

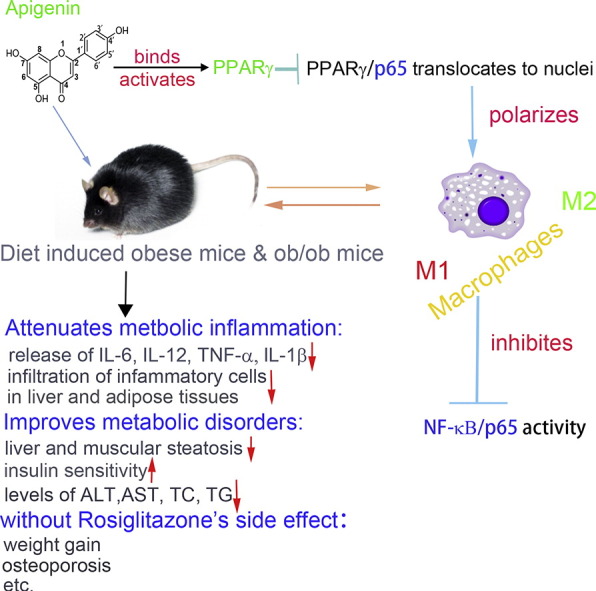
Abstract
PPARγ has emerged as a master regulator of macrophage polarization and is the molecular target of the thiazolidinedione drugs. Here we show that apigenin binds and activates PPARγ by acting as a modulator. Activation of PPARγ by apigenin blocks p65 translocation into nuclei through inhibition of p65/PPARγ complex translocation into nuclei, thereby decreasing NF-κB activation and favoringM2 macrophage polarization. In HFD and ob/ob mice, apigenin significantly reverses M1 macrophage into M2 and reduces the infiltration of inflammatory cells in liver and adipose tissues, as well as decreases the levels of pro-inflammatory cytokines, thereby alleviating inflammation. Strikingly, apigenin reduces liver and muscular steatosis, decreases the levels of ALT, AST, TC and TG, improving glucose resistance obviously. Unlike rosiglitazone, apigenin does not cause significant weight gain, osteoporosis et al. Our findings identify apigenin as a modulator of PPARγ and a potential lead compound for treatment of metabolic disorders.
Abbreviations: Api, apigenin; Rosi, rosiglitazone; HFD, high fat diet; ND, normal diet; IHC, Immunological Histological Chemistry; PPARγ, peroxisome proliferator activated receptor γ; Fizz1, found in inflammatory zone (FIZZ)1; Arg1, arginase; CD206, mannose receptor C type 1; TZD, thiazolidinedione; H&E, hematoxylin and eosin; AnV, Annexin V-FITC; PI, propidium iodide; ITC, isothermal titration calorimetry; ROS, reactive oxygen species; NO, nitric oxide; SPF, specific-pathogen-free; ALT, alanine aminotransferase; AST, aspartate aminotransferase; SEM, standard error of mean; EMSA, electrophoretic mobility shift assay; HFD, high fat diet; WAT, white adipose tissue; ATM, adipocyte tissue macrophages; NF-κB, nuclear factor kappa B; TC, total cholesterol; TG, triglycerides; ITTs, insulin tolerance tests; CT, computer tomography; SPPARMs, selective PPAR modulators; LBD, ligand binding domain; DBD, DNA binding domain
Keywords: Obesity-related inflammation, Macrophage polarization, Apigenin, PPARγ, NF-κB
Graphical Abstract
Highlights
-
•
Apigenin binds and activates PPARγ and significantly reverses the polarization of macrophages from M1 to M2 phenotype.
-
•
Activation of PPARγ by apigenin blocks p65 translocation through inhibiting p65/PPARγ complex translocation into nucleus.
-
•
Apigenin significantly attenuates metabolic inflammation and disorders without causing some side effects as TZD drugs do.
PPARγ is the molecular target of the thiazolidinedione drugs to treat type II diabetes. However, TZD drugs have some side effects including cardiovascular failure, liver toxicity, bone fractures and potential carcinogenesis, which have greatly limited their clinical use. Here, we find apigenin, a flavonoid molecule abundant in various fruits and vegetables, can control macrophage fate to inhibit inflammation and metabolic syndrome without causing some side effects as TZD drugs. Further study indicates that apigenin can target PPARγ with a range of beneficial effects and may represent a lead compound for developing new therapies against metabolic disorders.
1. Introduction
Nutrient excess and adiposity leads to chronic low-grade inflammation, which is referred to as obesity-related inflammation (Xu et al., 2003b). Obesity-related inflammation acts as a key pathogenic link between obesity and obesity-associated metabolic disorders, including insulin resistance (Xu et al., 2003a), type 2 diabetes (Duncan et al., 2003) and cancer (Howe et al., 2013). Thus, resolving the inflammation is one potential strategy to treat metabolic syndromes. Thus far, several drugs, such as metformin (Dinarello, 2010) and thiazolidinedione have been proven to restrain low-grade inflammation and therefore to treat insulin-resistance and correlated physiological functional disorders. However, further efforts are needed to develop newer and safer therapeutics to ameliorate obesity-related inflammation and reverse metabolic disorders.
PPARγ, which belongs to the PPAR family of ligand-inducible transcription factors, has been well documented to play a central role in adipogenesis and low-grade inflammation. PPARγ is implicated in the regulation of immunological events, playing an important role in mediating the differentiation and activation of immune cells, as well as modifying cytokine expression patterns and cell fates, thereby remodeling the immune balance (Cipolletta et al., 2012). In particular, PPARγ has been recognized as a pivotal anti-inflammatory regulator in atherosclerosis primarily through regulating the differentiation and functional polarization of macrophages (Bouhlel et al., 2007). Macrophages are heterogeneous and plastic, and there are at least two major macrophage populations: those in a predominantly M1-polarised pro-inflammatory state and those in a predominantly M2-polarised anti-inflammatory state (Chinetti-Gbaguidi and Staels, 2011). M1 cells are efficient producers of effector molecules (ROS and NO) and inflammatory cytokines (IL-1R, TNF, IL-6, etc.) and express typical phenotypic molecules, such as CD80 and CCR7. In contrast, the various forms of M2 cells share a distinct major signature with low IL-12, low IL-23, and high IL-10 and generally have high levels of scavenger, mannose, and galactose-type receptors, and arginine metabolism within these cells is shifted to production of ornithine and polyamines via arginase. M2 cells have been shown to express high levels of certain genes, such as chitinase-like Ym1, found in inflammatory zone (FIZZ)1 (Fizz1), arginase (Arg1) and mannose receptor C type 1 (CD206); these have become classical markers of M2 cells. M1 and M2 cells have distinct chemokine and chemokine receptor repertoires and therefore orchestrate different immune responses (Mantovani et al., 2004). Manipulation of M1/M2 homeostasis has been shown to be an effective strategy for clinical treatment of some inflammatory diseases. PPARγ activation can skew macrophages towards an anti-inflammatory M2 phenotype, resulting in inhibition of inflammation. Due to the role of PPARγ in macrophage polarization and anti-inflammation, PPARγ ligands have been used to treat metabolism-related inflammation and have shown significant anti-inflammatory therapeutic activity. Full agonist of PPARγ refers to a ligand which can bind to LBD domain with high-affinity and activate PPARγ thoroughly. For example, administration of pioglitazone, a full agonist of PPARγ, which can reduce the expression of IL-1β, IL-6, MCP-1, and TNF-α in peritoneal macrophages (Dasu et al., 2009), while rosiglitazone, another full agonist, upregulates the production of the anti-inflammatory molecule adiponectin, and thus decreases insulin resistance (Yang et al., 2002).
Ongoing work at developing ligands and modulators of PPARγ is focused on harnessing its anti-inflammatory properties. In recent years, some PPARγ agonists with anti–inflammatory effects (pioglitazone, Sitagliptin metformin/rosiglitazone combination) have already completed clinical trials. However, even with such promising therapeutic activity, the side effects of thiazolidinedione (TZD) drugs include cardiovascular failure, liver toxicity, bone fractures and potential carcinogenesis, these have greatly limited their clinical use (Lehrke and Lazar, 2005). Therefore, much attention has recently been paid to further optimization of the PPARγ ligands' structures to decrease or abrogate their side effects. In particular, exploration of natural compounds represents one promising strategy for developing new, safer ligands or modulators of PPARγ (Doshi et al., 2010). Apigenin (Api, 4,5,7-trihydroxyflavone) is a naturally occurring plant flavonoid abundant in various fruits and vegetables (Havsteen, 2002). It has lately gained attention as a beneficial and healthy compound because of its various biological effects and low intrinsic toxicity. Moreover, Api has been demonstrated to possess distinct anti-inflammatory activity in chronic inflammation (Choi et al., 2014) and skin inflammation (Byun et al., 2013). Api is also an inhibitor of NAD + ase CD38 and improves metabolic syndrome (Escande et al., 2013). In addition, Nicholas et al. has found that Api can specifically modulate NF-kB in macrophages by suppression the phosphorylation of p65 (Nicholas et al., 2007). It is noteworthy that Api might be a ligand of PPARγ via structure-based virtual screening (Salam et al., 2008a, Mueller et al., 2008). Hence, further study of Api and the underlying mechanisms related to the PPARγ pathway has potential therapeutic implications. In the current study, we identify Api as an effective ligand of PPARγ in macrophages. Importantly, Api can significantly attenuate obesity-related inflammation and metabolic disorders in high-fat diet-induced mice and ob/ob mice. Furthermore, unlike Rosi, a full ligand-type agonist of PPARγ, Api does not exhibit some adverse effects, such as obvious weight gain, osteoporosis, the increase of small adipocytes in white adipose tissue (WAT) and the accumulation of triglycerides in the serum of obese mice.
2. Materials and Methods Results
2.1. Reagents
Api (5,7-dihydroxyflavone, PubChem CID: 5281607, purity > 99%, chemical structure shown in Fig. 1a, purchased from Zelang biotechnology company (Nanjing, China) was dissolved in 100% DMSO. The final DMSO concentration in cell culture did not exceed 0.1% throughout the study. Rosi (PubChem CID: 77999) and GW9662 (PubChem CID 644213) were purchased from Sigma (St. Louis, MO). DMEM and RPMI1640 media were purchased from Gibco (Grand Island, NY). Penicillin and streptomycin, HRP-conjugated Goat Anti-Mouse IgG (H + L), FITC-coupled secondary antibody (be), Cy3-labeled Goat Anti-Mouse IgG (H + L), neutral red, CCK-8 and MTT are from Beyotime (Haimen, Jiangsu, China). TNF-α, IL-10, CCL2 and IL-1β ELISA assay kits were purchased from eBioscience (San Diego, CA). Alanine/aspartate aminotransferases (ALT/AST) assay kits, TC, TG, glucose and insulin kit were from Jiancheng Biology Institution (Nanjing, Jiangsu, China). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA). PPRE-Luc plasmid and dual-luciferase reporter assay systems were from Promega (Madison, WI, USA).
Fig. 1.

