

Abstract
Glycosaminoglycans (GAGs) bind all known amyloid plaques and help store protein hormones in (acidic) granular vesicles, but the molecular mechanisms underlying these important effects are unclear. Here we investigate GAG interactions with the peptide hormone salmon calcitonin (sCT). GAGs induce fast sCT fibrillation at acidic pH and only bind monomeric sCT at acidic pH, inducing sCT helicity. Increasing GAG sulfation expands the pH range for binding. Heparin, the most highly sulfated GAG, binds sCT in the pH interval 3–7. Small angle x-ray scattering indicates that sCT monomers densely decorate and pack single heparin chains, possibly via hydrophobic patches on helical sCT. sCT fibrillates without GAGs, but heparin binding accelerates the process by decreasing the otherwise long fibrillation lag times at low pH and accelerates fibril growth rates at neutral pH. sCT·heparin complexes form β-sheet-rich heparin-covered fibrils. Solid-state NMR reveals that heparin does not alter the sCT fibrillary core around Lys11 but makes changes to Val8 on the exterior side of the β-strand, possibly through contacts to Lys18. Thus GAGs significantly modulate sCT fibrillation in a pH-dependent manner by interacting with both monomeric and aggregated sCT.
Keywords: amyloid, fibril, glycosaminoglycan, heparin, hormone, salmon calcitonin
Introduction
Since their first description in 1855 (1), amyloid deposits have been extensively studied both in vivo and in vitro. Besides the major amyloidogenic protein component (e.g. amyloid-β, α-synuclein, or tau), amyloid deposits contain components like inflammatory molecules, metal ions, amyloid P, proteoglycans (PGs),2 and other proteins (2). PGs are present in both systemic (3), prion (4), and neuritic (5) amyloid plaques and appear directly involved in amyloid deposition (6). The precise role of PGs in amyloid deposition remains to be clarified. The core PG protein is mainly involved in protein and membrane binding (7). In contrast, PG activity in amyloid formation is associated with the anionic polysaccharide chains or glycosaminoglycans (GAGs) decorating the protein core (3, 5). GAGs are structurally diverse polysaccharides consisting of repeating disaccharide units with an amine, a carboxylic group, and widely varying sulfation levels. The GAG heparin has three sulfates in every disaccharide group, whereas chondroitin sulfate A (ChA), dermatan sulfate (DS), and heparan sulfate have lower overall degrees of sulfation (Fig. 1). Heparin is the GAG with the most consistent repetitive disaccharide composition, whereas other GAGs follow a less strictly repetitive sulfation pattern (8).
FIGURE 1.
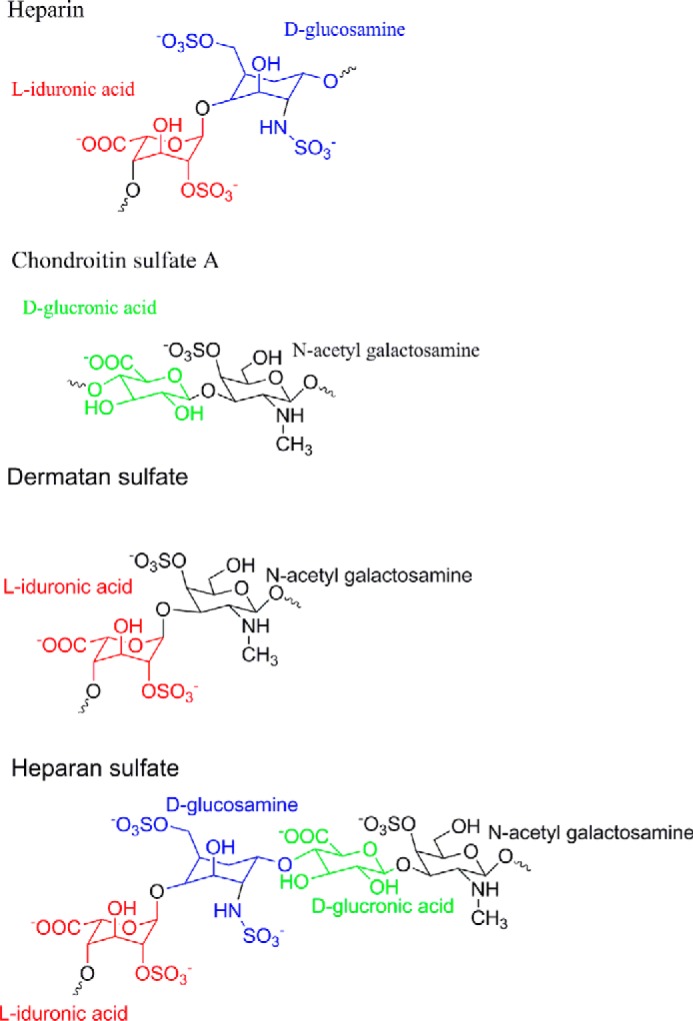
Schematic representation of typical disaccharide repeating units for heparin, ChA, DS, and heparan sulfate. Color coding represents different sugar types, highlighting differences in sulfation (8, 51). The triple sulfated disaccharide unit of heparin (top) makes up 80–90% of the polymer. Heparan sulfate is shown with three saccharides to indicate that the glyco acid is not solely iduronic acid. The glucuronic acid shown here has no sulfate. For the other GAGs, sulfation levels vary between different sources, and the model should be considered an average disaccharide unit.
PG functions range from cell signaling over proliferation (9) to structural roles. Aggrecan is the major constituent of connective tissue (9), and perlecan is a heparan sulfate PG that adds charges to the basement membrane in an asymmetric fashion (7). Heparin has been used as a model GAG in the study of the fibrillation of major disease-related amyloidogenic proteins such as amyloid-β (5, 10), α-synuclein (11), tau (12), human muscle acylphosphatase (13), and β2-microglobulin (14). Heparin commonly promotes aggregation by decreasing fibrillation times and affects fibril morphology. The mechanism for fibril enhancement and the resulting fibril morphology seems to be protein-specific (6). Besides promoting fibrillation, GAGs hinder cytotoxicity induced by prefibrillar oligomers of other proteins such as islet amyloid polypeptide (15) and amyloid-β (16). Furthermore, non-fibril-inducing GAG mimetics are a growing field of interest for therapeutic applications (17). Thus it remains unresolved whether GAGs primarily hinder or promote unwanted aggregation in vivo (17–19). Recently GAGs have been suggested to be involved in the storage of protein hormones in granular vesicles by promoting aggregation of protein hormones (20, 21). During granular vesicle formation, the pH decreases from 7.4 in the endoplasmic reticulum to 5.5 in mature granular vesicles (22), making it important to understand how pH affects protein-GAG interactions. Considering the central role played by GAGs in fibrillation, there is remarkably little information available on the molecular basis for these effects and whether and how GAGs incorporate into the highly repetitive cross-β amyloid structure. One of the few reports to address this is a study by Middleton and co-workers (23, 24) on heparin-amyloid β interactions where heparin binds on the outer surface of amyloid-β fibrils, interacting closely with the loop region and the flexible N terminus.
To address the role of heparin and other GAGs in the molecular steps leading to protein fibrillation, here we present a study of how GAGs affect fibrillation of the 32-residue peptide hormone salmon calcitonin (sCT) used to treat osteoporosis in postmenopausal women (25). sCT is more potent in this regard than human calcitonin (hCT) (26), partly attributed to its reduced fibrillation propensity (27). Nevertheless, spherical prefibrillar oligomers formed by sCT at pH 7.4 are neurotoxic (28) like other spherical prefibrillar oligomer-forming proteins (29). sCT spherical prefibrillar oligomers interact with the cell membrane in “raftlike” domains rich in ganglioside GM1 and cholesterol, leading to increased Ca2+ influx and cell death (30). Early fibrillation events of sCT (31) and hCT (32) both involve formation of a helical oligomeric complex. At neutral pH, hCT fibrillation is in part driven by hydrophobic interactions among Tyr12, Phe16, and Phe19 and a salt bridge between Asp15 and Lys18 (32). At low pH, hCT fibrillation is slowed by Asp15 protonation (32). The decreased fibrillation propensity of sCT is attributed to the increased number of polar amino acids compared with hCT (33). Our results show that this barrier to fibrillation can be overcome by introducing GAG as an electrostatic binding partner for excess positive charges. CD and small angle x-ray scattering (SAXS) were used to investigate the structural changes going from monomeric sCT to sCT·GAG binding complex and we used solid-state NMR spectroscopy to analyze structural details in the sCT fibrils with or without heparin.
Results
sCT and Heparin Form a Binding Complex with α-Helical Structure at Low pH
The basis for our studies is the fact that the interactions of calcitonin with GAGs in vivo take place over a pH range of several units (∼7.4–5.5). Over this pH interval, the charge of sCT will change by up to 1.3 units (see below), and this will affect interactions with the highly anionic GAG molecules. To investigate the role of charge in these interactions, we used the following three variants of sCT: 1) the natural form, which is C-terminally amidated (sCT-NH2) and goes from a net charge of approximately +3.9 at pH 5.5 to approximately +2.6 at pH 7.4 based on conventional pKa value calculations; 2) a form with a free C-terminal carboxylate group (sCT-OH), which will be anionic above approximately pH 4, going from a net charge of +2.9 at pH 5.5 to +1.6 at pH 7.4; and 3) the mutant His17 → Ala (sCTH17A-OH; with a free C-terminal carboxylate group), which removes the only side chain that titrates over the pH interval 7.4–5.5 so that the net charge of this mutant only decreases by 0.6 unit from +2.1 at pH 5.5 to +1.5 at pH 7.4.
