

Abstract
The evolution process includes genetic alterations that started with prokaryotes and now continues in humans. A distinct difference between prokaryotic chromosomes and eukaryotic chromosomes involves histones. As evolution progressed, genetic alterations accumulated and a mechanism for gene selection developed. It was as if nature was experimenting to optimally utilize the gene pool without changing individual gene sequences. This mechanism is called epigenetics, as it is above the genome. Curiously, the mechanism of epigenetic regulation in prokaryotes is strikingly different from that in eukaryotes, mainly higher eukaryotes, like mammals. In fact, epigenetics plays a significant role in the conserved process of embryogenesis and human development. Malfunction of epigenetic regulation results in many types of undesirable effects, including cardiovascular disease, metabolic disorders, autoimmune diseases, and cancer. This review provides a comparative analysis and new insights into these aspects.
Keywords: evolution, epigenetics, prokaryotes, eukaryotes, mammals, diseases
What is Epigenetics?
Epigenetics is a mechanism of gene transcription regulation that does not change the DNA sequence and is usually reversible. The reversibility of this phenomenon provides the opportunity for these cells to utilize the existing gene pool in different ways, as necessary, without permanently altering the content of the gene pool. During the course of development, from prokaryotes to mammals, a mechanism arose by which specific functions in terms of the regulation of gene expression could be performed. Thus, the epigenetic alterations are somewhat different in varying cell types and cells from different origins. The broad mechanisms by which these changes occur are methylation–demethylation, acetylation–deacetylation, and other modifications of histones, and also methylation–demethylation of specific regions in DNA on a finer scale. These interactions manipulate the affinity of histone binding and the topology of the DNA that winds around them. Consequently, the transcription of genes is affected by the ability or inability of proteins to bind to regulatory regions in open or condensed chromatin, respectively. Furthermore, the transcriptional regulation by epigenetic mechanisms is critical in the development in which changes determine the differentiation of cells into different cell types with specific functions.
Epigenetics and the Environment
The external world, inclusive of food, toxins, carcinogens, and many other day-to-day factors, has a significant impact on cellular regulation. These environmental factors have the potential to directly alter the DNA sequence, and they can also induce DNA methylation and histone modifications. Thus, exposure to particular environments has the potential to shift cellular equilibrium and create a microenvironment that is suitable for tumorigenesis or the development of other debilitating diseases. It has been shown that the activity of enzymes regulating the structure and activity of chromosomes is sensitive to environmental changes; thus, environmental agents may alter gene expression.1 This may result in widespread changes in cells that can propagate throughout the body by mitosis and evolve into a heritable epigenetic change. Natural selection may act on this chain of events and thus influence evolution.1
A common example of the epigenetic impact of the environment and the related health outcomes is lead exposure and its consequences. Recent studies suggest that lead exposure in children can change DNA methylation, histone modification, and miRNA expression, which may result in neurodegenerative diseases in adult life.2 Similarly, it is well known that carcinogen exposure can cause direct DNA damage and lead to cancer. However, carcinogens can also influence the microenvironment through epigenetic effects on epithelial cells, stromal cells, extracellular matrix constituents, and immune cells, resulting in tumor development.3,4 Tumor initiation is associated with the recruitment of these components as well as the production of cytokines. It has been shown that carcinogen exposure can result in gene methylation changes in stromal cells, resulting in breast, prostate, and some squamous cell carcinomas.5 In addition, exposure to nongenotoxic agents has been found to lead to methylome changes in the supporting cellular stroma. As an example, exposure to low-level benzene has been linked to both hypomethylation and hypermethylation of the p15 tumor suppressor gene promoter element in myeloid leukemia.5
Perhaps more surprisingly, epigenetic changes can result from the influence of nutrients and bioactive food components.6 These changes include DNA methylation and histone modifications. One study suggests that nutrition affects the aging process and cancer development, as well as adult obesity and the development of diabetes by these kinds of epigenetic changes.6
Prokaryotes and Bacteria
DNA adenine methylation plays a vital role in many bacterial cell processes, emphasizing epigenetic modifications as an important factor in bacterial survival (Fig. 1A and B). The methylated state of DNA during DNA replication affects the binding of proteins integral to the start of transcription and the rate at which it proceeds. SeqA is an example, which binds the hemimethylated DNA sequence GATC near the origin of replication and stops replication initiation—both sterically and by inhibiting the synthesis of the DnaA protein.7–14 Another example of adenine methylation playing a vital role in cell processes is DNA repair. Bacterial DNA repair is methylation dependent. The repair protein, MutH, recognizes the methylation state and edits the unmethylated daughter strand for repair to ensure that the parental strand is used as the template for repairing the DNA.15–20
Figure 1.

Differential DNA methylation patterns in prokaryotes, lower eukaryotes, and higher eukaryotes. (A) Pictorial representation of cytosine, 5-methylcytosine, 5-hydroxymethylcytosine, and N6-methyladenine; (B) prokaryote, (C) lower eukaryote, and (D) higher eukaryote DNA methylation is denoted in red on either adenine (A) or cytosine (C).
The restriction–modification (RM) system seems to have evolved as a method to combat pathogens invading the bacterial cell. The RM system is composed of a restriction enzyme and a methylase.21,22 The restriction enzyme cleaves foreign unmethylated DNA at the specific target site so as to preserve the methylated bacterial DNA.
DNA methylation of GATC is also utilized by the Escherichia coli bacteriophage P1 to package its DNA into capsids. The packing initiation site in P1 is known as the pac site, and it contains seven GATC sites within 162 base sequences.23 The P1 packaging enzyme only cleaves DNA into capsids at the pac site if a majority of these GATC sites are methylated on both strands. Thus, without methylation by DNA adenine methyltransferase (Dam) methylase, the phage DNA cannot be packed into capsids by the packaging enzyme. This has been demonstrated in Dam-negative mutant E. coli that was exposed to Dam-negative P1, showing that in the absence of methylation, there is a reduction in the number of P1 phage progeny compared to bacterial cells or phages with Dam methylase.24,25 The progeny produced in Dam methylase negative mutants do not include the pac sequences. An excellent review gives an in-depth description of epigenetic gene regulation in prokaryotes.25
Archaea are a domain and a kingdom of single-celled organisms distinct from bacteria and eukarya. Although their genome is prokaryotic, archaea share many similarities with eukaryotic organisms. Like bacteria, the archaeal genome may be either monoploid or polyploid. The ploidy state of an organism varies among different species and strains of archaea.26 Archaea are also known to exhibit epigenetic modifications to their genome. Certain species of archaea contain histone proteins that form tetrameric nucleosomes similar to eukaryotes.27 Archaeal histones are not as widespread as their eukaryotic orthologs and therefore may not play such an extensive role in DNA condensation.28 Furthermore, archaeal histones lack the N- and C-terminal tails that are present in eukaryotes, and therefore lack important sites for epigenetic modification.29 In addition to histones, some species of Archaea (specifically thermophiles and hyperthermophiles) contain the chromatin-binding protein Alba, which is also found in some eukaryotes.27 Alba binds double-stranded DNA, protecting it from nuclease degradation.30 Prokaryotic homologs of the histone deacetylase Sir2 (ssSir2) can interact with Alba. When Alba is deacetylated, its DNA-binding affinity increases, and it binds to repress transcription.31 This interaction resembles the way histone acetylation/deacetylation is used to regulate the expression of genes in eukaryotes. Like bacteria, several lineages of Archaea demonstrate the use of the CRISPR/Cas system of defense against foreign DNA, whereby invading DNA is incorporated into the CRISPR gene locus and used to generate small RNAs that interfere with and destroy the corresponding foreign DNA.32
Epigenetic Mechanisms in Lower Eukaryotes
Lower eukaryotes also harbor changes in methylation in adenine (Fig. 1C). However, the eukaryotic genome differs from prokaryotes because it is located within a membrane-bound nucleus and the DNA contains histones. This histone complex consists of an octamer of histone proteins, two each of H2A, H2B, H3, and H4, along with a single H1 protein (Fig. 2). DNA strands wind around the histone complex to form nucleosomes that are used to condense DNA into a tightly coiled and compact chromosome. In eukaryotes (and certain species of Archaea), gene expression may be regulated by modification to these histones, in addition to DNA. Among eukaryotes, histone proteins are highly conserved and thus play a critical role in DNA regulation and survival of the organism. Even the human H4 protein is 92% identical to its Saccharomyces cerevisiae ortholog.33 It is believed that the core histone proteins evolved from a common ancestral protein, due to the similarities in their C-terminal residues.34 However, with regard to epigenetic histone modification, it is known that the number of methylation sites on lysines of histones increase with the complexity of the organism, ie, fewer in lower eukaryotes, and more in higher eukaryotes.35 Enzymes that target repressive methylation sites on histones (H3K9, H3K27, and H4K20) are not found in S. cerevisiae; however, the first discovered histone acetyltransferase (HAT1) was cloned from S. cerevisiae.35,36 Finally, another conserved function is transcriptional regulation. For instance, yeast share transcriptional regulators with protozoa (Gcn5 protein ortholog), such as Thermus thermophile, and mammals (Rpd3, bovine ortholog). These examples directly exhibit conservation of transcriptional regulators from protozoa, to yeast, to mammals.36
Figure 2.