Api attenuates obesity-related inflammation. (a) The chemical structure of Api and Rosi. (b–c) HFD-fed mice treated with the vehicle (0.1% DMSO), indicated doses of Api for 21 days, the effects of Api or Rosi on body weight and the statistical analysis at 21st day, (n = 9). All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. **P < 0.01 compared with vehicle. (d) Representative H&E staining showed adipose tissue morphology of the HFD-fed mice treated with the vehicle (0.1% DMSO), indicated doses of Api for 21 days, original magnification × 400, n = 6. Arrows indicated the inflammatory cells in adipose tissue (n = 9). (e) Quantification of the infiltration of inflammatory cells into adipose tissue from the HFD-fed mice treated with the vehicle (0.1% DMSO), indicated doses of Api for 21 days, for five to eight sections/400 × field, five to six fields/gland/mouse, score according to the grade of lesion, slight (0.5), mild (1), moderate (2), severe (3), profound severe (4) and normal (0), (n = 6). All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. **P < 0.01 compared with vehicle. (f–i) Inflammatory cytokine IL-12, TNF-α, IL-6 and IL-1β in the serum of HFD-fed mice treated with the vehicle (0.1% DMSO), indicated doses of Api for 21 days were measured by ELISA assays according to manufacturer's instructions (n = 9). All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle. (j–l) 3 month years old male ob/ob mice were injected vehicle (0.1% DMSO) or 30 mg/kg Api for 21 days, the CCL2, IL-1β and IL-10 in the serum were measured by ELISA assays according to manufacturer's instructions (n = 6). All values are expressed as mean ± SEM. Statistical analysis is based on the Student's t-test. *P < 0.05, **P < 0.01 compared with vehicle.
2.2. Mice Treatment
Male C57BL/6J mice (3–4 weeks old) and male ob/ob mice were purchased from Animal Genetics Research Center of Nanjing University (Nanjing, China) and housed in a specific-pathogen-free (SPF) facility. Mice, starting at 3–4 weeks old, were randomly divided into four groups (n = 9 per group). Mice were fed for 16 weeks with either a normal chow diet (ND) consisting of 4.5% fat or a high-fat diet (HFD) (D12492, 60% fat, 20% carbohydrate, 20% protein, total 5.24 kcal/g; Research Diets Inc., New Brunswick, NJ). 19 weeks year-old HFD mice were grouped and injected with Api (10 mg/kg, 30 mg/kg or 50 mg/kg) (Liu et al., 2005, Dou et al., 2013), 10 mg/kg Rosi or vehicle alone (saline containing 0.1% DMSO) intraperitoneally daily for 21 days. Mice were weighed daily until sacrificed under anesthesia using diethylether. In addition, three month-old male C57BL/6J ob/ob mice were injected with 30 mg/kg Api or vehicle alone (saline containing 0.1% DMSO) intraperitoneally daily for 21 days. Animal welfare and experimental procedures were followed in accordance with the Guide for Care and Use of Laboratory Animals (National Institutes of Health, the United States) and the related ethical regulations of Nanjing University.
2.3. Isolation and Purification of Peritoneal Macrophages
Normal resident peritoneal cells of the Male C57BL/6J mice treated with Api (10 mg/kg, 30 mg/kg or 50 mg/kg), 10 mg/kg Rosi or vehicle alone for 21 days were obtained by peritoneal washing with 20 mL Dulbecco's PBS (D-PBS) containing 2% FBS and 40 mg/mL gentamicin. After centrifugation at 350g for 5 min, red blood cells were lysed in ACK buffer and mononuclear cells were re-suspended in complete medium (DMEM, 10% FBS, 2 mM l-glutamine, 100 units/mL penicillin and 100 units/mL streptomycin) and incubated at 37 °C for 2 h in plastic culture plates. Then, the non-adherent cells were removed, and the adherent cells were cultured in complete medium with different stimuli.
2.4. Isolation and Purification of Adipocyte Tissue Macrophages
Epididymis fat was excised and minced in Hanks' Balanced Salt Solution (HBSS; Invitrogen) containing calcium, magnesium and 0.5% BSA. Collagenase (Type II; Sigma-Aldrich, St Louis, MO) was added to a final concentration of 1 mg/mL and tissue suspensions were incubated at 37 °C for 20–30 min with constant shaking. The resulting cell suspensions were filtered through a 100-μm filter and centrifuged at 500g for 10 min to separate floating adipocytes from the SVC-containing pellet. The SVC was rinsed with PBS for two times and centrifuged at 500g for 10 min. The pellet cells resuspended in FACS buffer at a concentration of 7 × 106 cells/mL for fluorescence activated cells sorting or purification or for culture and live confocal microscopy studies. First the cells were labeled with mouse CD11b MicroBeads (MACS, 130-049-60) are purchased from Miltenyi Biotech. Then the cell suspension is loaded onto a MACS column. The magnetically labeled CD11b+ cells are retained on the column. The unlabeled cells run through and this cells fraction is depleted of CD11b+ cells. After removal of the Colum from the magnetic field, the magnetically retained CD11b+ cells can be eluted as the positively selected cell fractions. For RNA isolation, isolated cells were homogenized in QIAzol Lysis Reagent (Qiagen).
2.5. Hematoxylin and Eosin (H&E) and Immunological Histological Chemistry (IHC) Detection
After the mice were sacrificed, the livers, skeletal muscles and adipose tissues were removed and subsequently fixed in phosphate-buffered 10% formalin, and embedded in paraffin blocks. A section from each paraffin block was stained with hematoxylin and eosin (H&E) to examine the pathologic structures of the tissues and to score the inflammation cells infiltration for five to eight sections/400 × field, five to six fields/gland/mouse, score according to the grade of lesion, slight (0.5), mild (1), moderate (2), severe (3), profound severe (4) and normal (0), (n = 6). Images were obtained from fluorescence microscopy. The expression of CD68 (novus, A-5) was assessed by IHC staining of adipose tissue sections with antibody against the mouse CD68 antigen.
2.6. Cytokine Assay by ELISA
The TNF-α, IL-10, CCL2, IL-12 and IL-1β concentrations in 200 μL serum, the adiponectin concentration in 10 μL serum or the supernatants of 1 million macrophages were assessed using a standard sandwich ELISA according to the instruction manual.
2.7. Cell Viability Assay and Cell Death Assay
1.0 × 104 ANA-1 or RAW264.7 macrophage cells were seeded in 96-well plates, and the next day (at 65–75% confluence) cells were treated with various concentrations (1–100 μM) of Api for 24 h, in a set of four replicates including a control (0.1% DMSO). Adherent cells were assayed by MTT assay while suspension cells were assayed by the CCK-8 method. Api-induced cell death was determined by staining cells with Annexin V-FITC (AnV) and propidium iodide (PI). The cells were then collected and analyzed on a FACS Calibur cytometer using Cellquest software (Becton Dickinson) and a total of 10,000 cells per sample were analyzed in a diparametric plot (FL1 for log FITC and FL3 for log PI) to determine the percentage of phosphatidylserine (PS)-externalized AnV + PI − (high FITC/low PI) apoptotic cells and PI + (low FITC/high PI-plus-high FITC/high PI) necrotic cells.
2.8. Cell Models and Treatment
ANA-1 cells and RAW264.7 cells (China Center for Type Culture Collection) were cultured in the RPMI1640 and DMEM media, respectively, supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 units penicillin and 100 units streptomycin. In order to mimic the macrophage phenotype of obese mice in vivo, we established two cell models (M1 and M2) in vitro as shown below.
2.8.1. M1 Model
The M1 activation of macrophages was induced by LPS (500 ng/mL) for 24 h. Then the effect of Api on the function and the expression of M1 markers were assayed after treatment with Api (7.5 μM) or Rosi (10 μM) in the presence or absence of LPS.
2.8.2. M2 Model
The M2 macrophage cell model was induced by IL-4 (10 ng/mL) in cell lines and murine peritoneal macrophages for 24 h, in conjunction with treatment with Api (7.5 μM) or Rosi (10 μM) prior to various assays.
2.9. Co-culture Adipocyte and Macrophage
3T3-L1 adipocytes were cultured in DMEM and added 0.5 mM IBMX, 1 μM dexamethasone, 10 μg/mL insulin for 48 h to induce mature. Then macrophages were placed into a well containing mature adipocytes. 24 h after co-culturing, macrophages and adipocytes were collected separately for various assays.
2.10. Surface Marker Analysis
Primary peritoneal macrophages, ANA-1 cells and RAW264.7 cells were treated with IL-4 or LPS in the presence or absence of Api for 24 h. Cells were analyzed using Anti-mMGL1/2 PE conjugated Goat IgG (FAB4297P) are bought from R&D company. Anti-mouse CD80 (B7-1) PE (clone: 16-10A1), PE-conjugated anti-mouse CCR7 (clone: 4B12), anti-mouse MHC Class II (I-A) PE (cone: NIMR-4) and corresponding isotype controls were purchased from eBioscience (San Diego, CA).
2.11. Reactive Oxygen Species (ROS) Generation
ROS production was examined by DCFH-DA probe (Beyotime, China, noS0033). After the stimulation medium was removed, cells were washed twice with 2 mL warmed D-PBS, after which 1 mL D-PBS containing 10 μM DCFH-DA was added to the cells and incubated for 20 min at 37 °C. Cells were then washed with D-PBS and subjected to flow cytometry analysis.
2.12. Phagocytosis Assays
Phagocytosis assays were conducted using fluorescent red latex beads (1 μM diameter, L-2778, Sigma-Aldrich). Latex beads were opsonized with complete medium (10% FBS in PRMI 1640) for 1 h at 37 °C before the experiments. Opsonized beads were added to Api-treated cells at a ratio of 10:1 and incubated at 37 °C for 2 h. Phagocytosis was stopped with the addition of 1 mL ice-cold sterile PBS. Cells were harvested and washed in ice-cold PBS three times, and then subjected to the flow cytometry analysis. For each experiment, the data was analyzed on a FACS Calibur cytometer using Cellquest software (Becton Dickinson, Franklin Lakes, NJ, USA). Dead cells were excluded by forward and side scatter characteristics. Statistics presented are based on 10,000 events gated on the population of interest.
2.13. Detection of the Phagocytosis by Neutral Red Dye Uptake
Phagocytosis of macrophages was measured by the neutral red uptake method as described (Weeks et al., 1987). Briefly, the primary macrophages (1 × 106 cells/mL) from mice were incubated in a 96-well flat-bottomed microtiter plate 100 μL/well for 2 h at 37 °C in a 5% CO2 humidified incubator. After discarding the supernatant, 100 μL of 0.1% neutral red solution in PBS was added. Following 1 h incubation of the cell plates at 37 °C, each well was carefully rinsed three times with PBS and 100 μL cell lysis buffers [100% ethanol and 0.1 M acetic acid (1:1 v/v)] was added overnight at 4 °C. Optical density was measured at a wavelength of 490 nm. The absorbance represented the phagocytosis capacity of the macrophages.
2.14. Quantification of Nitrite in Cell Culture Supernatants
Cell culture supernatants from macrophages that had been cultured with or without the indicated stimuli were analyzed. The concentration of nitrites was assayed in duplicate by a standard Griess reaction (Guevara et al., 1998).