Initially we investigated how heparin affects the structure of monomeric sCT-OH at pH 5.5 and 7.4. Direct evidence for formation of a complex between heparin and sCT-OH at pH 5.5, but not pH 7.4, was obtained by asymmetric flow field flow fractionation (A4F) where large species elute after small species. We used fluorescein-labeled heparin (fl-hep) to detect the heparin component (fluorescence emission at 516 nm) and absorbance at 214 to monitor sCT-OH. At pH 5.5, a complex containing protein and fl-heparin elutes at 20 min, whereas sCT-OH without fl-heparin elutes around 8 min (Fig. 2A). At pH 7.4, there is no 20-min peak, and sCT-OH and heparin elute as separate peaks at 8 and 10 min, respectively (Fig. 2A).
FIGURE 2.

Heparin binds sCT only at low pH. A, A4F analysis of sCT-OH·heparin complex at pH 5.5 (red) and pH 7.4 (blue). sCT was detected using absorption at 214 nm for the sCT monomer (black) and sCT-OH·heparin (filled symbols). Heparin was detected using fluorescence (excitation at 485 nm and emission at 516 nm) (open symbols). B, CD spectra of 150 μg/ml heparin with 0.5 mg/ml sCT-NH2, sCT-OH, and sCTH17A-OH at pH 5.5 (filled symbols) or pH 7.4 (empty symbols). Neither heparin nor other GAGs investigated gave any significant signal at the relevant concentrations and buffer conditions used. Inset, CD spectra of sCT-NH2, sCT-OH, and sCTH17A-OH at pH 5.5 all show random coil structures. Same axis units as main graph. a.u., arbitrary units; deg, degrees; abs, absorbance.
The appearance of the binding complex at pH 5.5 was accompanied by a significant structural change according to far-UV CD. All three sCT peptides by themselves assume a random coil conformation with a far-UV CD minimum around 200 nm at both pH values (shown for pH 5.5 in Fig. 2B). This changes to two minima at 208 and 220 nm, implying a more α-helical structure, when heparin is added at pH 5.5 but not at pH 7.4. The helical structure was confirmed by the BeStSel deconvolution program (Table 1) (34). Because initially no pH difference was observed among the three sCT versions, we next carried out a more extensive pH titration of the interactions with heparin. Binding (measured in terms of the change in CD signal at a 200:220 nm signal ratio) was determined over the pH range 8–3 (Fig. 3). Binding showed a sigmoidal curve where the visually estimated midpoint of titration changed from ∼6.9 (sCT-NH2) to ∼6.5 (sCT-OH) and ∼5.9 (sCTH17A-OH). Thus, progressive addition of negative charge (from sCT-NH2 to sCT-OH) and removal of positive charge (from sCT-OH to sCTH17A-OH) shifted the midpoint down. This indicates that to bind heparin a higher level of protonation had to compensate for these electrostatic changes.
TABLE 1.
Structural content as percentage for different sCT variants calculated with BeStSel (34) from CD spectra of sCT versions with or without heparin at pH 5.5
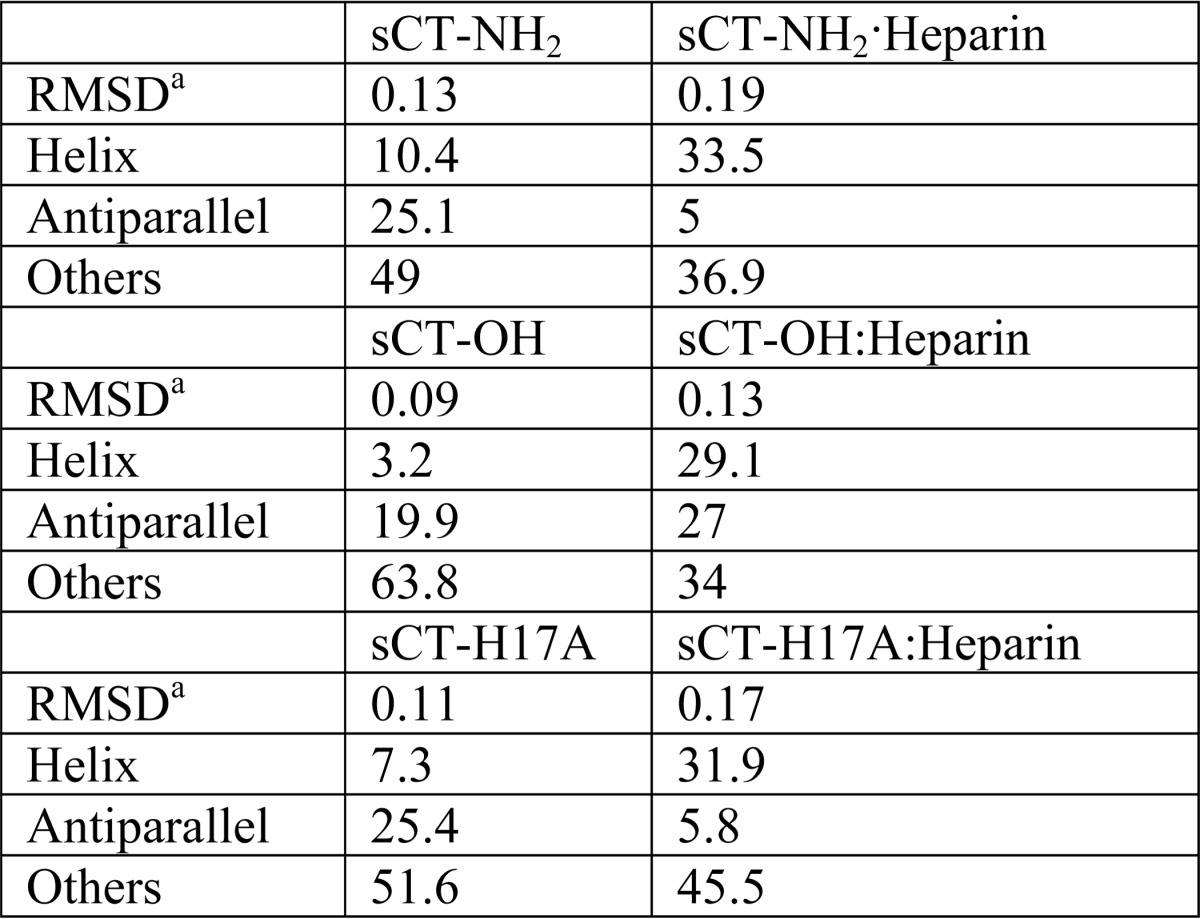
a Reported root mean square deviation (RMSD) for the fit of the entire model.
FIGURE 3.
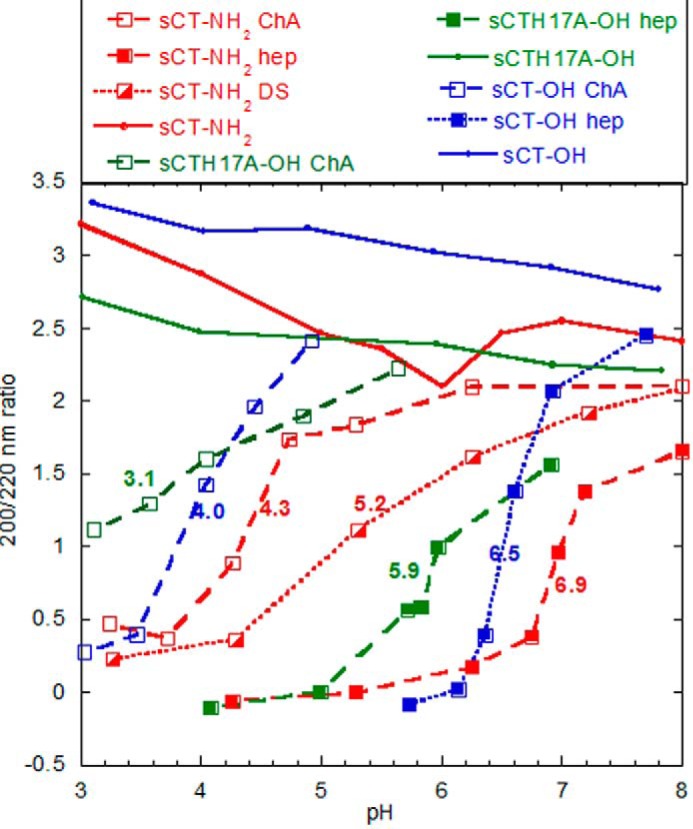
Charge and pH affect binding of GAGs to sCT. sCT binding to GAGs is shown as a function of pH monitored by CD. sCT alone had 200:220 ratios between 2.2 and 3.2 in the entire pH range (solid lines). Shown are sCT-NH2 in red, sCT-OH in blue, and sCTH17A-OH in green with heparin (filled symbols), DS (half-filled symbols), and ChA (open symbols). The samples contained 0.2 mg/ml sCT, 0.2 mg/ml GAG, and 20 mm citrate-phosphate buffer at the indicated pH values. The colored numbers in the graph are visual estimates of the pH midpoint of titration for the seven individual titration curves, which transition from random coil (high pH) to highly helical (low pH). The baseline values for the random coil are estimated from the GAG-free sCT variants at high pH, whereas the helix value is estimated to be around −0.2 based on the convergence of CD values at low pH. These values are merely used for comparative purposes (see main text).
