

Abstract
Background
ABP 501 is being developed as a biosimilar to adalimumab. Comprehensive comparative analytical characterization studies have been conducted and completed.
Objective
The objective of this study was to assess analytical similarity between ABP 501 and two adalimumab reference products (RPs), licensed by the United States Food and Drug Administration (adalimumab [US]) and authorized by the European Union (adalimumab [EU]), using state-of-the-art analytical methods.
Methods
Comprehensive analytical characterization incorporating orthogonal analytical techniques was used to compare products. Physicochemical property comparisons comprised the primary structure related to amino acid sequence and post-translational modifications including glycans; higher-order structure; primary biological properties mediated by target and receptor binding; product-related substances and impurities; host-cell impurities; general properties of the finished drug product, including strength and formulation; subvisible and submicron particles and aggregates; and forced thermal degradation.
Results
ABP 501 had the same amino acid sequence and similar post-translational modification profiles compared with adalimumab RPs. Primary structure, higher-order structure, and biological activities were similar for the three products. Product-related size and charge variants and aggregate and particle levels were also similar. ABP 501 had very low residual host-cell protein and DNA. The finished ABP 501 drug product has the same strength with regard to protein concentration and fill volume as adalimumab RPs. ABP 501 and the RPs had a similar stability profile both in normal storage and thermal stress conditions.
Conclusion
Based on the comprehensive analytical similarity assessment, ABP 501 was found to be similar to adalimumab with respect to physicochemical and biological properties.
Key Points
| ABP 501 is being developed by Amgen Inc. as a biosimilar to adalimumab. The primary and higher-order structure of ABP 501 drug product have been shown to be similar to adalimumab reference products (RPs). |
| ABP 501 behaved in a similar manner to adalimumab RPs in functional bioassays. |
| The high degree of similarity in structure and function provides assurance that ABP 501 will likely be similar to adalimumab RPs in the clinic, including pharmacokinetics, pharmacodynamics, efficacy, and safety. |
| Clinical similarity has been confirmed in human pharmacokinetics/pharmacodynamics (phase I) and clinical efficacy and safety (phase III) studies. |
Introduction
ABP 501 is being developed by Amgen Inc. (Thousand Oaks, CA, USA) as a biosimilar to adalimumab (Humira®), a recombinant monoclonal antibody (mAb) that binds tumor necrosis factor (TNF)-α, thus inhibiting engagement of TNF receptors and initiation of consequent proinflammatory signaling. Adalimumab is indicated for treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn′s disease, ulcerative colitis, plaque psoriasis, and hidradenitis suppurativa [1, 2].
A biosimilar is a biotherapeutic product that is similar in terms of quality, safety, and efficacy to an already licensed biotherapeutic reference product (RP) [3–5]. Guidelines describing quality considerations and non-clinical and clinical requirements for the development of biosimilar mAbs were issued by the European Medicines Agency (EMA) in 2012 and 2014 [6, 7] and the United States (US) Food and Drug Administration (FDA) issued final guidance for the regulatory review of biosimilars in 2015 [5, 8]. As of this writing, 19 biosimilars have been approved in Europe [9, 10] and in the USA the first biosimilar was approved in March 2015 [11].
Biotherapeutic products are structurally and functionally complex. Because the innovator’s manufacturing processes in producing the RP are proprietary, the development of a biosimilar involves reverse engineering of the RP to understand the critical quality attributes in order to design a new process that produces a similar molecule. Regulatory guidance recommends a totality of evidence approach based on a step-wise process for biosimilar product development; this begins with extensive structural analysis to understand the RP profile and to design the desired target product profile for the proposed biosimilar. Producing a biosimilar with a matching structure and functional properties is a challenging task. Differences in cell line and manufacturing processes are expected to result in minor analytical differences in a proposed biosimilar compared with RPs [12–19]. Nevertheless, biosimilars should match the biological functions of the RPs, including demonstration of expected functions without gain of new functions. Guidance documents, therefore, dictate the need for comprehensive and in-depth physicochemical and biofunctional characterization of proposed biosimilars [3–8, 20–22]. Observed analytical differences need to be characterized using orthogonal methods to cross-confirm the data, and to further understand differences. The potential impact of any differences on product safety and efficacy needs to be understood and justified in order to show these are not clinically relevant.
Here we report the analytical characterization of Amgen′s proposed biosimilar ABP 501, using comprehensive analytical techniques to determine its similarity with FDA-licensed adalimumab (adalimumab [US]) and European Union (EU)-authorized adalimumab (adalimumab [EU]), referred to as adalimumab RPs. The analytical methods used to evaluate the similarity of APB 501 with adalimumab RPs included those considered appropriate for lot release and stability testing to assess the purity, potency, strength, and identity of an mAb product, as well as characterization methods capable of detecting differences in the primary, secondary, and tertiary protein structures. The similarity assessment testing plan was designed to comprehensively assess physicochemical and functional similarity and ensure the detection and understanding of any differences between ABP 501 and adalimumab RPs according to US and EU regulatory guidelines [3, 5, 6, 8, 23].
Methods
The similarity testing plan, summarized in Table 1, assessed both the active ingredients and inactive ingredients in the drug products and provided a comprehensive structural and functional comparison. One ABP 501 reference standard lot produced from the same manufacturing process is used as assay standard for all analytical testing. All analyses were performed at Amgen.
Table 1.
Similarity testing plan and the analytical methods used for the physicochemical and functional characterization of the proposed biosimilar ABP 501 and adalimumab reference products
| Category | Attributes and analytical techniques |
|---|---|
| General properties | Protein concentration and deliverable volume Osmolality, pH, and surfactant concentration Appearance, color, and clarity |
| Primary structure | Molecular mass of intact whole protein Molecular mass of reduced and deglycosylated HC and LC Protein sequence by reduced peptide map Disulfide structure by non-reduced peptide map N-glycan map by HILIC HPLC Isoelectric point by capillary isoelectric focusing Extinction coefficient by amino acid analysis Identity by anti-idiotype ELISA |
| Higher-order structure | Secondary structure by FTIR Tertiary structure by near UV-CD Thermodynamic properties by DSC |
| Particles and aggregates | Subvisible particle concentrations by light obscuration Subvisible particle concentrations and morphology by MFI Submicron particle profile by DLS Submicron particle profile by FFF-LS Aggregate profile by AUC-SV Aggregate profile by SE-HPLC with light scattering |
| Product-related substances and impurities | Size variants by SE-HPLC, reduced and non-reduced CE-SDS Charge variants by CEX-HPLC |
| Host-cell impurities | Host-cell protein by ELISA, 2D-LC-MSE, and 2D precast SDS polyacrylamide Residual DNA by qPCR |
| Biological activity | TNFα-mediated apoptosis inhibition bioassay Effector function—ADCC and CDC sTNFα binding by ELISA FcRn binding by cell-based binding assay |
| Thermal forced degradation | Thermal stability at 25, 40, and 50 °C assessed by purity and potency assays |
2D two-dimensional, 2D-LC-MS E two-dimensional liquid chromatography coupled with online mass spectrometry involving data-independent MSE acquisition, ADCC antibody-dependent cell-mediated cytotoxicity, AUC-SV analytical ultracentrifugation sedimentation velocity, CDC complement-dependent cytotoxicity, CE-SDS capillary electrophoresis-sodium dodecyl sulfate, CEX-HPLC cation exchange-high-performance liquid chromatography, DLS dynamic light scattering, DSC differential scanning calorimetry, ELISA enzyme-linked immunosorbent assay, FcRn Fc neonatal receptor, FFF-LS field flow fractionation-light scattering, FTIR Fourier-transformed infrared, HC heavy chain, HILIC hydrophilic interaction liquid chromatography, HPLC high-performance liquid chromatography, LC light chain, MFI micro-flow imaging, qPCR quantitative polymerase chain reaction, SDS sodium dodecyl sulfate, SE-HPLC size exclusion-high-performance liquid chromatography, sTNFα soluble tumor necrosis factor-α, TNFα tumor necrosis factor-α, UV-CD ultraviolet circular dichroism
Materials
ABP 501 was manufactured by Amgen. Multiple lots of adalimumab (US) and adalimumab (EU) RPs were procured, stored according to the manufacturer′s instructions, and tested. For ABP 501, drug product was used for all analyses shown, with the exception of the analysis of host-cell impurities, for which ABP 501 drug substance was used.