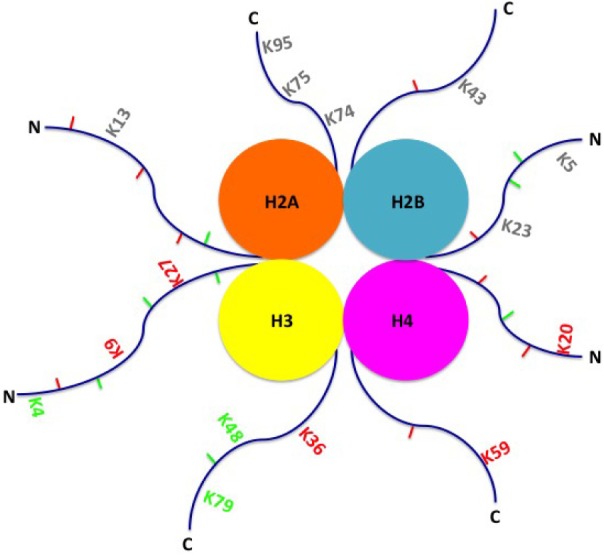
Activating and inhibitory histone modifications. Histone modifications include methylation and demethylation on lysine and arginine residues, ubiquitination, and sumoylation. Activating modifications (green), inhibitory modifications (red), and modifications with unknown function (gray) are shown on either the C-terminus (C) or N-terminus (N) of histone tails. Lysine methylations are marked with K and the number of the residue methylated.
In respect to the fungal genome, modifications to histones can be activating (H3K4me2) or silencing (H3K9me3, H3K27me3). Major proteins involved in this modification process are heterochromatin protein 1 (HP1) and histone methyltransferases KMT1 and KMT6. Species of fungi that interact with plants tend to have a more fluid genome with regions of high plasticity, manifesting as rapidly evolving sequences.37 This could potentially be a consequence of coevolution with the plant species with which these fungi interact. Evolution of these adaptations may have been conserved to allow fungi to interact with higher eukaryotes.37 A recently discovered family of histone demethylases known as Jumonji C (JmjC) domain proteins comprises 14 subfamilies that are found in plants, animals, and fungi.38 Five JmjC subfamilies, namely, KDM3, KDM5, JMJD6, PKDM11, and PKDM13, are found in plants, animals, and fungi, and phylogenetic analysis indicates that these families may have evolved from five ancestral genes in a common ancestor of these three kingdoms.38 Researchers found that three subfamilies, such as PKDM7, PKDM8, and PKDM9, are found in plants, but not in fungi or animals, indicating that these genes were lost by the common ancestor of animals and fungi after the plant kingdom diverged from their common ancestor. Likewise, fungi lack the subfamilies JARID2, KDM6, and PKDM12 that are found in animals, marking the divergence of the fungi and animal kingdoms from their common ancestor.38 Thus, yeast and fungi set the stage for both DNA and histone modifications, which are used by other eukaryotes and mammals.
While methylated cytosine (5mC) is the dominant form of DNA methylation in higher eukaryotes, some unicellular eukaryotes are known to use methylated adenine (6mA) as well as 5mC (Fig. 1C and D).39,40 6mA is used by prokaryotes to identify host DNA from foreign DNA to protect it from degradation. By contrast, 6mA in eukaryotes is believed to be an epigenetic regulator of development and gene regulation, even among higher eukaryotes.41–43 For example, 6mA is found in Chlamydomonas, where it serves as a marker of gene activation, and islands dense in 6mA are found clustered near transcription start sites.42 In Caenorhabditis elegans, 6mA is present in DNA, while 5mC is not, and 6mA tends to localize to GAGG and AGAA consensus sequences, marking the sites of active genes, as in Chlamydomonas.41
Conservation of methyltransferase and demethylase function is also observed throughout the evolutionary tree. Some examples include DNA N6-methyltransferase-1 (DAMT-1) and NMAD-1 in C. elegans,41 proteins ParB-MTase and TET/JBP in Chlamydomonas reinhardtii, methyltransferase CG14906 and demethylase DMAD in Drosophila melanogaster, DNA methyltransferase and methyl-CpG binding proteins associated with cytosine methylation in Platyhelminthes,44 cytosine methylation in Schistosoma mansoni,45 and methyltransferase-like 4 (METTL4) and demethylases TET1, TET2, and TET3 in Homo sapiens.46
Regeneration
Regeneration mechanisms provide key insights into how epigenetics has been utilized throughout evolutionary history to control an organism’s response to environmental stimuli. Interestingly, regenerative programming stems from highly conserved genes and cellular pathways,47,48 but few organisms possess a regenerative ability. This suggests that it is the expression of these pathways that are altered rather than the sequences themselves. Furthermore, without clear transmission of regenerative ability throughout evolutionary time, it is reasonable to infer that this may be an epigenetic process. On a more cellular level, cells are dedifferentiated during regeneration, but upon redifferentiation, the cells retain specification. This is an interesting phenomenon because it suggests a point in the gene regulatory network, which is not reversible, and also implies that downstream exists at this point of time, which maintains the differentiation state. We believe this maintenance to be epigenetically mediated. A vertebrate displaying regenerative capability is the zebrafish. Following fin amputation, a regenerative blastema is formed. The blastema utilizes differential gene expression to coordinate positioning and growth of the new limb. During the first hour postamputation, it has been found that the GCN5 gene is upregulated. This gene encodes many histone acetylases in zebrafish so it is no surprise that it was also found that H3K9K14 diacetylation increased.49 In addition to alterations in acetylation levels, several histone tails are differentially methylated during regeneration, such as H3K9me2 and H4K20me3.49 Thus, the histone modifications during regeneration of the zebrafish fin is another example of how epigenetic control of gene expression levels may be a key mechanism behind regenerative processes.
This regeneration process is not only present in vertebrates. Hydra have been vastly studied for their ability to regenerate polyps following separation into single cells.50,51 Upon wounding, epithelial cells dedifferentiate, forming stem cell like cells that direct the regrowth during the regeneration process. The newly dedifferentiated cells and some apoptotic cells then direct the regeneration process using signaling found in early embryo axis development, most significantly Wnt3 signaling, which is well known to be regulated by epigenetic mechanisms.51–53
It is important to mention that regeneration properties in single-celled organisms, invertebrates, and vertebrates do not appear consistently or linearly in the evolutionary time-line. Even within taxa of genetically similar organisms, regeneration is quite variable. There are hypotheses as to why this occurs, but little molecular data to support a universal hypothesis. Effectively, the role of epigenetics in regeneration may provide a comprehensive and universal hypothesis for this process and insight as to why some organisms retain the ability, while others do not.
Epigenetic Mechanisms in Mammals
Embryogenesis and development
Epigenetics, including DNA methylation and histone modification, plays a significant role from the start of fertilization, during embryogenesis, and throughout development. Essentially, modifications in histones determine open or closed chromatin conformation in specific regions, depending mainly on the acetylation–deacetylation of lysine residues on histone tails. More specific modifications are methylation and demethylation of lysine or arginine residues, which determines the conformational identity of a gene and its ability to be transcribed (Fig. 2). Finer modification controls are ubiquitination, sumoylation, and phosphorylation, which may be gene specific.54–58 Within the last two decades, a histone code model was proposed suggesting a particular combination of histone modifications, which could be responsible for chromatin conformation at specific sites of gene expression.59 Although novel, this model falls short of explaining the significant role of methylation at DNA CpG residues in this process.
CpG dinucleotides are found across the entire genome and are often clustered in islands where the cytosine is methylated. CpG DNA methylation significantly changes during embryogenesis. In the fertilized egg, first a global active demethylation occurs in the paternal DNA, which is independent of replication. Active demethylation has been shown to be mediated by conversion of methylated cytosine to hydroxymethylated cytosine by TET proteins.60 Complete demethylation is achieved by the removal of the hydroxymethylated cytosine residue. Differential expression of TET proteins influences this process.60 The demethylation process is followed by remethylation. After this phenomenon, the maternal genome is passively demethylated and remethylated.61 Most of these events are performed by the methylating–demethylating system present in the egg. DNA methylation is regulated by histone modification as H4R3 in the maternal oocyte recruits DNMT3a to the site of methylation (Fig. 3).62 DNMT3a and DNMT3b are the two enzymes that exhibit the methylation pattern in DNA during embryogenesis.61 This suggests that in the early events of fertilization and embryogenesis, the histones present in the egg dictate the events of DNA methylation and demethylation. As shown in Figure 3, inherited premade histone modifications present in the egg should determine the status of methylation during embryogenesis. Increased levels of H3R4 methylation in the maternal oocyte will recruit more DNMT3a, resulting in greater DNA CpG methylation (Fig. 3B). This methylation pattern will be distributed in all developing differentiated tissues and will be transmitted during replication. On the other hand, DNMT1 is mainly used for the methylation of the newly synthesized strand during replication in somatic cells.61 Interestingly, DNMT1 is also recruited at the site of methylation by histone modification H3K9me2/3 in association with UHRF1 to the site of methylation.63
Figure 3.