2.15. Western Blotting
Total protein concentration of serum was determined by Pierce BCA assay. The immunoblot experiments were performed as previously described (Bao et al., 2007). The total protein (50 μg/lane) was electrophoresed on 10% SDS-polyacrylamide gels and then electro-transferred onto a poly vinylidene fluoride membrane. Antibody against NF-κB p65 (Santa Cruz Biotechnology, Inc., no sc-8008), PPARγ (Santa Cruz Biotechnology, Inc., no sc-7273), IκBα (S36) polyclonal antibody (Bioworld Technology, Inc. no BS3601) and IκBα (phospho-S32/S36) polyclonal antibody (Bioworld Technology, Inc. no BS4105) p-p65 polyclonal antibody (Bioworld Technology, Inc. no BS4737) were used, and the intensities of the bands were quantified by Image J software.
2.16. ALT/AST Activity Assay
ALT and AST levels in serum from C57BL/6J mice, which are generally associated with hepatic steatosis and/or inflammation (Sheth et al., 1997), were assayed using ALT and AST activity assay kits. The absorbance at 510 nm was obtained with a micro-plate reader model 680 (Bio-Rad Laboratories, Hercules, CA, USA).
2.17. Isothermal Titration Calorimetry (ITC)
ITC experiments on the alpha subtype were performed at 25 °x using a MicroCal ITC200 microcalorimeter (MicroCal Inc., Northampton, MA, USA). Protein was extensively dissolved in a buffer of PBS (pH = 7.4) and the PBS buffer was used to dilute the Api stock solutions (185 mM in DMSO). DMSO was added to the protein solution in the same percentage of the ligand solution (below 0.05%). Protein solution (9.25 μM) was added to the sample cell and the ligand solution (10 times more concentrated than the protein) was injected into the cell in 19 aliquots of 20 μL for 4 s (the first injection was 0.4 μL for 0.8 s) with delay intervals between injections of 180 s. Reference titration of ligand into buffer was used to correct for heat of dilution. The syringe stirring speed was set at 1000 rpm. The thermodynamic data were processed with Origin 7.0 software provided by MicroCal. To correct for any discrepancies in the baseline outlined by the software, a manual adjustment was performed.
2.18. Quantitative Real-Time PCR
Total RNA was extracted from macrophages and reverse-transcribed to cDNA using the BioTeke supermoIII RT Kit (Bioteke Corporation, Beijing, China). The mRNA expression of mouse PPARγ, CD36 and Arg1 (both PPARγ-targeted genes), the M1 markers such as CCL3, IL-12β, NOS2, IL-6, IL-12, CCR2 and the M2 markers such as Arg1, CD206, Ym1, Fizz1 were detected by quantitative RT-PCR, which was performed with the iCycler thermocycler system and iQ5 optical system (Bio-Rad) using SYBR green I dye (Bio-Rad). Threshold cycle numbers were obtained using iCycler thermocycler system software version 1.0. PCR cycling conditions as follows: 1 cycle of 94 °C for 5 min followed by 40 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 45 s. The primers used are listed in Table s5. Relative mRNA expression of target genes was obtained by normalizing to the control group and the level of β-actin using the 2− ΔΔCt method (Livak and Schmittgen, 2001).
2.19. Construction of N-Domain Deletion Mutants of PPARγ Expression Plasmids
Vectors pcDNA3.1 (−)-PPARγ2 (aa1–505), pcDNA3.1 (−)-PPARγ2-DBD-AF2 (aa137–505), pcDNA3.1 (−)-PPARγ2-hinge-AF2 (aa211–505) and pcDNA3.1 (−)-PPARγ2-LBD (aa319–505) were constructed. The PCR primers are shown in Table s5.
2.20. Site-Directed Mutagenesis
The mutants K263R, K265R, L340F and S342T were generated using Plenti-hPPARγ plasmid as a template by polymerase chain reaction with a site-directed mutagenesis kit. The primers listed in Table s5 were used for Lys to Arg at positions 263 and 265, and Leu mutated to Phe, Ser mutated to Thr at positions 240 and 342 separately. The changes in the nucleotide bases are underlined. The site-specific mutations and absence of any spurious mutations were confirmed by DNA sequencing at Genscript Company.
2.21. Expression of Recombinant PPARγ-his
The cDNA coding sequence for the PPARγ protein (mouse, NCBI accession NP_035276) including SacI/Xbal was amplified by PCR, and then inserted into a pCzn1 plasmid in frame to the C-terminal of 6 × his tag. The inserted PPARγ in pCzn1-PPARγ was sequenced. PPARγ-his was expressed in Arctic Express™ transformed with the pCzn1-PPARγ expression plasmid. A culture was grown in Luria–Bertani (LB) medium containing 50 μg/mL ampicillin at 37 °C until the OD600 nm was 0.6–0.8, at which point isopropyl-β-d-thiogalactopyranoside (IPTG) (0.5 mM, final) was added to induce protein expression.
2.22. Purification of Recombinant PPARγ-his
Cells were harvested 12 h later and resuspended in Lysis buffer containing 20 mM Tris-HCL, 1 mM phenylmethylsulfonyl fluoride (PMSF) and bacteria protease inhibitor cocktail (pH 8.0), then sonicated 100 times for 12 s. Samples were centrifuged for 20 min at 10,000g and 4 °C. Precipitated material (inclusion bodies) containing PPARγ-his was washed three more times by resuspending the material in 20 mM Tris, 1 mM EDTA, 2 M Urea, 1 M NaCl, 1% Triton X-100 (pH 8.0), followed by centrifugation. Pellets from the final wash were resuspended in buffer A [20 mM Tris (pH 7.9), 5 mM DTT and 8 M Urea] and protein was extracted by overnight nutation at 4 °C. After centrifugation to remove the remaining insoluble material, samples containing PPARγ-his were aliquoted and stored at − 80 °C. PPARγ-his can be further purified by dialysis and Ni-NTA–agarose affinity chromatography. Recombinant PPARγ-his was eluted by 250 mM imidazole using the protocol as described elsewhere. The purification was identified by Coomassie Brilliant Blue staining or western blotting by using PPARγ antibody (Santa Cruz Biotechnology, Inc., no sc-7273).
2.23. Electrophoretic Mobility Shift Assay (EMSA)
The EMSA method was used to characterize the binding activities of PPARγ transcription factors in nuclear extracts using the LightShift™ Chemiluminescent EMSA Kit (Thermo Fisher Scientific, Waltham, MA, USA) as described by the manufacturer. Ana-1 cells were pretreated with Api (1 μM, 5 μM, 10 μM) or DMSO (10 μL/L) and 0.5 μg/mL LPS for 24 h and then the nuclear contents were extracted. Double-stranded oligonucleotides containing either the consensus transcription factor binding site for the PPRE sense (5′-CAAAACT AGGTCAAAGGTCA-3′) or the PPRE antisense (5′-TGACCTTTGACCT AGTTTTG-3′) labeled at the 3′-end with biotin were synthesized from Promega. The nuclear protein-biotin-labeled oligonucleotide complexes were separated from free biotin-labeled oligonucleotide by electrophoresis through 10% ploy-acrylamide gels and then transferred to a nylon membrane (Roche Applied Science). The biotin end-labeled DNA was detected using the Streptavidin-Horseradish Peroxidase Conjugate and Chemiluminescent Substrate. Cold competition was done by adding a 100-fold excess of specific unlabeled double-stranded probe to the reaction mixture.
The ALT/AST, TC, TG, glucose and insulin levels were assayed according to manufacturer's instructions. Arg1 activity was analyzed using the previously described method (Yang et al., 2012).
2.24. PPARγ Gene-Reporter Luciferase Assay
HEK293 cells were transfected with pIRES-hPPARγ/PPRE-Luc and pRL-control using Lipofectamine 2000 transfection reagent. As an internal control reporter, the plasmid pRL containing Renilla luciferase cDNA was used. After 24 h, luciferase activities were measured using the dual-luciferase reporter assay system. Renilla luciferase activity was normalized to firefly luciferase activity.
2.25. Molecular Modeling of Api in the PPARγ-Binding Site
The crystal structure of human PPARγ was obtained from the RCSB protein databank (PDB code: P37231). Autodock 4.2 and PyRx 0.5 programs were employed for virtual screening and the docked models were analyzed using PyMOL. We used Perl scripts to identify the polar and apolar interactions of the amino acid residue side chains with a distance of 4.0 Å between atoms.
2.26. RNA Extraction, Illumina Sequencing and Analysis
The peritoneal macrophages were isolated from the 19 weeks old ND and HFD mice firstly and lysed using Trizol reagent. RNA content and quality were assessed with an Agilent 2100 Bio analyzer, and RNA integrity was confirmed by electrophoresis on 1% agarose gels containing formaldehyde. Total RNA from three biological replicates were pooled to generate one lane for each cDNA library construction. After extracting the total RNA from the samples, mRNA extracts (∼ 20 μg RNA) were pooled and enriched using oligo (dT) magnetic beads. After adding the fragmentation buffer, the mRNA was cleaved into short fragments of about 200 bp. The first strand cDNA was synthesized by random hexamer-primer using the mRNA fragments as templates. Then the buffer, dNTPs, RNase H and DNA polymerase I were added to synthesize the second strand. The double strand cDNA was purified with QiaQuick PCR extraction kit and washed with EB buffer for end repair and single nucleotide A (adenine) addition. Finally, sequencing adaptors were ligated to the fragments. The required fragments were purified by agarose gel electrophoresis and enriched by PCR amplification. The library products were ready for sequencing analysis via Illumina HiSeq™ 2000, which was performed by “HuaDa Gene”. Clean reads were mapped to reference sequences using OAPaligner/SOAP2·The gene expression level was calculated by using RPKM (reads per exon kilo base per million mapped sequence reads) (Mortazavi et al., 2008). If there was more than one transcript for a gene, the longest one was used to calculate its expression level and coverage.
All reads were realigned to the NCBI m37 version of the mouse genome assembly using the Bowtie short read alignment program considering the 25 chromosome assemblies. Only uniquely mapping reads were used in this analysis. Differentially expressed genes (DEGs) between two samples were identified under the strict algorithm as described by Audic et al. (Audic and Claverie, 1997). The p-value corresponded to differential gene expression test. The False discovery Rate (FDR) method was used to determine the threshold of p-value in multiple tests. A FDR ≤ 0.001 and an absolute value of log2-Ratio ≥ 1 were used as the thresholds to judge the significance of gene expression difference.
2.27. Indirect Immunofluorescence and Microscopy
Cells were rinsed in ice-cold PBS followed by 15 min incubation with 4% paraformaldehyde at room temperature. Cell were then briefly rinsed with PBS, incubated for 5 min with 0.2% Triton-100, and washed 3 times, 5 min every time with PBS. Nonspecific binding sites were blocked with 5% BSA for 1 h. Primary antibodies PPARγ (81B8) Rabbit mAb (Cell Signaling Techn no#5468) and NFκB p65 (F-6) (Santa Cruz Biotechnology, Inc., no SC-8008) were diluted at 1:200, and incubated overnight at 4 °C. Following incubation in cells were washed 3 times, 5 min every time with PBST, and incubated with FITC-coupled secondary antibody (be) and Cy3-labeled Goat Anti-Mouse IgG (H + L) (Beytotime, China, diluted at 1:500) for 1 h at RT. Nuclei staining was performed by incubating the cells with DAPI (Beyotime, China) before mounting the processed coverslips onto ethanol-cleaned glass slides using Dabco mountant. The specimens were viewed with an inverted confocal microscope (Olympus, Japan). Image analysis was done with Image J software.
