

Abstract
Aims
We have observed that Bendavia, a mitochondrial-targeting peptide that binds the phospholipid cardiolipin and stabilizes the components of electron transport and ATP generation, improves cardiac function and prevents left ventricular remodeling in a 6 week rat myocardial infarction (MI) model. We hypothesized that Bendavia restores mitochondrial biogenesis and gene expression, suppresses cardiac fibrosis, and preserves sarco/endoplasmic reticulum (SERCA2a) level in the noninfarcted border zone of infarcted hearts.
Main methods
Starting 2 hours after left coronary artery ligation, rats were randomized to receive Bendavia (3 mg/kg/day), water or sham operation. At 6 weeks, PCR array and qRT-PCR was performed to detect gene expression. Picrosirius red staining was used to analyze collagen deposition.
Key findings
There was decreased expression of 70 out of 84 genes related to mitochondrial energy metabolism in the border zone of untreated hearts. This down-regulation was largely reversed by Bendavia treatment. Downregulated mitochondrial biogenesis and glucose & fatty acid (FA) oxidation related genes were restored by administration of Bendavia. Matrix metalloproteinase (MMP9) and tissue inhibitor of metalloproteinase (TIMP1) gene expression were significantly increased in the border zone of untreated hearts. Bendavia completely prevented up-regulation of MMP9, but maintained TIMP1 gene expression. Picrosirius red staining demonstrated that Bendavia suppressed collagen deposition within border zone. In addition, Bendavia showed a trend toward restoring SERCA2a expression.
Significance
Bendavia restored expression of mitochondrial energy metabolism related genes, prevented myocardial matrix remodeling and preserved SERCA2a expression in the noninfarcted border, which may have contributed to the preservation of cardiac structure and function.
Keywords: Myocardial infarction, Mitochondria, Bendavia, Cardioprotection
Introduction
Myocardial infarction (MI) results in substantial left ventricular damage, which may be followed by heart failure. Despite the considerable improvement in the treatment of acute MI in the past 20 years, MI mortality at 1 year still remains about 15%, and approximately 20% of patients with a first MI at ≥ 65 years of age will develop heart failure in 5 years1. The search for better therapies is one of the major challenges in cardiovascular disease. Emerging evidence shows that following MI, the heart becomes an energy-starved pump with decreased adenosine triphosphate (ATP) concentrations2, 3, reduced fatty acid (FA) oxidation rates4 and impaired mitochondrial biogenesis5. The modulation of mitochondrial function may be a promising new approach for the treatment of heart failure related to MI.
Mitochondria are the powerhouse of the cells and are responsible for transforming chemical energy into ATP in order to supply energy for the demands of cardiac muscle contraction. ATP is synthesized primarily by mitochondrial oxidative phosphorylation (OXPHOS) at the electron transport chain. Mitochondrial OXPHOS is composed of four complexes: I, II, III, and IV that are embedded in the inner mitochondrial membrane and the ATP synthase (complex V). The reductions in the expression level and activity of the respiratory chain complexes have been reported in animal models of heart failure post MI and in failing human hearts6, 7.
Bendavia, a cell-permeable peptide, is an analogue of Szeto-Schiller (SS)-peptides SS-31 and is also referred to as MTP-131 in the literature8. Bendavia selectively targets the inner mitochondrial membrane where it reduces levels of reactive oxygen species (ROS) and improves energetics through a cardiolipin-dependent mechanism9–14. Unlike mitochondrial-targeted antioxidant such as MitoQ, the uptake of Bendavia is independent of the mitochondrial membrane potential15. In a series of studies9, 10, we reported that Bendavia showed cardioprotective properties in the setting of acute ischemia/reperfusion injury models by enhancing mitochondrial energetics and reducing the production of cellular ROS levels. Recently, our laboratory has further demonstrated that chronic Bendavia therapy started 2 hours after myocardial infarction improves cardiac function and limits left ventricular remodeling in a 6 week rat MI model, without altering blood pressure or heart rate16.
In the present study, to further characterize the mitochondrial directed mechanism of Bendavia as it relates to improved cardiac function, we determined whether treatment with Bendavia restores mitochondrial function, suppresses cardiac fibrosis, preserves SERCA2a expression in a model of chronic left ventricular dysfunction following MI.
Methods
All experimental protocols were approved by the Institutional Animal Care and Use Committee, and performed in accordance with the “Guide for the Care and Use of Laboratory Animals” (National Academy Press, Washington DC, revised 2011, Eighth Edition). The Heart Institute at Good Samaritan Hospital was accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Experimental groups
Myocardial infarction was induced in female Sprague-Dawley rats as described previously16. The artery that we occlude is the proximal left coronary artery right below the level of the left atrial appendage. Starting 2 hours after left coronary artery ligation, rats were randomized to receive Bendavia (3 mg/kg/day) or water delivered by Alzet Osmotic Pump at 0.15 μl/hour (model 2006; 200 μl). Noninfarcted rats served as shams. At 6 weeks, hearts were rapidly removed and snap frozen in liquid nitrogen. Before freezing, left ventricles (LV) with MI were surgically separated into border zone (a strip of non-infarcted heart tissue about 2 mm in width surrounding the scar) and the remote nonischemic area (the segment from LV septum which is opposite of the infarct area). There are 5 groups that were studied: Sham = noninfarcted normal hearts, MI/BZ = noninfarcted border zone of water-treated infarcted hearts, MI/BZ + Bendavia = noninfarcted border zone of Bendavia-treated hearts, MI/R = remote noninfarcted area of water-treated hearts, and MI/R + Bendavia = remote noninfarcted area of Bendavia-treated hearts. Data on cardiac function and LV remodeling using this model were previously reported16.
