

Abstract
Conjugation of cancer targeting peptides (CTPs) with small molecular therapeutics has emerged as a promising strategy to deliver potent (but typically nonspecific) cytotoxic agents selectively to cancer cells. Here we report the engineered production of a CTP (NGR)-containing C-1027 and evaluation of its activity against selected cancer cell lines. C-1027 is an enediyne chromoprotein produced by Streptomyces globisporus, consisting of an apo-protein (CagA) and an enediyne chromophore (C-1027). NGR is a CTP that targets CD13 in tumor vasculature. S. globisporus SB1026, a recombinant strain engineered to encode CagA with the NGR sequence fused at its C-terminus, directly produces the NGR-containing C-1027 that is equally active as the native C-1027. Our results demonstrate the feasibility to produce CTP-containing enediyne chromoproteins by metabolic pathway engineering and microbial fermentation and will inspire efforts to engineer other CTP-containing drug binding proteins for targeted delivery.
Keywords: Enediyne, C-1027, CTP, Cancer, Streptomyces globisporus
1. Introduction
Cancer accounts for one in every seven deaths worldwide and is considered as an enormous global health burden. Traditional cancer chemotherapies usually kill fast-dividing cancer cells but also affect healthy cells, resulting in adverse side effects. It is therefore an important research goal to develop new cancer therapies with improved specificities. In the past two decades, cancer therapies have evolved using either monoclonal antibodies or small molecules that are designed specifically to target cancer cell surface receptors or penetrate the cell membrane and interact with cancer specific proteins.1 While the successes of antibodies and antibody-drug conjugates (ADCs) in targeted cancer therapies have clearly demonstrated that cancer cell surface receptors are uniquely promising targets, there are two major limitations in antibody-targeted therapy. First, the antibody is relatively large making it difficult to infiltrate the entire tumor mass, and second, the nonspecific uptake of the antibody into liver, spleen, and bone marrow may result in dose-limiting toxicity.2-4
Screening of phage-display peptide libraries identified cancer-targeting peptides (CTPs) that can be used as alternative targeting agents in cancer therapies. CTPs have several advantages over antibodies: (i) better tumor penetration because of smaller size; (ii) less nonspecific binding to the reticuloendothelial system; (iii) easy to be derivatized and manufactured; and (iv) proteolytic stability if D- or unnatural amino acids are used.5,6 For example, CTPs featuring an Arg-Gly-Asp (RGD) motif selectively bind to angiogenic blood vessel markers such as αvβ3 and αvβ5 integrins.7,8 In contrast, CTPs with an Asn-Gly-Arg (NGR) motif bind with lower affinity to integrin but exhibit high preference to aminopeptidase N, also known as CD13,9 a membrane-bound metalloprotease expressed at high levels in tumor vasculature and an important player in angiogenesis.10 Conjugation of RGD or NGR to short peptides11 or small molecule anticancer drugs12 have shown a remarkable improvement in cancer cell selectivity and specificity, demonstrating that CTP-small therapeutic conjugates may become promising targeted drugs for cancer treatment.5,6
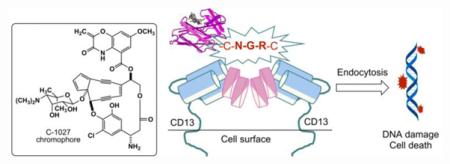
The chromoprotein family of enediyne antitumor antibiotics includes some of the most cytotoxic natural products known to date.13,14 As exemplified by C-1027,15-18 the chromoprotein is a noncovalent protein-small molecule complex consisting of an apo-protein (CagA) and an enediyne chromophore (C-1027) (Fig. 1). CagA serves to protect and transport the highly labile C-1027 chromophore. Upon release from CagA, the C-1027 chromophore readily undergoes a Bergman cyclization to generate a highly reactive benzenoid diradical (Fig. 1) that abstracts hydrogen atoms from DNA and causes both double-stranded breaks (DSBs) and interstrand cross-links (ICLs), hence the eventual cell death.19,20 While the natural enediynes have seen limited use as clinical drugs, mainly because of substantial general toxicity and lack of selectivity,21,22 both polymer-based delivery systems and ADCs have shown great clinical success or promise in cancer therapy.13,14
Figure 1.