2.28. Statistic Analysis
All of the quantification of the flow data was based on the analysis of at least three times from at least three mice. The data were expressed as the mean ± standard error of mean (SEM). The statistical analysis was performed by the Student's t-test when only two value sets were compared. A one-way ANOVA followed by a Dunnett's test was used when the data involved three or more groups, as indicated in the Figure legends. All of the statistical tests and graphical presentation were performed using the Prism 5.0 software (GraphPad, San Diego, CA). P < 0.05, P < 0.01 or P < 0.001 were considered statistically significant and indicated by *, ** or ***, respectively.
3. Results
3.1. Api Attenuates Obesity-Related Inflammation.
Accumulating evidence suggests that obesity causes chronic low-grade inflammation (Ouchi et al., 2011). In order to investigate the mechanisms for obesity-induced inflammation, 3–4 weeks male C57BL/6 J mice were fed with a high fat diet (HFD) for 16 weeks, consequently leading to a significant increase in body weight compared with control mice (Fig. S1a). The fat/body weight index has been significantly increased by HFD fed for 12 weeks compared with normal diet (ND) mice (Fig. S1b). Consistent with the increase in body weight, the level of the pro-inflammatory cytokine TNF-α (Fig. S1c) and the chemokine CCL2 (Fig. S1d) in the serum of HFD-fed mice was significantly increased in a time-dependent manner, suggesting that continual HFD feeding triggers sustained inflammation along with obesity development. Therefore, 16 weeks HFD-fed mice can be used to screen small molecules that may regulate the obesity-related inflammation.
Api, a natural plant flavonoid, has been reported to inhibit the secretion of IL-1β and TNF-α as well as regulate the synthesis of prostaglandin and NO in vitro (Ha et al., 2008). However, the in vivo effect of Api on obesity-related inflammation and its potential as an anti-inflammatory agent is still unknown. To address this question, the 16 weeks HFD male C57BL/6J mice was used, with or without intraperitoneal Api treatment. Meanwhile, Rosi (Rosi) was used as a positive control. As shown in Fig. 1b and c, the body weight of HFD mice in the Rosi-treated group increased, but was not affected by Api even at different doses. However, the fat/body weight index of HFD mice was not affected by Api or Rosi (Fig. S2a). Histological examination and IHC staining indicated that adipose tissues of HFD mice were infiltrated with inflammatory cells, and higher dosage of Api administration inhibited their infiltration (Fig. 1d–e, Fig. S3). The levels of pro-inflammatory cytokines IL-12, TNF-α, IL-6as well as IL-1β (Fig. 1f–i) were significantly reduced by 30 mg/kg or 50 mg/kg Api in a dosage-independent manner. In addition, the chemokine CCL2 in mouse serum (Fig. S2b) were all reduced by 30 mg/kg Api. Together, these results indicate that both 30 mg/kg and 50 mg/kg Api can efficiently inhibit HFD-induced inflammation in a dosage-independent manner, and 30 mg/kg dosage is required for the inhibition of inflammation. Moreover, the expression of IL-12, CCL2 as well as IL-1β in the adipose tissue of HFD mice were significantly reduced by Api (Fig. S4), suggesting that Api affected the production of inflammatory cytokines in adipose tissue.
To further confirm the anti-inflammatory effect of Api in another obesity-animal model, ob/ob mice were intraperitoneally treated with Api (30 mg/kg bw). Consistent with the results from HFD-fed mice, Api also caused a significant decrease in CCL2 and IL-1β levels and an increase in IL-10 concentration compared to the control group (Fig. 1j–l). Taken together with the above results, this suggests that Api can effectively attenuate obesity-related inflammation in vivo.
3.2. Api Restores the M1/M2 Status of Macrophages Both In Vivo and In Vitro
Macrophage infiltration into adipose tissue, which can be induced by increased adiposity, triggers local inflammation and insulin resistance. Macrophage M1/M2 status plays a fundamental role in this process (Olefsky and Glass, 2010). To elucidate the mechanism for Api attenuation of obesity-related inflammation, we first detected the macrophage phenotypes in the HFD-fed mice. As shown in Fig. S5a–b, the mRNA levels of IL-6 and TNF-α which are classical markers of M1 macrophages, exhibited sustained increases in the HFD-fed mice compared to the ND-fed group. The expression levels of CCR7 (Fig. S5c–d) were also significantly higher in HFD mice. Conversely, the transcription levels of classical M2 markers, including CD206 (Fig. S5e) and Fizz1 (Fig. S5f) were markedly decreased in HFD-fed mice compared to the control group. Consistently, the percentage of MGL1/2-positive macrophages significantly decreased from 34.8% to around 15.10% (Fig. S5g–h). These results suggest that polarization of macrophages is skewed to M1 rather than M2 differentiation during the development of obesity induced by a HFD, thereby leading to sustained inflammation.
Next, we investigated whether Api restrained inflammation via regulation of M1/M2 polarization of macrophages. When treated with Api, the increase in the expression of MHCII and CD80 that had been induced by HFD feeding was significantly abolished (Fig. 2a–b). Meanwhile, the expression of M2 markers, such as MGL1/2, was significantly up-regulated by Api (Fig. 2c). Furthermore, as shown in Fig. 2g–h, adipose tissue macrophages (ATMs) isolated from Api-treated HFD mice had reduced mRNA levels of CCL3 and CCL4 (M1 markers) but enhanced levels of Ym1 and Arg1 (M2 markers) when compared with non-Api treated mice, suggesting that Api restored the M1/M2 polarization of ATMs in HFD-fed mice. Similar results were obtained in ob/ob mice treated with Api (30 mg/kg) for 21 days (Fig. 2d–f, i–j). Together, Api regulated M1/M2 polarization of macrophages in vivo.
Fig. 2.

Api restores the M1/M2 status in obese mice. (a–c) The expression of surface markers MHCII, CD80 and MGL1/2 of mouse primary peritoneal macrophage isolated from HFD mice treated with the vehicle (0.1% DMSO), indicated doses of Api for 21 days was measured (left) and the flow data was quantified (right) by using on one-way ANOVA followed by a Dunnett's test (n = 9). All values are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ns, no significant difference compared with vehicle. (d–f) The expression of surface markers MHCII, CD80 and MGL1/2 of mouse primary peritoneal macrophage isolated from ob/ob mice treated with the vehicle (0.1% DMSO), 30 mg/kg Api for 21 days was measured (left) and the flow data was quantified (right) by using the Student's t-test (n = 9). All values are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle. (g–h) Relative mRNA expression of M1 and M2 macrophage markers in the adipose tissue macrophages (ATMs) from HFD mice treated with the vehicle (0.1% DMSO), indicated doses of Api for 21 days, CCL3, CCL4, Arg1 and Ym1 were measured by using qRT-PCR, and normalized to vehicle group and the level of β-actin. All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test (n = 9). *P < 0.05, **P < 0.01, ***P < 0.001, ns, no significant difference, compared with vehicle. (i–j) Relative mRNA expression of M1 macrophage markers, CCR2, CCL3, CCL4, TNF-α and M2 markers MMP-9, Arg1, Ym1 and CD206 in the ATMs from ob/ob mice treated with the vehicle (0.1% DMSO), 30 mg/kg Api for 21 days were measured by using qRT-PCR, and normalized to ob/ob (vehicle) group and the level of β-actin (n = 9). All values are expressed as mean ± SEM. Statistical analysis is based on the Student's t-test. *P < 0.05, **P < 0.01, ***P < 0.001 compared with vehicle.
In our in vitro cell models of M1 and M2 macrophages, we were able to confirm that Api treatment did not affect cell viability below 10 μ. But when the dosage was increased, the cell viability was significantly reduced by Api (Fig. S8a–c). Moreover, in M1 cell model, Api treatment significantly reduced the expression levels of CCR7 and CD80 (Fig. S6a) on the surface of macrophages, and the concentrations of IL-6 (Fig. S6b), IL-12 (Fig. S6c), TNF-α (Fig. S6d) and NO (Fig. S6e) in cell supernatant induced by LPS. However, in the M2 cell model, Api further up-regulated the concentration of IL-10 (Fig. S6f), Arg1 activity (Fig. S6g), the mRNA level of M2 genes (Fig. S6h) and the expression level of MGL1/2 (Fig. S6i) on the surface of macrophages.
To mimic the microenvironment of ATM, ana-1 macrophages were co-cultured with differentiated 3T3-L1 adipocytes, and subjected to Api treatment thereafter. We found that the M1 markers (Fig. S7a–c) were all decreased by Api and, in contrast, the M2 markers (Fig. S7d–f) were enhanced by Api treatment. Interestingly, in addition to the change in M1/M2 markers, the biologic functions of ATMs (phagocytic capability and ROS production) were depressed by Api in both HFD-fed mice (Fig. S8m) and ob/ob mice (Fig. S8n–q). The in vitro results were further confirmed by the results in the cell model (Fig. S8d–l). Together, these findings indicate that Api favors M2 polarization of ATMs and blocks the inflammatory functions of the macrophages, suppressing obesity-related inflammation in animal models of obesity.
3.3. Api Regulates M1/M2 Polarization via PPARγ.
Numerous studies have established the critical role of PPARγ in controlling macrophage M2 polarization (alternative activation) (Odegaard et al., 2007, Bouhlel et al., 2007). It has been suggested that Api itself is a ligand of PPARγ (Liang et al., 2001). We further examined whether Api regulates macrophage polarization through PPARγ. Upon stimulation with LPS, the percentage of MHCII-positive cells increased from 18.92% to 55.03%, but decreased to 35.40% when treated with Api simultaneously (Fig. S9a–b). However, exposure to a specific PPARγ antagonist, GW9662, which can inhibit the transcriptional activation of PPARγ, reversed the reduction of MHCII-positive cells by Api (Fig. S9a–b). Similarly, GW9662 treatment also inhibited the Api-induced decrease in the expression of CD80 as well as the mRNA levels of CCR2, TNF-α, CCL4 and IL-1β in M1 polarization model (Fig. S9c–e). Moreover, in the IL-4-induced M2 polarization model, the expected effect of Api-increased expression or transcription of M2 markers, including MGL1/2 (Fig. S9f–g), CD206, Arg1, Ym1 and IL-1r (Fig. S9h) was blocked by the PPARγ antagonist, GW9662. These results suggest that when the activity of PPARγ is inhibited by GW9662, the effects of Api on M1/M2 polarization are correspondingly suppressed.
To further confirm the role of PPARγ in Api-induced M1/M2 transition, the expression of PPARγ in Raw264.7 macrophages was knocked down by specific shRNA. At first, the efficacy of PPARγ shRNA was verified by western blot and qRT-PCR (Fig. S10a–b). When the expression of PPARγ was inhibited by PPARγ shRNA, the effects of Api on the transcription levels of both M1 (NOS2, TNF-α, CCL3 and CCL4) (Fig. 3a) and M2 (Ym1, CD163, CD206, Arg1) markers (Fig. 3b) were abolished. Moreover, PPARγ was also overexpressed in macrophages through constructing and transfecting PPARγ plasmids. The results indicate that the overexpression of PPARγ further enhanced the effects of Api in inhibiting the expression of M1 markers (Fig. 3c) while prompting the expression of M2 markers (Fig. 3d) in LPS-or IL-4-stimulated macrophages. Taken together, all these results suggest that regulation of macrophage M1/M2 polarization by Api is dependent on PPARγ.
Fig. 3.

