plasmids
A DNA sequence of human MTA1 (Uniprot: Q13330) corresponding to residues 449–715, with no tag (MTA1449‐715), cloned in the expression vector pcDNA3.1, was a kind gift from Prof. Gerd A. Blobel. A version of this construct including the mutation 678KRAARR to 678AAAAAA (MTA1449‐715 MUT) was obtained by site‐directed mutagenesis. Additionally, genes corresponding to human RBBP4 (full length; Uniprot: Q09028) with both FLAG and HA N‐terminal tags, and residues 440–550 of human MTA1 (MTA1440–550) with an N‐terminal FLAG tag, were cloned into pcDNA3.1.
Immunoprecipitation analysis
Combinations of constructs in pcDNA3.1 were cotransfected into suspension HEK Expi293F™ cells (Thermo Fisher) using Polyethylenimine (PEI) (Polysciences) and equimolar plasmid ratios. Prior to each cotransfection, a 1.9‐mL culture was grown to a density of 2 × 106 cells mL−1 in Expi293™ Expression Medium (Thermo Fisher). About 3.8 μg of the DNA mix was diluted in 205 μL of PBS (Thermo Fisher) and vortexed briefly; 7.6 μg of PEI was then added and the suspension was vortexed again and incubated for 20 min at room temperature before being added to the cells. Cells were collected in 1‐mL aliquots 65 h after transfection, and each of these lysed by sonication in 0.5 mL of buffer A [50 mM Tris pH 7.9, 150 mM NaCl, 1 mM PMSF, 1× Roche complete protease inhibitor, 0.2 mM DTT, 1% (v/v) Triton X‐100]. Cell lysates were incubated on ice for 30 min and then cleared by centrifugation. “Input” samples were collected at this stage. Cleared lysates were then mixed with 20 μL anti‐FLAG or anti‐HA Sepharose 4B beads (BioTool) overnight at 4°C. Next, the beads were washed five times with 1 mL of buffer B [50 mM Tris pH 7.5, 150 mM NaCl, 0.5% (v/v) Igepal], and the bound proteins eluted in 60 μL of buffer C [10 mM HEPES pH 7.5, 150 mM NaCl, and either 150 μg mL−1 3 × FLAG peptide or 2 mg mL−1 1 × HA peptide]. Samples were analysed by Western blot, using anti‐FLAG, anti‐HA or anti‐MTA1 antibodies (Cell Signalling Technologies).
Protein production
The FLAG‐RBBP4 and MTA1449–715 constructs were co‐transfected into suspension Expi293F™cells as described above, and the resulting sub‐complex purified also as described above. Briefly, a 150‐mL culture of cells was transfected with a mixture of 0.3 mg DNA, 16.7 mL PBS, and 0.6 mg PEI. A 50 mL of buffer A and 1 mL of FLAG beads were used, and each wash was done with 100 mL of buffer B. The subcomplex was eluted in 4 mL buffer C containing 150 μg mL−1 3 × FLAG peptide, and DTT was added to 1 mM final concentration.
Preparation of the RBBP4‐MTA1449‐715 subcomplex for crosslinking‐mass spectrometry (XL‐MS) analysis
The RBBP4‐MTA1449‐715 subcomplex was purified as described above using FLAG‐affinity chromatography. Prior to crosslinking, the excess 3× FLAG peptide used during the elution step was removed using Zeba Spin desalting gel filtration columns (7K MWCO; ThermoFisher Scientific), and the complex was exchanged into a buffer comprising 30 mM HEPES (pH 7.5) and 225 mM NaCl. For each crosslinking experiment, ∼15 µg of subcomplex at a concentration of ∼0.15 mg mL−1 was used. The purified samples were then prepared for XL‐MS essentially as described in Refs. 16 and 17 with some modifications. Briefly, for disuccinimidyl suberate (DSS) crosslinking, H12/D12‐DSS (1:1 ratio, 25 mM stock solution in anhydrous dimethylformamide; Creative Molecules) was added to a final concentration of 1 mM and incubated for 30 min at 37°C with constant mixing. The excess DSS was then quenched with 100 mM NH4HCO3 (50 mM final concentration) and further incubated at 37°C for 20 min. For adipic acid dihydrazide (ADH) crosslinking, H8/D8‐ADH (1:1 ratio, 100 mg mL−1 stock solution in 20 mM HEPES pH 7.4; Creative Molecules) and 4‐(4,6‐dimethoxy‐1,3,5‐triazin‐2‐yl)‐4‐methylmorpholinium chloride (DMTMM) (144 mg mL−1 stock solution in 20 mM HEPES pH 7.4; Sigma–Aldrich) were added to final concentrations of ∼8.3 mg mL−1 and 12 mg mL−1, respectively. The sample was then incubated for 1.5 h at 37°C with constant mixing. The excess DSS was then removed by using the Zeba Spin Desalting gel filtration columns (7K MWCO; ThermoFisher Scientific) into 20 mM HEPES pH 7.5, 150 mM NaCl. Both DSS and ADH crosslinked samples were then dried in a vacuum centrifuge.