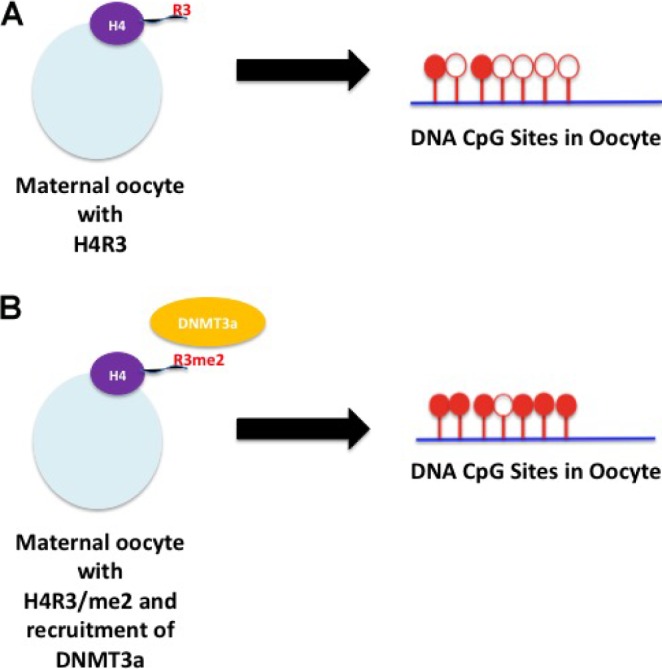
Differential methylation of H4R in the maternal oocyte regulates the methylation status of DNA during embryogenesis. (A) No maternal oocyte H4R3 methylation yields little DNA CpG site methylation. (B) Maternal oocyte H4R3 methylation yields more methylation of DNA CpG sites. Red indicates methylation and empty circles represent nonmethylation. DNMT3a denotes DNA methyltransferase 3a.
Recent studies show that histone modifications and CpG DNA methylation regulate each other and communicate during embryogenesis and the developmental process. H3K27me3 regulates tissue-specific gene expression by recruiting CCCTC binding factor (CTCF), which creates an insulation zone with cohesin. Essentially, this insulation zone inhibits enhancer/promoter interaction to bind RNA polymerase II for transcription initiation. While CpG methylation near the upstream promoter regions inhibits gene expression, enhancer methylation disrupts CTCF binding around the insulation zone to promote transcription.64
Thus, the process from fertilization to tissue development is tightly synchronized under control of epigenetic regulation. Aberration of this epigenetic balance in any developed tissue or organ may create an imbalance that may lead to disorders and diseases.
Aging
Aging is a universal process. It is characterized by the gradual deterioration of full physiological abilities, leading to impaired function and increased chance of death. Increasing age is associated with the development of cancer, diabetes, cardiovascular disorders, and neurodegenerative diseases. Several research studies implicate patterns of gene expression alterations in the aging process, such that a set of genes are downregulated while another set is upregulated. Recent studies have shown that drastic epigenetic alterations accumulate when people age. These alterations include both histone modifications and methylation of the CpG residues of DNA. Many researchers cite an epigenetic drift that is associated with aging, which is a gradual increase or decrease in methylation at specific loci.65 Additionally, alterations in histone methylation changes the life span of many species by regulating transcription.66
One of the few hallmarks of the aging process is telomere erosion. As somatic cells age, telomeres shorten.67 One study found that telomere dysfunction accelerates aging in mice and humans, while experimental stimulation of telomerase delayed aging in mice.67 There is contradictory research that telomere expression is regulated by epigenetic mechanisms.68 However, epigenetic drugs have a strong impact on the expression of telomeres; therefore, there is a strong possibility that telomeres are epigenetically regulated. It is important to note that the telomeres themselves do not undergo epigenetic modifications such as methylation and demethylation. Instead, adjacent subtelomeric DNA is heavily methylated and can be adjusted. A study showed that the methylation status of this subtelomeric DNA negatively correlates with telomere length, suggesting that telomere length can be influenced by epigenetic factors.68 The same study showed that treating human cancer cell lines with demethylating drugs results in hypomethylation of the subtelomeric region, increasing telomere recombination, which in turn results in increased telomere preservation.
In addition to changes in telomere length, aging has been linked to DNA methylation changes and, though less investigated, histone modification.69 Current research aims to investigate whether there are age-related epigenetic changes that occur in a predictable manner. Additionally, it is of interest to know whether these changes result in transcriptional alterations or whether they are nonfunctional and randomly accumulated. Jung et al69 studied human skin fibroblasts, combined genome-wide transcription, DNA methylation, and histone methylation to demonstrate a clear age-dependent decrease in expression of genes for protein involved in translation and ribosome function. They suggest a model with which age-dependent downregulation of these proteins contributed to extend the human life span. They invented a novel approach called “Three Component Analysis” to compare age-related epigenetic changes in fibroblasts. They found that an important aspect of aging is the downregulation of protein translation-related factors (eg, transcription factors and genes of ribosomal proteins) by epigenetic changes that act to extend span. Hofmann et al showed that mice that were Myc haploinsufficient (Myc+/−) had increased life spans.70 Jung et al observed that genes of protein translation were upregulated by Myc, suggesting that changing the expression of genes related to protein translation is linked to aging. They also found methylation changes in HOX gene clusters that have been previously linked to aging. Additionally, they found age-associated DNA hypermethylation at CpG islands proximal to promoters only.69
Another study suggesting that the downregulation of these proteins contributes to extend the human life span looked at epigenomic mechanisms that are potentially altered by age-associated differentially methylated regions (aDMRs) and by regulatory sequences in the promoters of age-associated genes (aGENs).71 This study found that several aDMRs and aGENs across several studies share a common Polycomb Repressive Complex 2 (PRC2) site, which is responsible for methylation and demethylation of other sites. The study found that PRC2 was associated with hypermethylation of aDMRs and other gene expression changes related to aging.71
Epigenetic Mechanisms in Human Disease
Metabolic syndromes and cardiovascular disorders
Somatic cells that accumulate epigenetic changes, like the ones stated above, are more susceptible to diseases. Metabolic syndromes, including diabetes and obesity, when uncontrolled are linked to many types of diseases. Most of these diseases are caused by nonfunctional pathways or by the activation of pathways that have adverse effects. For example, in type I diabetes, pancreatic beta cells do not produce insulin, whereas type II diabetics are insulin resistant. Diabetes is a debilitating disease that has many adverse effects on health when uncontrolled, including cardiovascular diseases, blindness, amputation of lower extremities, and renal failure.
Recent studies show a striking association of metabolic syndromes with epigenetic changes. Maternal and paternal nutrition is linked to epigenetic metabolic programming of offspring. Alteration of epigenetic pathways is associated with many risks involving type II diabetes, including abnormal insulin secretion and insulin action.72 Nutritional imbalance of parents affects the metabolic function of future generations by epigenetic alterations during embryogenesis and fetal development because epigenetic memory is heritable.72 These epigenetic alterations include DNA methylation, histone tail modifications, activity of small noncoding RNAs, and chromatin remodeling.73
The developmental origins of health and disease (DOHaD) hypothesis suggests that parental overconsumption or malnutrition affects the lifelong morbidity of offspring. Most of the current information in this regard comes either from animal studies or from the Dutch famine cohort study.74
One of the ways nutrients can affect epigenetic alterations is by the regulation of methyl donors.75 The enzymes that exchange methyl groups include S-adenosylmethionine-dependent DNA and protein methyl transferases, DNA demethylases, histone acetylases (lysine acetyl-transferases), and HDACs. Amino acids, including glycine, histidine, methionine, and serine, and vitamins, including B6, B12, and folate, are involved in methyl donation for both DNA and proteins. Therefore, any alterations in these amino acids and vitamins will affect methylation.
In a recent study, DNA methylation profiles of pancreatic islets were compared between type II diabetic and nondiabetic donors.76 Differential DNA methylation in 276 CpGs located in the promoters of 254 genes was observed. However, circulating blood cells from these two types of donors did not show many differences.