PPARγ is necessary for Api regulating M1/M2 status. (a) RAW264.7 cells transfected WT and PPARγ plasmids were treated with 500 ng/mL LPS or 500 ng/mL LPS + 7.5 μM Api simultaneously for 24 h, followed by qRT-PCR with indicated probes (below graphs). Data indicate fold induction (normalized by β-actin signal). All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001. (b) Cells in (a) were treated with 10 ng/mL IL-4 or 10 ng/mL IL-4 + 7.5 μM Api simultaneously for 24 h, and then subjected to qRT-PCR, the data analysis as (a). All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001. (c) RAW264.7 cells transfected scrambled or PPARγ shRNA were treated with 500 ng/mL LPS or 500 ng/mL LPS + 7.5 μM Api simultaneously for 24 h, followed by qRT-PCR with indicated probes (below graphs). Data indicate fold induction (normalized by β-actin signal). All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001. (d) Cells in (c) were treated with 10 ng/mL IL-4 or10 ng/mL IL-4 + 7.5 μM Api simultaneously for 24 h, then subjected to qRT-PCR, the data analysis as (c). All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001.
3.4. Api Binds to and Activates PPARγ
We investigated the underlying mechanism by which PPARγ contributes to Api-induced macrophage M1/M2 polarization. The mRNA levels of PPARγ were comparable between the Api-treated group and the vehicle control group (Fig. 4a), suggesting that Api does not affect the mRNA expression of PPARγ. Activation of PPARγ through the luciferase reporter assay indicated that the activity of PPARγ is significantly increased by Api when PPARγ is overexpressed (Fig. 4b), which is consistent with previous report (Liang et al., 2001). In order to detect whether Api can activate the endogenous PPARγ, the EMSA experiment was performed. The results of the EMSA assay further confirmed that transcriptional activity of endogenous PPARγ can be induced by Api (Fig. S10c). The mRNA level of PPARγ target gene CD36 is also enhanced by Api treatment. Since iNOS promoter activity is inhibited by PPARγ ligands (Ricote et al., 1998), the data shown in the Fig. 4c indicated that Api inhibited iNOS expression, suggesting that Api can activate PPARγ, and thus upregulate or downregulate the corresponding genes targeted by PPARγ.
Fig. 4.

Api binds to and activates PPARγ. (a) ANA-1 macrophages was treated with 10 ng/mL IL-4 or 10 ng/mL IL-4 plus 7.5 μM Api simultaneously for 24 h, the mRNA level PPARγ was measured by using qRT-PCR. Data indicate fold induction (normalized by β-actin signal). All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. ns, no significant difference. (b) Transcriptional activation of PPARγ in cells treated with the indicated dosages of Api or Rosi. HER293T cells were transfected with pIRES-mPPARγ/PPRE and pRL-control using Lipofectamine2000. Then cells were pre-treated with apigenin for 24 h. Luciferase activities were measured by using the dual-luciferase reporter assay system. All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 compared with 0 group. (c) ANA-1 macrophages was treated with the indicated group for 24 h, the mRNA level PPARγ activation related genes CD36 and iNOS was measured by using qRT-PCR. Data indicate fold induction (normalized by β-actin signal). All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. ns, no significant difference. (d) ITC data for binding of Api to PPARγ. The upper panels show the raw data, and the lower panels show the corresponding binding isotherm fitted according to the “one binding site” model. Reference titration of ligand into buffer was used to correct for heat of dilution. The thermodynamic parameters (K, ΔH, and ΔS) are indicated under the below. (e) The deletion mutant model. (f) HER293T cells were transfected with pIRES-mPPARγ truncated mutants/PPRE and pRL-control using Lipofectamine2000. Then cells were pre-treated with Api (1 μM, 10 μM) for 24 h. Luciferase activities were measured by using the dual-luciferase reporter assay system. All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 compared with 0 group. (g) Auto dock model of Api binding to the PPARγ. Hydrogen bonding was built between Api and the Lys263, Lys265, Leu340 and Ser 342 sites of PPARγ. (h) Mutants disturbed Api from activating PPARγ analyzed using the dual-luciferase reporter assay system. HER293T cells were transfected with pIRES-mPPARγ point mutants/PPRE and pRL-control using Lipofectamine2000. Then cells were pre-treated with Api (1 μM, 10 μM) for 24 h. Luciferase activities were measured by using the dual-luciferase reporter assay system. All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. *P < 0.05, **P < 0.01, ***P < 0.001 compared with 0 group.
Noeris K. Salam et al. have suggested that Api might be an agonist of PPARγ (Salam et al., 2008b). To explore whether Api can directly bind to PPARγ, we first expressed and purified the recombinant his6-tagged PPARγ (termed PPARγ-his) (Fig. S10d–f). Then qualitative method-ITC was employed to analyze the binding activity between PPARγ and Api at 298 K. The binding affinity and binding stoichiometry of PPARγ to Api were obtained via the one-site binding model fitting using integrated binding heat, which revealed 14 potential Api binding sites within PPARγ with moderate binding affinity (Ka = 44.900 ± 105,300 M− 1) (Fig. 4d).
PPARγ contains four domains, including the A/B domain, C (DNA binding domain, DBD), D (hinge) and F (ligand binding domain, LBD) domain. To identify the amino acid sequence of PPARγ that binds to Api, three different C-terminal deletion mutants (C/F, D/F, F) of PPARγ were constructed using an eukaryotic expression plasmid (plenty-v5-PPARγ) as described in Fig. 4e. Then the PPARγ activation assays were performed in 293T cells which expressed these deletion mutant plasmids. The results indicated that Api significantly activated the C/F (107aa–475aa) mutant, and partially activated the D/F (181–475aa) mutant (Fig. 4f). In contrast, the F (289–475aa) mutant could not be activated by 10 μM Api (Fig. 4f), suggesting that the sequence of 107–289 amino acids in PPARγ is responsible for binding to Api. To further determine the specific amino acids required for binding between Api and PPARγ, we used the molecule docking method, which suggested that four amino acids (Lys263, Lys265, Leu340, Ser342) of PPARγ form hydrogen bonds with Api (Fig. 4g). Then, site-directed mutants of these four amino acids of PPARγ were subsequently generated. As Fig. 4h shows, none of the single mutants (K263R, K265R, L340F and S342T) could be activated by Api, indicating that these four specific amino acids are required for binding and they might form a pocket for binding to Api. Interestingly, unlike Api, except K263-R mutant, the D/F, F trunk mutants and the other three point mutants had no effect on PPARγ activation by Rosi (10 μM) (Fig. S11), which further imply the different interaction mechanism between Api and Rosi with PPARγ.
3.5. Api Reduces NF-κB Signaling by Changing the PPARγ/p65 Complex in the Cytoplasm and Nucleus.
Nuclear factor kappa B (NF-κB), which is a key transcription factor regulating inflammation, has also been shown to play a central role in metabolic pathologies (Kiechl et al., 2013). Here, we performed RNA-Seq of the primary peritoneal macrophages from both HFD and ND mice. The sequencing quality and credibility were evaluated by the composition of raw reads of ND and HFD samples (Fig. S12a), the amount of clean reads (Fig. S12b), the number of reads (Fig. S12c), the gene expression levels (Fig. S12d) and the distribution of gene coverage (Fig. S12e). The data of the RNA-Seq was shown in Table s6. Among these, there were 246 genes up-regulated and 75 genes down-regulated in HFD mice (Fig. S10g). The results of sequencing was confirmed by qRT-PCR assay (Fig. S10h). The genes associated with the growth and survival of cells (Table s1), insulin resistance (Table s2), T cells activation (Table s3) and inflammation (Table s4) were all up-regulated in HFD mice. Among those, NF-κB signaling was activated and the mRNA level was significantly increased. Moreover, p65 expression in the peritoneal macrophages of HFD mice was significantly decreased by Api treatment (Fig. 5a). Furthermore, IκBα interacts with p65 to inhibit nuclear translocation, and activation of IκB kinase can lead to the degradation of IκBα and translocation of p65 into the nucleus. Our results show that Api reduces the phosphorylation level of IκBα in macrophages of HFD mice (Fig. 5b). Consistent with this finding, our EMSA showed that Api suppressed the activation of NF-κB (Fig. 5c) and our immunofluorescence assay and the quantification of the p65 fluorescence intensity showed that Api significantly inhibited the translocation of p65 from cytoplasm to nuclei (Fig. 5d–e).
Fig. 5.

Api regulates macrophage polarization via inhibiting the interaction between p65 and PPARγ. (a)The expression of p65 in the primary peritoneal macrophages of 19 weeks ND and HFD mice treated with 30 mg/kg Api for 21 days was assayed by western blotting for three times, 3 mice/time (top). And the quantification of the protein level by using image J software (bottom). All values are expressed as mean ± SEM. Statistical analysis is based on the Student's t-test. *P < 0.05 compared with ND or vehicle. (b)The impact of Api on IκBα, p-IκBα abundance were evaluated in the primary peritoneal macrophages of mice treated for 21 days with 30 mg/kg Api by western blotting for three times, 3 mice/group (top). And the quantification of the protein level by image J software (bottom). All values are expressed as mean ± SEM. Statistical analysis is based on the Student's t-test. **P < 0.01 compared with vehicle group. (c) The effect of 7.5 μM Api in LPS-induced ANA-1 on the activities of p65 was detected by EMSA assay. Figure shows one image from at least three independent experiments. (d) Using immunofluorescence, the p65 was evaluated in macrophage treated with Control, LPS (500 ng/mL), or LPS (500 ng/mL) and 7.5 μM Api simultaneously for 24 h. p65 is shown in red, and nuclei stained with DAPI. Original magnification is × 400. Figure shows one image from at least three independent experiments. (e) The nuclei/cytoplasm ratio of (d) at least three independent experiments was quantified by Image J software. All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. ***P < 0.001 compared with con or LPS group. (f) The p65/PPARγ complex in the ATM of mice treated with 30 mg/kg Api for 21 days was assayed by confocal microscopy. p65 is shown in red, PPARγ is shown in green and nuclei stained with DAPI. Original magnification is × 400. (g) Data from (F) at least three independent experiments was quantified by Image J software. All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. ***P < 0.001 compared with ND or HFD. (h) ANA-1 macrophages were treated with 500 ng/mL LPS, 10 ng/mL IL-4, 500 ng/mL LPS plus 7.5 μM Api simultaneously or 10 ng/mL IL-4 plus 7.5 μM Api for 24 h. The nuclei and cytoplasm protein was lysed and subjected to immunoprecipitation and western blotting (left). Data from at least three independent experiments was quantified by Image J software (right). All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. *P < 0.05, ***P < 0.001. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Previous studies have reported that PPARγ can suppress the activation of NF-κB in mouse macrophages (Liang et al., 1999) and inhibit inflammation through interacting with p65 and inducing its ubiquitination and degradation (Hou et al., 2012). It was also investigated that ligand-activated PPARγ can play a negative regulatory role in macrophage activation, resulting in lower NF-κB activity (Ricote et al., 1998) and the interaction between PPARγ and p65 dampens the pro-inflammatory signals (Sato et al., 2005). But, the mechanism of Api inhibition of the translocation of p65 remains unknown. Although PPARγ is a nuclear receptor, it is constitutively present in both the cytoplasm and nucleus. Here, we hypothesize that Api may change the level of p65/PPARγ complex in the cytoplasm and nucleus of macrophages. In fact, immunofluorescence assay and the quantification of the PPARγ/p65 fluorescence intensity showed that the higher level of the PPARγ/p65 complex in the nucleus of ATM of HFD mice was reduced by Api treatment (Fig. 5f–g). To further confirm this, we performed a co-immunoprecipitation assay in M1/M2 macrophages, and the result indicated that LPS treatment increased the interaction of PPARγ and p65 in the nucleus, while the effect was reduced by Api stimulation in the nucleus. In IL-4 stimulated macrophages, the PPARγ/p65 complex was reduced compared with non-treated cells and further reduced after IL-4 and Api co-treatment in the nucleus (Fig. 5h). Taken together, these results indicated that Api regulated M1/M2 status by changing the location of the PPARγ/p65 complex.