Comparisons of biophysical properties including near ultraviolet (UV)-circular dichroism (UV-CD), differential scanning calorimetry (DSC), analytical ultracentrifugation (AUC), and dynamic light scattering (DLS) required the dilution of all products into a common buffer at pH 5.2. Therefore, a buffer was prepared according to the formulation reported in the adalimumab (US) prescribing information [24], hereinafter referred to as adalimumab drug product buffer, and was used to dilute ABP 501 as well as adalimumab (US) and adalimumab (EU) drug product samples to concentrations at or below approximately 1 mg/mL prior to analysis.
Attributes and Methods
Intact and Reduced Deglycosylated Mass Analysis
Molecular masses of the intact molecules were determined by electrospray ionization time-of-flight mass spectrometer analysis. Samples were separated from buffer components and introduced to the mass spectrometer by reversed-phase high-performance liquid chromatography (HPLC) (Agilent 1100/1200; Agilent Technologies, Santa Clara, CA, USA). The resulting summed ion spectra were deconvoluted to produce molecular mass profiles and compared with theoretical mass values. For reduced and deglycosylated heavy chain (HC) and light chain (LC) mass analysis, samples were treated with peptide-N-glycosidase F (PNGase F) (Wako Chemicals, Richmond, VA, USA) to remove N-linked glycans, and were subsequently denatured with guanidine hydrochloride (Thermo Scientific, Somerset, NJ, USA) and the disulfides were reduced with dithiothreitol (Thermo Scientific). The disulfide-reduced and PNGase F-treated samples were separated from buffer components and introduced to the mass spectrometer via reversed-phase HPLC. Summed ion spectra were deconvoluted to produce molecular mass profiles and were compared with theoretical mass values.
Reduced and Non-Reduced Peptide Mapping
Reduced peptide mapping analysis was conducted by enzymatic digestion with trypsin (Roche Life Science, Indianapolis, IN, USA), followed by reduction with dithiothreitol and alkylation with sodium iodoacetic acid (Sigma-Aldrich, St Louis, MO, USA). The resulting cleavage fragments were separated by reverse-phase ultra-HPLC (Agilent 1290, Agilent Technologies; Acquity H-Class, Waters Corp, Milford, MA, USA) using an increasing gradient of acetonitrile in water. The eluted peaks were detected by UV absorbance (214 nm) and peptides identified by online liquid chromatography with tandem mass spectrometry (MS/MS) using an Orbitrap Velos mass spectrometer (Thermo Scientific). For non-reduced peptide mapping, samples were digested with endoprotease Lys-C (Wako Chemicals) under denaturing but non-reducing conditions, and the resulting peptides were analyzed in a similar manner. Peptides containing disulfide bonds were identified by comparison of peptides generated under reducing and non-reducing conditions.
Glycan Analysis
N-linked glycans were released with PNGase F and the released oligosaccharides were derivatized with the fluorescent label 2-aminobenzoic acid (Sigma-Aldrich) at the reducing terminal N-acetylglucosamine. The labeled oligosaccharides were separated by hydrophilic interaction liquid chromatography (HILIC) HPLC using acetonitrile mobile phase with an increasing gradient of ammonium formate in water and detected by fluorescence. Peak identification was performed using mass spectrometry by coupling the HILIC HPLC with an ion-trap mass spectrometer for verification against the expected glycan mass.
Isoelectric Points
Isoelectric points (pI) were determined by capillary isoelectric focusing (cIEF) analysis using a Beckman PA800 high-resolution capillary electrophoresis instrument equipped with a neutral-coated capillary. Samples were separated using a pH gradient and were detected by UV absorbance (280 nm). The pI of the sample peaks were determined using a linear regression between two pI marker peaks.
Extinction Coefficients
Extinction coefficients were determined experimentally by amino acid analysis. Samples were hydrolyzed and the resulting amino acids quantified by strong cation exchange (CEX) chromatography and post-column derivatization with ninhydrin using an amino acid analyzer. The UV absorbance at 280 nm was measured and the experimental extinction coefficients were derived.
Identity Assay
To determine if the drug products shared common complementarity determining region (CDR) structural characteristics, a dual anti-idiotype mAb sandwich enzyme-linked immunoassay (ELISA) identity test was used.
Fourier-Transformed Infrared Spectroscopy and Near Ultraviolet-Circular Dichroism Spectroscopy
Secondary structure was assessed by Fourier-transformed infrared (FTIR) spectroscopy. FTIR measurements were made at room temperature using a Bruker Vertex 70 FTIR spectrometer (Bruker Corporation, Billerica, MA, USA). To create a common sample matrix, ABP 501 samples were diluted 1:1 with the adalimumab drug product buffer, and adalimumab samples were diluted 1:1 with the ABP 501 formulation buffer. The spectrum of the buffer blank (the adalimumab drug product buffer and ABP 501 formulation buffer mixed at a 1:1 ratio) was recorded under identical conditions and was subtracted from the protein spectra. The second derivative spectrum was calculated using a 9-point smoothing of the original spectra. Spectral similarity was quantitatively determined using the Thermo OMNIC software QC compare function (Thermo Scientific).
Tertiary structure was assessed by near UV-CD spectroscopy. Near UV-CD measurements were made on an Applied Photophysics Chirascan spectropolarimeter (Applied Photophysics Ltd, Leatherhead, UK) at ambient temperature using cuvettes with a path length of 1 cm. All drug product samples were diluted to approximately 0.7 mg/mL with the adalimumab drug product buffer prior to measurements. The resulting spectra were corrected for concentration and contributions from buffer, and reported as circular dichroism ellipticity. Spectral similarity was quantitatively determined using the Thermo OMNIC software QC compare function.
Thermodynamic Properties by Differential Scanning Calorimetry
DSC measures thermal unfolding temperatures of specific domains of a protein. Thermal stability was assessed by DSC measurements that were made in triplicate using a MicroCal VP-Capillary DSC system (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). All samples were diluted to approximately 0.5 mg/mL with adalimumab drug product buffer prior to measurements.
Aggregates by Analytical Ultracentrifugation Sedimentation Velocity
Aggregates were analyzed by AUC sedimentation velocity (AUC-SV). AUC-SV measurements were made using a Beckman PCS ProteomeLab XL-1 analytical ultracentrifuge (Beckman Coulter Inc., Brea, CA, USA) at 45,000 rpm following the UV absorbance at 280 nm. All product samples were diluted to approximately 0.5 mg/mL with adalimumab drug product buffer before measurements. Scans were collected at 20 °C without delay between them. Data were analyzed using SEDFIT v9.4 [25], and for data analysis the frictional ratio, time invariant noise, and meniscus values were allowed to float during the non-linear least squares fit.
Aggregates by Size Exclusion-High-Performance Liquid Chromatography with Light Scattering Detection
Aggregates were analyzed by size exclusion-HPLC (SE-HPLC) with light scattering (LS) detection (SE-HPLC-LS). SE-HPLC measurements were made on an Agilent 1100 HPLC system with a Tosoh Bioscience (Minato, Tokyo, Japan) TSK-GEL G3000SWxl column (5 µm, 7.8 × 300 mm). The SE-HPLC system was equipped with an LS detector (DAWN® HELEOS™ II MALS; Wyatt Technology Corporation, Goleta, CA, USA), and a refractive index (RI) detector (Wyatt Optilab rEX RI; Wyatt Technology Corporation). Samples without dilution were injected into the HPLC system, and the levels of high molecular weight (HMW) species and monomer were quantified and their molecular weights calculated. A differential RI value of 0.185 mL/g was used for molecular weight calculation.