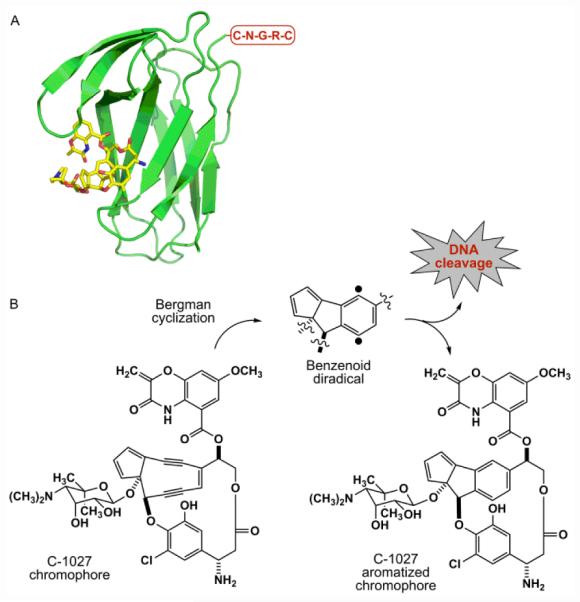
Schematic representation of an NGR-containing C-1027 chromoprotein. (A) The CagA-aromatized C-1027 chromophore complex structure (PDB code 1HZL), with its C-terminus depicted with the engineered NGR motif. (B) The C-1027 chromophore undergoing Bergman cyclization to yield a benzenoid diradical that causes DNA cleavage and cell death and the aromatized C-1027 chromophore.
Structures of both CagA and the CagA-aromatized C-1027 chromophore complex have been established.23 Since the C-1027 enediyne chromophore has been proposed to be in equilibrium with its benzenoid diradical form and is stabilized kinetically by CagA in the C-1027 chromoprotein, the CagA-aromatized C-1027 chromophore complex structure resembles that of the C-1027 chromprotein. Thus, the C-1027 enediyne chromophore is bound to a pocket of CagA formed by a four-stranded β-sheet and three loops by hydrophobic and electrostatic interactions (Fig. 1).23 CagA, serving as a natural drug carrier, therefore presents an opportunity to produce designer C-1027 chromoproteins by rational incorporation of targeting elements such as CTPs to increase selectivity and overcome nonspecific general cytotoxicity (Fig. 1).
Here we report the engineered production of an NGR-containing C-1027 in the C-1027 producing strain Streptomyces globisporus.24 The NGR motif, in the form of a CNGRC pentapeptide, is strategically fused to the C-terminus of CagA, which is distal to the C-1027 chromophore binding pocket, hence minimizing any potential adverse effects on formation of the CagA-C-1027 chromophore complex, and well exposed exteriorly, thus maximizing potential interaction with the CD13 target. The engineered NGR-containing C-1027 chromoprotein, produced directly by the recombinant S. globisporus SB1026 strain, is equally active as the native C-1027 chromoprotein against selected cancer cell lines. Our results demonstrate the feasibility to produce CTP-containing enediyne chromoproteins by metabolic pathway engineering and microbial fermentation and will inspire efforts to engineer other CTP-containing drug binding proteins for targeted delivery.
2. Results and Discussion
2.1. Engineering the S. globisporus SB1026 strain that produces NGR-containing C-1027
We have previously cloned and characterized the 75-kb C-1027 biosynthetic gene cluster from S. globisporus, and the cagA gene resides near one end of the cluster (Fig. S1).24 A two-step strategy was adopted to construct the SB1026 recombinant strain: (i) introduction of the ermE marker into or near cagA to facilitate gene replacement by homologous recombination and (ii) replacement of the mutated cagA allele with the engineered variant that encodes CagA with the designed NGR motif fused at its C-terminus (Fig. 2).
Figure 2.

Engineering the S. globisporus SB1026 strain that produces NGR-containing C-1027 chromoprotein. (A) Fusion of the NGR motif (red with the extra two Gly as linker in blue) at the C-terminus of CagA (only the C-terminus shown). (B) Constructions of the cagA mutant strain SB1024 and the NGR-containing C-1027 producing SB1026 strain. (C) Construction of the cagA mutant variant SB1025. AprR, apramycin resistant; AprS, apramycin sensitive; EryR, erythromycin resistant; EryS, erythromycin sensitive. (D) Southern analysis confirming the genotypes of SB1025 (lanes 2, 3) and SB1024 (lanes 4, 5) with the S. globisporus wild-type as control (lane 1). Genomic DNAs were digested with SacI, and the 4.3-kb SacI fragment was used as a probe. M, molecular weight standards.