GAG Sulfation Expands the pH Range of sCT Binding
Although the two titratable groups (C terminus and His17) have some impact on heparin binding as demonstrated above, all three sCT versions bound heparin at pH 5.5 but not pH 7.4. Therefore titration of His17 and the C terminus on their own could not explain why heparin binds sCT at pH 5.5 but not pH >7. To investigate whether other GAGs showed similar pH dependence, we compared sCT-NH2 binding with heparin (−4), DS (−3), and ChA (−2) (the average negative charge for a disaccharide unit is given in parentheses) using CD in the pH range 3–8 (Fig. 3). Decreasing the negative charge of the GAGs shifted the pH midpoint of titration downward from ∼6.9 (heparin) to ∼5.2 (DS) and ∼4.3 (ChA). Likewise, for sCT-OH and sCTH17A-OH, the ChA pH titration midpoints (4.0 and 3.1, respectively) were shifted down by approximately 2.5 pH units compared with heparin binding; sCT-OH had a midpoint at 6.5, and sCTH17A-OH had a midpoint at 5.9. The two phenomena appear to be additive in the sense that the pH dependence of binding of the three sCT peptides to ChA is similar to that of heparin, only shifted ∼2.5 units further down. Thus increased sulfation increases the pH range for GAG-sCT binding.
SAXS Reveals Heparin to Adopt a Less Extended Conformation When in Complex with sCT
Having established that heparin and sCT form a defined binding complex at pH 5.5, we used SAXS to obtain information about the shape of the complex. SAXS data for the complex (Fig. 4A) and free heparin (Fig. 4B) were transformed to pair distance functions (Fig. 4C), which revealed the complex to be more compact than heparin alone. We could model the sCT-OH·heparin complex using an ensemble-based method as a saccharide chain decorated with sCT-OH monomers in a coiled up structure with a radius of gyration (Rg) around 35 Å and a maximum diameter (Dmax) of 119 Å (Fig. 4A). Free heparin was modeled as a more flexible chain with Rg of 53Å and Dmax of 163Å (Fig. 4B).Thus CD, A4F, and SAXS all confirm formation of a relatively stable complex between sCT and heparin at pH 5.5 but not at pH 7.4.
FIGURE 4.

SAXS analysis of sCT·heparin complex and free heparin. A, SAXS spectra of purified sCT-OH·heparin complex in dots and model with Rg of 35 Å and Dmax of 119 Å in line. Inset, stick representation of modeled sCT·heparin complex. B, SAXS data of heparin in dots and model with radius of gyration of 53 Å and Dmax of 163 Å in line. Inset, stick representation of heparin generated from the SAXS data. C, comparison of p(r) functions of heparin alone (broken line) and sCT·heparin complex (full line). Error bars represent S.E.
Heparin Strongly Accelerates sCT Fibrillation Kinetics at Low pH and Increases the Elongation Rate at Neutral pH
To investigate how the formation of an sCT·hep binding complex affects sCT fibrillation, sCT and heparin were incubated with agitation at 37 °C. We observed that the binding complex eluting at 20 min in A4F disappears within around 45 min but is not replaced by free fl-heparin (Fig. 5B), indicating that both sCT and heparin are directly incorporated into larger species.
FIGURE 5.
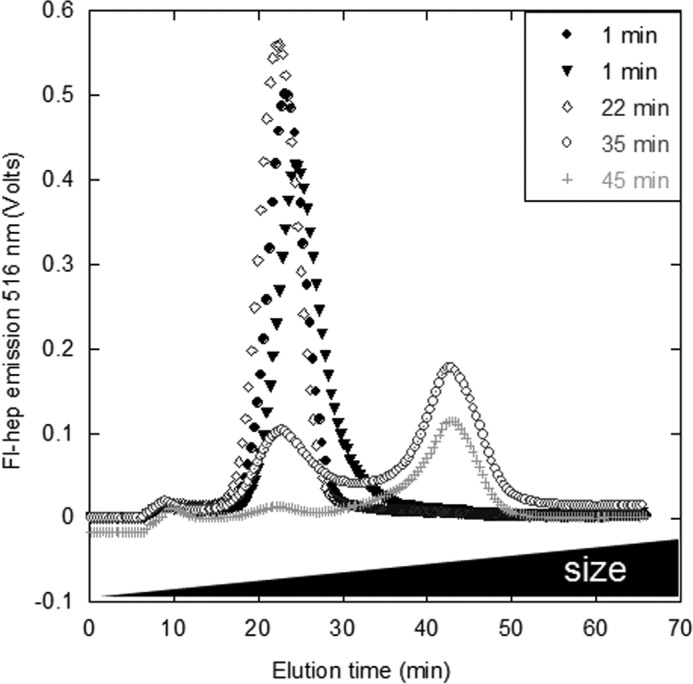
Heparin is not released from sCT-OH upon incubation. sCT-OH was incubated with fl-hep at pH 5.5 for 22–45 min (dark gray to light) or mixed immediately before injection (black). An sCT·hep binding complex (peak at 20 min) forms immediately and disappears as fibrillation proceeds. The larger complex at 40 min was not consistently detected.
sCT fibrillation kinetics at low pH in the presence of heparin could be followed using the amyloid-specific dye thioflavin T (ThT) (Fig. 6A). All fibrillation curves showed a short lag time followed by a steep elongation phase after which a plateau or end point level was reached within 2 h. The ThT end point levels obtained showed a sigmoidal relationship to heparin concentration (Fig. 7C). We were not able follow the aggregation of sCT in the absence of heparin by ThT fluorescence (we saw no or very low ThT signal at pH 5.5 and 7.4, respectively). However, using A4F analysis we observed sCT-OH to be monomeric after 5-h incubation at pH 5.5 without heparin (data not shown). After longer incubation times (days), we observed regularly twisted, long fibrils of sCT formed without heparin by transmission electron microscopy. CD spectra of the ThT-negative, heparin-free aggregates showed a broad minimum at 218 nm (data not shown). Thus the presence of an sCT·hep binding complex at pH 5.5 seems to induce faster fibrillation compared with heparin-free sCT fibrillation. To investigate how heparin affected sCT fibrillation at pH 7.4, we monitored the change in secondary structure by far-UV CD. We obtained robust fibrillation signals for sCT-NH2 (decreased levels of ellipticity at 206 nm) both in the presence and absence of heparin (Fig. 6B). Heparin did not alter lag times, but the apparent elongation rate (seen as the steepness of the growth phase after initiation of fibrillation) increases from 1.25 to 10 h−1. Because heparin-induced fibrillation of sCT-OH and sCTH17A-OH led to half-times ∼10 times longer than sCT-NH2 at pH 7.4 (Fig. 7A), it was not experimentally tractable to carry out CD experiments for these two sCT species. Taken together, our data suggest that heparin reduces the sCT fibrillation time at low pH, whereas at pH 7.4 heparin only increases the elongation rate and does not affect the fibrillation lag time.
FIGURE 6.

Heparin induces ThT-positive sCT fibrils and increases the elongation rate at pH 7.4. A, sCT fibrillation followed by ThT fluorescence. Shown are sCT-OH (blue), sCT-NH2 (red), and sCTH17A-OH (green) without heparin at pH 5.5 (open symbols) and with 1.5:1 hepdisacc:sCT (closed symbols). B, fibrillation of sCT-NH2 and sCT-NH2·heparin (1:1.6 molar ratio) at pH 7.4 followed by CD at 206 nm. The two conditions with/without heparin are each shown as duplicates to show the lag time variability. a.u., arbitrary units; mdeg, millidegrees.
FIGURE 7.

sCT fibrillation times are pH-dependent, and ThT signals scale with heparin concentration. A, fibrillation lag times for heparin-induced fibrillation of sCT-OH, sCT-NH2, and sCTH17A-OH. All samples contain 0.5 mg/ml sCT, 150 μg/ml heparin, 40 μm ThT, and 20 mm citrate-phosphate buffer. t½ values are a median of empirical sigmoidal fits to individual ThT fibrillation traces. B, heparin addition to preformed sCT fibrils with increase in ThT signal. The intercept is at ∼1.4 heparin disaccharide units (calculated using a molecular mass of 665.4 g/mol for a heparin disaccharide unit sodium salt) per sCT monomer. C, ThT mean end point intensity at pH 5.5 with varying heparin concentrations indicates an intercept at a ratio of 1.5 disaccharides per sCT monomer. a.u., arbitrary units. Error bars represent S.D.
Removal of Histidine in sCT Diminishes the Increasing Half-times Below pH 5
To follow up on the observed strong pH dependence of sCT·hep binding, we studied heparin-induced fibrillation over the pH range 3–8 for sCT-OH, sCT-NH2, and sCT-H17A (Fig. 7A). Assuming a standard His pKa value of ∼6, we expect His17 in sCT-OH and sCT-NH2 to be positively charged at pH <5.5, and this coincides with an increase in the half-time of fibrillation. In contrast, sCTH17A-OH, which lacks this positive charge, undergoes rapid fibrillation over the entire low pH range.
Heparin-free sCT Fibrils Are Able to Bind ThT When Titrated with Heparin
When ThT-negative sCT-OH fibrils were incubated with heparin, the fibrils bound ThT, leading to a ThT fluorescence level that increased hyperbolically with heparin concentration (Fig. 7B). Based on the intercept of linear regression fits of ThT end points at low and high concentration ranges, the binding stoichiometry of preformed sCT-OH fibrils was determined to be 1.4 disaccharide units per sCT-OH monomer. This ratio agrees well with the 1.5 disaccharide units per sCT-OH monomer found for heparin-induced sCT fibrillation from end point ThT levels (Fig. 7C). ThT fluorescence increased immediately after adding heparin (data not shown), most likely because binding of heparin to preformed sCT-OH fibrils makes the fibril surface much less positively charged and thus allows the positively charged ThT to bind.