PCR gene array
Total RNA was extracted using a Trizol reagent (Invitrogen). RNA was treated with RNase-free DNase and purified using RNase mini kit (Qiagen). Reverse transcription reaction was performed with 500ng of total RNA using RT2-first strand kit (SABioscience). Rat mitochondrial energy metabolism PCR array was performed to measure gene expression of the electron transport chain and oxidative phosphorylation complexes (Rat mitochondrial energy metabolism, PARN-008ZD. SABioscience) by using Bio-Rad CFX 96 touch real-time PCR detection system.
qRT-PCR
Total RNA was extracted using a Trizol reagent. RNA was treated with RNase-free DNase and purified using RNase mini kit (Qiagen). iScriptTM cDNA Synthesis Kit (Bio-Rad) was used for cDNA synthesis and quantitative RT-PCR was performed using a CFX96 touch real-time PCR system (Bio-Rad). Primers used for qPCR include: PGC1α forward GACCCTCCTCACACCAAAC, reverse GCGACTGCGGTTGTGTATG; β-actin forward CTGTGTGGATTGGTGGCTCT, reverse GCTCAGTAACAGTCCGCCTA; NRF1 forward CGCTCATCCAGGTTGGTACT, reverse TTCACCGCCCTGTAATGTGG; Tfam forward AGGGGGCTAAGGATGAGTC, reverse ATCACTTCGCCCAACTTCAG; ERRα forward AACGCCCTGGTGTCTCATC, reverse CTGATGGTGACCACTATCTC; PPARα forward CTCGGGGATCTTAGAGGCGA, reverse GCACCAATCTGTGATGACAACG; CD36 forward CTCACACAACTCAGATACTGCTG, reverse GCACTTGCTTCTTGCCAACT; GLUT4 forward TACCGTCTTCACGTTGGTCTC, reverse TAACTCATGGATGGAACCCGC; MMP9 forward GATCCCCAGAGCGTTACTCG, reverse GTTGTGGAAACTCACACGCC; Timp1 forward ACAGCTTTCTGCAACTCGGA, reverse AGCGTCGAATCCTTTGAGCA; and SERCA2a forward TTGTGGCCCGAAACTACCTG, reverse GGGCTGGAAGATGTGTTGCT.
Picrosirius red staining
After 6 weeks of treatment, the hearts were arrested in diastole by injecting intravenous potassium chloride and were pressure-fixed (13 cm water column) in 10% formalin. The formalin-fixed hearts were embedded in paraffin and 5μm thickness slides were staining with picrosirius red to estimate the extent of interstitial collagen using Image J. The collagen volume fraction was determined as the percentage of picrosirius red positive-stained area relative to total area.
Electron microscopy
All rats were assigned randomly to treatment with Bendavia or water for 6 weeks. Rats were anesthetized with an intraperitoneal injection of xylazine and ketamine. A catheter was inserted into the abdominal aorta toward the heart for perfusion fixation and a small nick was placed in the inferior vena cava to drain blood. Phosphate buffer solution was infused for 3 minutes to remove blood (pressure equal to the mean blood pressure at 122 cm H2O, 90 mm Hg); thereafter the heart was arrested in diastole by potassium chloride injection followed by 15 minutes perfusion with modified Karnovsky solution. The heart was excised and ~1 mm × 1 mm × 2 mm thick slices were cut from the border zone and remote area, immersed in modified Karnovsky’s fixative overnight at 4°C for further fixation and then processed for ultrastructural analysis using a transmission electron microscope (JEOL JEM-2100 at 100kV). Quantitative analysis was performed blindly from 10 images per sample (3000x magnification). The following quantitative measurements were obtained using Image J: mitochondrial size (μM2), mitochondrial number/10 μM2, width (μM), length (μM) and ratio of width to length. At least 300 mitochondria in the border zone and 200 mitochondria in the remote area of each rat were measured for quantitative assessment.
Cardiolipin analysis
Cardiolipin analysis was carried out by Miao Wang at Sanford-Burnham Medical Research Institute, Orlando, FL. Heart tissue was pulverized into a fine powder by a stainless steel biopulverizer at the temperature of liquid nitrogen. The tissue powders of 10 to 20 mg were weighed and homogenized in 0.5 mL 10x diluted PBS in 2.0 ml cryogenic vials (Corning Life Sciences, Tewksbury, MA) by using a digital sonifier (Branson 450, Danbury, CT). Protein assay on the homogenates was performed by using a bicinchoninic acid protein assay kit (Thermo Scientific, Rockford, IL) with bovine serum albumin as standards. The determined cardiolipin levels were normalized to the protein content of individual samples. Individual homogenate of the heart samples (equal ~ 0.8 mg protein amount) was accurately transferred into a disposable glass culture test tube. Cardiolipin internal standard was added prior to lipid extraction. Lipid extraction was performed by using a modified Bligh and Dyer procedure as described previously17.
Each lipid extract was resuspended into a volume of 200 μL of chloroform/methanol (1:1, v/v) per mg of protein and flushed with nitrogen, capped, and stored at −20 °C for lipid analysis. For electrospray ionization (ESI) direct infusion analysis, lipid extract was further diluted to a final concentration of ~500 fmol/μL by CHCl3/MeOH/isopropanol (1/2/4, v/v/v), and the mass spectrometric analysis was performed on a Q-Exactive mass spectrometer (Thermo Scientific, San Jose, CA) equipped with an automated nanospray device (TriVersa NanoMate, Advion Bioscience Ltd., Ithaca, NY) and operated with Xcalibur software. Identification and quantification of lipid molecular species were performed using an automated software program18.
Statistical Analysis
All results are expressed as means +/− SEM and analyzed using student’s t-test or 1 way ANOVA as appropriate. Statistically significant differences were established at p <0.05.