Methods of metabolizing lipids and glucose can also be a contributing factor to the onset of diabetes. The end products of fermentation, especially short-chain fatty acids, are involved in epigenetic alterations by interacting with free fatty acid receptors (FFARs).76 For example, the short-chain fatty acid, butyric acid, is a well-known inhibitor of Class II HDACs. Methylation of the FFAR3 promoter region and long interspersed nuclear element 1 (LINE1) was determined in the microbiota of type II diabetics compared to normal individuals. The diversity of microbiota was lower in diabetic patients, FFAR3 methylation was higher. Methylation of LINE1, a marker of global methylation did not alter significantly. These studies showed a correlation between microbiota and small fatty acids with diabetes.76
As diabetes and obesity are linked to cardiovascular diseases, epigenetics has also been found to be associated with the onset of cardiovascular diseases.77 For example, hypomethylation by the accumulation S-adenosylhomocysteine (SAH) and methyltransferase inhibitors reduces glutathione peroxidase 1 (GPx-1) expression to promote oxidative stress. This causes upregulation of cell adhesion molecules in endothelial cells recruiting leukocytes and resulting in inflammatory response.77
Micronutrients also play a role in epigenetic regulation. The selenium complex affects redox balance by interacting with the methionine–homocysteine cycle. In a mouse model, selenium supplementation reduced DNA methyl transferase (DNMT) activity and overall methylation level, lowering oxidative stress. However, although there was no myocardial hypertrophy, diastolic dysfunction and myocardial fibrosis were still observed. These manifestations were associated with significant changes in myocardial gene expression due to the reduction in DNMT activity.78
These studies suggest that epigenetic changes are involved in metabolic syndromes and cardiovascular health. Future studies will reveal the role of epigenetic in regulating metabolic balance and cardiovascular function.
Neurological disorders
Neurological cells are one of the most specialized cell types and have one of the lowest division rates. Many neurological disorders are characterized by inheritable genetic alterations, such as Down syndrome and Tay-Sachs disease. Recent research, however, demonstrates that many disorders and diseases, such as brain tumors, initiate with epigenetic alterations at the level of histone modifications and DNA methylation in somatic cells. Several excellent reviews are available on this topic.79–81 An important example of a neurological disorder caused by epigenetic alterations is Rett syndrome, which is a leading form of severe mental retardation in females.82 The MeCP2 gene is mutated in 95% of individuals with this disorder. Mutation in this gene alters the association of the MeCP2 protein and HDACs around the methylated CpG islands to inhibit transcription of specific genes.83,84 Additionally, in Huntington’s disease patients, the Huntingtin protein is mutated and can thus enter the nucleus and affect histones. In this disease, Huntingtin protein reduces acetylation on histones H3 and H4 and increases H3K4me3.85 In contrast, Alzheimer’s disease appears to be regulated by histone acetylation,86 and it was observed that neuron-specific expression of HDAC2 results in synaptic plasticity and memory formation.87 Interestingly and in accordance with this observation, mouse model experiments showed that treatment with class I and II HDAC inhibitors enhances memory.88
Autoimmunity
Autoimmunity is the loss of self-tolerance. Currently, the mechanisms causing autoimmunity are not well understood. However, the evolution of epigenetic research has provided insight toward the attainment of autoimmune disorders by bridging genetic and environmental factors.89 It is believed that autoimmunity is developed when genetically predisposed individuals encounter an environmental agent, which causes epigenetic changes that trigger development of the disease.90 The number of genetic loci whose gene expression is modified by these epigenetic mechanisms is growing rapidly.
It has been suggested that the combined action of epigenetic modifications (of both target genes and transcription factor genes) and transcription factors contributes to the pathogenesis of various autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, type 1 diabetes, and multiple sclerosis (MS).91 A recent study showed that there was only 20% concordance in homozygotic twins for systemic lupus erythematosus.91 This suggests that epigenetics and environmental factors played a role in the development of the disease.
MS is believed to be a T-cell-mediated autoimmune disorder. A recent case–control study compared DNA methylation changes associated with MS in CD8+ T-cells between relapsing MS patients and healthy individuals. A total of 79 CpGs showed differential methylation patterns in the MS patients versus the healthy individuals.92 Among the differentially methylated genes, the most significant decrease in methylation in the patient cohort was seen in the ferritin light chain (FTL) gene promoter. FTL is a subunit of ferritin. Defects in FTL are commonly associated with neurodegenerative diseases due to accumulation of iron in the brain, as seen in MS patients. Therefore, the misregulation of the FTL gene due to epigenetic changes could be an important factor in MS disease pathology.92
Viruses
An interesting example of environmental alteration of the epigenetic status in humans is viral infection. As an example, the Epstein–Barr virus (EBV) affects or has affected close to 95% of the adult population.93 EBV is typically asymptomatic in infants and young children, but it leads to mononucleosis in adolescents and young adults, a disease that causes the patient to experience fever, lymphadenopathy, sore throat, and splenomegaly, among other symptoms.93 Later in life, EBV is associated with the development of carcinomas and lymphomas.93 This is because EBV results in a lifelong infection of epithelial and B-cells in the oral cavity.94 When these cells are infected, they exhibit a distinct EBV infection pattern that is regulated by epigenetic modifications, including methylation and alteration to chromatin structure. The virus reprograms the host’s epigenetic machinery, which permanently affects the host cells.94 One study has demonstrated that cells that have been exposed to EBV experience hypermethylation at CpG islands when compared to cells that have not been exposed to the virus.94 Some of these methylations affect the transcription of genes, which may increase the tumorigenic capabilities of the host cells and lead to the development of cancer.
Human immunodeficiency virus (HIV) is another virus that uses epigenetic mechanisms to thrive in its host cells. HIV is an extremely efficacious virus because it is able to effectively hide from the host’s immune system. It accomplishes this through the use of HDACs.95 HDACs regulate the transcription of viral DNA by maintaining closed chromatin conformation around the viral DNA.95 By this mechanism, HIV remains latent in the host, but undetectable by the host’s immune system. Treatment with HDAC inhibitors reverses this negative regulation and reactivates the latent virus to allow the host cells to initiate cell death and eliminate infected cells.95
Cancer
Cancer is caused by both inherited genetic aberrations and genetic aberrations accumulated in somatic cells. It was previously thought that cancer was caused by environmental effects that alter cells in an irreversible manner, causing them to replicate more. It was believed then that genetic alterations may not play any significant role in carcinogenesis. The link between genetics and cancer was shown by Rous’s classical experiments, demonstrating that viruses can cause leukemia.96 In the early 1970s, the first oncogene was discovered from the study of Rous sarcoma virus.97 The following decades showed a tremendous advance in the understanding of cancer at the molecular level.
Now we know that cancer, whether inherited or not, is caused by genomic instability mediated by alterations in DNA, including chromosomal abnormalities, mutations, changes in gene sequence, and activation and deactivation of oncogenes and tumor suppressor genes.98 Research from the past few decades made this theme of genetic alteration an accepted dogma of how cancer manifests. This phenomenon was eloquently described in the paper, “The Hallmarks of Cancer”, which discusses six specific pathways by which cancer progresses and metastasizes.99 Different types of cancers are produced by a combination of these six pathways, each of which has well-defined steps. These steps are the products of individual genes that comprise the entire signal transduction network inside the cell. Some of the growth-promoting genes of these signal transduction pathways can become constitutively activated by mutations (e.g., Src, Ras), copy number increases (HER2 in breast cancer), or chromosomal trans-locations (BCR-Abl in chronic myeloid leukemia). In contrast, tumor suppressor genes are either silenced by deletion (p53, Rb) or by mutation (p53).61 Recent studies show that the development of cancer results from the accumulation of a large number of mutations, most of which are passive and only a small percentage of which actually drive cancer development. These diverse mutations can originate even within one type of cancer, creating a tremendously heterogeneous population. A stepwise mutation model was proposed by Vogelstein and Kinzler100 in the progression of colon cancer. However, Sarkar et al argued that the tremendous diversity of mutations in specific tumor types suggests that initiation of cancer cannot be random.61,101 These authors hypothesize that the formation of cancer progenitor cells is an epigenetic event that silence tumor suppressor genes and may also upregulate growth promoting genes by methylation.101–105 As was discussed in the section “Embryogenesis”, support for this hypothesis can be found in the study of enhancer methylation, which disrupts the insulation zone created by CTCF mediated by di- or trimethylation of H3K27.104 Traditional therapies do not kill cancer progenitor cells and drug-resistant cancer cells as they do not inhibit epigenetic alterations present in cancer cells. This supports the hypothesis that epigenetic alterations are involved in the initial stages of cancer initiation and progression.106 Elevated methylation levels are observed in leukemia cells. Following traditional chemotherapy, and after remission, the level of methylation was not decreased.107 In a systems biology study, it was recently shown that DNMT1 is allosterically activated at the sites of genes it hypermethylates for silencing in leukemia cells.108 The maintenance of hypermethylation even after remission is a potential cause of leukemia relapse. This could be due to the formation of new progenitor cells and maintenance of the drug-resistant cancer cells. Thus, it is reasonable to believe that the combination of epigenetic drugs with traditional therapies should reduce cancer relapse by sensitizing those regions methylated in the cancer cells and subsequently killing cancer progenitor cells and drug-resistant cells.61,101–104,106,109 A cohort study with lung cancer patients showed that pretreatment with epigenetic drugs reduced relapse.110 The reduction in relapse may possibly involve two events: cancer progenitor cell death and sensitization of drug-resistant cancer cells. Indeed, treatment with epigenetic drugs sensitized drug-resistant ovarian cancer cells to the platinum drugs.111
Though genetic alterations and epigenetic events appear to be two separate phenomena during carcinogenesis, it is possible that epigenetics may also regulate gene copy number variation. Recent studies have shown that ERBB2 (HER2) copy number increases (17q21.3) in many breast cancer patients, while breast cancer 1 (BRCA1) is reciprocally deleted from chromosome 17.112 Interestingly, RIPK2 and MYC on chromosome 8 show this same pattern of duplication and deletion. Chromosome 17 alterations are important for both basal type (triple-negative) breast cancers and ovarian cancers of epithelial origin. This upregulation in growth-promoting gene copy number and downregulation in cell cycle regulating gene copy numbers raises the possibility that these alterations contribute to carcinogenesis.112 This phenomenon could be achieved by several diverse mechanisms, the discussion of which is beyond the scope of this article. In short, it could be achieved both during meiosis and/or postfertilization. In this way, upregulation of some genes and downregulation of others could be inherited as well as generated in adult somatic cells. It has been observed that some cells in somatic tissues acquire this trait, whereas others are normal. This suggests that epigenetic modifications may also be involved in this process.