To further establish that heparin incorporates into mature sCT fibrils, fibrils were made at different heparin:sCT-OH ratios and pelleted to enable visualization of the soluble and insoluble fractions, respectively, using toluidine blue on SDS-polyacrylamide gels (Fig. 8A, purple shades). At low ratios (1.5 heparindisacc:sCT), heparin localizes in the pellet fraction together with sCT-OH fibrils. Heparin is only found in the supernatant when added in excess (15 heparindisacc:sCT) as expected from the estimated binding ratios (Fig. 7, B and C).
FIGURE 8.

Heparin is incorporated into sCT fibrils. A, SDS-PAGE showing exhaustive trypsin digestion of sCT-OH fibrils prepared with 0:1, 1.5:1, and 15:1 hepdisacc:sCT. s, postfibrillation soluble fraction; p, postfibrillation pellet fraction (the postfibrillation pellet was trypsin-treated); ST, soluble fraction post-trypsin treatment; PT, pellet fraction post-trypsin treatment. Heparin stains as the purple diffuse bands, intact sCT-OH stains as the upper blue band, and the N-terminal sCT peptide stains as the lower blue band. B, AFM of sCT-OH·hep (15:1 hepdisacc:sCT; post-trypsin treatment; trypsin-digested fibrils visualized on Tricine gel in A). C, Tricine gels of trypsin-treated sCT-NH2 fibrils, preformed sCT-NH2 fibrils with heparin added at 5:1 hepdisacc:sCT post-fibrillation (sCT-NH2 + hep), and sCT-NH2·heparin fibrils formed at 5:1 hepdisacc:sCT. Trypsin concentrations were 0, 0.145, 0.725, and 1.45 μm in lanes 0–3, respectively. Due to the ultracentrifugation step, the protein concentrations vary in each lane. Gel images are exposure- and brightness-corrected for improved visualization.
We next investigated whether sCT-NH2·heparin fibrils are more stable against proteases than heparin-free sCT-NH2 fibrils. Trypsin released a ∼2-kDa fragment from both sCT-NH2·heparin and heparin-free sCT-NH2 fibrils according to Tricine acrylamide gels (Fig. 8A). MS analysis showed the fragment to be the N-terminal part of sCT, i.e. residues Cys1–Arg24 containing uncleaved Lys11 and Lys18 trypsin cleavage sites. The sCT(1–24) peptide is present in the insoluble fraction, and fibrillar structures were observed by AFM (Fig. 8B). However, for fibrils formed in the presence of heparin, protease treatment also leaves a detectable amount of full-length sCT (Fig. 8C, bottom panel, upper band in lanes 1–3). We also investigated sCT fibrils formed without heparin where heparin was added to the fibrils before protease treatment. These fibrils show intermediate levels of protection (Fig. 8C, middle panel, lanes 1–3), showing more full-length sCT than heparin-free fibrils (Fig. 8C, top panel, lanes 1–3) but less than the fibrils formed in the presence of heparin (Fig. 8C, bottom panel, lanes 1–3). When treating soluble sCT with trypsin, no peptide was retained in the gel (data not shown), most likely reflecting the small size of tryptic products sCT(1–11), sCT(12–18), sCT(19–24), and sCT(25–32). For sCT-OH, trypsin digestion of fibrils gave results identical to those presented here for sCT-NH2 (data not shown). We conclude that heparin is incorporated into mature sCT fibrils, resulting in partial protection of Arg24 against trypsin cleavage.
Solid-state NMR Suggests That sCT Makes Up the Fibrillating Core whereas Heparin Interacts with Residues on the Outer Side of a β-Strand
We obtained atomic level detail about the interactions between heparin and sCT in the mature fibrils using solid-state NMR. An sCT-NH2 peptide that contained three 13C- and 15N-labeled amino acids (Arg24, Lys11, and Val8) was synthesized. Fibrils of the labeled peptide were made either in the absence of heparin or at a 5:1 heparindisacc:sCT molar ratio. Two-dimensional 13C-13C and 15N-13C correlation spectra (Fig. 9A) allowed us to unambiguously assign chemical shifts for all 13C and 15N in Val8 and Lys11 for both samples, whereas only the backbone nuclei for Arg24 were visible in the sCT-NH2·heparin fibril sample, and no signals from Arg24 were found in the sCT-NH2 fibrils formed without heparin. The NMR spectra showed relatively narrow lines, indicating well ordered fibrils, and each labeled site gave rise to only a single signal, which indicates a symmetric packing of the fibrils (35).
FIGURE 9.

NMR of sCT and sCT·heparin fibrils. A, two-dimensional solid-state NMR 13C-13C and 15N-13C correlation spectra for sCT·heparin fibrils with assignments of the individual peaks included. B and C, 13C solid-state NMR spectra acquired using CP (blue) to identify rigid parts of the samples and INEPT (red) to excite flexible parts for sCT·heparin fibrils (B) and sCT fibrils (C).
The assigned chemical shift values are compared with random coil values in Table 2. A secondary chemical shift analysis, which exploits the relationship between chemical shift values and secondary structure (36), shows that Val8 and Lys11 are in a β-sheet (for both samples), whereas the analysis remains inconclusive for Arg24 in sCT-NH2·hep. There are no signals for Arg24 in heparin-free sCT-NH2 fibrils.
TABLE 2.
Chemical shifts for isotope-labelled side chains in sCT and sCT·heparin fibrils obtained by ssNMR
Secondary structure class is assigned based on comparison with random coil (r.c.) shifts, Differences between sCT·heparin and sCT fibrils >1 ppm marked in yellow for Val8.a
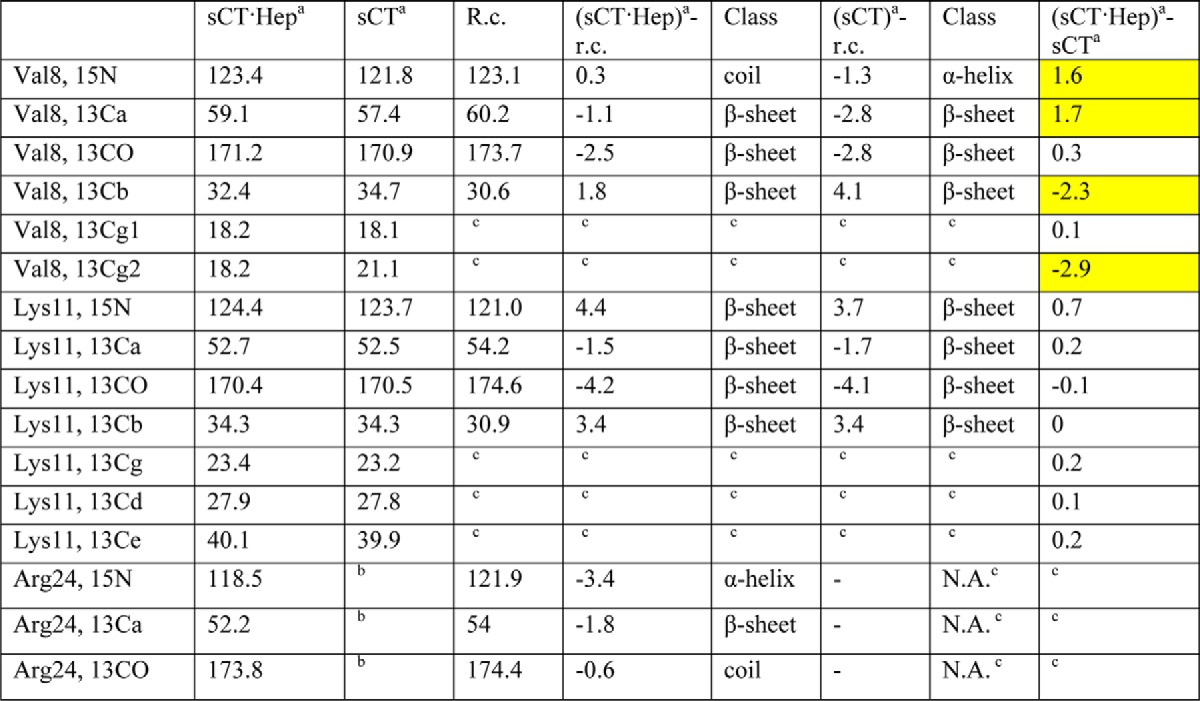
a Fibrils formed at pH 5.5 and 37 °C with orbital shaking.
b Peaks could not be assigned.
c Not applicable.
Differences between the fibrils formed with and without heparin may be identified from differences in chemical shift values between the two samples (Table 2). Val8 is influenced by heparin because the chemical shifts differ by more than 1 ppm, whereas Lys11 is unaffected with only minor chemical shift differences. The missing Arg24 signals may be explained by an inhomogeneous, unstructured, or flexible C terminus. To resolve this, we acquired one-dimensional cross-polarization (CP) spectra, which show only rigid parts of the samples, and INEPT spectra, which only give signals from flexible parts. The two-dimensional spectra mentioned above are initiated with CP elements and thus only reveal the rigid parts of the molecules. With strong signals from Val8 and Lys11 in the two-dimensional spectra, the central part of the peptide must be rigid and part of the fibril core. The INEPT spectrum of sCT-NH2·heparin contains no signals (Fig. 9B), and the signals from the side chain of Arg24 in the fibrils containing heparin are broadened beyond detection. This can be explained by an unspecific interaction between Arg24 and heparin or by an inhomogeneous C terminus. Conversely, for sCT-NH2, INEPT signals from both Val8 and Arg24 are seen (Fig. 9C), whereas Lys11 gives no INEPT signals. These findings indicate that Lys11 is buried inside the fibril, whereas Arg24 is situated in a flexible C terminus (Fig. 10C). Lys18 and Arg24 are hatched to highlight the expected heparin binding site (Fig. 10C). The basic residues Lys18 and Arg24 could coordinate acid and sulfate groups of heparin, whereas chemical shift differences for Val8 in sCT-NH2 compared with sCT-NH2·heparin fibrils indicate that heparin affects this residue.