Results
Bendavia protects mitochondrial energy metabolism gene expression
We determined whether Bendavia protects the gene expression related to mitochondrial energy metabolism, including genes that code for all 5 mitochondrial electron transport complexes. Heat map analysis showed a decrease in mitochondrial energy metabolism gene expression (70 out of 84 genes) in MI/BZ vs sham. The reduction was largely reversed by administering Bendavia as shown in MI/BZ + Bendavia group (Figure 1A). The volcano plot identified that the expression of the majority of genes was reversed toward normal with 5 genes reaching a significant difference in MI/BZ+Bendavia compared with MI/BZ (Figure 1B). The identity of these 5 genes belonging to mitochondrial complex I and IV is summarized in the table 1. There were no group differences in gene expressions in the remote nonischemic areas.
Figure 1.

Bendavia protects genes involved in mitochondrial energy metabolism and restores transcripts associated with complexes I and IV. (A) Heatmap of 84 mitochondrial energy metabolism gene expressions. Red and green colors indicate increased and decreased gene expression, respectively (N=4 in sham, N=5 in the rest of groups). The top row represents gene expression in non-ischemic sham hearts. The second row represents gene expression within the non-infarcted border zone of water-treated rats (MI/BZ). Note that the green stripe represents down-regulated genes. The third row from the top represents non-infarcted border zone of the Bendavia-treated rats. Note that many of the genes that were down-regulated in the water group are up-regulated in the Bendavia group (MI/BZ+Bendavia). There were no changes with Bendavia in the non-ischemic remote areas of the LV (MI/R + Bendavia) compared to the water control rats (MI/R). (B) A ‘volcano’ plot of mitochondrial energy metabolism. Each point represents the average MI/BZ + Bendavia: MI/BZ ratio (fold change, x axis, log scale) for all 84 genes, versus the statistical significance for the difference in MI/BZ + Bendavia vs MI/BZ expression (y axis, log scale). Some of the notable genes that were upregulated by Bendavia included genes involved in mitochondrial complex I: Ndufb3, Ndufa7, Ndufc2 and Ndufa5 and mitochondrial complex IV: Cox6c. P values <0.05 are considered statistically significant (N=4 in sham, N=5 in the rest of groups).
Table 1.
Mitochondrial energy metabolism related gene expression with significant difference in MI/BZ + Bendavia vs MI/BZ
| Symbol | Name | Fold change | p-value |
|---|---|---|---|
| Complex I | |||
| Ndufb3 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex 3 | 1.41 | 0.0222 |
| Ndufa7 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 7 | 1.26 | 0.0412 |
| Ndufc2 | NADH dehydrogenase (ubiquinone) 1, subcomplex unknown, 2 | 1.30 | 0.0443 |
| Ndufa5 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 5 | 1.26 | 0.0471 |
|
| |||
| Complex IV | |||
| Cox6c | Cytochrome c oxidase, subunit VIc | 1.41 | 0.0107 |
Bendavia restores mitochondrial biogenesis and regulates the gene expression of glucose and fatty acid oxidation
In addition to the analysis described above we also determined whether Bendavia could restore mitochondrial biogenesis in this chronic MI rat model. Peroxisome proliferator-activated receptor gamma coactivator-1α (PGC1α) and its target genes were examined. PGC1α is a master regulator of mitochondrial biogenesis, which can co-activate nuclear-encoded respiratory proteins (NRF) to regulate the expression of mitochondrial transcription factor A (Tfam). Tfam is responsible for both the replication and transcription of mitochondrial DNA19, 20. Our data showed that PGC1α, NRF1 and Tfam were decreased by 42%, p=0.007, 29%, p=0.022 and 30%, p=0.059, respectively, in the MI/BZ group compared with sham. Bendavia completely prevented down-regulation of PGC1α and its target genes (Figure 2A, 2B and 2C). There were no group differences in gene expressions in the remote areas (Figure 2D, 2E and 2F).
Figure 2.

Bendavia restores gene expression associated with mitochondrial biogenesis in the border zone. Real time PCR analysis for genes of (A) PGC1α in border zone, (B) NRF1 in border zone, (C) Tfam in border zone, (D) PGC1α in remote area, (E) NRF1 in remote area and (F) Tfam in remote area, at 6 weeks after myocardial infarction. All real time PCR data were normalized to β-actin and presented relative to the sham group. * p<0.05 vs. sham, # p<0.05 vs. MI/BZ (N=7 in each group).
In parallel to impaired mitochondrial biogenesis, the altered regulation of FA and glucose utilization is also believed to contribute to cardiac dysfunction. We, therefore, measured the gene expression level of peroxisome proliferator-activated receptor (PPARα), estrogen-related receptor (ERRα), glucose transporter 4 (GLUT4) and fatty acid transporter (CD36). Bendavia upregulated the expression of ERRα and PPARα in MI/BZ+Bendavia compared with MI/BZ (Figure 3A and 3B), although, these findings did not achieve statistical significance. The expressions of CD36 and GLUT4 were significantly reduced in the MI/BZ group compared to sham and Bendavia completely prevented the downregulation of these two genes (Figure 3C and 3D). There were no significant changes in the remote areas (Figure 3E, 3F, 3G and 3H).
Figure 3.

Bendavia normalizes genes associated with glucose and fatty acid oxidation. Real time PCR analysis for genes of (A) PPARα in border zone, (B) ERRα in border zone, (C) GLUT4 in border zone, (D) CD36 in border zone, (E) PPARα in remote area, (F) ERRα in remote area, (G) GLUT4 in remote area and (H) CD36 in remote area, at 6 weeks after myocardial infarction. All real time PCR data were normalized to β-actin and presented relative to the sham group. * p<0.05 vs. sham, # p<0.05 vs. MI/BZ (N=7 in each group).