Since we cannot predict a priori if cagA confers major self-resistance in S. globisporus, meaning a cagA deletion mutant would be lethal, two versions of cagA mutants were constructed. As depicted in Fig. 2B and 2C, we inserted ermE into either the BstXI site, placing ermE in the middle of cagA, disrupting its function, to afford pBS1138 or the XhoI site, placing ermE downstream of cagA, hence retaining a functional copy of cagA, to afford pBS1139. Both pBS1138 and pBS1139 were introduced into S. globisporus, and the desired double-crossover homologous recombination mutants were selected on the basis of an apramycin sensitive/erythromycin resistant phenotype, affording mutant strains SB1024 and SB1025, respectively; the genotypes of both strains were confirmed by Southern analysis (Fig. 2D).
Both SB1024 and SB1025 were fermented, with the S. globisporus wild-type as a control, and bioassayed against Micrococcus luteus as described previously to examine their ability to produce C-1027.24-27 We were surprised that C-1027 production was undetected in both strains (Fig. 3A), as it was not apparent why insertion of ermE downstream of cagA as in SB1025 would abolish C-1027 production. While we cannot exclude the possibility of polar effect, upon insertion of ermE, on downstream genes, resulting in the abolishment of C-1027 biosynthesis in SB1024 and SB1025, it is also possible that the labile C-1027 enediyne chromophore could not be biosynthesized in sufficient quantities, or even if biosynthesized, undergoes rapid Bergman cyclization into the inactive aromatized form, in the absence of CagA, therefore never constituting a real threat to the producing strains. Only SB1024 was moved forward to construct the SB1026 strain.
Figure 3.
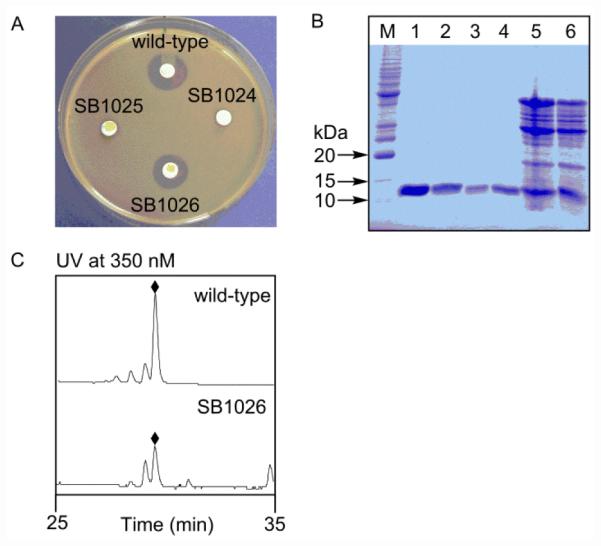
Production of NGR-containing C-1027 chromoprotein by S. globisporus SB1026. (A) C-1027 chromoprotein production as evaluated by bioassay against M. luteus. (B) CagA production as evaluated by 15% SDS-PAGE analysis. M, molecular weight standards. Crude (lane 5) and purified NGR-containing CagA (lanes 1, 2) from SB1026; crude (lane 6) and purified native CagA (lanes 3, 4) from wild-type. The predicted molecular weights of native CagA and NGR-containing CagA are 10,501.5 Da and 11,149.2 Da, respectively. (C) C-1027 chromophore (◆) production as evaluated by HPLC analysis.
Finally, we designed pBS1142 by fusing the NGR motif to the C-terminus of CagA (Fig. 2A, 2B). We preferred the C-terminus of CagA on the basis that: (i) it is distal to the C-1027 chromophore binding pocket, minimizing potential adverse impact on formation of the CagA-C-1027 chromophore complex and (ii) it is well exposed exteriorly, maximizing potential interaction of the resultant NGR-containing C-1027 chromoprotein with the CD13 target. The fact that the C-terminus is less structured also eases any concern that engineered NGR motif will impact the structural integrity of the resultant NGR-containing C-1027 chromoprotein (Fig. 1A). Thus, pBS1142 was introduced into SB1024, and the desired double-crossover homologous recombination mutant was selected on the basis of an apramycin sensitive/erythromycin sensitive phenotype, affording the mutant strain SB1026 (Fig. 2D), whose genotype was confirmed by DNA sequencing. SB1026 was similarly fermented, with the S. globisporus wild-type strain as a control.24-27 Bioassay against M. luteus supported that NGR-containing C-1027 was restored, with a titer comparable to that of the native C-1027 chromoprotein in the wild-type strain (Fig. 3A).