3.6. Api Attenuates Metabolic Syndrome
Obesity causes excess fat accumulation and chronic low-grade inflammation in various tissues, especially in insulin-responsive organs such as skeletal muscle, liver and adipose tissue, thereby contributing to the development of metabolic abnormalities (Despres and Lemieux, 2006). In our model, both ALT and AST were increased up to 3-fold or 50-fold in the serum of HFD mice (Fig. S13a–b). In addition, the levels of total cholesterol (TC), triglycerides (TG), glucose, and carbonylational proteins were also significantly increased in HFD mice (Fig. S13c–f), indicating that a HFD induced liver injury and metabolic disorder. Treatment with Api markedly reduced the levels of ALT, AST, TC and TG in the serum of HFD mice, with effects similar to Rosi, which is a specific PPARγ agonist (Fig. 6e–f). The histological examination of liver and muscle sections further demonstrated that both Api and Rosi efficiently attenuated the derangement of cell structures, excessive lipid accumulation and pathological status induced by a HFD (Fig. 6a and c) and the score showed that Api obviously attenuated their steatosis (Fig. 6b and d). In addition to the HFD mouse model, we also investigated the ability of Api to relieve metabolic syndrome in ob/ob mice. Similarly, the levels of ALT, AST, TC, TG and glucose in the serum of ob/ob mice were all decreased significantly by Api administration (Fig. 6e–h). All these results suggest that Api can efficiently attenuate the syndromes of metabolic abnormality in both HFD and ob/ob animal models.
Fig. 6.

Api attenuates the metabolic disorders of obese mice. (a-c) Representative H&E staining showed liver/muscular morphology from 16-week-old HFD mice (n = 9) treated with the vehicle indicated doses of Api for 21 days and (b–d) the Quantification of hepatic/muscular steatosis for five to eight sections/400 × field, five to six fields/gland/mouse, score according to the grade of lesion, slight (0.5), mild (1), moderate (2), severe (3), profound severe (4) and normal (0), n = 6, original magnification × 400. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. ***P < 0.001 compared with vehicle. (e–h) the activities of ALT (e) and AST (f), The levels of TC (G) and TG (H) in serum of HFD and ob/ob mice were significantly reduced by Api or Rosi, n = 9 or 6. All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test or based on the Student's t-test for comparing two groups. ***P < 0.001 compared with vehicle.
3.7. Api Exhibits No Adverse Effects Found in Thiazolidinediones
Thiazolidinediones (TZDs), which are specific ligands for PPARγ, have been used as anti-diabetic drugs and act as insulin sensitizers in type 2 diabetes mellitus. According to our above results, Api might also act as an insulin sensitizer since it is also a specific ligand for PPARγ. To test this, we treated HFD mice with 30 mg/kg Api or 10 mg/kg Rosi for 21 days. The data shown in Fig. 7a indicate that mice treated with Api had improved glucose tolerance with an approximately 40–50% reduction in the AUC of glucose, whereas Rosi treatment induced an approximately 20–30% reduction. Insulin tolerance tests (ITTs) revealed that Api treatment has no significant improving on insulin sensitivity in obese mice compared with the vehicle group (Fig. S14). Mice treated with Api or Rosi both had reduced fasting insulin levels and glucose levels (Fig. 7b and c). In addition, adiponectin, a classical marker of insulin sensitivity, was enhanced by both Api as well as Rosi (Fig. 7d). These results suggest that Api acts as an insulin sensitizer, similar to Rosi.
Fig. 7.

Api treatment improves insulin sensitivity and glucose tolerance. (a) glucose tolerance tests and the total area under the curve (AUC) in HFD mice treated with vehicle, Api or rogiglatazone, n = 9. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. Ns, no significant difference, *p < 0.05 compared with vehicle group. (b) The levels of glucose and (c) insulin in the serum of HFD mice treated with vehicle, Api or rogiglatazone, n = 9. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. *p < 0.05 compared with vehicle group. (d) Adiponetin in the serum of mice was detected by ELISA, n = 9. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. **p < 0.01 compared with vehicle group. (e) The size of adipocyte was quantified by the microscope micrometers at the 100 × light microscope. Count the numbers of adipocytes in the measurement unit area (25 mm2), unit: numbers/25 mm2/100 ×, n = 6. All values are expressed as mean ± SEM. Statistical analysis is based on one-way ANOVA followed by a Dunnett's test. **p < 0.01 compared with vehicle group.
However, the clinical use of TZDs in type 2 diabetes mellitus is limited by adverse effects, including body-weight gain, osteoporosis and the increase of small adipocytes in WAT (Schwartz, 2006), leaving room for development of new and safer PPARγ-modulating drugs. To evaluate the potential of Api as new an anti-diabetic drug, we also assessed Api's safety and adverse effects. In contrast to TZDs, apigenin treatment does not induce an obvious change in body weight (Fig. 1b–c). Previous reports have indicated that TZDs also increase the number of small adipocytes in WAT, potentially by promoting differentiation (Okuno et al., 1998), but our histological examination demonstrated that WAT from Api-treated mice exhibited reduced adipocyte size (Fig. 7e). In addition, different from TZDs, Api reduced the accumulation of triglycerides in the serum of both HFD and ob/ob mice (Fig. 6h). Another undesirable side effect of TZDs is osteoporosis, and recent studies have reported that TZDs cause osteoporotic fractures in rats, mice and humans. We therefore employed the method of computer tomography (CT) to detect the bone intensity of Api and Rosi-treated mice. As Table 1 shows, there was no significant bone loss in the Api-treated group compared to controls. In contrast, Rosi treatment induced obvious bone loss. Although there was no obvious improvement in the loss of femoral bone, Api markedly increased the volume ratio of trabecular bone from 14.123 to 26.164 while Rosi significantly reduced it to 2.366. Taken together, Api appears to be a safer modulator of PPARγ than TZDs, as it is able to induce insulin sensitization without the same adverse effects.
Table 1.
Density of trabecular bone and femoral bone analysis by using CT scan.
| Femoral (g/cm2) | Trabecular (g/cm2) | Trabecular BV% | |
|---|---|---|---|
| Vehicle | 1.106 ± 0.0954a | 0.1501 ± 0.0012a | 14.123 ± 1.202a |
| 10 mg/kg | 1.233 ± 0.039a | 0.1401 ± 0.0024a | 15.523 ± 1.091a |
| 50 mg/kg | 1.328 ± 0.058a | 0.1820 ± 0.0013b | 26.164 ± 1.532b |
| Rosi-10 mg/kg | 1.003 ± 0.018a | 0.1208 ± 0.0016c | 2.366 ± 0.195c |
Values without a common letter in their superscripts in the same column differ (P < 0.05).
4. Discussion
PPARγ is an attractive pharmacological target for the development of drugs to treat metabolic disorders such as insulin resistance (Xu et al., 2003a), type II diabetes (Saltiel and Olefsky, 1996) and chronic inflammation (Buckingham, 2005). As potent full agonists of PPARγ, thiazolidinediones (TZDs), as a class of antidiabetic drugs that includes Rosi and pioglitazone (Marciano et al., 2014), have been widely used for treating the above mentioned disorders. However, side effects of TZDs have greatly limited their therapeutic use. Although not yet unequivocally demonstrated, it is likely that most side effects of TZDs are associated with their high binding affinities for PPARγ and the resultant over-activation of the classical PPARγ pathway. The fact that ligand binding induced conformational changes in PPARγ offers the opportunity for specific ligand-selective regulation of PPARγ transcriptional activity (Higgins and Depaoli, 2010), prompting researchers to seek and discover some effective modulators of PPARγ, and giving rise to the concept of selective PPAR modulators (SPPARMs). Identifying efficient SPPARMs, which can partially activate PPARγ but without the severe side effects of full PPARγ agonists (Higgins and Mantzoros, 2008, Gregoire et al., 2009), is a promising approach to development of new and safer drugs.
Here, we report that Api, a natural PPARγ ligand, can significantly curb obesity-related inflammation, as well as markedly ameliorate the metabolic abnormalities and insulin resistance induced by obesity. Api treatment does not exhibit some of the severe adverse effects of Rosi. Our results suggest that Api may act as a potential SPPARM of PPARγ that is useful for treating metabolic diseases. At the transcriptional level, Api only shows moderate PPARγ transactivation activity, with approximately 50% of the Rosi-induced effect, suggesting that Api partially activates PPARγ. Liang et al. have utilized in vitro studies to show that Api is an allosteric effector of PPARγ and is able to bind to PPARγ (Guevara et al., 1998). Another study by Salam et al. using the induced-fit docking method suggests that Api induces PPARγ conformational change and that the binding of Api to PPARγ is different from Rosi (Salam et al., 2008a). In agreement with these studies, our results confirm that Api binds to PPARγ. Furthermore, we found that both the hinge and LBD domain of PPARγ are important for Api binding, which are different from the domains bound by Rosi. By performing mutation experiments, we have identified the four amino acids (K263, K265, L340 and S342) of PPARγ-located at the hinge (181–289aa) and LBD (289–475aa) domains respectively—necessary for Api binding. Additionally, our auto-docking assay indicates that these four amino acids interact with Api through hydrogen bonds and form a PPARγ-binding pocket. Different from Api, Rosi only binds to the LBD domain of PPARγ (Willson et al., 2001), and such a difference in binding may explain the differences in activity between Api and Rosi. Importantly, our in vivo experiments demonstrate that Api improved the glucose tolerance and insulin resistance induced by a high-fat diet without any adverse effects, including the weight gain, hepatic lipid accumulation and osteoporosis seen with Rosi. Herein, we propose that Api is a promising SPPARM of PPARγ.
Macrophages are now regarded as prominent players in metabolic disorder and associated diseases. Their sub-populations (M1 and M2 macrophages) may have either deleterious or protective functions towards inflammatory regulation depending on certain conditions of various inflammatory microenvironments (Chinetti-Gbaguidi and Staels, 2011). M1/M2 status of macrophages represents one of the pivotal characteristics of the inflammatory state and is crucial in rendering them amenable to anti-inflammatory manipulation (Fujisaka et al., 2009). Other studies have indicated that M2 activation mediated by PPARγ or M1 depression mediated by knocking out CD11c + ameliorates obesity-related inflammation and related insulin resistance (Odegaard et al., 2007, Patsouris et al., 2008). In addition, drugs such as statins, GW7845, and others have been used successfully in clinical therapy for glomerulonephritis and atherosclerosis by targeting M1/M2 status (Li et al., 2000). Hence, modification of the balance of the M1/M2 steady state has shown efficacy in therapeutic applications.