Usually during carcinogenesis, the methylation level around certain genes is increased, but overall methylation of the genome is low. Cells have a natural tendency to normalize methylation via an active demethylation mechanism that utilizes TET proteins to convert methylcytosines to hydroxymethylcytosines.113,114 This is followed by the deletion of the methylated region, which is accomplished by cytosine deaminases that act on both methylated cytosines and hydroxymethylcytosines to invoke the base excision repair system that creates a lesion in the DNA.115 Figure 4 shows a simplistic model of how this may result in the deletion of tumor suppressor genes. When 5hmC is acted upon by cytosine deaminases, it results in a base mismatch. DNA glycosylase enzymes will remove the former 5hmC, resulting in an apyrimidine (AP) site. The phosphate backbone around the AP site is excised by AP endonuclease and phosphodiesterase enzymes, resulting in a single-strand break. If this occurs simultaneously on opposite ends of the gene, it may result in a double-stranded break (dsB) at different locations. This break could possibly occur on both strands at the beginning and at the end of a gene. This break may result in DNA ligation that attaches the two ends of the tumor suppressor gene together, resulting in the loss of the gene (Fig. 4). In contrast, oncogenes such as HER2 show copy number increase in cancer cells as described above. This may be an extension of the same mechanism that results in gene loss through DNA repair mechanisms. The increase in copy number still requires excision of DNA as described in Figure 4, but the actual process of copy number increase may follow the same mechanism of antigen receptor or antibody class switch recombination (CSR), which is directed by the chromosomal loop domain mediated by CTCF and cohesion.115 This is a new concept and further studies are necessary to test this hypothesis.
Figure 4.
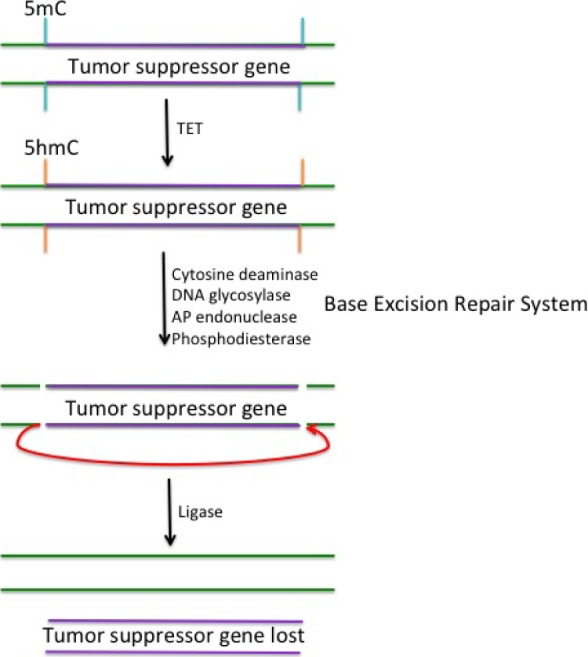
Gene deletion due to base excision of hydroxymethylcytosine. Demethylation of 5-methylctyosine (5mC) is catalyzed by the TET enzymes. TET converts 5-methylcytosine (indicated in blue) to 5-hydroxymethylcytosine (5hmC; indicated in orange). 5hmC may subsequently be acted upon by cytosine deaminase enzymes, resulting in a base mismatch that can be targeted by the base excision repair system to correct the mismatch. DNA glycosylase enzymes act to remove the base, creating an AP site. AP endonuclease and phosphodiesterase enzymes act on the AP site to excise the phosphate backbone where the 5mC used to be, creating a lesion in the DNA to be filled in by DNA polymerase. However, if both ends of the gene have bases excised at the same time and on both strands, it is possible for the DNA to religate, bringing the opposite ends of the gene together, and removing the gene from the genome. This model depicts a tumor suppressor gene (indicated in purple) being lost from the rest of the DNA (green).
Future Study
It is interesting to observe that though epigenetics has evolved as a method of adaptation for a broad spectrum of prokaryotes and eukaryotes, the mechanisms and requirements for epigenetics have remained relatively conserved in individual cell types. The evolution of epigenetics is characterized by the shift of epigenetic regulation of prokaryotes, to lower eukaryotes, to higher eukaryotes, to humans. For example, in prokaryotes, adenine is the most important methylation target that functions to protect DNA from restriction digestion. On the other hand, mammalian modifications are centered on cytosine methylation of CpG residues, which are mainly clustered in CpG islands. The purpose of this methylation is very different from that of prokaryotes. These modifications often correlate with histone modifications, thus regulating each other. Meanwhile, the epigenetic mechanisms of lower eukaryotes incorporate modification found in both prokaryotes and mammalian cells.
Research over the past few decades has elucidated the importance of epigenetic regulation for human embryogenesis and development. Any tissue-specific alteration in this regulation may cause a vast array of diseases. As these alterations are not permanent, do not change the DNA sequences, and are reversible in nature, there is tremendous opportunity to manipulate epigenetic alterations to manage and cure many disease types. As mammalian systems are more complex compared to lower eukaryotes and prokaryotes, the regulation of CpG DNA methylation by histone modifications, and reversely the regulation of histone modifications by gene products regulated by CpG DNA methylation, will uncover many mysteries not yet understood. For example, Figure 3 demonstrates how modified histones already present in egg cells may possibly influence the CpG DNA methylation of the progeny. It is already known that during imprinting that is regulated by lncRNA, even the production of lncRNA is regulated by CpG DNA methylation in many circumstances.116,117 Histone modification enzymes could be regulated similarly.
Another area that warrants further investigation is the regeneration process previously described. There is sufficient indication that the regeneration process is regulated by epigenetic mechanisms, but the exact mechanisms in different types of organisms have not been determined. The study of the simple model systems that were described in the regeneration section would have great benefit in understanding the balance between pluripotency of stem cells and differentiation.
Figure 4 illustrates a model of how cancer cells may dispose of tumor suppressor genes by hijacking existing DNA excision repair mechanisms that are essentially involved in active demethylation. We further describe the possible involvement of chromosomal loop domains in increasing the copy number of oncogenes in the cancer cell, which is possibly a modified mechanism of antigen receptor recombination. The loss of tumor suppressor genes and the gain of oncogene copy number is a known phenomenon. There is tremendous potential to determine how exactly epigenetic regulation is involved in mediating this process.
In short, the reversible nature of epigenetic alterations generates a tremendous potential that histone modifications regulate DNA methylation, and DNA methylation in turn regulates histone modifications, which constitute a tight cycle, like many of the well-defined cycles present in biochemical pathways. These cycles should be conserved during normal development constituting an epigenetic switch, which should be conserved for a particular tissue development or a particular function. When the switch is altered, the imbalance may cause situations that initiate disease or disorders.