Dried, crosslinked protein samples were resuspended in 50 µL of 8 M urea and the proteins were reduced (5 mM TCEP, 37°C, 30 min) and alkylated (10 mM iodoacetamide, 20 min, room temperature in the dark). The samples were then diluted to 6 M urea using 200 mM Tris‐HCl pH 8 and Trypsin/Lys‐C mix (Promega) was added to an enzyme:substrate ratio of 1:25 (w/w). The solution was incubated at 37°C for 4 h. Following this digestion step, the sample was further diluted to 0.75 M urea using 50 mM Tris‐HCl pH 8 and additional Trypsin (Promega) was added at an enzyme:substrate ratio of 1:50 (w/w). The sample was then incubated at 37°C overnight (∼16 h). Following overnight digestion, the samples were acidified by addition of formic acid to a final concentration of 2% (v/v) and centrifuged at 16,000 g for 10 min. The supernatant was then desalted using 50‐mg Sep‐Pak tC18 cartridges (Waters), eluted in 50:50:0.1 acetonitrile:water:formic acid (v/v/v), and dried in a vacuum centrifuge.
For size exclusion chromatography fractionation (SEC), the dried desalted peptides were resuspended in 150 µL of SEC mobile phase [acetonitrile:water:trifluoroacetic acid, 70:30:0.1 (v/v/v)] and loaded onto a Superdex Peptide HR 10/30 column connected to a Biologic DuoFlowTM system (Bio‐Rad) with a BioFracTM fraction collector (Bio‐Rad). A flow rate of 0.5 mL min−1 was used and the separation was monitored by UV absorption at 215, 225, 254, and 280 nm using a Biologic QuadTecTM UV/Vis detector (Bio‐Rad). Fractions were collected as 2‐min windows over 1.25 column volumes (30 mL). Based on the UV absorption traces, fractions of interest (retention volumes ∼12–17 mL) for mass spectrometry were dried in a vacuum centrifuge.
Mass spectrometry
For LC‐MS/MS, peptides were resuspended in 3% (v/v) acetonitrile, 0.1% (v/v) formic acid and loaded onto a 20 cm × 75 µm inner diameter column packed in‐house with 1.9‐µm C18AQ particles (Dr Maisch GmbH HPLC) using an Easy nLC‐1000 nanoHPLC (Proxeon). Peptides were separated using a linear gradient of 5–30% Buffer B over 120 min at 200 nL min−1 at 55°C (Buffer A = 0.1% (v/v) formic acid; Buffer B = 80% (v/v) acetonitrile, 0.1% (v/v) formic acid). Mass analyses were performed using a Q‐Exactive mass spectrometer (Thermo Scientific). Following each full‐scan MS1 at 70,000 resolution at 200 m/z (300–1750 m/z; 3 × 106 AGC; 100 ms injection time), up to 10 most abundant precursor ions were selected for MS/MS (17,500 resolution; 1 × 105 AGC; 60 ms injection time; 32 normalized collision energy; 2 m/z isolation window; 1.7 × 105 intensity threshold; minimum charge state of +3; dynamic exclusion of 20 s).
Peak lists were generated using the msConvert tool26 and submitted to the database search program Mascot (Matrix Science). The data was searched with oxidation (M) and carbamidomethyl (C) as variable modifications using a precursor‐ion and product‐ion mass tolerance of ±15 ppm and ±0.02 Da, respectively. The enzyme specificity specified was trypsin with up to two missed cleavages and all taxonomies in the Swiss‐Prot database (March 2016; 550,740 entries) were searched. A decoy database of reversed sequences was used to estimate the false discovery rates. To be considered for further analysis, identified peptides had to be top‐ranking and statistically significant (P < 0.05) according to the Mascot expect metric.