Reduced and Non-Reduced Capillary Electrophoresis-Sodium Dodecyl Sulfate
Capillary electrophoresis-sodium dodecyl sulfate (CE-SDS) is used for separation of denatured protein size variants under non-reduced or reduced conditions. For non-reduced CE-SDS (nrCE-SDS), drug product samples of ABP 501 and adalimumab RPs were denatured with sodium dodecyl sulfate (Beckman Coulter, Jersey City, NJ, USA) at 60 °C for 5 min. For reduced CE-SDS (rCE-SDS), β-mercaptoethanol (Thermo Scientific) was added to the protein denaturation step to reduce the disulfide bonds. After denaturation, both non-reduced and reduced samples were injected onto a bare, fused silica capillary (Beckman Coulter, Jersey City, NJ, USA) and separated based on hydrodynamic size resulting from an applied electric field in which migration of smaller-sized proteins is inversely related to overall size. Analytes were monitored by UV absorbance at 220 nm, and purity evaluated by determining the peak area of each species as a percentage of the total peak area.
Submicron Particles by Dynamic Light Scattering and Field Flow Fractionation
The presence and distribution of submicron particles were assessed by DLS. DLS measurements were made in triplicate on a Wyatt Dynapro (Wyatt Technology Corporation) DLS plate reader at 25 °C using a 386-well glass bottom plate (20 μL per sample). Samples were diluted to approximately 1 mg/mL with the adalimumab drug product buffer prior to measurements. Solvent viscosity was calculated by the application program (DYNAMICS® software 7.0; Wyatt Technology Corporation) based on buffer composition, and was used for deriving particle hydrodynamic radius and distribution.
The presence and distribution of submicron particles were further assessed by field flow fractionation (FFF) with LS detection (FFF-LS). FFF-LS measurements were made using an Eclipse™ Flow-FFF system coupled with a DAWN HELEOS II LS detector (Wyatt Technology Corporation). The resulting FFF-LS profiles were used to detect the presence of submicron particles and to analyze their distribution. Polystyrene particle standards of 100, 200, and 500 nm were used as positive controls and buffer blank was used as a negative control.
Subvisible Particles by Light Obscuration and Micro-Flow Imaging
Subvisible particles were assessed by light obscuration (LO). LO measurements were made with an HIAC 9703+ liquid particle counting system (Beckman Coulter Life Sciences, Brea, CA, USA) equipped with an HRLD 150 sensor (Hach Company, Loveland, CO, USA). For each sample test, the particle concentration results were reported as cumulative particle counts per mL for ≥2, ≥5, ≥10, and ≥25 μm size ranges.
Subvisible particles were further assessed by micro-flow imaging (MFI). MFI measurements were made with an MFI 5200 flow microscope equipped with a 100 µm/1.6 mm/SP3 flow cell (ProteinSimple, San Jose, CA, USA). For each sample test, particle concentration results were reported as cumulative particle counts per mL for ≥2, ≥5, ≥10, and ≥25 μm size ranges, as well as ≥5 μm non-spherical particles with an aspect ratio of <0.70.
Cation Exchange-High-Performance Liquid Chromatography
Charged isoforms in drug product samples of ABP 501 and adalimumab RPs were separated on a CEX-HPLC Pro Pac® WCX-10 analytical column, 4.0 × 250 mm column (Dionex Corp., Sunnyvale, CA, USA), eluted using a salt gradient and monitored by UV absorbance. Purity was evaluated by determining the peak area of each charged isoform group (main, acidic, and basic) that eluted separately as a percentage of the total peak area.
Host-Cell Impurities
The residual host-cell protein (HCP) in ABP 501, adalimumab (US), and adalimumab (EU) were characterized and compared using three orthogonal methods. An HCP ELISA method was developed specifically for the ABP 501 cell line and process. This method provides quantitative measurement of total HCP detected through the use of an anti-HCP polyclonal antibody reagent. To further characterize the type of HCPs present in each product, a reverse-phase two-dimensional (2D) liquid chromatography (2D-LC) coupled with online mass spectrometry involving data-independent (MSE) acquisition (2D-LC-MSE) method was used to identify and quantify HCPs based on a mass spectrometric technique. Finally, a 2D differential in-gel electrophoresis (2D-DIGE) method comparing ABP 501 and adalimumab (US) was performed to confirm the absence of new species in a qualitative manner.
Residual DNA was analyzed by quantitative polymerase chain reaction and fluorescence detection, as previously described [26].
Functional Bioassays
The potency of ABP 501 and of adalimumab RPs was assessed in an apoptosis inhibition bioassay that utilizes the human histiocytic lymphoma cell line U-937. Induction of antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) were evaluated using Chinese hamster ovary (CHO) target cells (CHO M7) (Amgen) expressing a non-cleavable membrane-expressed form of TNFα [27]. Briefly, for ADCC, target cells were loaded with calcein-acetoxymethyl (AM), and were opsonized with a concentration range of drug product test article prior to co-incubation with effector cells (NK92-M1 cells stably transduced with human CD16). Calcein released into the media as a result of the cytotoxicity reaction was measured using a microplate reader. For CDC, calcein-AM-loaded CHO M7 target cells were opsonized with a concentration range of antibody, followed by addition of baby rabbit complement to bring about the cytotoxicity. The supernatant was removed and fluorescence was measured using a microplate reader. Test sample cytotoxicity relative to the ABP 501 reference standard was determined and reported as percent relative cytotoxicity.
Relative binding of drug product test article to Fc neonatal receptor (FcRn) and soluble TNFα (sTNFα) was assessed using binding assays. For FcRn binding, fluorescently labeled recombinant immunoglobulin G (IgG), type I-Fc (Amgen) is used to compete for binding with the test article to 293T cells (a variant of the human embryonic kidney cell line, 293, expressing the SV40 large T antigen) (American Type Culture Collection, Manassas, VA, USA) and is engineered to express FcRn on their surface.
Relative binding to sTNFα is a solid-phase ELISA. Recombinant human sTNFα is coated onto the wells of a microtiter ELISA plate. Receptor binding activities are calculated based on the ratio of EC50 (half maximal effective concentration) values of the reference standard curve relative to the test sample. Binding units are defined as the number of units of binding activity per 600 ng/mL of reference protein.
Stability and Degradation at Accelerated Conditions
The thermal stability and degradation profiles of ABP 501 drug product, adalimumab (US), and adalimumab (EU) were determined at 25, 40, and 50 °C. Since there are differences between product formulations for ABP 501 and FDA-licensed and EU-authorized adalimumab, potential differences in degradation rates or profiles due to formulation buffers were examined by comparing ABP 501 in its own buffer and in a buffer representative of adalimumab formulation buffer. Subsequent characterization of the degradation samples were conducted using SE-HPLC, CEX-HPLC, rCE-SDS, nrCE-SDS, and apoptosis inhibition bioassay.
Results and Discussion
The similarity testing plan (Table 1) to compare ABP 501with adalimumab (US) and adalimumab (EU) RPs assessed both the active ingredients and inactive ingredients in the ABP 501 and adalimumab RP drug products, and provided a comprehensive structural and functional comparison. Where applicable, state-of-the-art and orthogonal methods were used to further analyze product attributes.