2.2. Production of NGR-containing C-1027 chromoprotein by S. globisporus SB1026
A scale-up fermentation of S. globisporus SB1026 was carried out, with the wild-type as a control, to produce and purify the NGR-containing C-1027 chromoprotein for structural confirmation, following essentially identical reported procedures.24-27 We first subjected the crude chromoprotein preparations to SDS-PAGE, showing comparable productions of the NGR-containing CagA from SB1026 and the native CagA from the wild-type strain (Fig, 3B), a finding that is consistent with the bioassay results (Fig. 3A). We next purified both the NGR-containing CagA and the native CagA protein to homogeneity (Fig. 3B) and subjected them to electrospray ionization mass spectroscopy (ESI-MS) (Fig. S2), yielding [M + H]+ ions at m/z 10,496 for the native CagA (calculated molecular weight 10,501.5) and 11,142 for the NGR-containing CagA (calculated molecular weight 11,149.2), respectively, confirming the predicted molecular species. Finally, we isolated the C-1027 chromophore from both the native and NGR-containing C-1027 chromprotein complexes and confirmed its identity by HPLC (Fig. 3C) and MS analysis, unambiguously establishing that fusion of the NGR motif to the C-terminus of CagA had no adverse effect on the production of the holo-NGR-containing C-1027 chromoprotein in SB1026 (i.e., fully loaded with the C-1027 chromophore in titers similar to those from the wild-type).
2.3. Determination of the expression levels of CD13 in selected breast cancer cell lines
The NGR motif is known to bind CD13 with high affinity.9 We first assayed five breast cancer cell lines MDA-MB-468, MDA-MB-231, SK-BR-3, MCF-7, and T47D for CD13 expression by flow cytometry, using a CD13-specific monoclonal antibody. While CD13 expression was not detected in MDA-MB-231, SK-BR-3 and MCF-7 and only a low level of CD13 expression was observed in T47D (0.5-log-unit signal displacement), a high level of CD13 expression (2-log-unit signal displacement) was found for MDA-MB-468 (Fig. S3). Collectively, these cell lines therefore provide an opportunity to evaluate and compare the antitumor activity of the native and NGR-containing C-1027 chromoproteins in these CD13-positive and CD13-negative cancer cell lines.
2.4. The effects of native and NGR-containing C-1027 chromoproteins on tumor cell viability
We carried out cytotoxicity evaluation (MTS assay) of the native and NGR-containing C-1027 chromoproteins against three selected cell lines: MDA-MB-468 (CD13-positive) and MDA-MB-231 and SK-BR-3 (both CD13-negative). The cells were incubated for 24 h with different concentrations of the C-1027 chromoprotein before the MTS assay was developed. The IC50 values of the native and NGR-containing C-1027 were 0.081 nM and 0.033 on MDA-MB-468 cells, 0.035 nM and 0.18 nM on MDA-MB-231 cells, and 0.17 nM and 0.072 nM on SK-BR-3 cells, respectively (Table 1 and Fig. S4A). Both the native and NGR-containing C-1027 chromoproteins showed rapid and complete cell killing in sub-nanomolar ranges, confirming that fusion of the NGR motif to the C-terminus of CagA did not impair formation between CagA and the C-1027 chromophore into functional chromoprotein complex. However, no apparent NGR-mediated selective killing on CD13-positive cancer cells was observed under the extended incubation time (24 h).
Table 1.
The cytotoxicity of native and NGR-containing C-1027 chromoproteins on selected CD13-positive and CD13-negative cancer cell lines
| IC50 (nM)a |
||
|---|---|---|
| cell lines | native C-1027 | NGR-containing C-1027 |
| MDA-MB-468 | 0.081 ± 0.006 | 0.033 ± 0.002 |
| MDA-MB-231 | 0.35 ± 0.01 | 0.18 ± 0.01 |
| SK-BR-3 | 0.17 ± 0.01 | 0.072 ± 0.005 |
IC50s were determined by curve fitting of the dose-response data (see Fig. S4A)
Since it was reported previously that an NGR-containing liposomal doxorubicin preparation was able to bind to the cell membrane and internalize within CD13-expressing cells in 15 minutes,12,28 we next tested the native and NGR-containing C-1027 chromoproteins under shorter incubation times to differentiate CD13-mediated cell killing. Thus, we incubated the cells with the C-1027 chromoproteins for 15 min, washed the cells with PBS, and further incubated the cells with drug-free complete medium for 72 h, before the MTS assay was developed. However, both the native and NGR-containing C-1027 showed little cytotoxicity on the CD13-negative MDA-MB-231 and SK-BR-3 cells at the concentrations (up to 10 nM) tested (Fig. S4B). Although a modest cytotoxicity was observed on the CD13-positive MDA-MB-468 cells, with the NGR-containing C-1027 killing ~40% of MDA-MB-468 cells at 1.1 nM, the difference between the native and NGR-containing C-1027 chromoproteins is too small to draw any definitive conclusion if the subtle difference observed truly resulted from NGR-mediated targeting. Due to the rapid internalization rate and extreme cytotoxicity of C-1027, we may not be able to differentiate NGR-mediated selective killing under the conditions tested.