Bendavia has no effect on myocardial ultrastructure
The electron micrographs were taken from tissue samples in the noninfarcted border zone and remote areas of water and Bendavia-treated hearts at 6 weeks. The mitochondrial cristae appeared intact and compact. We did not observe swollen mitochondria and separation or disruption of the cristae in either border zone or remote area at 6 weeks in this model. The mitochondria at the border zone (Figure 4A and 4C) appeared to be rounder and smaller in shape and occurred more in pools than between the myofilaments compared to the remote nonischemic area (Figure 4B and 4D). These mitochondrial clusters suggest mitochondrial pathology at the border zone21, 22. However, there was no qualitative difference in myocardial ultrastructure between water and Bendavia-treated groups. The overall number and size of the mitochondria were comparable between water and Bendavia-treated group within the border zone and remote area (Table 2). Therefore, quantitative data from water and Bendavia group were pooled for analysis. The overall number of interfibrillar mitochondria significantly increased in the border zone compared with the remote area, and the average length of the mitochondria significantly decreased in the border zone reflecting a more rounded morphology (Table 3). We therefore examined the quantity and type of cardiolipin, the molecular target for Bendavia, in these mitochondria.
Figure 4.

Representative electron micrograph in border zone (A) and remote area (B) at 3000x magnification. The basic internal mitochondrial structure appears normal with intact cristae and no swelling. The green line was drawn around and across mitochondria. The mitochondria at the border zone appear to be rounder and smaller in shape and occur more in pools compared to mitochondria in the remote nonischemic area. Representative high-resolution electron micrograph in border zone (C) and remote area (D) at 8000x magnification.
Table 2.
Mitochondrial ultrastructure in border zone and remote area
| Border zone | Remote area | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Water (n=5) | Bendavia (n=6) | p | Water (n=5) | Bendavia (n=6) | p | |
| Size (μM2) | 0.357 ± 0.034 | 0.319 ± 0.04 | 0.495 | 0.365 ± 0.024 | 0.387 ± 0.016 | 0.447 |
| Number/10 μM2 | 7.314 ± 0.627 | 8.424 ± 0.997 | 0.394 | 5.862 ± 0.234 | 5.925 ± 0.309 | 0.880 |
| Width (W) (μM) | 0.448 ± 0.017 | 0.452 ± 0.050 | 0.954 | 0.450 ± 0.028 | 0.444 ± 0.011 | 0.843 |
| Length (L) (μM) | 0.848 ±0.049 | 0.807 ± 0.054 | 0.600 | 0.879 ±0.032 | 0.945 ± 0.024 | 0.122 |
| Ratio of W/L | 0.560 ± 0.022 | 0.554 ± 0.028 | 0.871 | 0.535 ± 0.038 | 0.491 ± 0.024 | 0.338 |
Table 3.
Mitochondrial ultrastructure in border zone and remote area.
| Border zone (n=11) | Remote area (n=11) | p | |
|---|---|---|---|
| Size (μM2) | 0.336 ± 0.026 | 0.377 ± 0.014 | 0.181 |
| Number/10 μM2 | 7.919 ± 0.611 | 5.896 ± 0.190 | 0.005* |
| Width (W) (μM) | 0.450 ± 0.027 | 0.447 ± 0.013 | 0.917 |
| Length (L) (μM) | 0.826 ±0.036 | 0.915 ± 0.021 | 0.044* |
| Ratio of W/L | 0.557 ± 0.017 | 0.511 ± 0.022 | 0.118 |
p <0.05 border zone vs. remote area
Effect of Bendavia on cardiolipin
Cardiolipin is a phospholipid that is exclusively expressed on the inner mitochondrial membrane where it is essential for cristae structure and supercomplex formation. Cardiolipin species have four fatty acyl chains (Tetra-acyl cardiolipin) and the (18:2)4 cardiolipin is dominant species in heart. Cardiolipin depletion and structure change have been reported in a variety of pathological settings including heart failure23, 24. Recently, it has been demonstrated that Bendavia selectively interacts with cardiolipin to protect the structure of mitochondrial cristae and promote oxidative phosphorylation11. Our data demonstrated that there was a trend (not statistically significant) for the depleted total cardiolipin, (18:2)4 cardiolipin species and monolysocardiolipin (MLCL) in the border zone and the remote area to be restored in the Bendavia group (n=9) compared with the water group (n=10) (Figure 5).
Figure 5.

Bendavia show a trend of increasing the content of total cardiolipin, (18:2)4 cardiolipin and monolyso cardiolipin (MLCL) in border zone (A), (B) and (C) and remote area (D), (E) and (F).
Bendavia modulates postischemic LV remodeling
MMPs and TIMPs play an important role in LV remodeling.25 qRT-PCR analysis showed that MMP9 and TIMP1 gene expression were significantly increased by 7.6 fold, p=0.026 and 4.4 fold, p=0.016, respectively, in the MI/BZ vs sham. Bendavia completely prevented up-regulation of MMP9, but maintained TIMP1 gene expression (Figure 6A and 6B). There were no significant differences in the remote areas (Figure 6C and 6D). Next, we assessed Picrosirius red staining to directly quantify collagen deposition in the border zone and remote area, respectively. As shown in Figure 6E, Bendavia significantly suppressed collagen deposition comparing the MI/BZ + Bendavia (11 ± 1% of the area of border zone) with MI/BZ (15 ± 1%, p=0.03). Bendavia also showed a trend to decrease collagen deposition in the remote area (Figure 6F). This is likely a downstream effect of Bendavia on mitochondrial energy metabolism including restoration of healthy ATP levels and reduction of the pathological production of ROS.
Figure 6.

Bendavia suppresses cardiac fibrosis by regulating gene expression of MMP9 and TIMP1. Real time PCR analysis for genes of (A) MMP9 in border zone, (B) TIMP1 in border zone, (C) MMP9 in remote area and (D) TIMP1 in remote area, at 6 weeks after myocardial infarction. All real time PCR data were normalized to β-actin and presented relative to the sham group. * p<0.05 vs. sham, # p<0.05 vs. MI/BZ (N=7 in each group). (E) Collagen deposition in border zone assessed by Picrosirius red staining. * p<0.05 vs. MI/BZ (N=26 in MI/BZ group, N=28 in MI/BZ+Bendavia group). (F) Collagen deposition in remote area assessed by Picrosirius red staining. (N=26 in MI/R group, N=28 in MI/R+Bendavia group).