Primary Structure
Similarity for the primary structures between ABP 501 and adalimumab (US) and adalimumab (EU) was investigated using several complementary characterization methods, including intact protein mass analysis, reduced and deglycosylated HC and LC mass analyses, reduced and non-reduced peptide mapping, glycan mapping and species characterization, comparison of pI, extinction coefficients, and confirmation of identity using an anti-idiotypic immunoassay. Results of intact mass analysis, reduced peptide mapping, and glycan mapping and characterization are shown in Fig. 1a–c, respectively.
Fig. 1.

Primary structure: comparison of ABP 501 (red line), adalimumab (US) (black line), and adalimumab (EU) (blue line) intact molecular mass profile (a), reduced peptide map (b), and glycan map (c). Adalimumab (EU) European Union-authorized adalimumab, adalimumab (US) United States Food and Drug Administration-licensed adalimumab, AU absorbance units, LU luminescent units. See text for definition of Peaks A-D
As shown in Fig. 1a, the predominant species were consistent with the presence of two core-fucosylated complex N-linked glycans with 0, one, or two terminal galactose residues, depicted as G0F, G1F, and G2F, respectively. Peaks A, B, C, and D were identified as the predominant forms with molecular weights consistent with a structure that contains two glycans comprising G0F:G0F (peak A); G0F:G0F+K (peak B); G0F:G1F (peak C); and A2G1F:A2G1F or A2G0F:A2G2F (peak D). The intact molecular mass of all predominant species matched the theoretical mass and were similar among the three products as shown in Table 2. Additionally, the molecular masses of reduced and deglycosylated drug product provided further assurance that the polypeptide compositions were similar and as expected (Table 2).
Table 2.
Summary of attributes for APB 501, adalimumab (US), and adalimumab (EU)
| Analytical testing/attributes | ABP 501 [range (n)] | Adalimumab (US) [range (n)] | Adalimumab (EU) [range (n)] |
|---|---|---|---|
| Protein concentration (mg/mL) | 47.9–52.6 (10) | 48.1–52.3 (23) | 49.6–53.7 (18) |
| Volume (mL) | 0.79–0.81 (12) | 0.80–0.81 (14) | 0.79–0.81 (10) |
| Intact molecular weight (Da) | |||
| A: Glycosylation—G0F:G0F | 148,083–148,084 (4) | 148,083–148,083 (4) | 148,083–148,084 (4) |
| B: Glycosylation—G0F:G0F (+K) | 148,208–148,212 (4) | 148,212–148,212 (4) | 148,211–148,212 (4) |
| C: Glycosylation—G0F:G1F | 148,244–148,245 (4) | 148,244–148,245 (4) | 148,244–148,245 (4) |
| D: Glycosylation—G1F:G1F or G0F:G2F | 148,407–148,407 (4) | 148,406–148,407 (4) | 148,405–148,407 (4) |
| Reduced and deglycosylated heavy chain (Da) | 49,200.4–49,200.7 (4) | 49,200.4–49,200.6 (4) | 49,200.4–49,200.5 (4) |
| Reduced and deglycosylated light chain (Da) | 23,412.2–23,412.4 (4) | 23,412.2–23,412.4 (4) | 23,412.2–23,412.4 (4) |
| Glycosylation at Asn 301 (%) | 99.0–99.4 (10) | 97.5–98.3 (24) | 97.7–98.8 (18) |
| Glycan map (%) | |||
| Galactosylation | 19.9–26.5 (7) | 17.6–21.6 (24) | 17.7–21.5 (18) |
| High mannose | 5.0–8.5 (7) | 7.0–9.7 (24) | 6.8–8.6 (18) |
| Afucosylation | 1.6–2.2 (7) | 1.1–1.7 (24) | 1.2–1.7 (18) |
| Total afucosylation | 6.6–10.8 (7) | 8.9–12.4 (24) | 9.0–10.9 (18) |
| Sialylation | 0.5–0.8 (7) | 0.1–0.3 (24) | 0.2–0.3 (18) |
| FTIR/spectral similarity (%) | |||
| US RP | 99.3–99.9 (6) | 99.0–99.9 (6) | 99.2–99.8 (6) |
| EU RP | 99.4–99.9 (6) | 98.9–99.8 (6) | 99.2–99.9 (6) |
| Near UV-CD/spectral similarity (%) | |||
| US RP | 97.4–99.3 (6) | 98.8–99.4 (6) | 98.8–99.4 (6) |
| EU RP | 97.4–99.2 (6) | 98.8–99.3 (6) | 98.2–99.5 (6) |
| DSC (°C) | |||
| T m1 | 71.7–72.1 (6) | 71.7–72.1 (6) | 71.7–72.1 (6) |
| T m2 | 82.2–82.7 (6) | 82.5–83.0 (6) | 82.4–82.9 (6) |
| AUC-SV/monomer (%) | 98.4–99.9 (6) | 97.4–99.8 (6) | 97.5–99.5 (6) |
| SE-HPLC-LS MW (kDa) | |||
| Monomer | 145–145 (3) | 145–146 (3) | 145–146 (3) |
| Dimer | 318–322 (3) | 307–320 (3) | 310–318 (3) |
| LO/particles-size (particles/mL) | |||
| ≥2 µm | 5140–23,748 (10) | 4560–31,000 (7) | 9447–15,820 (7) |
| ≥5 µm | 1000–7630 (10) | 1057–13,600 (7) | 3577–7587 (7) |
| ≥10 µm | 93–1525 (10) | 107–3727 (7) | 570–2284 (7) |
| ≥25 µm | 0–14 (10) | 4–97 (7) | 3–60 (7) |
| MFI/non-spherical particles-size ≥5 µm (particles/mL) | 24–172 (10) | 18–139 (7) | 7–183 (7) |
| CHO cell protein by ELISA (ppm) | 0–46 (10) | 129–168 (3) | 87–171 (3) |
Adalimumab (EU) European Union-authorized adalimumab, adalimumab (US) United States Food and Drug Administration-licensed adalimumab, Asn asparagine, AUC-SV analytical ultracentrifugation sedimentation velocity, CHO Chinese hamster ovary, DSC differential scanning calorimetry, ELISA enzyme-linked immunoassay, EU European Union, FTIR Fourier-transformed infrared, LO light obscuration, MFI micro-flow imaging, MW molecular weight, n number of batches, RP reference product, SE-HPLC-LS size exclusion-high performance liquid chromatography with light scattering detection, US United States, UV-CD ultraviolet circular dichroism
The reduced peptide map overlays (Fig. 1b) showed similar peak profiles for the three products, and the differences between the observed masses and the theoretical masses for all tryptic peptides were within the instrument precision of 15 ppm for all three products. The same post-translational modifications were detected between ABP 501 and adalimumab RPs, and no new species were detected by UV detection in the tryptic peptide maps between the three products. Examination of the tryptic peptides revealed a single consensus site for N-linked glycosylation located on the HC at N301 in both ABP 501 and adalimumab RP. Consistent with N301 being the only site glycosylated, cleavage by endopeptidase trypsin generated a single glycopeptide, H23, within the peptide map of both ABP 501 and adalimumab RP. The peptide map liquid chromatography and mass spectrometry (LC-MS) data showed the prevalent glycan structures to be core fucosylated biantennary complex structures consistent with the glycan map results, as discussed later in this section. Five low-abundance peptides were identified to contain modifications and were observed in all ABP 501 and adalimumab RP lots. The elution positions and MS/MS spectrum of these peptides were the same for all three products. These variants included low levels of peptides lacking the expected N-linked glycan and unprocessed C-terminal lysine of the HC. Occupancy at the expected N301 glycosylation site was further quantified by the rCE-SDS method in which non-glycosylated and glycosylated HCs are well-resolved. ABP 501 has greater than 99 % glycosylation, while adalimumab has greater than 97 % glycosylation at the expected N301 site (Table 2).