FIGURE 10.

Models of sCT·heparin structures and fibrillation paths. A, helical wheel projection of sCT(8–24) (VLGKLSQELHKLQTYPR) using the European Bioinformatics Institute online tool. Hydrophobic residues are shown as blue squares, basic residues are represented as black circles, and polar and anionic side chains are shown as red diamonds. B, proposed model for GAG-inducing fibrillation of sCT at pH 5.5 and 7.4. At pH 5.5, monomeric sCT binds heparin, reducing fibrillation times and leading to heparin-containing fibrils. At pH 7.4, monomeric sCT does not bind heparin, but after sCT oligomerization, heparin interacts with either prefibrillar aggregates or the growing fibrils, forming heparin-containing fibrils. C, schematic representation of sCT in a β-strand conformation with isotope-labeled (black) (NMR) and basic/acidic residues (gray) at the indicated positions. The schematic represents a view down the fibril axis with hatched sides of possible heparin binding sites along the fibril. A disaccharide unit is drawn to scale with the β-strands to visualize size relations.
Serendipitously we also synthesized a sCT-OH Gly10 → Lys/Lys11 → Gly mutant. This mutant underwent a structural change upon heparin addition, similar to that seen for the other sCT peptides (data not shown). This indicates that the exact position of Lys11 is not crucial for heparin binding. However, the sCTG10K/K11G-OH double mutant does not fibrillate within 60 h (sCT-OH fibrillation is observed within 2 h). In practice, this double mutant moves Lys11 from one side of a β-strand to the opposite site. These findings nicely agree with the high level of Lys protection against trypsin digestion and NMR studies, indicating Lys11 to be similar for both sCT-NH2 and sCT-NH2·heparin fibrils. We suggest that Lys11 is not crucial for heparin binding but is most likely central to sCT-NH2 fibrillation and part of the fibrillating core. In a β-strand structure, Lys11 resides on the same side of the strand as Glu15, allowing sCT-NH2 to form salt bridges between Lys11 and Glu15 on opposing sCT-NH2 monomers in the core of a fibril strand (Fig. 10C). Val8, whose chemical shifts differ in sCT-NH2 and sCT-NH2·heparin, is on the opposing side of a sCT-NH2 β-strand compared with Lys11. However, Val8 is on the same side as Lys18, which could be a possible electrostatic binding partner for heparin in sCT-NH2 fibrils (Fig. 10C).
Discussion
Our data clearly show that sCT fibrillation is affected by GAGs in a complex manner with pH having a large impact on sCT-GAG interactions. The immediate formation of a binding complex at low pH indicates that under these conditions GAGs must affect the formation of spherical prefibrillar oligomers and hence might affect their toxicity (30). Also our data show that chondroitin sulfate A and less sulfated GAGs are not merely less active versions of heparin but have altered pH binding profiles. Understanding these effects is a prerequisite for understanding the delicate balance between the beneficial storage of hormonal peptides in granular vesicles versus disease-related amyloid formation. Our data from a broad range of techniques show that sCT under mildly acidic conditions (pH 5.5), but not under neutral conditions (pH 7.4), forms a semistable binding complex with heparin. There is no evidence to suggest that this binding complex dissociates before it starts to form amyloid-like fibrils when agitated at 37 °C. These fibrils show strong ThT fluorescence, twisted rod-like structures with thickness of 5–8 nm, and a strong CD β-sheet signal. We have multiple lines of evidence for heparin incorporation into sCT fibrils. Nevertheless, ssNMR data indicate that sCT-NH2 and sCT-NH2·heparin fibrils have similar fibrillating cores, providing evidence that Lys11 is in the fibrillating core and experiences similar environments in the two fibril types. NMR data further suggest that Val8 points outward from the strand and that Arg24 is present in the flexible C terminus supported by trypsin/MS data showing Arg24 to be accessible for trypsin cleavage. Both Val8 and Arg24 are affected by the presence of heparin in sCT-NH2 fibrils.
Charges Determine the pH Range for sCT GAG Binding
We reasoned that His17 might serve as a pH sensor for sCT binding to GAGs through its positive charge. This is indeed the case because sCTH17A-OH CD titration curves (Fig. 3) are shifted down to lower pH compared with sCT-OH. This means that that half-binding is achieved around pH 5.9 for sCTH17A-OH in contrast to sCT-OH (pH 6.5) and sCT-NH2 (pH 6.9). Thus to compensate for the loss of a positive charge in sCTH17A-OH, it is necessary to decrease the pH to achieve heparin binding. For amyloid β, a more dramatic effect is seen in that a His → Ser mutant completely loses its heparin binding abilities both at pH 8 and pH 4 (37). Similarly, the reduction in negative charge in the GAG ChA compared with heparin means that electrostatic interactions with sCT are weak, and therefore the positive charge of sCT has to be increased by lowering the pH to achieve efficient binding.
Combining CD and SAXS Data Provides Structural Information about the Binding Complex
The α-helical structure suggested by CD combined with results from SAXS inspired us to suggest that sCT monomers bind through electrostatic interactions between the basic amino acids present on one side of helical sCT and the negative heparin surface (Fig. 10A), leaving an exposed hydrophobic patch on sCT as a possible interaction site for other sCT monomers in the same conformation. sCT-sCT hydrophobic interactions, which are known to occur in GAG-free sCT dimers (31), may also occur along the heparin surface and would lead to a denser and less elongated structure of the binding complex as suggested here by SAXS. Upon incubation, this complex converts into heparin-containing fibrils. At pH 7.4, heparin interacts with sCT-NH2 after self-nucleation, again leading to heparin-containing fibrils.
sCT-NH2 Nucleation Is Heparin-independent at pH 7.4
Our data also provide mechanistic insight into how GAGs affect sCT fibrillation. At pH 7.4, monomeric sCT-NH2 does not bind heparin and fibrillates rapidly in the absence of heparin. Remarkably, heparin does not affect the fibrillation lag time at pH 7.4 but increases the elongation rate. The simplest interpretation of this is that at neutral pH heparin does not interfere with the ability of sCT-NH2 to form fibrillation nuclei but rather promotes the subsequent fibril extension process, very likely by binding to the growing fibrils and stabilizing them through interactions with cationic side chains as indicated by our ssNMR data (Fig. 9). This is consistent with our observation that heparin does not interact with monomeric sCT and that fibrillation times do not correlate with GAG concentration at pH 7.4 (data not shown) but that the ThT intensity nonetheless follows GAG concentration in a saturable manner (data not shown). The same considerations likely apply to sCT-OH and sCTH17A-OH. The shorter fibrillation times of sCT-NH2 compared with sCT-OH indicate that sCT self-nucleation at pH 7.4 is more efficient when the C terminus is amidated, most likely due to the removal of electrostatic repulsions between the C termini. These results agree with our studies of a fragment of human islet amyloid polypeptide where C-terminal amidation also affected fibrillation kinetics (38). As a further corroboration, an inspection of the calculated net charge of the three sCT species as a function of pH (assuming random coil pKa values for the titratable side chains) reveals that the fibrillation times for sCTH17A-OH and sCT-OH start to increase when the overall charge drops below +2 (pH 6 and 6.8, respectively). In contrast, the sCT-NH2 charge remains above +2 in the pH range investigated (up to pH 8) and does not diminish its fibrillation kinetics over this range.
sCT Fibrillation Is Slowed by His Protonation under Acidic Conditions
To understand the lag time increase at low pH seen for sCT-OH and sCT-NH2, we turn to the structure of the fibrils of sCT-NH2 deduced from ssNMR and complementary experiments. sCT-NH2 and sCT-NH2·heparin fibrils give rise to similar chemical shifts for Lys11, and CP and INEPT experiments reveal the residue to be highly immobilized in both fibril types, suggesting that the side chain is positioned on the inside of the β-structure. In that case, the β-strand structure constrains other uneven numbered amino acids to be placed on the inside of the β-strand. Our trypsin digest data reveal that Lys18 is highly protected from cleavage; hence we suggest it to be located in, or close to, the β-strand, leaving His17 most likely placed in the interior of the β-structure just like Lys11. Lys11 has a possible salt bridge partner in Glu15, leaving His17 to pair up with Leu9, a hydrophobic residue not likely to favor a positively charged His. These structural speculations rationalize why (heparin-induced) sCT fibrillation times increase at low pH for sCT-OH and sCT-NH2 but not for sCTH17A-OH. At pH 5.5, 76% of His17 will be protonated (assuming a pKa of 6.0). Fibrillation times start to increase from pH 5.3 where 86% of His17 is protonated.