Bendavia preserves SERCA2a expression
Depressed SERCA2a has been implicated as a major factor in the failing heart.26 To determine whether Bendavia preserves SERCA2a level, SERCA2a gene expression was examined. As shown in Figure 7A, SERCA2a expression was markedly decreased by 44%, p<0.05 in the MI/BZ group vs sham and Bendavia restored SERCA2a gene expression toward normal. There were no group differences in the nonischemic remote area (Figure 7B).
Figure 7.
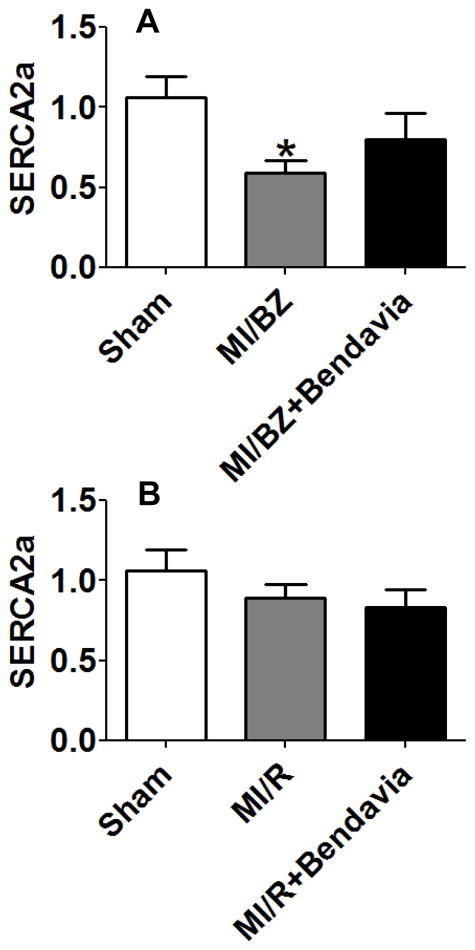
Bendavia preserves SERCA2a expression. Real time PCR analysis for genes of (A) SERCA2a in border zone and (B) SERCA2a in remote area, at 6 weeks after myocardial infarction. All real time PCR data were normalized to β-actin and presented relative to the sham group. * p<0.05 vs. sham (N=10 in each group).
Discussion
Early coronary reperfusion via thrombolytic therapy or percutaneous coronary intervention remains the only established intervention for reducing myocardial infarct size and improving survival rate in humans. Nevertheless, delay of the door to balloon or needle time requires new adjunctive therapy for additional improvements in morbidity and mortality of post-infarction heart failure.27 In addition, a recent study suggests that despite earlier “door to balloon time”, mortality rates have plateaued and new therapies are needed to further improve survival rates28.
Our previous study demonstrated that Bendavia restored the regulators and mediators mitochondrial-related gene expression in the noninfarcted border zone16. In this study, we further found that post-infarction chronic therapy of Bendavia restored gene expression of mitochondrial energy metabolism, including all 5 mitochondrial complexes, promoted mitochondrial biogenesis and regulated glucose & fatty acid oxidation related gene expression. Furthermore, we demonstrated that Bendavia preserved SERCA2a expression and reduced cardiac fibrosis, indicating that this compound represents a novel strategy to improve cardiac function post-MI.
The energy demands of the heart are immense and require a highly active mitochondrial system. Dysregulation of mitochondrial function occurs in many human diseases including post-MI heart failure6. In recent years, several mitochondrial-targeting strategies have emerged as potential treatment paradigms. One example is MitoQ, a ubiquinone moiety conjugated to triphenylphosphonium cations (TPP+) that freely crosses membranes and accumulates in the mitochondrial matrix. MitoQ has been shown to reduce mitochondrial ROS and protect the heart against ischemia-reperfusion injury29. However, mitochondrial uptake of MitoQ is membrane potential-dependent30 and studies showed that this compound inhibited mitochondrial bioenergetics13, 31 which may limit its therapeutic potential in the failing heart. Clearly, development of additional novel cell-permeable and mitochondrial-targeted compounds is needed.
Bendavia can easily cross cell membranes and selectively targets the inner mitochondrial membrane as a result of its interaction with the phosophilipid cardiolipin, which resides exclusively in this location. Importantly, this localization of Bendavia is independent of the mitochondrial membrane potential15. Numerous studies have demonstrated that Bendavia reduces the levels of ROS, decreases lipid peroxidation, inhibits mitochondrial permeability transition in pathological states and protects mitochondrial cristae9, 12, 13, 32.
In the failing heart, there is a reduced activity of electron transport chain complexes and decreased capacity for oxidative phosphorylation, which resulted in a significant decrease in myocardial ATP and phosphocreatine content33–35, Using a Rat Mitochondrial Energy Metabolism RT2 Profiler PCR Array that profiles the expression of 84 key genes involved in mitochondrial respiration, including genes encoding components of electron transport chain complexes, our study showed for the first time that Bendavia treatment restored the majority of mitochondrial energy metabolism related gene expression within the noninfarcted border zone. In particular, genes involved in complex I (NADH-coenzyme Q reductase): Ndufb3, Ndufa7, Ndufc2 and Ndufa5 and complex IV (Cytochrome C Oxidase): Cox6c were significantly upregulated by Bendavia treatment in the noninfarcted border zone. Our previous data demonstrated that the activity of complex I and complex IV were markedly reduced in the border zone and Bendavia significantly increased complex I and IV activity to normal levels16. These findings suggest that downregulation of mitochondrial energy metabolism related gene expression and reduction of complex IV activity in MI/BZ group may indicate a state of cell energy deprivation. Bendavia’s ability to restore mitochondrial bioenergetics may be beneficial in treating heart failure post-MI or other ischemic and nonischemic impairments in cardiac function.