After the removal of unprocessed C-terminal lysine by carboxypeptidase B (CPB) treatment, ABP 501 had similar levels of acidic and basic variants compared with adalimumab (US) and adalimumab (EU) after CPB treatment (Fig. 2a). In addition, peptide containing deamidation at N388 and deamidation at N393 were observed in all three products. Oxidation was observed on peptide at position M432. All of the modifications were observed in ABP 501, adalimumab (US), and adalimumab (EU) at similarly low levels. No sequence variants were observed in ABP 501 and adalimumab RPs within the detection limit of the method. Additionally, the non-reduced peptide map confirmed that disulfide structure for ABP 501, adalimumab (US), and adalimumab (EU) were consistent with the expected disulfide structure for IgG1. All 16 cysteine-containing residues were confirmed for all three products. The observed molecular masses for the disulfide-linked peptides in the non-reduced peptide maps and the cysteine-containing peptides in the reduced peptide maps were all within 15 ppm of their theoretical masses for peptides lower than 10,000 Da (using monoisotopic mass) and within 100 ppm for peptides greater than 10,000 Da (using average mass).
Fig. 2.

Charge profiles: comparison of ABP 501 (red line), adalimumab (US) (black line), and adalimumab (EU) (blue line) for carboxypeptidase B-treated cation exchange-high performance liquid chromatography profiles (a) and intact capillary isoelectric focusing profiles (b). Adalimumab (EU) European Union-authorized adalimumab, adalimumab (US) United States Food and Drug Administration-licensed adalimumab, AU absorbance units, pI isoelectric points, mAU milli absorbance units
The apparent pI was evaluated using cIEF, which resolved several charged species that are present in all three products at similar levels (Fig. 2b). ABP 501 has similar pI profiles to adalimumab (US) and adalimumab (EU). Additionally, the experimentally determined extinction coefficients by amino acid analysis also confirmed that ABP 501, adalimumab (US), and adalimumab (EU) have similar extinction coefficient values, and a dual anti-idiotype mAb sandwich ELISA identity test confirmed that ABP 501, adalimumab (US), and adalimumab (EU) share common CDR structural and conformational characteristics (data not shown).
Figure 1c shows the overlay of glycan maps for ABP 501, adalimumab (US), and adalimumab (EU). ABP 501, adalimumab (US), and adalimumab (EU) were shown to have similar profiles and all major glycans were present, with minor differences observed at low abundance. For example, ABP 501 has a similar level of Man-5 to that of adalimumab (US) and adalimumab (EU), but a lower level of Man-6 glycan which elutes at approximately 16 min. More than 25 glycan species were identified and quantified individually. Four glycan groups, which included terminally galactosylated glycans, high mannose glycans, afucosylated glycans, and sialylated glycans, were evaluated as part of the similarity assessment.
Galactosylation is important for the subcomponent of complement C1 (C1q) binding to IgG1, which is a key step in the activation of IgG1-mediated cell lysis via CDC. The galactosylation group includes all complex and hybrid glycan structures that contain at least one terminal galactose. The level of galactosylation in the ABP 501 lots was similar to the levels observed in adalimumab (US) and adalimumab (EU). This is also supported by no differences in CDC activities being observed for any ABP 501 lot compared with adalimumab (EU) and adalimumab (US) (Table 3).
Table 3.
Summary of functional assays for APB 501, adalimumab (US), and adalimumab (EU)
| Bioassay | ABP 501 [range, % (n)] | Adalimumab (US) [range, % (n)] | Adalimumab (EU) [range, % (n)] |
|---|---|---|---|
| TNF-mediated apoptosis inhibition | 98–110 (10) | 95–114 (21) | 91–122 (18) |
| ADCC | 53–103 (10) | 41–127 (17) | 61–114 (15) |
| CDC | 94–105 (10) | 84–105 (17) | 82–104 (15) |
| Soluble TNFα binding | 96–121 (10) | 99–128 (10) | 103–122 (10) |
| FcRn binding | 86–101 (10) | 91–114 (15) | 81–116 (14) |
Adalimumab (EU) European Union-authorized adalimumab, adalimumab (US) United States Food and Drug Administration-licensed adalimumab, ADCC antibody-dependent cell-mediated cytotoxicity, CDC complement-dependent cytotoxicity, FcRn Fc neonatal receptor, n number of batches, TNF tumor necrosis factor, TNFα tumor necrosis factor-α
High mannose glycan types are naturally occurring in human serum IgGs and could impact pharmacokinetics via differential clearance through binding to mannose-binding receptors [28] and ADCC function through binding to FcγRIIIa [29, 30]. The high mannose group includes all high mannose glycans, M5, M6, M7, and M8. The level of high mannose was similar in ABP 501, adalimumab (US), and adalimumab (EU), suggesting there would be no impact on pharmacokinetics. Indeed, the results of a human pharmacokinetics similarity study conducted in healthy adults showed ABP 501 exposure and pharmacokinetic profiles to be similar to those for adalimumab (US) and adalimumab (EU) following administration of a single 40 mg subcutaneous dose [31].
Absence of the core fucose moiety in the Fc N-linked glycan can significantly enhance the FcγRIIIa binding affinity of IgG1, resulting in increased ADCC [30, 32–34]. The N-linked glycans missing core fucose moiety, afucosylated glycans, include complex-type, hybrid-type, and terminal high mannose glycans [35]. The total afucosylation glycans in ABP 501 was similar to that in adalimumab (US) and adalimumab (EU) (Table 2). The results are further supported by the similar ADCC activity observed between ABP 501 and adalimumab (US) and adalimumab (EU) (Table 3). The levels of sialylated glycans in ABP 501 were slightly higher than those in adalimumab (US) and adalimumab (EU); however, the levels were all close to or below 1 % (Table 2) and no difference in ADCC was observed between ABP 501 and adalimumab (US) and adalimumab (EU) (Table 3).
Higher-Order Structure
The higher-order structure of ABP 501, adalimumab (US), and adalimumab (EU) was characterized using multiple biophysical techniques. The secondary structure was characterized by FTIR spectroscopy, the tertiary structure by near UV-CD spectroscopy, and the thermal stability and overall conformation by DSC.
FTIR and UV-CD spectra for ABP 501 and adalimumab RPs showed similar secondary and tertiary structures (Fig. 3a, b). Figure 3a shows the overlaid FTIR profiles, which are visually similar. The spectra exhibit a strong β-sheet band at around 1639 cm-1 together with a β-sheet band at 1689 cm-1, indicating the presence of predominantly antiparallel β-sheet structure typical of antibodies. The FTIR spectral similarities of all individual ABP 501 lots compared with adalimumab RPs are greater than 95 %—the precision of the technology (Table 2). Figure 3b shows the overlaid near UV-CD profiles, which are visually similar. The spectra contain signals with peaks from tryptophan, tyrosine, and phenylalanine, superimposed on the broad disulfide signal from 250 to 280 nm. The intensity of these features reflects the native structure of the mAb molecule and demonstrates that the disulfide bonds and aromatic amino acids are in the expected environment due to the folding of the proteins. The near UV-CD spectral similarities of all individual ABP 501 lots compared with adalimumab RPs are greater than 95 %—the precision of the technology (Table 2). Furthermore, the thermodynamic properties of the products were assessed and compared by DSC. The DSC scans were visually similar for ABP 501 and adalimumab RPs (Fig. 3c), and the thermal melting temperatures Tm1 and Tm2 were also similar among the three products (Table 2), indicating similar thermodynamic properties. The results of these three tests demonstrate the higher-order structure of ABP 501 was highly similar to adalimumab RPs.
Fig. 3.