Abbreviations
- aDMRs
associated differentially methylated regions
- aGEN
age-associated gene
- BRCA1
breast cancer 1
- CD8
cluster of differentiation 8
- CGI4906
kinase-binding protein 4906
- CpG
cytosine-phosphate-guanine dinucleotide
- CSR
class switch recombination
- CTCF
CCCTC binding factor
- Dam
DNA adenine methyltransferase
- DAMT-1
DNA N6-methyltransferase-1
- DMAD
dimethylaminomethylene diphosphonate
- DNMT1
DNA methyltransferase 1
- DNMT3a
DNA methyltransferase 3a
- DNMT3b
DNA methyltransferase 3b
- dsB
double-stranded break
- EBV
Epstein–Barr virus
- ERBB2
Erb-B2 receptor tyrosine kinase 2
- FFAR
free fatty acid receptor
- FTL
ferritin light chain
- GCN5
general control of amino acid synthesis protein 5
- GPx-1
glutathione peroxidase 1
- H1
histone 1
- H2A
histone 2A
- H2B
histone 2B
- H3
histone 3
- H4
histone 4
- H3K4me2
dimethylation of lysine 4 of histone 3
- H3K9
lysine 9 of histone 3
- H3K9me2
dimethylation of lysine 9 of histone 3
- H3K9me3
trimethylation of lysine 9 of histone 3
- H3K27
lysine 27 of histone 3
- H3K27me3
trimethylation of lysine 27 of histone 3
- H4K20
lysine 20 of histone 4
- HDAC
histone deacetylase
- HER2
Erb-b2 receptor tyrosine kinase 2
- HIV
human immunodeficiency virus
- HOX
homeobox
- HP1
heterochromatin protein 1
- JARID2
Jumonji AT rich interactive domain 2
- JmjC
Jumonji C
- JMJD6
Jumonji domain-containing protein 6
- KDM3
lysine demethylase 3
- KDM5
lysine demethylase 5
- KDM6
lysine demethylase 6
- KMT1
lysine methyltransferase 1
- KMT6
lysine methyltransferase 6
- LINE1
long interspersed nuclear element 1
- METTL4
methyltransferase-like 4
- miRNA
microRNA
- MS
multiple sclerosis
- MTase
methyltransferase
- Myc
V-Myc avian myelocytomatosis viral oncogene homolog
- NMAD-1
DNA N6-methyltransferase-1
- p15
cyclin-dependent kinase 4 inhibitor 2
- PKDM7
protein lysine demethylase 7
- PKDM8
protein lysine demethylase 8
- PKDM9
protein lysine demethylase 9
- PKDM11
protein lysine demethylase 11
- PKDM12
protein lysine demethylase 12
- PKDM13
protein lysine demethylase 13
- PRC2
polycomb repressive complex 2
- RIPK2
receptor-interacting serine–threonine kinase 2
- RM
restriction–methylation
- SAH
S-adenosylhomocysteine
- TAD
topologically associated domains
- TET
ten-eleven translocation
- UHRF1
ubiquitin-like with PHD and ring finger domains 1
- Wnt3
wingless-type MMTV integration site family 3
- 5mC
methylated cytosine
- 6mA
methylated adenine
Footnotes
ACADEMIC EDITOR: Christian Bronner, Editor in Chief
PEER REVIEW: Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1007 words, excluding any confidential comments to the academic editor.
FUNDING: AW, ML, SH, and KL were supported by UROP, and BU. ML was supported by MSSRP, BUSM. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert single-blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived the concept for this paper: SS. Developed the outline: SS and AW. All the authors (AW, ML, MG, CT, SH, KL, KH, and SS) contributed equally to the writing of this paper.
REFERENCES
- 1.Turner BM. Epigenetic responses to environmental change and their evolutionary implications. Philos Trans R Soc Lond B. 2009;364:1534. doi: 10.1098/rstb.2009.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eid A, Zawia N. Consequences of lead exposure, and its emerging role as an epigenetic modifier in the aging brain. Neurotoxicology. 2016 doi: 10.1016/j.neuro.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Casey SC, Vaccari M, Al-Mulla F, et al. The effect of environmental chemicals on the tumor microenvironment. Carcinogenesis. 2015;36:S160–S183. doi: 10.1093/carcin/bgv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chai H, Brown RE. Field effect in cancer: an update. Ann Clin Lab Sci. 2009;39(4):331–337. [PubMed] [Google Scholar]
- 5.Bollati V, Baccarelli A, Hou L, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67:876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- 6.Choi SW, Claycombe KJ, Martinez JA, Frisco S, Schalinske KL. Nutritional epigenomics: a portal to disease prevention. Adv Nutr. 2013;4:5. doi: 10.3945/an.113.004168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bogan JA, Helmstetter CE. DNA sequestration and transcription in the oriC region of Escherichia coli. Mol Microbiol. 1997;26:889–896. doi: 10.1046/j.1365-2958.1997.6221989.x. [DOI] [PubMed] [Google Scholar]
- 8.Campbell JL, Kleckner N. E. coli oriC and the dnaA gene promoter are sequestered from dam methyltransferase following the passage of the chromosomal replication fork. Cell. 1990;74:967–979. doi: 10.1016/0092-8674(90)90271-f. [DOI] [PubMed] [Google Scholar]
- 9.Han JS, Kang S, Lee H, Kim HK, Hwang DS. Sequential binding of SeqA to paired hemi-ethylated GATC sequences mediates formation of higher order complexes. J Biol Chem. 2003;278:34983–34989. doi: 10.1074/jbc.M304923200. [DOI] [PubMed] [Google Scholar]
- 10.Kang S, Lee H, Han JS, Hwang DS. Interaction of SeqA and Dam methylase on the hemimethylated origin of Escherichia coli chromosomal DNA replication. J Biol Chem. 1999;274:11463–11468. doi: 10.1074/jbc.274.17.11463. [DOI] [PubMed] [Google Scholar]
- 11.Klungsoyr HK, Skarstad K. Positive supercoiling is generated in the presence of Escherichia coli SeqA protein. Mol Microbiol. 2004;54:123–131. doi: 10.1111/j.1365-2958.2004.04239.x. [DOI] [PubMed] [Google Scholar]
- 12.Lu M, Campbell JL, Boye E, Kleckner N. SeqA: a negative modulator of replication initiation in E. coli. Cell. 1994;77:413–426. doi: 10.1016/0092-8674(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 13.Molina F, Skarstad K. Replication fork and SeqA focus distributions in Escherichia coli suggest a replication hyperstructure dependent on nucleotide metabolism. Mol Microbiol. 2004;52:1597–1612. doi: 10.1111/j.1365-2958.2004.04097.x. [DOI] [PubMed] [Google Scholar]
- 14.Taghbalout A, Landoulsi A, Kern R, et al. Competition between the replication initiator DnaA and the sequestration factor SeqA for binding to the hemimethylated chromosomal origin of E. coli in vitro. Genes Cells. 2000;5:873–884. doi: 10.1046/j.1365-2443.2000.00380.x. [DOI] [PubMed] [Google Scholar]
- 15.Au KG, Welsh K, Modrich P. Initiation of methyl-directed mismatch repair. J Biol Chem. 1992;267:12142–12148. [PubMed] [Google Scholar]
- 16.Bakker M, Smith DW. Methylation of GATC sites is required for precise timing between rounds of DNA replication in Escherichia coli. J Bacteriol. 1989;4(10):5738–5742. doi: 10.1128/jb.171.10.5738-5742.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhagwat AS, Lieb M. Cooperation and competition in mismatch repair: very short-patch repair and methyl-directed mismatch repair in Escherichia coli. Mol Microbiol. 2002;44:1421–1428. doi: 10.1046/j.1365-2958.2002.02989.x. [DOI] [PubMed] [Google Scholar]
- 18.Modrich P. Methyl-directed DNA mismatch correction. J Biol Chem. 1989;264:6597–6600. [PubMed] [Google Scholar]
- 19.Schlagman SL, Hattman S, Marinus MG. Direct role of the Escherichia coli Dam DNA methyltransferase in methylation-directed mismatch repair. J Bacteriol. 1986;165:896–900. doi: 10.1128/jb.165.3.896-900.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith DW, Garland AM, Herman G, Enns RE, Baker TA, Zyskind JW. Importance of state of methylation of oriC GATC sites in initiation of DNA replication in Escherichia coli. EMBO J. 1985;4:1319–1326. doi: 10.1002/j.1460-2075.1985.tb03779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arber W, Dussoix D. Host specificity of DNA produced by Escherichia coli. I. Host controlled modification of bacteriophage lambda. J Mol Biol. 1962;5:18–36. doi: 10.1016/s0022-2836(62)80058-8. [DOI] [PubMed] [Google Scholar]
- 22.Dussoix D, Arber W. Host specific of DNA produced by Escherichia coli. II. Control over acceptance of DNA from infecting phage lambda. J Mol Biol. 1962;5:37–49. doi: 10.1016/s0022-2836(62)80059-x. [DOI] [PubMed] [Google Scholar]
- 23.Yarmolinski MB, Sternberg N. Bacteriophage P1. Bacteriophages. 1988;1:291–292. Chapter 9. [Google Scholar]
- 24.Sternberg N, Coulby J. Cleavage of the bacteriophage P1 packaging site (pac) is regulated by adenine methylation. Proc Natl Acad Sci U S A. 1990;87(20):8070–8074. doi: 10.1073/pnas.87.20.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casadesus J, Low D. Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev. 2006;70(3):830–856. doi: 10.1128/MMBR.00016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliverio AM, Katz LA. The dynamic nature of genomes across the tree of life. Genome Biol Evol. 2014;6(3):482–488. doi: 10.1093/gbe/evu024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wardleworth BN, Russell RJM, Bell SD, Taylor GL, White MF. Structure of Alba: an archaeal chromatin protein modulated by acetylation. EMBO J. 2002;21(17):4654–4662. doi: 10.1093/emboj/cdf465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sandman K, Reeve JN. Structure and functional relationships of archaeal and eukaryal histones and nucleosomes. Arch Microbiol. 2000;173(3):165–169. doi: 10.1007/s002039900122. [DOI] [PubMed] [Google Scholar]