Mitochondrial biogenesis is a process responsible for mitochondrial component synthesis and assembly that correlates with energy metabolism. In the healthy heart, mitochondrial biogenesis matches cardiac growth and cardiac work. Recently, a considerable amount of data suggests that mitochondrial biogenesis disorders play a critical role in cardiac dysfunction and heart failure36–38. PGC1α, a master regulator of mitochondrial biogenesis, is preferentially expressed in the heart serving as transcriptional coactivator. Expression levels of PGC1α are intricately linked to the maintenance of heart function. Spiegelman et al39 reported that there was reduced mitochondrial enzymatic activities and pronounced decrease in ATP concentrations with genetic ablation of the PGC1α mouse heart. Our present study demonstrated that chronic Bendavia therapy completely prevented down-regulation of mitochondrial biogenesis genes within the border zone, which may represent a mechanism by which it preserved cardiac function.
PGC1α also can directly coactivate peroxisome proliferator-activated receptors (PPARs) and estrogen-related receptors (ERRs) regulating glucose and FA oxidation. There are 3 PPAR isoforms and PPARα is highly expressed in heart and its activation increases fatty acid oxidation40. The ERR family includes ERRα, ERRβ, and ERRγ. ERRα cooperates with PPARα and NRF regulating glucose and FA utilization. Glucose and FA uptake are mainly mediated by GLUT4 and the FA transporter CD36 in healthy adult heart. Moreover, these two genes are also PGC1α downstream target genes. In general, oxidation of FA is reduced in the failing heart41, 42. However, the alteration in glucose utilization in the failing heart is variable as decreased, unchanged or increased glucose oxidation. Our result showed that myocardial Glut4 was 51 % lower in the noninfarcted border zone of infarcted hearts indicating a decreased capacity for glucose uptake. Two previously published papers reported that CD36 was up-regulated in acute ischemic brain and Bendavia attenuated the expression of CD3643, 44. In our chronic MI model, we found that Bendavia prevented the downregulation of CD36. These findings suggest that Bendavia has an ability to bring CD36 toward normal level whether CD36 is up or down-regulated in pathologic condition. In addition, Kelly DP et al reported that the failing heart exhibited a decrease in PPARα and ERRα content, correlated with FA oxidation gene down-regulation and reduced fatty acid utilization45. Our results are generally consistent with studies in the literature that showed a decrease in the expression of glucose and FA utilization related genes within the border zone. Importantly, these decreases in gene expression were reversed by chronic Bendavia therapy. Therefore, these findings suggest that impaired myocardial glucose and FA uptake may contribute to the progression of heart failure post-MI and Bendavia plays a beneficial role in regulating mitochondrial substrate utilization. Our data are also in line with a recent study46, showing that myocardial expression of PGC1α and PPARα was blunted in renovascular hypertension, but improved in Bendavia-treated pigs.
Mitochondrial and sarcoplasmic reticulum (SR) are the two organelles that coordinate energy production and calcium control. Any perturbation in mitochondrial or cytosolic Ca2+ homeostasis will have a detrimental effect on cell function. In the ischemic heart, the decreased ATP leads to elevation in cytosolic calcium level. Abnormal calcium cycling is widely attributed to decreased SERCA expression47. For this reason, a number of experimental and clinical efforts have aimed to improve cardiac function in heart failure by increasing SERCA using gene therapy48, 49. Our present study demonstrated that chronic Bendavia administration preserved SERCA2a expression in the border zone, suggesting that improved mitochondria function is also beneficial for cellular Ca2+ homeostasis.
Alterations in the balance of MMPs and TIMPs are involved in myocardial matrix remodeling. MMPs are expressed at very low levels in normal myocardium but markedly increased expression and activity of MMPs have been demonstrated in human and animal hearts during the remodeling process after MI50, 51. Rohde et al52 demonstrated that pharmacological MMP inhibitor attenuated left ventricular enlargement and increased fractional shortening after experimental MI in mice. In addition, the particular importance of MMP9 has been identified in LV remodeling. Ducharme et al53 reported that targeted deletion of MMP9 attenuated left ventricular enlargement and collagen accumulation in mice post-MI. TIMP1 is an important endogenous inhibitor of myocardial MMPs. In a mouse model post-MI, overexpression of adenoviral TIMP1 resulted in decreased collagen deposition and prevention of cardiac rupture54. Our present result showed that Bendavia completely prevented up-regulation of MMP9, but maintained TIMP1 gene expression suggesting a reduction of MMP activity and less pronounced ventricular remodeling in the Bendavia treatment group.
A published paper reported that mitochondrial cristae quality and density significantly declined in the transverse aortic constriction (TAC)-induced model of cardiac hypertrophy and it was ameliorated by Bendavia55. This pressure-overload-induced heart failure model showed mitochondrial swelling and disrupted cristae in saline-TAC hearts. However, in our 6-week rat MI model, we detected no evidence of this type of visual mitochondrial injury at an ultrastructural level in the control water-treat group. This lack of morphologic damage to the mitochondria may explain why we did not detect the difference between water and Bendavia-treated groups. Perhaps, if we had extended our study to 6 months (at a time where the heart is more dilated, ventricular function has further deteriorated and mitochondria structure may have exhibited more damage), we may have been able to identify a protective effect of Bendavia upon mitochondrial structure. The trend toward preservation of cardiolipin content and type further suggests that mitochondrial structural improvements with Bendavia may follow with more chronic therapy.