Analysis of XL‐MS data
Analysis of the XL‐MS data was performed with the pLink software27 pLink search parameters that differ from the default settings were as follows: precursor mass tolerance ±15 ppm, product‐ion mass tolerance ±20 ppm, variable modifications of oxidation (M) and carbamidomethyl (C), enzyme specificity of trypsin with up to two missed cleavages per chain. H12/D12‐DSS crosslinker settings were: crosslinking sites were Lys and protein N‐terminus, isotope shift 12.075 Da, xlink mass‐shift 138.068 Da, monolink mass‐shift 156.079 Da. For the H8/D8‐ADH crosslinker settings: crosslinking sites were Asp, Glu, and protein C‐terminus, isotope shift 8.050 Da, xlink mass‐shift 138.090 Da, monolink mass‐shift 156.100 Da. For the DMTMM crosslinker (side reaction in the ADH crosslinking) settings: crosslinking sites were Asp, Glu and protein C‐terminus to Lys, xlink mass‐shift ‐18.0106 Da, monolink mass‐shift 0 Da. The protein database used for searching consisted of all the NuRD components (CHD4, MTA1, MTA2, MTA3, HDAC1, HDAC2, HDAC3, GATAD2A, GATAD2B, RBBP4, RBBP7, MBD2, MBD3) plus the top 10 contaminants identified (from the Mascot search) in the samples. The default FDR of 5% was used and only peptides with scores ≤1 × 10−4 were considered for further analysis. Finally, all spectra were also manually verified; only crosslinks with at least four fragment ions on both the alpha‐ and beta‐chain each were retained for modelling.
Sucrose density gradient ultracentrifugation
Sucrose density gradients 2–25% (w/v) in 50 mM HEPES‐KOH pH 8.2, 150 mM NaCl were prepared in 12 mL ultracentrifugation tubes (Beckman Coulter) using the GraFix protocol,18 with 0.15% (v/v) glutaraldehyde in the 25% (w/v) sucrose buffer as the cross‐linking agent. Prepared gradients were left standing at 4°C for at least 1 h prior to usage. A glutaraldehyde‐free cushion of 600 µL of the 2% (w/v) sucrose buffer was placed on top of the gradients. The sample (200 µL) was then layered on top of the cushion and ultracentrifuged (186,000g, 4°C, 18 h). The gradients were fractionated as 200‐µL aliquots collected from the top, and each of these mixed with 20 µL of 1 M Tris pH 8.0 to deactivate remaining glutaraldehyde.
EM sample preparation
Samples of the RBBP4‐MTA1449‐715 subcomplex recovered from GraFix density gradient centrifugation had the sucrose diluted to an approximate final concentration of 0.1% (w/v) by buffer exchange using a Vivaspin 2 device (CTA, 20,000‐Da MWCO) (Sartorius Stedim), previously blocked with 0.1% (v/v) Tween‐20. The exchange buffer used was 50 mM HEPES KOH pH 8.2, 150 mM NaCl, 1 mM DTT. Finally, the sample was centrifuged at 5,000 g for 5 min to remove possible aggregates. Final concentration of the FLAG‐RBBP4/MTA1449–715 subcomplex was estimated to be ∼7 ng µL−1.
Negative‐stain EM
The RBBP4‐MTA1 complex (10 μL) was applied to a glow‐discharged, carbon‐coated 400‐mesh copper grid (GSCu400CC—ProSciTech). After an incubation time of 5 min, the grid was blotted and washed with five drops of distilled water, blotted again and subsequently stained with a 2% (w/v) uranyl acetate solution for one minute. Excess stain was then blotted away and the grid allowed to dry under air at ambient conditions. Images were acquired using a Tecnai T12 TEM operated at 120 kV and equipped with a Direct Electron LC‐1100 (4k × 4k) lens‐coupled CCD camera. Images were recorded at a nominal magnification of 52,000× and were binned by a factor of 2, resulting in a pixel size of 5.58 Å at the specimen level. Defocus values ranged from ‐1 to ‐2.5 μm.
EM image processing and 3D reconstruction
Data were processed using the RELION software package.19 Four hundred micrographs were recorded, a subset of which were used for manual picking of an initial 1730 particles. These were used to generate templates for autopicking, which yielded a dataset of 12,114 particles. Initial, manual inspection of the dataset (removal of obvious false positives) reduced this to 9000 particles, which were used to generate 90 reference free 2D classes. The same 9000 particles were used for initial 3D classification. For this a starting model was built by placing two copies of the monomeric RBBP4 crystal structure (PDB 4PBY) alongside each other in a way that they were touching end on. The model was sampled at 40 Å and low pass filtered in RELION to 50 Å. Ten 3D classes were generated.