- 29.White MF, Bell SD. Holding it together: chromatin in the Archaea. Trends Gen. 2002;18(12):621–626. doi: 10.1016/s0168-9525(02)02808-1. [DOI] [PubMed] [Google Scholar]
- 30.Lurz R, Grote M, Dijk J, Reinhardt R, Dobrinski B. Electron microscopic study of DNA complexes with proteins from the Archaebacterium Sulfolobus acidocaldarius. EMBO J. 1986;5(13):3715–3721. doi: 10.1002/j.1460-2075.1986.tb04705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bell SD, Botting CH, Wardleworth BN, Jackson SP, White MF. The interaction of Alba, a conserved Archaeal chromatin protein, with Sir2 and its regulation by acetylation. Science. 2002;296(5565):148–151. doi: 10.1126/science.1070506. [DOI] [PubMed] [Google Scholar]
- 32.Terns MP, Terns RM. CRISPR-based adaptive immune systems. Curr Opin Microbiol. 2011;14(3):321–327. doi: 10.1016/j.mib.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marino-Ramirez L, Jordan IK, Landsman D. Multiple independent evolutionary solutions to core histone gene regulation. Genome Biol. 2006;7(12):R122. doi: 10.1186/gb-2006-7-12-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reeck GR, Swanson E, Teller DC. The evolution of histones. J Mol Evol. 1978;10:4. doi: 10.1007/BF01734220. [DOI] [PubMed] [Google Scholar]
- 35.Trojer P, Reinberg D. Histone lysine demethylases and their impact on epigenetics. Cell. 2006;125:2. doi: 10.1016/j.cell.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 36.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism, and beyond. Nat Rev Mol Biol. 2015;16(4):258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- 37.Soyer JL, Rouxel T, Fudal I. Chromatin-based control of effector gene expression in plant-associated fungi. Curr Opin Plant Biol. 2015;26:51–56. doi: 10.1016/j.pbi.2015.05.025. [DOI] [PubMed] [Google Scholar]
- 38.Qian S, Wang Y, Ma H, Zhang L. Expansion and functional divergence of Jumonji c-containing histone demethylases: significance of duplications in ancestral angiosperms and vertebrates. Plant Physiol. 2015;168(4):1321–1337. doi: 10.1104/pp.15.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo G, Blanco MA, Greer EL, He C, Shi Y. DNA N-methyladenine: a new epigenetic mark in eukaryotes? Mol Cell Biol. 2015;16(12):705–710. doi: 10.1038/nrm4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hattman S, Kenny C, Berger L, Pratt K. Comparative study of DNA methylation in three unicellular eucaryotes. J Bacteriol. 1978;135(3):1156–1157. doi: 10.1128/jb.135.3.1156-1157.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greer EL, Blanco MA, Gu L, et al. DNA methylation on N6-adenine in C. elegans. Cell. 2015;161(4):868–878. doi: 10.1016/j.cell.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu Y, Luo GZ, Chen K, et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell. 2015;161(4):879–892. doi: 10.1016/j.cell.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang G, Huang H, Liu D, et al. N6-methyladenine DNA modification in drosophila. Cell. 2015;161:4. doi: 10.1016/j.cell.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 44.Geyer KK, Chalmers IW, Mackintosh N, et al. Cytosine methylation is a conserved epigenetic feature found throughout the phylum Platyhelminthes. BMC Genomics. 2013;14:462. doi: 10.1186/1471-2164-14-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geyer KK, Rodríguez López CM, Chalmers IW, et al. Cytosine methylation regulates oviposition in the pathogenic blood fluke Schistosoma mansoni. Nat Commun. 2011;2:424. doi: 10.1038/ncomms1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heyn H, Esteller M. An adenine code for DNA: a second life for N6- methyladenine. Cell. 2015;161(4):710–713. doi: 10.1016/j.cell.2015.04.021. [DOI] [PubMed] [Google Scholar]
- 47.Garza-Garcia AA, Driscoll PC, Brockes JP. Evidence for the local evolution of mechanisms underlying limb regeneration in salamanders. Integr Comp Biol. 2010;50(4):528–535. doi: 10.1093/icb/icq022. [DOI] [PubMed] [Google Scholar]
- 48.Brockes JP, Kumar A, Velloso CP. Regeneration as an evolutionary variable. J Anat. 2001;199:3–11. doi: 10.1046/j.1469-7580.2001.19910003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saxena S, Purushothaman S, Meghah V, et al. Role of annexin gene and its regulation during zebrafish caudal fin regeneration. Wound Rep Reg. 2016;24(3):551–559. doi: 10.1111/wrr.12429. [DOI] [PubMed] [Google Scholar]
- 50.Bosch TCG. Why polyps regenerate and we don’t: towards a cellular and molecular framework for Hydra regeneration. Dev Biol. 2007;303(2):421–433. doi: 10.1016/j.ydbio.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 51.Fujisawa T. Hydra regeneration and epitheliopeptides. Dev Dynamics. 2003;226(2):182–189. doi: 10.1002/dvdy.10221. [DOI] [PubMed] [Google Scholar]
- 52.Chera S, Ghila L, Dobretz K, et al. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell. 2009;17(2):279–289. doi: 10.1016/j.devcel.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 53.Hobmayer B, Rentzsch F, Kuhn K, et al. WNT signalling molecules act in axis formation in the diploblastic metazoan Hydra. Nature. 2000;407(6801):186–189. doi: 10.1038/35025063. [DOI] [PubMed] [Google Scholar]
- 54.Matsuzaki H, Okamura E, Takahashi T, et al. De novo DNA methylation through the 5′-segment of the H19 ICR maintains its imprint during early embryogenesis. Development. 2015;142(22):3833–3844. doi: 10.1242/dev.126003. [DOI] [PubMed] [Google Scholar]
- 55.Messerschmidt DM. A twist in zygotic reprogramming. Nat Cell Biol. 2016;18(2):139–140. doi: 10.1038/ncb3304. [DOI] [PubMed] [Google Scholar]
- 56.Gunesdogan U, Jackle H, Herzig A. A genetic system to assess in vivo the functions of histones and histone modifications in higher eukaryotes. EMBO Rep. 2010;11(10):772–776. doi: 10.1038/embor.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szyf M. Nongenetic inheritance and transgenerational epigenetics. Trends Mol Med. 2014;21(2):134–144. doi: 10.1016/j.molmed.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 58.Sanz LA, Kota SK, Feil R. Genome-wide DNA demethylation in mammals. Genome Biol. 2010;11(3):110. doi: 10.1186/gb-2010-11-3-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chi P, Allis CD, Wang GG. The histone code hypothesis. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams K, Christensen J, Helin K. DNA methylation: TET proteins—guardians of CpG islands? EMBO Rep. 2012;13(1):28–35. doi: 10.1038/embor.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sarkar S, Horn G, Moulton K, et al. Cancer development, progression, and therapy: an epigenetic overview. Int J Mol Sci. 2013;14:10. doi: 10.3390/ijms141021087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao Q, Rank G, Tan YT, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3a, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol. 2009;16(3):304–311. doi: 10.1038/nsmb.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dowen JM, Fan ZP, Hnisz D, et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159(2):374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang H, Maurano MT, Qu H, et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012;22(9):1680–1688. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Issa J. Aging and epigenetic drift: a vicious cycle. J Clin Invest. 2014;124(1):24–29. doi: 10.1172/JCI69735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCauley BS, Dang W. Histone methylation and aging: lessons learned from model systems. Biochim Biophys Acta. 2014;1839(12):1454–1462. doi: 10.1016/j.bbagrm.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vera E, Canela A, Fraga MF, Estellar M, Blasco MA. Epigenetic regulation of telomeres in human cancer. Oncogene. 2008;27(54):6817–6833. doi: 10.1038/onc.2008.289. [DOI] [PubMed] [Google Scholar]
- 69.Jung M, Jin S, Zhang X, et al. Longitudinal epigenetic and gene expression profiles analyzed by three-component analysis reveal down-regulation of genes involved in protein translation in human aging. Nucl Acids Res. 2015;43(15):e100. doi: 10.1093/nar/gkv473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hofmann JW, Zhao X, De Cecco M, et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell. 2015;160(3):477–488. doi: 10.1016/j.cell.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dozmorov MG. Polycomb repressive complex 2 epigenomic signature defines age-associated hypermethylation and gene expression changes. Epigenetics. 2015;10(6):484–495. doi: 10.1080/15592294.2015.1040619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ong T, Ozanne SE. Developmental programming of type 2 diabetes: early nutrition and epigenetic mechanisms. Curr Opin Clin Nutr Metabol Care. 2015;18(4):354–360. doi: 10.1097/MCO.0000000000000177. [DOI] [PubMed] [Google Scholar]