Higher-order structure: comparison of ABP 501 (red line), adalimumab (US) (black line), and adalimumab (EU) (blue line) Fourier-transformed infrared spectra (a), ultraviolet circular dichroism spectra (b), and differential scanning calorimetry profile (c). Adalimumab (EU) European Union-authorized adalimumab, adalimumab (US) United States Food and Drug Administration-licensed adalimumab, CD circular dichroism, Cp specific heat capacity
Particles and Aggregates
Particles and aggregates of ABP 501 and adalimumab RPs were assessed using a combination of test methods that are capable of detecting the presence and/or quantifying the amounts of particles and aggregates of various sizes and shape (spherical or non-spherical). Subvisible particles were evaluated quantitatively by LO and MFI. Particle concentrations for ≥2, ≥5, ≥10, and ≥25 μm sizes were similar for ABP 501 and adalimumab RPs (Table 2). The subvisible particles primarily consisted of silicon oil droplets with low levels of non-spherical particles that are presumed to be proteinaceous, and the proteinaceous particle concentrations were similar between ABP 501 and adalimumab RPs. Submicron particle presence was qualitatively assessed by DLS and FFF (Fig. 4a, b), and no submicron particle presence above the assays′ sensitivities was detected for any of the products. Additionally, aggregates assessed by AUC-SV were similar between ABP 501 and adalimumab RPs (Fig. 4c). SE-HPLC-LS analysis indicated that ABP 501 and adalimumab RPs had similar types and amounts of aggregates (Table 2). These results demonstrate that ABP 501 has similar aggregate and particle levels to adalimumab RPs.
Fig. 4.

Particle and aggregates: comparison of ABP 501 (red line), adalimumab (US) (black line), and adalimumab (EU) (blue line) submicron particle profiles as assessed by dynamic light scattering (a) and field flow fractionation (b), and aggregates as assessed by analytical ultracentrifugation-sedimentation velocity (c). Adalimumab (EU) European Union-authorized adalimumab, adalimumab (US) United States Food and Drug Administration-licensed adalimumab, C(S) sedimentation coefficient
Biological Activity
The biological activities comparing TNFα-mediated apoptosis inhibition, induction of ADCC and CDC, and the relative binding to FcRn and sTNFα are summarized in Table 3. Ten ABP 501 lots were compared with at least ten and up to 21 lots of adalimumab (US) and adalimumab (EU). ABP 501 had similar apoptosis inhibition, TNFα binding, ADCC, CDC, and FcRn binding activities to that of adalimumab RPs, and all ten ABP 501 lots were within the activity ranges observed in adalimumab for these biological functions. For apoptosis inhibition bioassay, the average relative potency for ABP 501 lots, adalimumab (US) lots, and adalimumab (EU) lots were 104, 106, and 103 %, respectively. Further biological and pharmacological characterization was performed to demonstrate that ABP 501 is highly functionally similar to adalimumab, and details of the biological characterization including binding affinity and specificities of the target and Fc function are discussed in a companion article (Velayudhan et al. [36]).
Product-Related Substances and Impurities
Product-related substances and impurities, including size and changed product variants of ABP 501, adalimumab (US), and adalimumab (EU) were assessed using a battery of separation methods. Size variants were assessed under native conditions by SE-HPLC (Fig. 5a), and under denaturing conditions by rCE-SDS and nrCE-SDS (Fig. 5b) to identify covalently associated aggregates and incomplete disulfide-linked fragments and clips. ABP 501 had a slightly lower level of non-glycosylated HC, indicating a higher level of complete occupancy at the asparagine (Asn) 301 glycosylation site, consistent with the reduced peptide map data. Charge variants were assessed by cIEF and CEX-HPLC, and ABP 501 was found to have a lower level of basic variants relative to adalimumab (US) and adalimumab (EU). After treatment with CPB, ABP 501, adalimumab (US), and adalimumab (EU) were found to be similar in the level of acidic and basic variants (Fig. 2a, b).
Fig. 5.
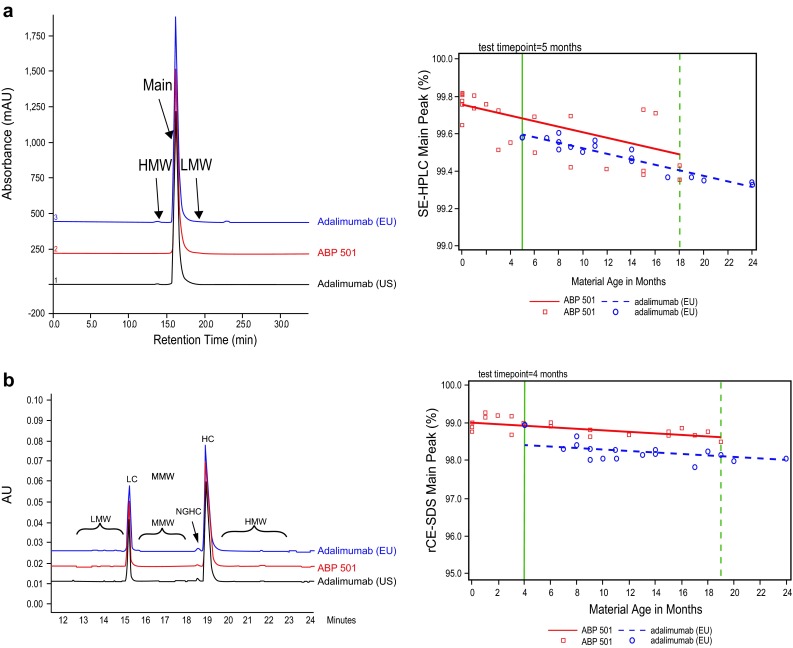
Physicochemical properties of size variants, as assessed by SE-HPLC (a) and rCE-SDS (b) for ABP 501 (red line), adalimumab (US) (black line), and adalimumab (EU) (blue line). Adalimumab (EU) European Union-authorized adalimumab, adalimumab (US) United States Food and Drug Administration-licensed adalimumab, AU absorbance units, HC heavy chain, HMW high molecular weight, LC light chain, LMW low molecular weight, mAU milli absorbance units, MMW medium molecular weight, NGHC non-glycosylated heavy chain, rCE-SDS reduced capillary electrophoresis-sodium dodecyl sulfate, SE-HPLC size exclusion-high-performance liquid chromatography
Many of the product-related substances and impurities could be degradation products and the levels may change over the shelf-life, and hence any similarity assessment of the quantitative levels requires consideration of material age at the time of testing. ABP 501 and adalimumab (US) and adalimumab (EU) were found to have similar levels of size and charge variants for materials of the same shelf-life age.
Host-Cell Impurities
To demonstrate similarity for product safety, including immunogenicity, analyses of relevant host-cell impurities, including HCP and host-cell DNA, were conducted. The residual HCPs were quantified and characterized by orthogonal methods, including HCP ELISA, 2D-DIGE, and 2D-LC-MSE. ABP 501 had lower levels of residual HCP than adalimumab (US) and adalimumab (EU) as measured by HCP ELISA (Table 2), and this result is supported by 2D-LC-MSE characterization in which few CHO proteins were identified in adalimumab (US) and adalimumab (EU) at the levels of 5–50 ppm. The residual DNA was found to be below the method limit of quantification for ABP 501 and adalimumab.
General Properties
Similarity assessment of ABP 501, adalimumab (US), and adalimumab (EU) general properties included product strength via protein concentration and volume, as well as other dosage form properties such as osmolality, pH, appearance, color, clarity, and surfactant concentration. The protein concentration and fill volume in each pre-filled syringe was similar in the three drug products (Table 2). Additionally, although ABP 501 is formulated differently from adalimumab (US) and adalimumab (EU), osmolality, pH, appearance, color, clarity, and surfactant concentration all met the pre-specified criteria for analytical similarity.