As previously reported, Bendavia interacts with cardiolipin and improves electron transport and reduces ROS level in stressed cardiomyocytes. In the present study there was a non-significant trend supporting the concept that Bendavia may have normalized cardiolipin levels in the border zone; however our n values may have been too low in the present study to show a statistically significant improvement in this model.
Conclusion
Bendavia is a compound that targets mitochondria and improves energy production and decreases the generation of ROS. Our data showed for first time that Bendavia’s action of improving cardiac function and preventing adverse LV remodeling may be through its impact on primary and downstream pathways related to mitochondrial function and energetics. Bendavia reversed the down-regulation of mitochondrial energy metabolism gene expression, promoted mitochondrial biogenesis, regulated glucose & fatty acid oxidation related gene expression, preserved SERCA2a and reduced cardiac fibrosis in the noninfarcted border zone.
Limitation
The gene expressions need to be further confirmed at protein levels in future studies.
Acknowledgments
Grant support: This study was supported by a grant from Stealth BioTherapeutics, Inc., Newton, Mass.
Footnotes
Conflict of interest statement: Dr. Kloner and Dr. Brown receive research support and are consultants from Stealth BioTherapeutics, Inc. The other authors report no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedrich J, Apstein CS, Ingwall JS. 31P nuclear magnetic resonance spectroscopic imaging of regions of remodeled myocardium in the infarcted rat heart. Circulation. 1995;92:3527–38. doi: 10.1161/01.cir.92.12.3527. [DOI] [PubMed] [Google Scholar]
- 3.Murray AJ, Lygate CA, Cole MA, Carr CA, Radda GK, Neubauer S, Clarke K. Insulin resistance, abnormal energy metabolism and increased ischemic damage in the chronically infarcted rat heart. Cardiovasc Res. 2006;71:149–57. doi: 10.1016/j.cardiores.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 4.Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovasc Res. 2011;90:202–9. doi: 10.1093/cvr/cvr038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol. 2013;61:599–610. doi: 10.1016/j.jacc.2012.08.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–35. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- 7.Kalsi KK, Smolenski RT, Pritchard RD, Khaghani A, Seymour AM, Yacoub MH. Energetics and function of the failing human heart with dilated or hypertrophic cardiomyopathy. Eur J Clin Invest. 1999;29:469–77. doi: 10.1046/j.1365-2362.1999.00468.x. [DOI] [PubMed] [Google Scholar]
- 8.Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clinical pharmacology and therapeutics. 2014;96:672–83. doi: 10.1038/clpt.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown DA, Hale SL, Baines CP, Rio CL, Hamlin RL, Yueyama Y, Kijtawornrat A, Yeh ST, Frasier CR, Stewart LM, Moukdar F, Shaikh SR, Fisher-Wellman KH, Neufer PD, Kloner RA. Reduction of early reperfusion injury with the mitochondria-targeting Peptide bendavia. J Cardiovasc Pharmacol Ther. 2013;19:121–32. doi: 10.1177/1074248413508003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, Koomalsingh KJ, Gorman JH, 3rd, Sloan RC, Frasier CR, Watson CA, Bostian PA, Kypson AP, Brown DA. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective Peptide. J Am Heart Assoc. 2012;1:e001644. doi: 10.1161/JAHA.112.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, Szeto HH. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. Journal of the American Society of Nephrology: JASN. 2013;24:1250–61. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting Mitochondrial Cardiolipin and the Cytochrome C/Cardiolipin Complex to Promote Electron Transport and Optimize Mitochondrial Atp Synthesis. Br J Pharmacol. 2013 doi: 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szeto HH. First-In-Class Cardiolipin Therapeutic to Restore Mitochondrial Bioenergetics. Br J Pharmacol. 2013 doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, Kavanagh TJ, Szeto HH, Rabinovitch PS, Marcinek DJ. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging cell. 2013;12:763–71. doi: 10.1111/acel.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–90. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 16.Dai W, Shi J, Gupta RC, Sabbah HN, Hale SL, Kloner RA. Bendavia, a mitochondria-targeting peptide, improves postinfarction cardiac function, prevents adverse left ventricular remodeling, and restores mitochondria-related gene expression in rats. Journal of cardiovascular pharmacology. 2014;64:543–53. doi: 10.1097/FJC.0000000000000155. [DOI] [PubMed] [Google Scholar]
- 17.Christie WHX. Lipid Analysis: Isolation, Separation, Identification and Lipidomic Analysis. 4. The Oily Press; Bridgwater, England: 2010. [Google Scholar]
- 18.Yang K, Cheng H, Gross RW, Han X. Automated lipid identification and quantification by multidimensional mass spectrometry-based shotgun lipidomics. Analytical chemistry. 2009;81:4356–68. doi: 10.1021/ac900241u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005;115:547–55. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rowe GC, Jiang A, Arany Z. PGC-1 coactivators in cardiac development and disease. Circ Res. 2010;107:825–38. doi: 10.1161/CIRCRESAHA.110.223818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Vos KJ, Allan VJ, Grierson AJ, Sheetz MP. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Current biology: CB. 2005;15:678–83. doi: 10.1016/j.cub.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 22.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–26. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA, Rees ML, Maxey ML, McCune SA, Moore RL. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. Journal of lipid research. 2007;48:1559–70. doi: 10.1194/jlr.M600551-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. American journal of physiology Cell physiology. 2007;292:C33–44. doi: 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- 25.Spinale FG. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res. 2002;90:520–30. doi: 10.1161/01.res.0000013290.12884.a3. [DOI] [PubMed] [Google Scholar]