Residues 11–18 Form the Central Core of sCT Fibrils
Results from ssNMR spectra, ThT binding assays, fibrillation studies, and proteolysis data indicate that heparin-induced fibrils consist of an sCT-NH2 fibrillating core with heparin decorating the surface of the otherwise positively charged protein surface. Because Lys11 and Lys18 are not cleaved by trypsin when sCT-NH2 is in the fibrillar state and the sCT(1–24) peptide remains in a fibrillar/insoluble form after digestion, we suggest that the central part of sCT-NH2 constitutes the β-strand, whereas the C-terminal part containing Arg24 is accessible for proteolytic attack. This agrees well with the NMR data where Arg24 was found to be in a flexible state in sCT-NH2 fibrils but was not detectable in sCT-NH2·heparin fibrils, possibly because unordered heparin interactions lead to line broadening beyond detection. In both fibril types, Lys11 is rigid, whereas Val8 is somewhat flexible in sCT-NH2 fibrils and rigid in sCT-NH2·heparin fibrils. We suggest that Val8 is positioned on the outside of the fibril core, just like Lys18, so that it is affected by heparin binding to the fibril exterior. Similarly, for heparin-free hCT fibrils, the C-terminal part was found to be flexible, whereas the central part was found in a β-sheet (32). Thus for both sCT-NH2 and hCT, the central part of the peptide constitutes the fibrillation core. We suggest Lys11 to be part of the fibril interior, possibly stabilized by Glu15, whereas Lys18 may act as the heparin binding partner on the exterior side of a β-strand. This is in contrast to what is suggested for hCT where Lys18 is thought to interact with Asp15 (32). If such an interaction is also in the fibril interior it would lead to a face-to-back (39) arrangement assuming identical strands. We suggest sCT-NH2 to have Lys11 placed in the fibril interior in a face-to-face arrangement (pairing β-stands are found in the same conformation and arranged with the same residues in the interior of the structure) stabilized by a salt bridge to Glu15.
Biological Implications
sCT is known to be less fibrillation-prone than hCT because of the increased polarity of sCT (33). Lowering the pH increases the polarity of the peptide due to His protonation, and hence both hCT and sCT are expected to be less fibrillation-prone at low pH. We observed GAGs to have the most dramatic effect on fibrillation propensity at low pH compared with the more subtle kinetic effect observed at pH 7.4. In CT-producing cells, the protein is packed into granular vesicles under increasingly acidic conditions, reaching pH 5.5 in the mature granular vesicles. The reduced pH could lead to increased binding (and subsequent aggregation) of sCT to highly sulfated GAGs such as heparan sulfate, which is also known to be present in mature granular vesicles (40). Our study is in good agreement with earlier reports on heparin-promoting fibrillation in numerous other proteins including amyloid-β (10), α-synuclein (11), tau (12), and β2-microglobulin (14) and provides new insight into how GAG-protein interactions can be manipulated simply by altering the pH.
Experimental Procedures
Materials
sCT-NH2 was from Polypeptide (Limhamn, Sweden). Heparin sodium salt and chondroitin sulfate A and were from Sigma-Aldrich. Heparan sulfate and dermatan sulfate were from Iduron (Cheshire, UK). NHS-fluorescein was from Thermo Scientific (Slangerup, Denmark). 13C,15N-Labeled Val, Arg, and Lys were from Sigma-Aldrich.
Peptide Synthesis
All peptides were synthesized utilizing the SPPS Fmoc/tert-butyl methodology (41) on an ABI433A peptide synthesizer (Applied Biosystems). Global deprotection/cleavage was conducted by gently agitating the dried down peptidyl resin in 95% TFA,2.5% triisopropylsilane, 2.5% H2O for 2 h. The crude linear peptide was collected by precipitation with ice-cold tert-butylmethyl ether, dissolved in 20% aqueous acetonitrile, and lyophilized. Formation of the disulfide bridge was effected using trans-[Pt(en)2Cl2]2+ as described by Shi and Rabenstein (42). Briefly, the crude linear crude peptide was dissolved in pH 7 phosphate buffer (0.1 m) at a concentration of approximately 0.1 mm. The platinate complex (∼1.2 eq) was added, and the cyclization was followed by HPLC. Upon completion, the crude cyclized peptide was subjected to purification by means of semipreparative HPLC. Analytical HPLC and semipreparative HPLC were performed on a Agilent 1200 or 1260 system (Agilent Technologies) using eluent A (H2O, 0.1% TFA) and eluent B (acetonitrile, 0.1% TFA). Semipreparative purification was performed on a Phenomenex Luna C18(2) (10 × 250 mm, 5-μm particle size, 100-Å pore size) column (Phenomenex, Vaerloese, Denmark) using a gradient of 10–60% B over 40 min at a flow rate of 5 ml/min (detection at 280 nm).
trans-[Pt(en)2Cl2]2+
To a solution of 0.5 g of dichlorobis(en)Pt(II) in 10 ml, concentrated HCl was slowly added under stirring followed by 10 ml of H2O2 (∼30%). The suspension was stirred for 1 h and then filtered. The filtrate was concentrated and dried in vacuo.
sCT-NH2 with 13C and 15N Labeling at Val8, Lys11, and Arg24
The peptide was synthesized on a 50-μmol scale using Tentagel XV RAM resin (0.21 mmol/g). Fmoc-amino acids (5 eq) were coupled for 30 min using PyOxim (5 eq) and N,N-diisopropylethylamine (10eq) in DMF (3 ml) expect for the labeled Fmoc-amino acids (3 eq), which were coupled for 60 min. Fmoc removal was effected with 20% piperidine, DMF (2 × 3 min). Semipreparative HPLC yielded the peptide in >95% purity (15 mg). High resolution MS: C12813C17H241N3715N7O48S2+ [M + H+]; calculated, 3454.76, found, 3454.77. RP-HPLC: tR = 7.24 min (Phenomenex Aeris WP C18 (4.6 × 150 mm, 3.6 μm, 100 Å); 10–60% B over 10 min at 2 ml/min.
sCTH17A-OH
The peptide was synthesized on a 100-μmol scale using Tentagel H-Pro-Trt resin (0.21 mmol/g). Fmoc-amino acid couplings were performed using 1H-benzotriazolium 1-[bis(dimethylamino)methylene]-5-chlorohexafluorophosphate (1-),3-oxide (5 eq), N,N-diisopropylethylamine (10 eq) in DMF (3 ml) for 10 min. Fmoc removal was conducted with 20% piperidine, DMF (2 × 3 min). Semipreparative HPLC yielded the peptide in >95% purity (25 mg). High resolution MS: C142H238N41O49S2+ [M + H+]; calculated, 3365.68; found, 3365.71. RP-HPLC: tR = 7.96 min (Phenomenex Aeris C18 (4.6 × 150 mm, 3.6 μm, 100 Å); 5–90% B over 15 min at 2 ml/min).
sCT-OH
The peptide was synthesized on a 200-μmol scale using a H-Pro-2CT PS resin (0.7 mmol/g). Couplings were performed using diisopropylcarbodiimide (10 eq) and 6-chloro-1-hydroxybenzotriazole (5 eq) in DMF (3 ml) for 20 min. Fmoc removal was conducted with 20% piperidine, DMF (2 × 3 min). Semipreparative HPLC yielded the peptide in >95% purity (54 mg). MALDI-TOF MS: C145H240N43O49S2+ [M + H+]; calculated, 3431.7; found, 3431.6. RP-HPLC: tR = 9.60 min (Phenomenex Aeris C18 column (4.6 × 150 mm, 3.6 μm, 100 Å); 5–90% B over 15 min at 2 ml/min).
sCT-OH(G10L/L11G)
The peptide was synthesized on a 100-μmol scale using a Tentagel H-Pro-Trt PS resin (0.17 mmol/g). Couplings were conducted using PyOxim (5 eq) and N,N-diisopropylethylamine (10 eq) in DMF (3 ml) for 20 min. Fmoc removal was conducted with 20% piperidine, DMF (2 × 3 min). Semipreparative HPLC yielded the peptide in >95% purity. MALDI-TOF MS: C145H240N43O49S2+ [M + H+]; calculated, 3431.7; found, 3429.8. RP-HPLC: tR = 13.7 min (Phenomenex Kinetex C18 (4.6 × 150 mm, 2.6 μm, 100 Å); 40–80% B over 15 min at 1 ml/min).
Prior to use, all sCT versions were dissolved in Milli-Q water. Protein concentration was determined via absorption at 280 nm using a molecular mass of 3434 Da and an extinction coefficient of 1615 m−1 cm−1.
Fibrillation Assays
Peptide fibrillations were performed in a 96-well clear bottom plate (catalog number 265301, Thermo Fischer Nunc) loaded with one glass bead of 3–4-mm diameter in each well (Glaswarenfabrik Karl Hecht, Sondheim, Germany). The total volume in each well was 150 μl. The buffers used were 20 mm acetate, pH 5.5, with 15 mm NaCl or 20 mm sodium phosphate, pH 7.4, with 15 mm NaCl. For the fibrillation assay spanning pH 2.8–8, the buffer system was 20 mm citrate-phosphate mixed to obtain the desired pH. All samples were mixed in Eppendorf tubes, checked for pH (room temperature), and stored on ice. ThT was added to a final concentration of 40 μm from a 1.2 mm stock in ethanol (final ethanol concentration, 3.3 volume %). Protein concentration was 0.5 mg/ml, and GAG concentration was 150 μg/ml unless otherwise indicated. Fibrillation was performed in a Genius Pro plate reader (Tecan, Männedorf, Switzerland) with the following settings: 37 °C; excitation, 448 nm; emission, 485 nm; gain, 40; and 40-μs integration time. For each measurement, planar orbital shaking for 3 min at 2.5-mm amplitude was applied every 5 min. 10 reads were averaged for each well. ThT traces were fitted to a sigmoidal function using nonlinear least square regression in KaleidaGraph (v4.0; Synergy Software, Reading, PA).