Stability and Degradation at Accelerated Conditions
As part of the analytical similarity assessment, thermal stability and degradation studies were performed at 25, 40, and 50 °C to aid in the comparison of ABP 501 with adalimumab (US) and adalimumab (EU) drug products. The conditions were selected to provide quantitative comparisons of degradation profiles. These thermal studies evaluated potential differences in structure and impurities and served as the primary mode for comparison of stress stability profiles of ABP 501 with those of adalimumab (US) and adalimumab (EU). Because ABP 501 is formulated differently from adalimumab, ABP 501 thermal degradation was studied using both the ABP 501 formulation and the adalimumab formulation. When formulated in the adalimumab formulation buffer, ABP 501 exhibited an increased rate of higher-order aggregate formation at 50 °C (Fig. 6a) compared with ABP 501 in ABP 501 formulation buffer (Fig. 6b). The formulation difference did not impact the rate of formation of low molecular weight species in ABP 501 (Fig. 5). Subsequent characterization of the higher-order aggregates at 50 °C by SE-HPLC confirmed these species to be large oligomers in the range of 800–3500 kDa, compared with the main peak of 147 kDa of monomeric antibody. The increased formation of HMW species of ABP 501 in the adalimumab formulation buffer is consistent with the degradation behavior of adalimumab (US) and adalimumab (EU) under thermal stress at 50 °C.
Fig. 6.
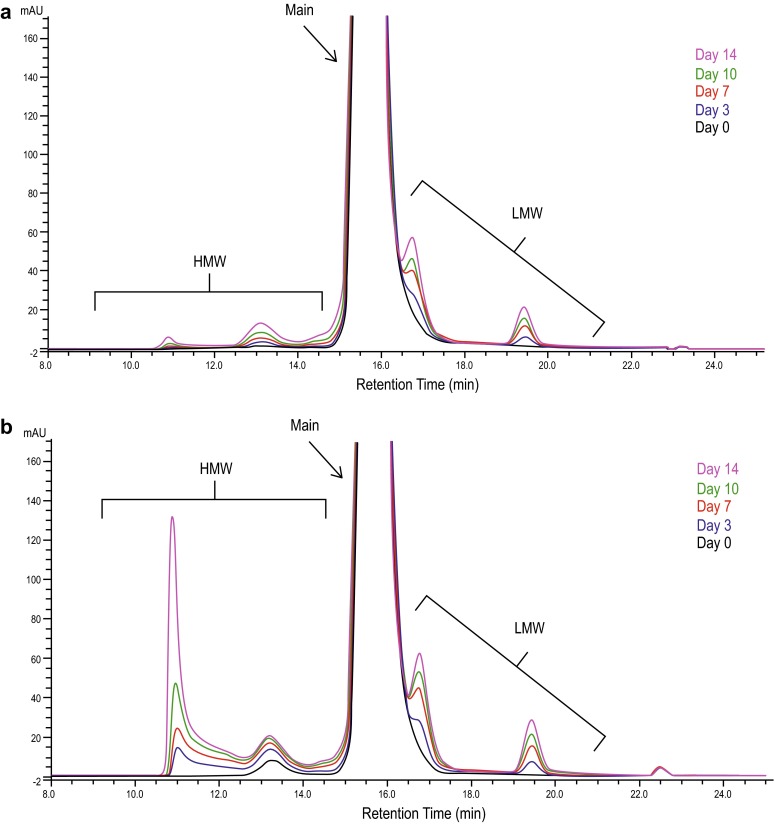
Timecourse size exclusion-high-performance liquid chromatography profiles of ABP 501 incubated at 50 °C in ABP 501 formulation buffer (a) and adalimumab formulation buffer (b). HMW high molecular weight, LMW low molecular weight, mAU milli absorbance units
Conclusions
ABP 501 is being developed by Amgen as a biosimilar product to the RP adalimumab (Humira®). A comprehensive analytical strategy using state-of-the-art, sensitive, and orthogonal methods was developed and implemented to assess analytical and functional similarity between ABP 501 and the innovator RPs. We have evaluated primary structure, higher-order structure, particles and aggregates, product-related substances and impurities, host-cell impurities, biological activities, thermal stability and degradation studies, and general properties. The results support the conclusion that ABP 501 is analytically similar to the RPs.
The primary and higher-order structure of ABP 501, as well as size and charge variants, have been shown to be similar to adalimumab (US) and adalimumab (EU) RPs; the aggregate and particle levels between ABP 501 and adalimumab RPs are similar; and the residual host-cell impurities, including HCP and DNA, in ABP 501 are below the levels observed in adalimumab RPs. Furthermore, functional biological characterization, including apoptosis inhibition bioassay, effector functions such as ADCC and CDC, sTNFα and FcRn binding assays revealed that ABP 501 has equivalent biological activities to adalimumab (US) and adalimumab (EU). ABP 501 formulated into ABP 501 formulation buffer shows a non-inferior stability profile compared with ABP 501 formulated in adalimumab drug product formulation buffer. A comprehensive physiochemical characterization and preliminary biofunctional characterization are important steps in generating the totality of evidence for biosimilarity during the step-wise development of a biosimilar. The results presented here demonstrate that ABP 501 is analytically similar to adalimumab (US) and adalimumab (EU) RPs. Further biological and functional characterization of ABP 501 relative to the two adalimumab RPs has been undertaken and the results of the biological functional tests are the subject of a companion article (Velayudhan et al. [36]). Taken together, our results demonstrate that ABP 501 is similar to adalimumab RPs, both analytically and functionally.
Acknowledgments
The authors would like to acknowledge the technical contributions of R. Sivendran, Q. Luo, H. Chung, K. Lawson, A. Ip, L. Chu, J. Wen, G. Svitel, J. Pollastrini, N. Jiao, A. Rohrbach, G. Maher, Y.-F. Chen, Q. Zhang, J. Rattan, Y. Jiang, J. Velayudhan, C. Pastula, R. Burdick, and P. Husak for their technical contributions to this work. Medical writing and editorial support was provided by Joyce Willetts, PhD, consultant to MedVal Scientific Information Services, LLC, Skillman, NJ, USA, under the guidance of Monica Ramchandani, PhD, Amgen Inc.
Author contributions
Jennifer Liu is the lead author and is responsible for the overall design and execution of the study. Tamer Eris is the subject expert for biochemical analysis and he contributed to the authoring and reviewing of this manuscript. Cynthia Li is the subject expert for higher-order structure analysis and she contributed to the authoring and reviewing of this manuscript. Shawn Cao is the subject expert for particles and aggregates analysis and he contributed to the authoring and reviewing of this manuscript. Scott Kuhns is the subject expert for biological activity analysis and he contributed to the authoring and reviewing of this manuscript.
Compliance with Ethical Standards
Amgen Inc. conducted and funded all analyses. Amgen Inc. also provided funding for medical writing and editorial support to MedVal Scientific Information Services, LLC, Skillman, NJ, USA. This article does not contain any studies with human participants or animals performed by any of the authors. This paper was prepared according to the International Committee of Medical Journal Editors′ (ICMJE) Uniform Requirements and the International Society for Medical Publication Professionals′ Good Publication Practice for Communicating Company-Sponsored Medical Research: The GPP2 Guidelines.
Conflict of interest
Jennifer Liu, Tamer Eris, Shawn Cao, and Scott Kuhns are employees and stockholders of Amgen, Inc. Cynthia Li is an employee of Amgen Inc.
References
- 1.AbbVie . Humira (adalimumab) injection [prescribing information] North Chicago: AbbVie Inc; 2015. [Google Scholar]
- 2.Tebbey PW, Varga A, Naill M, Clewell J, Venema J. Consistency of quality attributes for the glycosylated monoclonal antibody Humira® (adalimumab) MAbs. 2015;7(5):805–811. doi: 10.1080/19420862.2015.1073429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.European Medicines Agency. Guideline on similar biological medicinal products. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf. Accessed 7 July 2016.