Intriguingly, an NGR-containing C-1027 preparation was shown previously to improve CD13-mediated antitumor activity.29 However, in that study, (i) the NGR motif was fused to the N-terminus of CagA, (ii) the NGR-containing CagA was overproduced in E. coli, (iii) the holo-NGR-containing C-1027 chromoprotein complex was reconstituted using the NGR-containing CagA purified from E. coli and the C-1027 chromophore purified from S. globisporus wild-type, (iv) only 65% of the reconstituted NGR-containing C-1027 chromoprotein complex was loaded with the C-1027 chromophore, and (v) human fibrosarcoma HT-1080 cell line (CD13-positive) was used.29 While it remains to be sorted out if these differences account for the discrepancies seen between the two studies, the early findings do support the wisdom of engineering CTP-containing C-1027 chromoproteins to explore their potential as a targeted anticancer therapy.
3. Conclusion
CTPs identified through screening of phage-display peptide libraries provide promising targeting agents in cancer therapies.5,6 The enediyne family of antitumor antibiotics includes some of the most potent natural products known to date, and members of the enediyne family of natural products have been developed into efficacious anticancer drugs when their extremely potent cytotoxicity is delivered to tumor cells.13,14 Engineering of CTPs into the apo-proteins of the chromoprotein family of enediynes therefore represents an innovative strategy to develop the enediynes into a targeted anticancer therapy. We demonstrated in this study the production of an NGR-containing C-1027 chromoprotein, which is as active as the native C-1027 from the wild-type producer, by the engineered, recombinant S. globisporus SB1026 and by direct microbial fermentation. This strategy should be applicable to the engineered production of other CTP-containing C-1027 chromoproteins or any other members of the chromoprotein family of enediyne natural products. Since other drug binding proteins are known, such as BlmA21,30 for the bleomycin family of anticancer drugs and MtmC21,31 for the anticancer drug mitomycin, variations of our strategy could be envisaged to engineer CTPs into other drug binding proteins for targeted delivery.
4. Experimental section
4.1. Bacterial strains, plasmids and culture conditions
Strains and plasmids used in this study are summarized in Table S1. E. coli strains carrying plasmids were grown in Luria-Bertani medium and were selected with appropriate antibiotics. S. globisporus wild-type and recombinant strains were cultivated at 30 °C in tryptic soy broth (TSB) for growth of mycelia, on ISP4 media for sporulation, and on ISP4 media supplemented with 0.05% yeast extract and 0.1% tryptone for conjugation.32 All media for Streptomyces growth were prepared according to standard protocols.32
4.2. Biochemicals, chemicals, media, and cell culture
Common biochemicals and chemicals were from standard commercial sources. ISP4 and TSB were from Difco Laboratories (Detroit, MI). Restriction enzymes and other molecular biology reagents were from New England Biolabs (Ipswich, MA). Human breast cancer cells MDA-MB-468, MDA-MB-231, SK-BR-3, MCF-7, and T47D cells (American Type Culture Collection) were maintained in 10% (v/v) FBS in DMEM medium supplemented with 100 μg/mL streptomycin and 100 U/mL penicillin (all Life Technologies, Carlsbad, CA) at 37 °C in a humidified atmosphere of 5% CO2.
4.3. DNA isolation and manipulation
All plasmid subcloning experiments were performed in E. coli DH5α using standard protocols.33 Plasmid extractions and DNA purifications were carried out by using commercial kits (Qiagen, Valenica, CA). Genomic DNAs were isolated according to the literature protocol.32 The digoxigenin-11-dUTP labeling and detection kit (Roche Diagnostics Corp, Indianapolis, IN) was used for preparation of DNA probes, Southern hybridization, and detection according to the protocols provided by the manufacturer. PCR amplifications were performed using Taq DNA polymerase or Pfx high fidelity DNA polymerase.