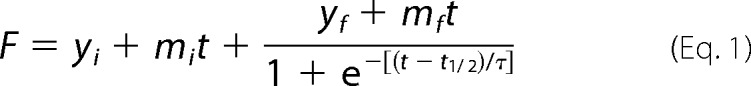 |
where F is the measured fluorescence intensity; yi and yf are initial and final ThT intensity levels, respectively; t½ is the time at which ThT intensity reached yf/2; mi and mf are the slopes of the initial and final baselines; and τ is the elongation rate. ThT end point intensity was taken as the mean data points after the end of the elongation phase.
Circular Dichroism
CD spectra were recorded on a J-810 spectrometer (Jasco) in a 1-mm quartz cuvette from 250 to 190 nm with 0.2-nm data pitch, 1-s integration time, 1-s bandwidth, and six accumulations. Only data points with detector voltages below 600 V were used. To record spectra of soluble species at pH 5.5 and 7.4, sCT was dissolved to 0.2–0.4 mg/ml and measured in the appropriate buffer. GAGs were present at concentrations mentioned in the text. Aggregated samples were pelleted at 11,000 × g for 30 min at 4 °C. The supernatant was gently removed from each sample and analyzed for monomer content using SDS-PAGE together with an sCT calibration series. The insoluble pellet fraction was resuspended in the appropriate buffer without ThT or GAG. The sCT concentration in the aggregated sample was calculated by subtracting the measured sCT supernatant concentration from the initial concentration. For the pH 2.8–8 binding assay, GAGs were added in a 5:1 heparindisacc:sCT ratio. Negligible CD signals were obtained from GAGs and buffers. For sCT-NH2, GAG binding at pH 3–8 structural conversion is reported as the ratio of ellipticities at 200 and 220 nm. sCT-NH2 fibrillation spectra were recorded at 5-min intervals from 250 to 195 nm with 0.1-nm data pitch, 2-nm bandwidth, 4-s response, and 50 nm/min scanning speed at 37 °C. Between reads, the cuvette was incubated in a Vortemp 56EVC (Tehtnica, Železniki, Slovenija) Eppendorf thermoshaker modified to hold cuvettes at 59 rpm and 37 °C. Protein concentration was 0.5 mg/ml, and heparin concentration was 150 μg/ml.
Fibril Imaging by Atomic Force Microscopy
sCT fibrils were twice pelleted and resuspended in double distilled H2O to avoid salt precipitates. The washed and diluted fibrils were dried on freshly cleaved mica and analyzed by atomic force microscopy on a 5100 AFM with PicoView 1.12 (Agilent Technologies) using a silicon cantilever (AppNano, ACSTA-50) in tapping mode with a resolution of 1024 × 1024 points in a 3 × 3-μm frame, scan speed of 1.5 μm/s, and integral/proportional gain of 3%. No morphological difference was observed between washed and unwashed fibrils. The height profiles of the fibrils were extracted using the software Gwyddion (v2.30; Free Software Foundation), and all heights reported on twisted fibrils were measured on the highest points in the repetitive profile along a fibril axis.
Fibril Trypsin Digestion and Analysis
sCT-NH2 and sCT-NH2·hep (2:1, 3:1, or 1:5 sCT:heparindisacc) fibrils were formed at pH 5.5 in a plate reader assay, pelleted at 136,000 × g, washed, and resuspended in phosphate buffer at pH 7.4. Trypsin was added to a final concentration of 0, 0.145, 0.725, and 1.45 μm, respectively, and incubated at 37 °C for 1 h with 20 rpm shaking in a Vortemp 56EVC Eppendorf thermoshaker. In all cases, trypsin activity was inhibited by addition of 1 mm phenylmethylsulfonyl fluoride (PMSF), and samples were reduced and treated with iodoacetamide. 145 μm sCT-OH, 1.5 mg/ml heparin, pH 7.4, fibrils for AFM were pelleted, washed as above, and treated with 1 μm trypsin for 2 h at pH 7.4. Trypsin activity was inhibited by addition of 2 μm PMSF. The digested sCT supernatants and fibrils were analyzed by Tricine SDS-PAGE using a 16% separating gel, 10% overlay gel, and 4% sample gel as described previously (43). Gels were stained with Coomassie Brilliant Blue and toluidine blue for peptide and heparin visualization, respectively.
In-gel Digestion and Micropurification
For each fibril experiment, two bands were subjected to Lys-C in-gel digestion, namely the non-trypsin-treated sCT band and the lower sCT peptide band observed at the highest trypsin concentration. Gel bands were incubated 2 × 15 min in 50% acetonitrile, dehydrated in neat acetonitrile for 15 min, and equilibrated in 0.1 m NH4HCO3 for 5 min before an equal volume of acetonitrile was added and further incubated for 15 min. Supernatants were removed, and the gel pieces were lyophilized for 10 min. In-gel digestions were performed by incubating the gel pieces with 50 ng of sequencing grade Lys-C (Promega) in 50 mm NH4HCO3 at 37 °C for 16 h. The resulting peptides were desalted using homemade microcolumns (P10 tips plugged with C18 material) and stored at −20 °C before LC-MS/MS analysis.
Parallel Reaction Monitoring-MS
Parallel reaction monitoring (PRM) analyses were performed using an EASY-nLC II system (Thermo Fisher Scientific) connected to a TripleTOF 5600+ mass spectrometer (AB SCIEX) equipped with a NanoSpray III source (AB SCIEX) and operated under Analyst TF 1.6.0 control. Lys-C-digested and micropurified samples were dissolved in 0.1% formic acid, injected, and trapped on an in-house-packed trap column (2 cm × 100 μm inner diameter) using RP ReproSil-Pur C18-AQ 3-μm resin (Dr. Maisch GmbH). Peptides were eluted from the trap column and separated on a 15-cm analytical column (75-μm inner diameter) pulled and packed in-house with RP ReproSil-Pur C18-AQ 3-μm resin and sprayed directly into the mass spectrometer. Peptides were eluted at a flow rate of 250 nl/min using a 20-min gradient from 5 to 35% solvent B (solvent A, 0.1% formic acid; solvent B, acetonitrile, 0.1% formic acid). The parallel reaction monitoring acquisition method was generated by adding an inclusion list to a standard information-dependent acquisition method and by increasing the MS/MS-triggered cps threshold to 3,000,000 cps, thereby preventing MS/MS of m/z values not found in the inclusion list (supplemental Table S1). The number of precursor ions to be selected for MS/MS in each cycle was adjusted to the number of m/z values in the inclusion list, and the mass range of the survey scan was set to ±50 Da of the highest and lowest precursor ion in the inclusion list.
LC-MS/MS Data Processing
Parallel reaction monitoring-MS files were converted to Mascot generic format (MGF) using the AB SCIEX MS Data Converter Beta 1.3 and the “protein pilot MGF” parameters. The peak lists were used to match the calcitonin sequence using an in-house database by searching in Mascot 2.5.0 (Matrix Science, London, UK). Either Lys-C or trypsin was selected as the digestion enzyme, allowing one missed cleavage; carbamidomethyl was entered as a fixed modification; and C-terminal amidation and oxidation of methionine were selected as variable modifications. The data were searched with a mass tolerance of the precursor and product ions of 10 ppm and 0.2 Da, respectively, using electrospray ionization quadrupole (ESI-QUAD)-TOF as the instrument setting. The significance threshold (p) was set at 0.01, and the ion score expect cutoff value was set at 0.005. All data were imported and processed in MS Data Miner v.1.3 (44) (supplemental Tables S2 and S3).
Fluorescein Labeling of Heparin
Heparin labeling was done as described previously (45). Unreacted NHS-fluorescein was removed by dialysis (Specra-Por Float-A-Lyzer G2, 3.5–5 kDa). The degree of labeling of heparin was determined using a fluorescein extinction coefficient of 10,600 m−1 cm−1 at 477 nm and was estimated to be ∼1 fluorescein to 5 disaccharide units. The ability of heparin to induce sCT-OH fibrillation at pH 5.5 was not significantly different from unlabeled heparin. Analysis using an A4F system (AF2000, Postnova Analytics GmbH, Germany) verified the presence of a single heparin peak with a fluorescence emission at 516 nm by excitation at 485 nm.
Protein Separation by Asymmetric Flow Field Flow Fractionation
A4F was used instead of size exclusion chromatography to avoid sCT-OH fibrillation in the size exclusion chromatography column and because of its flexible separation range. A4F separation was performed using an A4F system on a 1-kDa regenerated cellulose membrane using the aforementioned buffers. The samples were focused for 5 min, and elution started with 5 min of constant perpendicular flow at 3 ml/min after which the perpendicular flow was reduced by 0.11 ml/min in a linear fashion. Sample load was 100 μl of 0.5 mg/ml sCT-OH and 150 μg/ml GAG.
Size Exclusion Chromatography Purification of sCT-OH·Heparin Binding Complex for Small Angle X-ray Scattering
sCT-OH·heparin binding complex was purified on a Superdex 200 10/300 GL column (GE Healthcare) in pH 5.5 buffer as described above. 250 μl of 1 mg/ml sCT-OH with 0.5 mg/ml heparin was injected. sCT-OH monomer eluted in one peak at 18 ml. The sCT-OH·heparin complex eluted after 9 ml just after the exclusion limit (8 ml) and was concentrated from 1 ml to 100 μl and 0.5 mg/ml using a spin filter (3 kDa; Amicon).