A second round of 3D classification was performed, using a smaller but higher quality particle dataset. Particles falling into the 68 best classes from the initial 2D classification were grouped and subjected to a further, more stringent round of manual inspection, with poor quality particles (e.g., obvious aggregates, small particles) removed, leaving the 4000 best particles to be taken forward for 3D classification and refinement. This time the initial reference model was generated by refitting the RBBP4 monomers into the best 3D model from the first round of 3D classification, which again was converted to a MRC formatted map at 40‐Å resolution and low pass filtered to 50 Å in RELION. Four 3D classes were generated by unsupervised 3D classification. Class 1 contained the most particles (1709 particles) and the model generated from this class was refined further to obtain a final 3D envelope that was used for subsequent modeling of the complex [Fig. 4(C,D)]. All single particle image processing was performed with no symmetry assumed or imposed. No absolute hand determination was performed and, for subsequent model fitting, the handedness of the map was flipped as this appeared to give a fit more consistent with the data. The EM map will be deposited in the EMDB prior to publication.
Modeling of the RBBP4‐MTA1449‐715 complex
The RBBP4‐MTA1449‐715 model was generated by fitting the RBBP4 crystallographic dimer from 4PBY15 into the output EM envelope using the “fit in map” function of the CHIMERA software.28 A series of bioinformatics tools were used to analyse the MTA1491‐499 sequence and guide modeling. This included: a multiple sequence alignment (PRALINE22) across a range of taxa, secondary structure prediction (PORTER21), and coiled coil analysis using PAIRCOIL2.24 Predicted helices and short connecting loops were modeled in PYMOL and COOT.29 Crosslink distances were assessed on the basis of adhering to the following approximate distance restraints: ADH = 22 to 26 Å, DSS = 29 to 30 Å, and ZLXL = 12 to 14 Å.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
MJL and LB acknowledge the Australian Microscopy and Microanalysis Research Facility (AMMRF) for access to their facilities as part of this work.
References
- 1. Nguyen VQ, Ranjan A, Stengel F, Wei D, Aebersold R, Wu C, Leschziner AE (2013) Molecular architecture of the ATP‐dependent chromatin‐remodeling complex SWR1. Cell 154:1220–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tosi A, Haas C, Herzog F, Gilmozzi A, Berninghausen O, Ungewickell C, Gerhold CB, Lakomek K, Aebersold R, Beckmann R, Hopfner KP (2013) Structure and subunit topology of the INO80 chromatin remodeler and its nucleosome complex. Cell 154:1207–1219. [DOI] [PubMed] [Google Scholar]
- 3. Grune T, Brzeski J, Eberharter A, Clapier CR, Corona DF, Becker PB, Muller CW (2003) Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol Cell 12:449–460. [DOI] [PubMed] [Google Scholar]
- 4. Horton JR, Elgar SJ, Khan SI, Zhang X, Wade PA, Cheng X (2007) Structure of the SANT domain from the Xenopus chromatin remodeling factor ISWI. Proteins 67:1198–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yamada K, Frouws TD, Angst B, Fitzgerald DJ, DeLuca C, Schimmele K, Sargent DF, Richmond TJ (2011) Structure and mechanism of the chromatin remodelling factor ISW1a. Nature 472:448–453. [DOI] [PubMed] [Google Scholar]
- 6. Yoshida T, Hazan I, Zhang J, Ng SY, Naito T, Snippert HJ, Heller EJ, Qi X, Lawton LN, Williams CJ, Georgopoulos K (2008) The role of the chromatin remodeler Mi‐2beta in hematopoietic stem cell self‐renewal and multilineage differentiation. Genes Dev 22:1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. dos Santos RL, Tosti L, Radzisheuskaya A, Caballero IM, Kaji K, Hendrich B, Silva JC (2014) MBD3/NuRD facilitates induction of pluripotency in a context‐dependent manner. Cell Stem Cell 15:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo J, Su F, Chen D, Shiloh A, Gu W (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408:377–381. [DOI] [PubMed] [Google Scholar]
- 9. Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, Mansour AA, Caspi I, Krupalnik V, Zerbib M, Maza I, Mor N, Baran D, Weinberger L, Jaitin DA, Lara‐Astiaso D, Blecher‐Gonen R, Shipony Z, Mukamel Z, Hagai T, Gilad S, Amann‐Zalcenstein D, Tanay A, Amit I, Novershtern N, Hanna JH (2013) Deterministic direct reprogramming of somatic cells to pluripotency. Nature 502:65–70. [DOI] [PubMed] [Google Scholar]