4.4. Construction of the two cagA mutant strains S. glopisporus SB1025 and SB1024
A 4.3-kb SacI fragment from pBS1005,25 containing sgcA1, cagA, sgcA4, and the truncated sgcC3 and sgcA5, was subcloned into the same site of pUC18 to yield pBS1135. Upon digestion with KpnI, a 1.43-kb KpnI fragment containing the ermE gene was isolated from pIJ4026, blunt-ended with the DNA polymerase I, and ligated into the XhoI site (downstream of cagA) and BstXI site (within the cagA) of pBS1135, which were treated with DNA polymerase I and shrimp alkaline phosphatase, to generate pBS1137 and pBS1136, respectively. The 5.76-kb SacI fragments from pBS1137 and pBS1136, were then ligated into the EcoRV site of pKC1139, resulting in pBS1138 and pBS1139, which were introduced into S. globisporus by E. coli-S. globisporus conjugation according to the literature procedure with the following modifications.25 S. globisporus spores were suspended in TSB medium and heat-shocked at 50 °C for 10 min, followed by incubation at 30 °C for 6 hr. Germinated spores (as conjugation recipients) were mixed with E. coli ET12567/pUZ8002 harboring pBS1138 or pBS1139 (as conjugation donor) and spread onto modified ISP4 plates freshly supplemented with 20 mM MgCl2. After incubation at 30 °C for 16 to 20 h, each plate was overlaid with 1 mL of sterilized water containing apramycin (50 μg/mL) and nalidixic acid (50 μg/mL). Incubation continued at 28 °C until exconjugants appeared. Since pKC1139 bears a temperature sensitive replicon functioning only at temperature below 34 °C, double crossover mutants (AprSEryR), named SB1025 (insertion at XhoI site) and SB1024 (insertion at BstXI site), were selected by incubation at 39 °C.
4.5. Construction of the S. globisporus SB1026 strain that produces the NGR-containing C-1027
The following pair of primers were designed to clone by PCR the fragment encoding the NGR-containing CagA: cagA-FP (5 ’-CTGTGCCTCCGCAGCCGCCTCAGTGCCGCGAGtgatcaACC CCCTCCTC-3’) and cagA-RP (5 ’-GTctcgagTCAGCAACGGCCGTTGCAGCCGCCGCCGAAGG TCAGAGCCACG-3’) (the BclI and XhoI sites are shown in lower case, and underlined letters represent the reverse complement coding sequence of CNGRC, which is located at the C-terminus of CagA). The resulting PCR product was cloned into pGEM-T Easy (Promega, Madison, WI) to yield pBS1140. Upon digestion with BclI and XhoI, the 0.9-kb fragment was ligated into the same sites of pBS1135 to generate pBS1141. The 4.3-kb EcoRI and HindIII fragment from pBS1141 was then cloned into the same sites of pKC1139 to give pBS1142, which was introduced into SB1024 by conjugation from E. coli ET12567/pUZ8002. The spores of the exconjugants were spread onto ISP4 plates containing 50 µg/mL of apramycin and incubated at 39 °C, from which single crossover mutants (AprREryR) were obtained. The spores of the single crossover mutants were then spread onto ISP4 plates without antibiotics and incubated at 39 °C, from which double crossover mutant (AprSEryS), SB1126, was obtained.
4.6. Production, isolation and analysis of native and NGR-containing C-1027 chromoproteins
S. globisporus wild-type and recombinant strains were fermented using a two-stage fermentation process as described previously.24-27 Briefly, a spore suspension (5 μL) of each S. globisporus strain was first inoculated into 50 mL of seed medium in a 250-mL baffled flask and incubated at 28 °C and 250 rpm for 2 days. The resulting seed inoculum was used to inoculate the production medium (5 mL into 50 mL of production medium in a 250-mL flask) and incubated at 28 °C and 250 rpm for an additional 4 days. Both seed and production media consisted of 2% glycerol, 2% dextrin (sterile stock solution added after autoclaving), 1% fish meal, 0.5% peptone, 0.2% (NH4)2SO4, and 0.2% CaCO3 (pH 7.0). C-1027 production was detected by assaying its antibacterial activity against M. luteus.44 The fermentation supernatant (20 μL) was added to sterile 3M paper discs placed on LB agar plate seeded with an overnight M. luteus culture (0.01% [v/v]).