In the present study, we demonstrate the link between Api and macrophage polarization in obesity-related inflammation regulation. Api modifies the balance between M1 and M2 polarization by binding and activating PPARγ, thereby exerting its biological actions in obesity-induced inflammation. The efficacy of Api on macrophage is similar with another flavonoid compound, chrysin, which can significantly inhibit obesity-related inflammation via regulating macrophage polarization by activating PPARγ in our previous study (Feng et al., 2014). In fact, many reports have investigated the underling mechanisms shared by flavonoids such as flavone, kaempferol, quercetin (Welton et al., 1986) and quercetin glucuronides (Kawai et al., 2008, De Whalley et al., 1990, Liang et al., 1999), and they can target macrophages, suppress the transcription activity of cyclooxygenase (COX)-2 and iNOS and so on. But for the detailed and exact mechanism are required to further investigate. Furthermore, it is interesting that the efficacy of Api as a SPPARM of PPARγ also impacts insulin resistance and glucose tolerance in obese mice without the obvious adverse effects shown by TZDs, suggesting an advantage for using a natural PPARγ ligand in metabolic disorder therapy.
Although the above results establish that Api increases M2 macrophages by binding to and activating PPARγ and thereby attenuates inflammation, the underlying molecular mechanisms remain elusive. PPARγ agonists exert their antagonistic effects on inflammatory responses through promoter-specific repression of NF-κB target genes (Pascual et al., 2005, Straus and Glass, 2007), providing a clue that the NF-kB pathway might be involved in the effects of PPARγ modulators. Accordingly, we performed an RNA-Seq analysis and verification assay to investigate whether NF-κB signaling is involved in the M1/M2 shift mediated by Api. As expected, the results indicate that NF-κB signaling is activated in macrophages of HFD mice. The nuclear form of the NF-κB transcription factor binds to DNA as a heterodimer of p50 and p65 polypeptide and p65 subunit is responsible for initiating transcription (Schmitz and Baeuerle, 1991). Administration with Api disturbs the translocation of p65 to the nucleus and thus inhibits NF-κB activation. Moreover, PPARγ can interact with the p65 subunit of NF-κB in vitro (Chung et al., 2000). Our further study found that in M1 cells, PPARγ/p65 complex are mainly located in the nuclei while in M2 cells, PPARγ/p65 complex are mainly located in the cytoplasm. After Api treatment, its binding with PPARγ inhibited p65 translocation to nuclei, thereby impeding the formation of nuclei PPARγ/p65 complex and leading to the prolonged retention of the PPARγ/p65 complex in the cytoplasm. This suggests that Api binding with PPARγ inhibited p65 translocation to nuclei and changed the location of PPARγ/p65 complex along with macrophage polarization. Above results may represent a new strategy to develop a PPARγ modulator to control obesity related inflammation and related diseases via regulation macrophage polarization. Phosphorylation of p65 helps stabilized NF-κB in the nucleus for gene transcription and thereby is widely used as an indicator of NF-κB activation (Hu et al., 2004). In addition, previous studies have reported that p65 phosphorylation induced macrophage polarization (Nicholas et al., 2007, Hu et al., 2004, Lee et al., 2014). Here, we detected the phosphorylation of p65 in M1 and M2 macrophages to investigate if the phosphorylation of p65 participates in Api/PPARγ regulating p65 translocation. The data showed in the Fig. S15 indicated that after the cells treated for 2 h, the phosphorylation of p65 was increased in M1 cells while Api reversed this effect, which is consistent with previous study that Api inactivated NF-κB by the suppression of p65 phosphorylation in M1 cells (Nicholas et al., 2007). Moreover, the reduction of phosphorylation of p65 in M2 cells was further reduced by Api treatment, indicating that phosphorylation of p65 participated in Api/PPARγ regulating p65 translocation. Thus, Api binding with PPARγ inhibited p65 translocation to nuclei via regulating phosphorylation of p65 in macrophage polarization. In addition, it is possible that except from PPARγ we found, some other proteins may also be involved in the process of Api regulating inflammation as the Arango D et al. reported that there are 160 high-confidence candidate apigenin targets in the human (Arango et al., 2013).
In summary, Api is a promising natural modulator of PPARγ that ameliorates obesity-related inflammation efficiently without any of the known side effects of TZDs. We found that Api partially activates PPARγ via binding to certain amino acid residues in the LBD and hinge domains of PPARγ. Moreover, we suggest that the possible mechanism underlying the anti-inflammatory action of Api is through modification of macrophage functional polarization via regulation of the location of p65/PPARγ. These finding indicate that Api is a potential drug candidate for the treatment of obesity-related inflammation and related comorbidities, thereby providing a reasonable strategy and explanation for developing more SPPARγ modulators that shift macrophage polarization.
Author contributions
F.X.J, Z.F.F, Q.H.H, Z.J.F, Y.W, C.J.J and Y.N.F performed the experiments for this work. S.P.P, F.X.J designed the research and analyzed the data. F.X.J, W.D and S.P.P wrote the manuscript. F.H.J and L.J.X. performed the auto-docking experiment and analyzed the data. C.D.H participated in the IF analysis. Owen. Y. and T.R.X. provided scientific suggestions and contributed to the manuscript revision. S.P.P supervised the project.
Conflicts of Interest
The authors declare no conflict of interest.
Acknowledgements
The work was supported by the National Natural Science Foundation of China under Grant 81273527, 81421091, 81503082 and 81473220, the Natural Science Foundation of Jiangsu Province of China under Grants BK20150575, Postdoctoral Science Foundation of China under Grants 2015M570437.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2016.06.017.
Appendix A. Supplementary data
Supplementary material.
References
- Arango D., Morohashi K., Yilmaz A., Kuramochi K., Parihar A., Brahimaj B., Grotewold E., Doseff A.I. Molecular basis for the action of a dietary flavonoid revealed by the comprehensive identification of apigenin human targets. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E2153–E2162. doi: 10.1073/pnas.1303726110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audic S., Claverie J.M. The significance of digital gene expression profiles. Genome Res. 1997;7:986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Bao X., Cui J., Wu Y., Han X., Gao C., Hua Z., Shen P. The roles of endogenous reactive oxygen species and nitric oxide in triptolide-induced apoptotic cell death in macrophages. J. Mol. Med. (Berl) 2007;85:85–98. doi: 10.1007/s00109-006-0113-x. [DOI] [PubMed] [Google Scholar]
- Bouhlel M.A., Derudas B., Rigamonti E., Dievart R., Brozek J., Haulon S., Zawadzki C., Jude B., Torpier G., Marx N., Staels B., Chinetti-Gbaguidi G. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Buckingham R.E. Thiazolidinediones: pleiotropic drugs with potent anti-inflammatory properties for tissue protection. Hepatol. Res. 2005;33:167–170. doi: 10.1016/j.hepres.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Byun S., Park J., Lee E., Lim S., Yu J.G., Lee S.J., Chen H.Y., Dong Z.G., Lee K.W., Lee H.J. Src kinase is a direct target of apigenin against UVB-induced skin inflammation. Carcinogenesis. 2013;34:397–405. doi: 10.1093/carcin/bgs358. [DOI] [PubMed] [Google Scholar]
- Chinetti-Gbaguidi G., Staels B. Macrophage polarization in metabolic disorders: functions and regulation. Curr. Opin. Lipidol. 2011;22:365–372. doi: 10.1097/MOL.0b013e32834a77b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.S., Islam M.N., Ali M.Y., Kim E.J., Kim Y.M., Jung H.A. Effects of C-glycosylation on anti-diabetic, anti-Alzheimer's disease and anti-inflammatory potential of apigenin. Food Chem. Toxicol. 2014;64:27–33. doi: 10.1016/j.fct.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Chung S.W., Kang B.Y., Kim S.H., Pak Y.K., Cho D., Trinchieri G., Kim T.S. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. J. Biol. Chem. 2000;275:32681–32687. doi: 10.1074/jbc.M002577200. [DOI] [PubMed] [Google Scholar]
- Cipolletta D., Feuerer M., Li A., Kamei N., Lee J., Shoelson S.E., Benoist C., Mathis D. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–553. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasu M.R., Park S., Devaraj S., Jialal I. Pioglitazone inhibits Toll-like receptor expression and activity in human monocytes and db/db mice. Endocrinology. 2009;150:3457–3464. doi: 10.1210/en.2008-1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Whalley C.V., Rankin S.M., Hoult J.R., Jessup W., Leake D.S. Flavonoids inhibit the oxidative modification of low density lipoproteins by macrophages. Biochem. Pharmacol. 1990;39:1743–1750. doi: 10.1016/0006-2952(90)90120-a. [DOI] [PubMed] [Google Scholar]
- Despres J.P., Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881–887. doi: 10.1038/nature05488. [DOI] [PubMed] [Google Scholar]
- Dinarello C.A. Anti-inflammatory agents: present and future. Cell. 2010;140:935–950. doi: 10.1016/j.cell.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi L.S., Brahma M.K., Bahirat U.A., Dixit A.V., Nemmani K.V. Discovery and development of selective PPAR gamma modulators as safe and effective antidiabetic agents. Expert Opin. Investig. Drugs. 2010;19:489–512. doi: 10.1517/13543781003640169. [DOI] [PubMed] [Google Scholar]
- Dou W., Zhang J.J., Zhang E.Y., Sun A.N., Ding L.L., Chou G.X., Wang Z.T., Mani S. Chrysin ameliorates chemically induced colitis in the mouse through modulation of a PXR/NF-kappa B signaling pathway. J. Pharmacol. Exp. Ther. 2013;345:473–482. doi: 10.1124/jpet.112.201863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan B.B., Schmidt M.I., Pankow J.S., Ballantyne C.M., Couper D., Vigo A., Hoogeveen R., Folsom A.R., Heiss G. Low-grade systemic inflammation and the development of type 2 diabetes — the atherosclerosis risk in communities study. Diabetes. 2003;52:1799–1805. doi: 10.2337/diabetes.52.7.1799. [DOI] [PubMed] [Google Scholar]
- Escande C., Nin V., Price N.L., Capellini V., Gomes A.P., Barbosa M.T., O'neil L., White T.A., Sinclair D.A., Chini E.N. Flavonoid apigenin is an inhibitor of the NAD + ase CD38: implications for cellular NAD + metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. 2013;62:1084–1093. doi: 10.2337/db12-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X., Qin H., Shi Q., Zhang Y., Zhou F., Wu H., Ding S., Niu Z., Lu Y., Shen P. Chrysin attenuates inflammation by regulating M1/M2 status via activating PPARgamma. Biochem. Pharmacol. 2014;89:503–514. doi: 10.1016/j.bcp.2014.03.016. [DOI] [PubMed] [Google Scholar]