- 10. Scarsdale JN, Webb HD, Ginder GD, Williams DC, Jr. (2011) Solution structure and dynamic analysis of chicken MBD2 methyl binding domain bound to a target‐methylated DNA sequence. Nucleic Acids Res 39:6741–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hong W, Nakazawa M, Chen YY, Kori R, Vakoc CR, Rakowski C, Blobel GA (2005) FOG‐1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA‐1. Embo J 24:2367–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, Bunker RD, Wirth U, Bouwmeester T, Bauer A, Ly‐Hartig N, Zhao K, Chan H, Gu J, Gut H, Fischle W, Muller J, Thoma NH (2011) Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell 42:330–341. [DOI] [PubMed] [Google Scholar]
- 13. Murzina NV, Pei XY, Zhang W, Sparkes M, Vicente‐Garcia J, Pratap JV, McLaughlin SH, Ben‐Shahar TR, Verreault A, Luisi BF, Laue ED (2008) Structural basis for the recognition of histone H4 by the histone‐chaperone RbAp46. Structure 16:1077–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, Fairall L, Schwabe JW (2013) Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell 51:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alqarni SS, Murthy A, Zhang W, Przewloka MR, Silva AP, Watson AA, Lejon S, Pei XY, Smits AH, Kloet SL, Wang H, Shepherd NE, Stokes PH, Blobel GA, Vermeulen M, Glover DM, Mackay JP, Laue ED (2014) Insight into the architecture of the NuRD complex: structure of the RbAp48‐MTA1 subcomplex. J Biol Chem 289:21844–21855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leitner A, Joachimiak LA, Unverdorben P, Walzthoeni T, Frydman J, Forster F, Aebersold R (2014) Chemical cross‐linking/mass spectrometry targeting acidic residues in proteins and protein complexes. Proc Natl Acad Sci USA 111:9455–9460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leitner A, Reischl R, Walzthoeni T, Herzog F, Bohn S, Forster F, Aebersold R (2012) Expanding the chemical cross‐linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol Cell Proteom 11:M111 014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stark H (2010) GraFix: stabilization of fragile macromolecular complexes for single particle cryo‐EM. Methods Enzymol 481:109–126. [DOI] [PubMed] [Google Scholar]
- 19. Scheres SH (2012) RELION: implementation of a Bayesian approach to cryo‐EM structure determination. J Struct Biol 180:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK (2005) Coupled folding and binding with alpha‐helix‐forming molecular recognition elements. Biochemistry 44:12454–12470. [DOI] [PubMed] [Google Scholar]
- 21. Pollastri G, McLysaght A (2005) Porter: a new, accurate server for protein secondary structure prediction. Bioinformatics 21:1719–1720. [DOI] [PubMed] [Google Scholar]
- 22. Simossis VA, Heringa J (2005) PRALINE: a multiple sequence alignment toolbox that integrates homology‐extended and secondary structure information. Nucleic Acids Res 33:W289–W294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kloet SL, Baymaz HI, Makowski M, Groenewold V, Jansen PW, Berendsen M, Niazi H, Kops GJ, Vermeulen M (2015) Towards elucidating the stability, dynamics and architecture of the nucleosome remodeling and deacetylase complex by using quantitative interaction proteomics. FEBS J 282:1774–1785. [DOI] [PubMed] [Google Scholar]
- 24. McDonnell AV, Jiang T, Keating AE, Berger B (2006) Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics 22:356–358. [DOI] [PubMed] [Google Scholar]
- 25. Smits AH, Jansen PW, Poser I, Hyman AA, Vermeulen M (2013) Stoichiometry of chromatin‐associated protein complexes revealed by label‐free quantitative mass spectrometry‐based proteomics. Nucleic Acids Res 41:e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, Hoff K, Kessner D, Tasman N, Shulman N, Frewen B, Baker TA, Brusniak MY, Paulse C, Creasy D, Flashner L, Kani K, Moulding C, Seymour SL, Nuwaysir LM, Lefebvre B, Kuhlmann F, Roark J, Rainer P, Detlev S, Hemenway T, Huhmer A, Langridge J, Connolly B, Chadick T, Holly K, Eckels J, Deutsch EW, Moritz RL, Katz JE, Agus DB, MacCoss M, Tabb DL, Mallick P (2012) A cross‐platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 30:918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang B, Wu YJ, Zhu M, Fan SB, Lin J, Zhang K, Li S, Chi H, Li YX, Chen HF, Luo SK, Ding YH, Wang LH, Hao Z, Xiu LY, Chen S, Ye K, He SM, Dong MQ (2012) Identification of cross‐linked peptides from complex samples. Nat Methods 9:904–906. [DOI] [PubMed] [Google Scholar]
- 28. Yang Z, Lasker K, Schneidman‐Duhovny D, Webb B, Huang CC, Pettersen EF, Goddard TD, Meng EC, Sali A, Ferrin TE (2012) UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J Struct Biol 179:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Cryst D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information