- 4.WHO Expert Committee on Biological Standardization. Guidelines on evaluation of similar biotherapeutic products (SBPs). WHO Technical Report Series no. 977. Annex 2. 2013. http://who.int/biologicals/publications/trs/areas/biological_therapeutics/TRS_977_Annex_2.pdf. Accessed 7 July 2016.
- 5.U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product. Guidance for industry. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291134.pdf. Accessed 7 July 2016.
- 6.European Medicines Agency, Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. Accessed 7 July 2016.
- 7.European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). http://www.ebe-biopharma.eu/newsroom/download/58/document/ema:chmp:bwp:247713:2012.pdf. Accessed 7 June 2016.
- 8.U.S. Department of Health and Human Services. Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed 7 July 2016.
- 9.European Medicines Agency. European public assessment reports. European Medicines Agency. http://www.ema.europa.eu/ema/index.jsp?curl=pages%2Fmedicines%2Flanding%2Fepar_search.jsp&mid=WC0b01ac058001d124&searchTab=searchByAuthType&alreadyLoaded=true&isNewQuery=true&status=Authorised&keyword=Enter+keywords&searchType=name&taxonomyPath=&treeNumber=&searchGenericType=biosimilars&genericsKeywordSearch=Submitudy. Accessed 25 Apr 2015.
- 10.Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs. 2013;5(5):621–623. doi: 10.4161/mabs.25864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.US Food and Drug Administration. FDA approves first biosimilar product Zarxio [press release]. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm436648.htm. Accessed 7 July 2016.
- 12.Jung SK, Lee KH, Jeon JW, Lee JW, Kwon BO, Kim YJ, et al. Physicochemical characterization of Remsima. MAbs. 2014;6(5):1163–1177. doi: 10.4161/mabs.32221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beck A, Dimer H, Ayoub D, Debaene F, Wagner-Rousset E, Carapito D, et al. Analytical characterization of biosimilar antibodies and Fc-fusion proteins. Trends Analyt Chem. 2013;48:81–95. doi: 10.1016/j.trac.2013.02.014. [DOI] [Google Scholar]
- 14.Xie H, Chakraborty A, Ahn J, Yu YQ, Dakshinamoorthy DP, Gilar M, et al. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs. 2010;2(4):379–394. doi: 10.4161/mabs.11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen SL, Wu SL, Huang LJ, Huang JB, Chen SH. A global comparability approach for biosimilar monoclonal antibodies using LC-tandem MS based proteomics. J Pharm Biomed Anal. 2013;80:126–135. doi: 10.1016/j.jpba.2013.02.040. [DOI] [PubMed] [Google Scholar]
- 16.Cao J, Sun W, Gong F, Liu W. Charge profiling and stability testing of biosimilar by capillary isoelectric focusing. Electrophoresis. 2014;35(10):1461–1468. doi: 10.1002/elps.201300471. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Fu T, Liu T, Guo H, Guo Q, Xu J, et al. Characterization of alanine to valine sequence variants in the Fc region of nivolumab biosimilar produced in Chinese hamster ovary cells. MAbs. 2016;8(5):951-60. [DOI] [PMC free article] [PubMed]
- 18.Lopez-Morales CA, Miranda-Hernandez MP, Juarez-Bayardo LC, Ramirez-Ibanez ND, Romero-Diaz AJ, Pina-Lara N, et al. Physicochemical and biological characterization of a biosimilar trastuzumab. Biomed Res Int. 2015;2015:427235. doi: 10.1155/2015/427235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pike MC, Peters RK, Cozen W, Probst-Hensch NM, Felix JC, Wan PC, et al. Estrogen-progestin replacement therapy and endometrial cancer. J Natl Cancer Inst. 1997;89(15):1110–1116. doi: 10.1093/jnci/89.15.1110. [DOI] [PubMed] [Google Scholar]
- 20.Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11(7):527–540. doi: 10.1038/nrd3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Damen CW, Chen W, Chakraborty AB, van OM, Mazzeo JR, Gebler JC, et al. Electrospray ionization quadrupole ion-mobility time-of-flight mass spectrometry as a tool to distinguish the lot-to-lot heterogeneity in N-glycosylation profile of the therapeutic monoclonal antibody trastuzumab. J Am Soc Mass Spectrom. 2009;20(11):2021–2033. doi: 10.1016/j.jasms.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 22.Vanhoenacker G, Vandenheede I, David F, Sandra P, Sandra K. Comprehensive two-dimensional liquid chromatography of therapeutic monoclonal antibody digests. Anal Bioanal Chem. 2015;407(1):355–366. doi: 10.1007/s00216-014-8299-1. [DOI] [PubMed] [Google Scholar]
- 23.Gahoual R, Burr A, Busnel JM, Kuhn L, Hammann P, Beck A, et al. Rapid and multi-level characterization of trastuzumab using sheathless capillary electrophoresis-tandem mass spectrometry. MAbs. 2013;5(3):479–490. doi: 10.4161/mabs.23995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Humira (adalimumab) [prescribing information]. North Chicago: Abbott Laboratories; 2005.
- 25.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J. 2000;78(3):1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verardo ML, Carvalho JG, Delgado DN, Kuhns ST. Accuracy and sensitivity of residual DNA detection by QPCR is not predicted by target copy number. Biotechnol Prog. 2012;28(2):428–434. doi: 10.1002/btpr.731. [DOI] [PubMed] [Google Scholar]
- 27.Arora T, Padaki R, Liu L, Hamburger AE, Ellison AR, Stevens SR, et al. Differences in binding and effector functions between classes of TNF antagonists. Cytokine. 2009;45(2):124–131. doi: 10.1016/j.cyto.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 28.Goetze AM, Liu YD, Zhang Z, Shah B, Lee E, Bondarenko PV, et al. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology. 2011;21(7):949–959. doi: 10.1093/glycob/cwr027. [DOI] [PubMed] [Google Scholar]
- 29.Li P, Jiang N, Nagarajan S, Wohlhueter R, Selvaraj P, Zhu C. Affinity and kinetic analysis of Fcgamma receptor IIIa (CD16a) binding to IgG ligands. J Biol Chem. 2007;282(9):6210–6221. doi: 10.1074/jbc.M609064200. [DOI] [PubMed] [Google Scholar]
- 30.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277(30):26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 31.Kaur PP, Chow V, Zhang N, Moxness M, Markus R. Pharmacokinetic equivalence of ABP 501 relative to adalimumab: results from a randomized, single-blind, single-dose, parallel group study in healthy subjects [abstract] Arthritis Rheum. 2014;66(10 suppl):S661–S662. doi: 10.1136/annrheumdis-2015-208914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278(5):3466–3473. doi: 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- 33.Niwa R, Sakurada M, Kobayashi Y, Uehara A, Matsushima K, Ueda R, et al. Enhanced natural killer cell binding and activation by low-fucose IgG1 antibody results in potent antibody-dependent cellular cytotoxicity induction at lower antigen density. Clin Cancer Res. 2005;11(6):2327–2336. doi: 10.1158/1078-0432.CCR-04-2263. [DOI] [PubMed] [Google Scholar]
- 34.Ferrara C, Stuart F, Sondermann P, Brunker P, Umana P. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J Biol Chem. 2006;281(8):5032–5036. doi: 10.1074/jbc.M510171200. [DOI] [PubMed] [Google Scholar]
- 35.Yu M, Brown D, Reed C, Chung S, Lutman J, Stefanich E, et al. Production, characterization, and pharmacokinetic properties of antibodies with N-linked mannose-5 glycans. MAbs. 2012;4(4):475–487. doi: 10.4161/mabs.20737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Velayudhan J, Chen Y, Rohrbach A, Pastula C, Maher G, Thomas H, Brown R, Born TL. Demonstration of functional similarity of proposed biosimilar ABP 501 to adalimumab. BioDrugs. 2016. doi:10.1007/s40259-016-0185-2. [DOI] [PMC free article] [PubMed]