- Fujisaka S., Usui I., Bukhari A., Ikutani M., Oya T., Kanatani Y., Tsuneyama K., Nagai Y., Takatsu K., Urakaze M., Kobayashi M., Tobe K. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;58:2574–2582. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoire F.M., Zhang F., Clarke H.J., Gustafson T.A., Sears D.D., Favelyukis S., Lenhard J., Rentzeperis D., Clemens L.E., Mu Y., Lavan B.E. MBX-102/JNJ39659100, a novel peroxisome proliferator-activated receptor-ligand with weak transactivation activity retains antidiabetic properties in the absence of weight gain and edema. Mol. Endocrinol. 2009;23:975–988. doi: 10.1210/me.2008-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guevara I., Iwanejko J., Dembinska-Kiec A., Pankiewicz J., Wanat A., Polus A., Golabek I., Bartus S., Malczewska-Malec M., Szczudlik A. Determination of nitrite/nitrate in human biological material by the simple Griess reaction. Clin. Chim. Acta. 1998;274:177–188. doi: 10.1016/s0009-8981(98)00060-6. [DOI] [PubMed] [Google Scholar]
- Ha S.K., Lee P., Park J.A., Oh H.R., Lee S.Y., Park J.H., Lee E.H., Ryu J.H., Lee K.R., Kim S.Y. Apigenin inhibits the production of NO and PGE(2) in microglia and inhibits neuronal cell death in a middle cerebral artery occlusion-induced focal ischemia mice model. Neurochem. Int. 2008;52:878–886. doi: 10.1016/j.neuint.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Havsteen B.H. The biochemistry and medical significance of the flavonoids. Pharmacol. Ther. 2002;96:67–202. doi: 10.1016/s0163-7258(02)00298-x. [DOI] [PubMed] [Google Scholar]
- Higgins L.S., Depaoli A.M. Selective peroxisome proliferator-activated receptor gamma (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation. Am. J. Clin. Nutr. 2010;91:267S–272S. doi: 10.3945/ajcn.2009.28449E. [DOI] [PubMed] [Google Scholar]
- Higgins L.S., Mantzoros C.S. 2008. The Development of INT131 as a Selective PPAR. Modulator: Approach to a Safer Insulin Sensitizer. (PPAR Research). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y., Moreau F., Chadee K. PPARgamma is an E3 ligase that induces the degradation of NFkappaB/p65. Nat. Commun. 2012;3:1300. doi: 10.1038/ncomms2270. [DOI] [PubMed] [Google Scholar]
- Howe L.R., Subbaramaiah K., Hudis C.A., Dannenberg A.J. Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin. Cancer Res. 2013;19:6074–6083. doi: 10.1158/1078-0432.CCR-12-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Nakano H., Sakurai H., Colburn N.H. Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF-kappaB activation and transformation in resistant JB6 cells. Carcinogenesis. 2004;25:1991–2003. doi: 10.1093/carcin/bgh198. [DOI] [PubMed] [Google Scholar]
- Kawai Y., Nishikawa T., Shiba Y., Saito S., Murota K., Shibata N., Kobayashi M., Kanayama M., Uchida K., Terao J. Macrophage as a target of quercetin glucuronides in human atherosclerotic arteries: implication in the anti-atherosclerotic mechanism of dietary flavonoids. J. Biol. Chem. 2008;283:9424–9434. doi: 10.1074/jbc.M706571200. [DOI] [PubMed] [Google Scholar]
- Kiechl S., Wittmann J., Giaccari A., Knoflach M., Willeit P., Bozec A., Moschen A.R., Muscogiuri G., Sorice G.P., Kireva T., Summerer M., Wirtz S., Luther J., Mielenz D., Billmeier U., Egger G., Mayr A., Oberhollenzer F., Kronenberg F., Orthofer M., Penninger J.M., Meigs J.B., Bonora E., Tilg H., Willeit J., Schett G. Blockade of receptor activator of nuclear factor-kappaB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat. Med. 2013;19:358–363. doi: 10.1038/nm.3084. [DOI] [PubMed] [Google Scholar]
- Lee J.Y., Jeong H.J., Kim M.K., Wee W.R. Bone marrow-derived mesenchymal stem cells affect immunologic profiling of interleukin-17-secreting cells in a chemical burn mouse model. Korean J. Ophthalmol. 2014;28:246–256. doi: 10.3341/kjo.2014.28.3.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrke M., Lazar M.A. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- Li A.C., Brown K.K., Silvestre M.J., Willson T.M., Palinski W., Glass C.K. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J. Clin. Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.C., Huang Y.T., Tsai S.H., LIN-Shiau S.Y., Chen C.F., Lin J.K. Suppression of inducible cyclooxygenase and inducible nitric oxide synthase by apigenin and related flavonoids in mouse macrophages. Carcinogenesis. 1999;20:1945–1952. doi: 10.1093/carcin/20.10.1945. [DOI] [PubMed] [Google Scholar]
- Liang Y.C., Tsai S.H., Tsai D.C., Lin-Shiau S.Y., Lin J.K. Suppression of inducible cyclooxygenase and nitric oxide synthase through activation of peroxisome proliferator-activated receptor-gamma by flavonoids in mouse macrophages. FEBS Lett. 2001;496:12–18. doi: 10.1016/s0014-5793(01)02393-6. [DOI] [PubMed] [Google Scholar]
- Liu L.Z., Fang J., Zhou Q., Hu X., Shi X., Jiang B.H. Apigenin inhibits expression of vascular endothelial growth factor and angiogenesis in human lung cancer cells: implication of chemoprevention of lung cancer. Mol. Pharmacol. 2005;68:635–643. doi: 10.1124/mol.105.011254. [DOI] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T) (− Delta Delta C) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Marciano D.P., Chang M.R., Corzo C.A., Goswami D., Lam V.Q., Pascal B.D., Griffin P.R. The therapeutic potential of nuclear receptor modulators for treatment of metabolic disorders: PPARgamma, RORs, and Rev-erbs. Cell Metab. 2014;19:193–208. doi: 10.1016/j.cmet.2013.12.009. [DOI] [PubMed] [Google Scholar]
- Mortazavi A., Williams B.A., Mccue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Mueller M., Lukas B., Novak J., Simoncini T., Genazzani A.R., Jungbauer A. Oregano: a source for peroxisome proliferator-activated receptor gamma antagonists. J. Agric. Food Chem. 2008;56:11621–11630. doi: 10.1021/jf802298w. [DOI] [PubMed] [Google Scholar]
- Nicholas C., Batra S., Vargo M.A., Voss O.H., Gavrilin M.A., Wewers M.D., Guttridge D.C., Grotewold E., Doseff A.I. Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-kappaB through the suppression of p65 phosphorylation. J. Immunol. 2007;179:7121–7127. doi: 10.4049/jimmunol.179.10.7121. [DOI] [PubMed] [Google Scholar]
- Odegaard J.I., Ricardo-Gonzalez R.R., Goforth M.H., Morel C.R., Subramanian V., Mukundan L., Red Eagle A., Vats D., Brombacher F., Ferrante A.W., Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno A., Tamemoto H., Tobe K., Ueki K., Mori Y., Iwamoto K., Umesono K., Akanuma Y., Fujiwara T., Horikoshi H., Yazaki Y., Kadowaki T. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J. Clin. Investig. 1998;101:1354–1361. doi: 10.1172/JCI1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky J.M., Glass C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Ouchi N., Parker J.L., Lugus J.J., Walsh K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G., Fong A.L., Ogawa S., Gamliel A., Li A.C., Perissi V., Rose D.W., Willson T.M., Rosenfeld M.G., Glass C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsouris D., Li P.P., Thapar D., Chapman J., Olefsky J.M., Neels J.G. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8:301–309. doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M., Li A.C., Willson T.M., Kelly C.J., Glass C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Salam N.K., Huang T.H., Kota B.P., Kim M.S., Li Y., Hibbs D.E. Novel PPAR-gamma agonists identified from a natural product library: a virtual screening, induced-fit docking and biological assay study. Chem. Biol. Drug Des. 2008;71:57–70. doi: 10.1111/j.1747-0285.2007.00606.x. [DOI] [PubMed] [Google Scholar]
- Salam N.K., Huang T.H.W., Kota B.P., Kim M.S., Li Y.H., Hibbs D.E. Novel PPAR-gamma agonists identified from a natural product library: a virtual screening, induced-fit docking and biological assay study. Chem. Biol. Drug Des. 2008;71:57–70. doi: 10.1111/j.1747-0285.2007.00606.x. [DOI] [PubMed] [Google Scholar]
- Saltiel A.R., Olefsky J.M. Thiazolidinediones in the treatment of insulin resistance and type II diabetes. Diabetes. 1996;45:1661–1669. doi: 10.2337/diab.45.12.1661. [DOI] [PubMed] [Google Scholar]
- Sato N., Kozar R.A., Zou L., Weatherall J.M., Attuwaybi B., Moore-Olufemi S.D., Weisbrodt N.W., Moore F.A. Peroxisome proliferator-activated receptor gamma mediates protection against cyclooxygenase-2-induced gut dysfunction in a rodent model of mesenteric ischemia/reperfusion. Shock. 2005;24:462–469. doi: 10.1097/01.shk.0000183483.76972.ae. [DOI] [PubMed] [Google Scholar]
- Schmitz M.L., Baeuerle P.A. The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J. 1991;10:3805–3817. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz A.V. 2006. Diabetes, TZDs, and Bone: a Review of the Clinical Evidence. (PPAR Research). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth S.G., Gordon F.D., Chopra S. Nonalcoholic steatohepatitis (vol 126, pg 137, 1997) Ann. Intern. Med. 1997;127:658. doi: 10.7326/0003-4819-126-2-199701150-00008. [DOI] [PubMed] [Google Scholar]
- Straus D.S., Glass C.K. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Weeks B.A., Keisler A.S., Myrvik Q.N., Warinner J.E. Differential uptake of neutral red by macrophages from three species of estuarine fish. Dev. Comp. Immunol. 1987;11:117–124. doi: 10.1016/0145-305x(87)90013-9. [DOI] [PubMed] [Google Scholar]
- Welton A.F., Tobias L.D., Fiedler-Nagy C., Anderson W., Hope W., Meyers K., Coffey J.W. Effect of flavonoids on arachidonic acid metabolism. Prog. Clin. Biol. Res. 1986;213:231–242. [PubMed] [Google Scholar]
- Willson T.M., Lambert M.H., Kliewer S.A. Peroxisome proliferator-activated receptor gamma and metabolic disease. Annu. Rev. Biochem. 2001;70:341–367. doi: 10.1146/annurev.biochem.70.1.341. [DOI] [PubMed] [Google Scholar]
- Xu H., Barnes G.T., Yang Q., Tan G., Yang D., Chou C.J., Sole J., Nichols A., Ross J.S., Tartaglia L.A., Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.Y., Barnes G.T., Yang Q., Tan Q., Yang D.S., Chou C.J., Sole J., Nichols A., Ross J.S., Tartaglia L.A., Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.S., Jeng C.Y., Wu T.J., Tanaka S., Funahashi T., Matsuzawa Y., Wang J.P., Chen C.L., Tai T.Y., Chuang L.M. Synthetic peroxisome proliferator-activated receptor-gamma agonist, rosiglitazone, increases plasma levels of adiponectin in type 2 diabetic patients. Diabetes Care. 2002;25:376–380. doi: 10.2337/diacare.25.2.376. [DOI] [PubMed] [Google Scholar]
- Yang W.W., Lu Y., Xu Y.C., Xu L.Z., Zheng W., Wu Y.Y., Li L., Shen P.P. Estrogen represses hepatocellular carcinoma (HCC) growth via inhibiting alternative activation of tumor-associated macrophages (TAMs) J. Biol. Chem. 2012;287:40140–40149. doi: 10.1074/jbc.M112.348763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.