Isolation of the C-1027 chromoprotein, HPLC analysis of C-1027 chromophore, and SDS-PAGE of CagA were carried out following previously reported precedures.24-27 Briefly, (NH4)2SO4 was added to the 250 mL fermentation supernatant of relevant strains to 90% saturation and then adjusted to pH 4.0 with 0.1 N HCl. The precipitated C-1027 chromoprotein was dissolved in 15 mL of 0.1 M potassium phosphate (pH 8.0). The supernatant was then extracted with 50 mL of ethyl acetate, concentrated in vacuum, and re-dissolved in 250 μL of methanol. The cleared sample (25 μL) was subjected to HPLC on an Agilent C18 column (5 μm, 150 × 4.6 mm), eluted isocratically with 20 mM potassium phosphate (pH 6.86)/ CH3CN (50:50, [v/v]) at a flow rate of 1.0 mL/min and detected by monitoring UV absorbance at 350 nm. A C-1027 chromophore standard for HPLC analysis was confirmed by ESI-MS.
The (NH4)2SO4 precipitated C-1027 chromoprotein was further purified via an ion exchange column HiTrap Q XL (GE Healthcare, Little Chalfont, UK), using 20 mM N-methyl piperazine (pH 4.8) as start buffer and 20 mM N-methyl piperazine (pH 4.8) containing 1 M NaCl as elution buffer. The active fractions were combined, concentrated and subjected to a size exclusion column Superdex Tm200 (Hiload 16/60) previously equilibrated with 5mM Tris HCl (pH 8.5) for further purification. The single protein peak was pooled and lyophilized for SDS-PAGE and ESI-MS analysis. ESI-MS was performed with an Agilent 1100 HPLC-MSD SL ion trap mass spectrometer (Agilent Technologies, Santa Clara, CA).
4.7. Flow cytometry
Breast cancer cell lines MDA-MB-468, MDA-MB-231, SK-BR-3, MCF-7, and T47D cells were harvested with TrypLE Express (Life Technologies), washed with PBS once, resuspended in ice-cold 0.5% (w/v) BSA in PBS (flow cytometry buffer), and aliquots of 50 µL containing 5 × 105 cells were distributed into a V-bottom 96-well plate (Corning). The cells were incubated with 1 µg of PE-conjugated anti-human CD13 antibody (Biolegend, San Diego, CA) for 30 min on ice. After washing twice with ice-cold flow cytometry buffer, the cells were resuspended in 200 µL of flow cytometry buffer and analyzed using an LSR II Flow Cytometer (Becton-Dickinson). Data were analyzed using FlowJo software (Tree Star).
4.8. Cell viability assay
The effects of native and NGR-containing C-1027 chromoproteins on tumor cell viability were assessed using CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (Promega, Madison, WI). Cells were plated in 96-well plates at 5000 cells/well and allowed to adhere overnight at 37°C in a humidified atmosphere of 5% CO2. Medium was then removed and replaced by fresh culture medium containing graded doses of native and NGR-containing C-1027 chromoproteins. The cells were treated for 15 min, then the drugs were washed away and the cells were further incubated for 72 h in drug-free complete medium before the MTS assay was developed. Alternatively, the cells were incubated with drugs for 24 h before the MTS assay was developed.
Supplementary Material
Acknowledgments
We thank Dr. Y. Li, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences, Beijing, China, for the S. globisporus wild-type strain. This work was supported in part by a German Research Foundation (DFG) postdoctoral fellowship (to I.C) and the US National Institutes of Health grant CA78747 (to B.S.).