- 26.Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–7. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz Longacre L, Kloner RA, Arai AE, Baines CP, Bolli R, Braunwald E, Downey J, Gibbons RJ, Gottlieb RA, Heusch G, Jennings RB, Lefer DJ, Mentzer RM, Murphy E, Ovize M, Ping P, Przyklenk K, Sack MN, Vander Heide RS, Vinten-Johansen J, Yellon DM. New horizons in cardioprotection: recommendations from the 2010 National Heart, Lung, and Blood Institute Workshop. Circulation. 2011;124:1172–9. doi: 10.1161/CIRCULATIONAHA.111.032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menees DS, Peterson ED, Wang Y, Curtis JP, Messenger JC, Rumsfeld JS, Gurm HS. Door-to-balloon time and mortality among patients undergoing primary PCI. The New England journal of medicine. 2013;369:901–9. doi: 10.1056/NEJMoa1208200. [DOI] [PubMed] [Google Scholar]
- 29.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–95. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 30.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–56. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 31.Reily C, Mitchell T, Chacko BK, Benavides G, Murphy MP, Darley-Usmar V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013;1:86–93. doi: 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sloan RC, Moukdar F, Frasier CR, Patel HD, Bostian PA, Lust RM, Brown DA. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol. 2012;52:1009–18. doi: 10.1016/j.yjmcc.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 33.Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C, Kostler H, von Kienlin M, Harre K, Hahn D, Neubauer S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40:1267–74. doi: 10.1016/s0735-1097(02)02160-5. [DOI] [PubMed] [Google Scholar]
- 34.Jarreta D, Orus J, Barrientos A, Miro O, Roig E, Heras M, Moraes CT, Cardellach F, Casademont J. Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy. Cardiovasc Res. 2000;45:860–5. doi: 10.1016/s0008-6363(99)00388-0. [DOI] [PubMed] [Google Scholar]
- 35.Buchwald A, Till H, Unterberg C, Oberschmidt R, Figulla HR, Wiegand V. Alterations of the mitochondrial respiratory chain in human dilated cardiomyopathy. Eur Heart J. 1990;11:509–16. doi: 10.1093/oxfordjournals.eurheartj.a059743. [DOI] [PubMed] [Google Scholar]
- 36.Russell LK, Finck BN, Kelly DP. Mouse models of mitochondrial dysfunction and heart failure. J Mol Cell Cardiol. 2005;38:81–91. doi: 10.1016/j.yjmcc.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 37.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94:525–33. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 38.Shi J, Jiang B, Qiu Y, Guan J, Jain M, Cao X, Bauer M, Su L, Burkly LC, Leone TC, Kelly DP, Liao R. PGC1alpha plays a critical role in TWEAK-induced cardiac dysfunction. PLoS One. 2013;8:e54054. doi: 10.1371/journal.pone.0054054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–71. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Ferre P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53(Suppl 1):S43–50. doi: 10.2337/diabetes.53.2007.s43. [DOI] [PubMed] [Google Scholar]
- 41.Razeghi P, Young ME, Abbasi S, Taegtmeyer H. Hypoxia in vivo decreases peroxisome proliferator-activated receptor alpha-regulated gene expression in rat heart. Biochem Biophys Res Commun. 2001;287:5–10. doi: 10.1006/bbrc.2001.5541. [DOI] [PubMed] [Google Scholar]
- 42.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–42. doi: 10.1161/01.cir.94.11.2837. [DOI] [PubMed] [Google Scholar]
- 43.Kim EH, Tolhurst AT, Szeto HH, Cho SH. Targeting CD36-mediated inflammation reduces acute brain injury in transient, but not permanent, ischemic stroke. CNS neuroscience & therapeutics. 2015;21:385–91. doi: 10.1111/cns.12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007;282:4634–42. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- 45.Sack MN, Kelly DP. The energy substrate switch during development of heart failure: gene regulatory mechanisms (Review) Int J Mol Med. 1998;1:17–24. doi: 10.3892/ijmm.1.1.17. [DOI] [PubMed] [Google Scholar]
- 46.Eirin A, Williams BJ, Ebrahimi B, Zhang X, Crane JA, Lerman A, Textor SC, Lerman LO. Mitochondrial targeted peptides attenuate residual myocardial damage after reversal of experimental renovascular hypertension. Journal of hypertension. 2014;32:154–65. doi: 10.1097/HJH.0b013e3283658a53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–77. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- 48.Lipskaia L, Chemaly ER, Hadri L, Lompre AM, Hajjar RJ. Sarcoplasmic reticulum Ca(2+) ATPase as a therapeutic target for heart failure. Expert Opin Biol Ther. 2010;10:29–41. doi: 10.1517/14712590903321462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Periasamy M, Huke S. SERCA pump level is a critical determinant of Ca(2+)homeostasis and cardiac contractility. J Mol Cell Cardiol. 2001;33:1053–63. doi: 10.1006/jmcc.2001.1366. [DOI] [PubMed] [Google Scholar]
- 50.Cleutjens JP, Kandala JC, Guarda E, Guntaka RV, Weber KT. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27:1281–92. doi: 10.1016/s0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- 51.Tyagi SC, Kumar SG, Haas SJ, Reddy HK, Voelker DJ, Hayden MR, Demmy TL, Schmaltz RA, Curtis JJ. Post-transcriptional regulation of extracellular matrix metalloproteinase in human heart end-stage failure secondary to ischemic cardiomyopathy. J Mol Cell Cardiol. 1996;28:1415–28. doi: 10.1006/jmcc.1996.0132. [DOI] [PubMed] [Google Scholar]
- 52.Rohde LE, Ducharme A, Arroyo LH, Aikawa M, Sukhova GH, Lopez-Anaya A, McClure KF, Mitchell PG, Libby P, Lee RT. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation. 1999;99:3063–70. doi: 10.1161/01.cir.99.23.3063. [DOI] [PubMed] [Google Scholar]
- 53.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–42. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 55.Dai DF, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L, Beyer RP, Crispin DA, Shulman NJ, Szeto HH, Tian R, MacCoss MJ, Rabinovitch PS. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail. 2013;6:1067–76. doi: 10.1161/CIRCHEARTFAILURE.113.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]