- 73.Alfredo Martinez J, Milagro FI, Claycombe KJ, Schalinske KL. Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv Nutr. 2014;5(1):71–81. doi: 10.3945/an.113.004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prescott SL, Allen K, Armstrong K, et al. The establishment of DOHaD working groups in Australia and New Zealand. J Dev Orig Health Dis. 2016:1–7. doi: 10.1017/S2040174416000167. [DOI] [PubMed] [Google Scholar]
- 75.Niculescu MD, Zeisel SH. Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine, and choline. J Nutr. 2002;132(8):2333S–2335S. doi: 10.1093/jn/132.8.2333S. [DOI] [PubMed] [Google Scholar]
- 76.Remely M, Aumueller E, Merold C, et al. Effects of short chain fatty acid producing bacteria on epigenetic regulation of FFAR3 in type 2 diabetes and obesity. Gene. 2014;537(1):85–92. doi: 10.1016/j.gene.2013.11.081. [DOI] [PubMed] [Google Scholar]
- 77.Xiao Y, Su X, Huang W, et al. Role of S-adenosylhomocysteine in cardiovascular disease and its potential epigenetic mechanism. Int J Biochem Cell Biol. 2015;67:158–166. doi: 10.1016/j.biocel.2015.06.015. [DOI] [PubMed] [Google Scholar]
- 78.Metes-Kosik N, Luptak I, DiBello PM, et al. Both selenium deficiency and modest selenium supplementation lead to myocardial fibrosis in mice via effects on redox-methylation balance. Mol Nutr Food Res. 2012;56(12):1812–1824. doi: 10.1002/mnfr.201200386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Qureshi IA, Mehler MF. Understanding neurological disease mechanisms in the era of epigenetics. JAMA Neurol. 2013;70(6):703–710. doi: 10.1001/jamaneurol.2013.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huynh JL, Casaccia P. Epigenetic mechanisms in multiple sclerosis: implications for pathogenesis and treatment. Lancet Neurol. 2013;12(2):195–206. doi: 10.1016/S1474-4422(12)70309-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Masliah E, Dumaop W, Galasko D, Desplats P. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics. 2013;8(10):1030–1038. doi: 10.4161/epi.25865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shahbazian MD, Zoghbi HY. Rett Syndrome and MeCP2: linking epigenetics and neuronal function. Am J Hum Genet. 2002;71(6):1259–1272. doi: 10.1086/345360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 84.Jones PL, Veenstra GJ, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19(2):187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 85.Steffan JS, Kazantsev A, Spasic-Boskovic O, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97(12):6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim HS, Kim EM, Kim NJ, et al. Inhibition of histone deacetylation enhances the neurotoxicity induced by the C-terminal fragments of amyloid precursor protein. J Neurosci Res. 2004;75(1):117–124. doi: 10.1002/jnr.10845. [DOI] [PubMed] [Google Scholar]
- 87.Guan JS, Haggarty SJ, Giacometti E, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459(7243):55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kilgore M, Miller CA, Fass DM, et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s Disease. Neuropsychopharmacology. 2010;35(4):870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aslani S, Mahmoudi M, Karami J, et al. Epigenetic alterations underlying autoimmune diseases. J Autoimmun. 2016;49(2):69–83. doi: 10.3109/08916934.2015.1134511. [DOI] [PubMed] [Google Scholar]
- 90.Hewagama A, Richardson B. The genetics and epigenetics of autoimmune disease. J Autoimmun. 2009;33(1):3–11. doi: 10.1016/j.jaut.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu H, Zhao M, Yoshimura A, Chang C, Lu Q. Critical link between epigenetics and transcription factors in the induction of autoimmunity: a comprehensive review. Clin Rev Allergy Immunol. 2016;50(3):333–344. doi: 10.1007/s12016-016-8534-y. [DOI] [PubMed] [Google Scholar]
- 92.Maltby VE, Graves MC, Lea RA, et al. Genome-wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clin Epigenetics. 2015;7:118. doi: 10.1186/s13148-015-0152-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cohen JI. Optimal treatment for chronic active Epstein-Barr virus disease. Pediatr Transplant. 2009;13(4):393–396. doi: 10.1111/j.1399-3046.2008.01095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Birdwell CE, Queen KJ, Kilgore PC, et al. Genome-wide DNA methylation as an epigenetic consequence of Epistein-Barr virus infection of immortalized keratinocytes. J Virol. 2014;88(19):11442–11458. doi: 10.1128/JVI.00972-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shirakawa K, Chavez L, Hakre S, Calvanese V, Verdin E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013;21(6):277–285. doi: 10.1016/j.tim.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Van Epps HL. Peyton Rous: father of the tumor virus. J Exp Med. 2005;201(3):320. doi: 10.1084/jem.2013fta. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martin GS. The road the Src. Oncogene. 2004;23(48):7910–7917. doi: 10.1038/sj.onc.1208077. [DOI] [PubMed] [Google Scholar]
- 98.Weitzel JN, Blazer KR, MacDonald DJ, Culver JO, Offit K. Genetics, genomics, and cancer risk assessment: state of the art and future directions in the era of personalized medicine. Cancer J Clin. 2011;61(5):327–359. doi: 10.3322/caac.20128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 100.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 101.Byler S, Sarkar S. Do epigenetic drug treatments hold the key to killing cancer progenitor cells? Epigenomics. 2014;6(2):161–165. doi: 10.2217/epi.14.4. [DOI] [PubMed] [Google Scholar]
- 102.Byler S, Goldgar S, Heerboth S, et al. Genetic and epigenetic aspects of breast cancer progression and therapy. Anticancer Res. 2014;34(3):1071–1077. [PubMed] [Google Scholar]
- 103.Sarkar S, Goldgar S, Byler S, Rosenthal S, Heerboth S. Demethylation and re-expression of epigenetically silenced tumor suppressor genes: Sensitization of cancer cells by combination therapy. Epigenomics. 2013;5(1):87–94. doi: 10.2217/epi.12.68. [DOI] [PubMed] [Google Scholar]
- 104.Heerboth S, Housman G, Leary M, et al. EMT and tumor metastasis. Clin Transl Med. 2015;4:6. doi: 10.1186/s40169-015-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Heerboth S, Lapinska K, Snyder N, Leary M, Rollinson S, Sarkar S. The use of epigenetics drugs in diseases: an overview. Gen Epigenetics. 2014;6:9–19. doi: 10.4137/GEG.S12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Longacre M, Snyder NA, Housman G, et al. A comparative analysis of genetic and epigenetic events of breast and ovarian cancer related to tumorigenesis. Int J Mol Sci. 2016;17(5):E759. doi: 10.3390/ijms17050759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Agrawal S, Unterberg M, Koschmieder S, et al. DNA methylation of tumor suppressor genes in clinical remission predicts the relapse risk in acute myeloid leukemia. Cancer Res. 2007;67(3):1370–1377. doi: 10.1158/0008-5472.CAN-06-1681. [DOI] [PubMed] [Google Scholar]
- 108.Samorodnitsky E, Ghosh E, Mazumder S, Sarkar S. Methylation by DNMT1 is more efficient in chronic lymphocytic leukemia cells than in normal cells. J Proteomics Bioinform. 2014 doi: 10.4172/jpb.S10-004. [DOI] [Google Scholar]
- 109.Housman G, Byler S, Heerboth S, et al. Drug resistance in cancer: an overview. Cancers (Basel) 2014;6(3):1769–1792. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Juergens RA, Wrangle J, Vendetti FP, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cacan E, Ali MW, Boyd NH, Hooks SB, Greer SF. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS One. 2014;9(1):e87455. doi: 10.1371/journal.pone.0087455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Inaki K, Menghi F, Woo XY, et al. Systems consequences of amplicon formation in human breast cancer. Genome Res. 2014;24(10):1559–1571. doi: 10.1101/gr.164871.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ko M, An J, Pastor WA, Koralov SB, Rajewsky K, Rao A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol Rev. 2015;263(1):6–21. doi: 10.1111/imr.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nabel CS, Jia H, Ye Y, et al. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol. 2012;8(9):751–758. doi: 10.1038/nchembio.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hu J, Zhang Y, Zhao L, et al. Chromosomal loop domains direct the recombination of antigen receptor genes. Cell. 2015;163(4):947–959. doi: 10.1016/j.cell.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.He Y, Meng XM, Huang C, et al. Long noncoding RNAs: novel insights into hepatocelluar carcinoma. Cancer Lett. 2014;344(1):20–27. doi: 10.1016/j.canlet.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 117.Song MA, Tiirikainen M, Kwee S, Okimoto G, Yu H, Wong LL. Elucidating the landscape of aberrant DNA methylation in hepatocellular carcinoma. PLoS One. 2013;8(2):e55761. doi: 10.1371/journal.pone.0055761. [DOI] [PMC free article] [PubMed] [Google Scholar]