Footnotes
Supplementary Material
Supplementary material associated with this article can be found, in the online version, at http://
References and notes
- 1.Abramson R. 2015 http://www.mycancergenome.org/content/molecular-medicine/overview-of-targeted-therapies-for-cancer/
- 2.Chari RV. Acc Chem Res. 2008;41:98. doi: 10.1021/ar700108g. [DOI] [PubMed] [Google Scholar]
- 3.Ducry L, Stump B. Bioconjug Chem. 2010;21:5. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 4.Srinivasarao M, Galliford CV, Low PS. Nat. Rev. Drug Discov. 2015;14:203. doi: 10.1038/nrd4519. [DOI] [PubMed] [Google Scholar]
- 5.Aina OH, Sroka TC, Chen ML, Lam KS. Biopolymers. 2002;66:184. doi: 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]
- 6.Aina OH, Liu R, Sutcliffe JL, Marik J, Pan CX, Lam KS. Mol Pharm. 2007;4:631. doi: 10.1021/mp700073y. [DOI] [PubMed] [Google Scholar]
- 7.Pasqualini R, Koivunen E, Ruoslahti EJ. Cell Biol. 1995;130:1189. doi: 10.1083/jcb.130.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koivunen E, Wang B, Ruoslahti E. Biotechnology. 1995;13:265. doi: 10.1038/nbt0395-265. [DOI] [PubMed] [Google Scholar]
- 9.Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, Ashmun RA, Shapiro LH, Arap W, Ruoslahti E. Cancer Res. 2000;60:722. [PMC free article] [PubMed] [Google Scholar]
- 10.Wickström M, Larsson R, Nygren P, Gullbo J. Cancer Sci. 2011;102:501. doi: 10.1111/j.1349-7006.2010.01826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qifan W, Fen N, Ying X, Xinwei F, Jun D, Ge Z. Tumour Biol. 2016 doi: 10.1007/s13277-016-4961-x. doi:10.1007/s13277-016-4961-x. [DOI] [PubMed] [Google Scholar]
- 12.Garde SV, Forté AJ, Ge M, Lepekhin. E. A.; Panchal CJ, Rabbani SA, Wu JJ. Anticancer Drugs. 2007;18:1189. doi: 10.1097/CAD.0b013e3282a213ce. [DOI] [PubMed] [Google Scholar]
- 13.Van Lanen SG, Shen B. Curr. Topics Med. Chem. 2008;8:448. doi: 10.2174/156802608783955656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang Z-X. Nat. Prod. Rep. 2010;27:499. doi: 10.1039/b908165h. [DOI] [PubMed] [Google Scholar]
- 15.Zhen YS, Ming XY, Yu B, Otani T, Saito H, Yamada Y. J Antibiot. 1988;42:1294. doi: 10.7164/antibiotics.42.1294. [DOI] [PubMed] [Google Scholar]
- 16.Minami Y, Yoshida K, Azuma R, Saeki M, Otani T. Tetrahedron Lett. 1993;34:2633. [Google Scholar]
- 17.Yoshida K, Minami Y, Azuma R, Saeki M, Otani T. Tetrahedron Lett. 1993;34:2637. [Google Scholar]
- 18.Iida K, Fukuda S, Tanaka T, Hirama M, Imajo S, Ishiguro M, Yoshida K, Otani T. Tetrahedron Lett. 1996;37:4997. [Google Scholar]
- 19.Kennedy DR, Gawron LS, Ju J, Liu W, Shen B, Beerman T,A. Cancer Res. 2007;67:773. doi: 10.1158/0008-5472.CAN-06-2893. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy DR, Ju J, Shen B, Beerman TA. Proc Natl Acad Sci USA. 2007;104:17632–1767. doi: 10.1073/pnas.0708274104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nicolaou KC, Smith AL, Yue EW. Proc Natl Acad Sci USA. 1993;90:5881. doi: 10.1073/pnas.90.13.5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Chem Rev. 2005;105:739. doi: 10.1021/cr030117g. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka T, Fukuda-Ishisaka S, Hirama M, Otani T. J Mol Biol. 2001;309:267. doi: 10.1006/jmbi.2001.4621. [DOI] [PubMed] [Google Scholar]
- 24.Liu W, Christenson SD, Standage S, Shen B. Science. 2002;297:1170. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]
- 25.Liu W, Shen B. Antimicrob. Agents Chemother. 2000;44:382. doi: 10.1128/aac.44.2.382-392.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Yin M, Horsman GP, Huang S, Shen B. J. Antibiot. 2010;63:482. doi: 10.1038/ja.2010.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Yin M, Horsman GP, Shen B. J. Nat. Prod. 2011;74:420. doi: 10.1021/np100825y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Hensbergen Y, Broxterman HJ, Elderkamp YW, Lankelma J, Beers JC, Heijn M, Boven E, Hoekman K, Pinedo HM. Biochem Pharmacol. 2002;63:897. doi: 10.1016/s0006-2952(01)00928-5. [DOI] [PubMed] [Google Scholar]
- 29.Zheng YB, Shang BY, Li Y, Zhen YS. Biomed Pharmacother. 2013;67:164. doi: 10.1016/j.biopha.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Galm U, Wendt-Pienkowski E, Wang L, Huang S-X, Unsin C, Tao M, Coughlin JM, Shen B. J. Nat. Prod. 2011;74:526. doi: 10.1021/np1008152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheldon PJ, Johnson DA, August PR, Liu H-W, Sherman DH. J. Bacteriol. 1997;179:1796. doi: 10.1128/jb.179.5.1796-1804.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kieser T, Bibb MJ, Chater KF, Buttner MJ, Hopwood D. John Innes Foundation; Norwich, UK: 2000. Norwich. [Google Scholar]
- 33.Sambrook J, Fritsch EF, Maniatis T. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.