SAXS Measurements
The filtrate from the concentration step was used as a reference. Heparin dissolved in this reference buffer was used as a control. SAXS was measured in 2-mm capillaries on a modified Bruker NanoSTAR instrument (46). A pure water measurement was used to convert the scattering to absolute scale. The sCT-OH·heparin sample and its reference were measured for 1 h. Pure heparin dissolved in filtrate buffer was measured at concentrations of 2, 0.5, and 0.2 g/liter. Only the 0.2 g/liter sample was used because the others showed clear concentration effects. The 0.2 g/liter heparin sample and its reference were measured for 4 h each. The data are displayed as I(q) versus q where q is the size of the momentum transfer q = 4π sin(θ)/λ and 2θ is the scattering angle. The pair distance distribution functions p(r) were found using GNOM (47). The modeling was performed using the Ensemble Optimization Method (EOM) (48) on the basis of up to 10 blocks, each containing one copy of sCT (modified from Protein Data Bank code 2GLH) and three heparin disaccharides, each of which consists of N,O6-disulfoglucosamine (SGN) and 2-O-sulfo-α-l-idopyranuronic acid (IDS), described as (SGN × IDS)3. The building block was put together in PyMOL (The PyMOL Molecular Graphics System, version 1.7.4, Schrödinger, LLC). In the case of modeling the heparin alone, the basic building block consisted of Cys × (SGN × IDS)3 × Pro where the amino acid caps (cysteine and proline) allow the program to run.
Fibril Structural Analysis Using Solid-state NMR
sCT-NH2 with uniform 13C- and 15N-labeled Val8, Lys11, and Arg24 was incubated at pH 5.5 with heparin at a 5:1 heparindisacc:sCT ratio overnight at 37 °C with one glass bead and orbital shaking (900 rpm, TS100 Biosan Thermoshaker) to form sCT-NH2·heparin fibrils. CD showed a strong 218-nm minimum, and AFM verified the presence of fibrils. Fibrils were pelleted at 11,000 × g, and no residual sCT-NH2 was detected in the supernatant by CD or AFM. Labeled sCT-NH2 was also incubated without heparin at pH 5.5 in a plate reader assay for 4 days. CD and AFM confirmed fibril content, and fibrils were pelleted by centrifugation at 136,000 × g for 30 min.
The NMR experiments were carried out on 700-MHz Avance III and 500-MHz Avance III Bruker spectrometers (Rheinstetten, Germany) equipped with 4-mm triple resonance Bruker probes. One-dimensional 13C spectra were acquired using CP and INEPT techniques with 3072 points, 2048 repetitions, and a relaxation delays of 3 s. The 1H-13C CP experiments used an 80–100% ramp for 1H and square pulses for 13C with average values of 54 (1H) and 42 kHz (13C) and mixing times of 2.5 ms. The INEPT spectra used 180° pulses of 7.6 μs. 13C-13C correlations were obtained using dipolar assisted rotational resonance mixing (49) with mixing times of 20 and 200 ms, 512 points for spectral widths of 200 ppm in the indirect dimensions, 80 repetitions, and s relaxation delays of 3 s. Two-dimensional adiabatic double CP spectra (500 MHz) with 8-ms mixing were used for acquiring 15N-13C correlations with average rf fields of 41/32 kHz (13C/15N) and 100-kHz continuous wave 1H decoupling during mixing. The 15N dimension used a spectral width of 120 ppm and 128 increments, and 224 repetitions per increment were applied. All experiments used magic angle spinning with spinning rates of 12 kHz (700 MHz) and 9 kHz (500 MHz) and ∼100 kHz SPINAL-64 (50) 1H decoupling during direct and indirect acquisition periods. The samples were kept around 4 °C using a precooled gas flow. All spectra are referenced to tetramethylsilane at 0 ppm using an external adamantane reference sample.
Author Contributions
K. G. M and D. E. O. planned experiments. K. G. M, E. H. T. N, M. B., E. T. P., C. M. J., and G. C. performed experiments and communicated experimental methods and results. D. E. O., J. S. P., T. V., T. S., and J. J. E. supervised experiments. K. G. M wrote the manuscript. D. E. O. edited the manuscript.
Supplementary Material
Acknowledgments
We thank Line F.B. Christensen, Brian S. Vad, and Ove Lillelund for fruitful discussions and helpful advice.
This work was supported by Danish Research Foundation inSPIN Grant DNRF59 and Danish Ministry of Higher Education and Science Grant AU-2010-612-181. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1–S3.
- PG
- proteoglycan
- AFM
- atomic force microscopy
- ChA
- chondroitin sulfate A
- DS
- dermatan sulfate
- GAG
- glycosaminoglycan
- hCT
- human calcitonin
- hep
- heparin
- SAXS
- small angle x-ray scattering
- sCT
- salmon calcitonin
- ThT
- thioflavin T
- GM1
- monosialotetrahexosylganglioside
- fl-hep
- fluorescein-labeled heparin
- A4F
- asymmetric flow field flow fractionation
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- CP
- cross-polarization
- INEPT
- insensitive nuclei enhanced by polarization transfer
- NHS
- N-hydroxysuccinimide
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- DMF
- N,N-dimethylformamide
- RP
- reversed phase
- ssNMR
- solid-state NMR
- en
- ethylenediamine.
References
- 1. Virchow R. (1855) Zur cellulose-frage. Arch. Pathol. Anat. 8, 140–144 [Google Scholar]
- 2. Atwood C. S., Martins R. N., Smith M. A., and Perry G. (2002) Senile plaque composition and posttranslational modification of amyloid-β peptide and associated proteins. Peptides 23, 1343–1350 [DOI] [PubMed] [Google Scholar]
- 3. Castillo G. M., Cummings J. A., Yang W., Judge M. E., Sheardown M. J., Rimvall K., Hansen J. B., and Snow A. D. (1998) Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan's enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes 47, 612–620 [DOI] [PubMed] [Google Scholar]
- 4. Snow A. D., Wight T. N., Nochlin D., Koike Y., Kimata K., DeArmond S. J., and Prusiner S. B. (1990) Immunolocalization of heparan sulfate proteoglycans to the prion protein amyloid plaques of Gerstmann-Straussler syndrome, Creutzfeldt-Jakob disease and scrapie. Lab. Invest. 63, 601–611 [PubMed] [Google Scholar]
- 5. Castillo G. M., Lukito W., Wight T. N., and Snow A. D. (1999) The sulfate moieties of glycosaminoglycans are critical for the enhancement of β-amyloid protein fibril formation. J. Neurochem. 72, 1681–1687 [DOI] [PubMed] [Google Scholar]
- 6. Malmos K. G., and Otzen D. (2014) Glycosaminoglycans and fibrillar polymorphism, in Bio-nanoimaging: Protein Misfolding and Aggregation, pp. 281–290, Elsevier, Amsterdam [Google Scholar]
- 7. Bernfield M., Kokenyesi R., Kato M., Hinkes M. T., Spring J., Gallo R. L., and Lose E. J. (1992) Biology of the syndecans: a family of transmembrane heparan sulfate proteoglycans. Annu. Rev. Cell Biol. 8, 365–393 [DOI] [PubMed] [Google Scholar]
- 8. Beaty N. B., and Mello R. J. (1987) Extracellular mammalian polysaccharides: glycosaminoglycans and proteoglycans. J. Chromatogr. 418, 187–222 [DOI] [PubMed] [Google Scholar]
- 9. Iozzo R. V. (2000) Proteoglycans: Structure, Biology, and Molecular Interactions, pp. 1–60, CRC Press, Boca Raton, FL [Google Scholar]
- 10. Bruinsma I. B., te Riet L., Gevers T., ten Dam G. B., van Kuppevelt T. H., David G., Küsters B., de Waal R. M., and Verbeek M. M. (2010) Sulfation of heparan sulfate associated with amyloid-β plaques in patients with Alzheimer's disease. Acta Neuropathol. 119, 211–220 [DOI] [PubMed] [Google Scholar]
- 11. Lehri-Boufala S., Ouidja M. O., Barbier-Chassefière V., Hénault E., Raisman-Vozari R., Garrigue-Antar L., Papy-Garcia D., and Morin C. (2015) New roles of glycosaminoglycans in α-synuclein aggregation in a cellular model of Parkinson disease. PLoS One 10, e0116641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu H. L., Fernández C., Fan J. B., Shewmaker F., Chen J., Minton A. P., and Liang Y. (2010) Quantitative characterization of heparin binding to Tau protein: implication for inducer-mediated Tau filament formation. J. Biol. Chem. 285, 3592–3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Motamedi-Shad N., Garfagnini T., Penco A., Relini A., Fogolari F., Corazza A., Esposito G., Bemporad F., and Chiti F. (2012) Rapid oligomer formation of human muscle acylphosphatase induced by heparan sulfate. Nat. Struct. Mol. Biol. 19, 547–554 [DOI] [PubMed] [Google Scholar]
- 14. Borysik A. J., Morten I. J., Radford S. E., and Hewitt E. W. (2007) Specific glycosaminoglycans promote unseeded amyloid formation from β2-microglobulin under physiological conditions. Kidney Int. 72, 174–181 [DOI] [PubMed] [Google Scholar]
- 15. De Carufel C. A., Nguyen P. T., Sahnouni S., and Bourgault S. (2013) New insights into the roles of sulfated glycosaminoglycans in islet amyloid polypeptide amyloidogenesis and cytotoxicity. Biopolymers 100, 645–655 [DOI] [PubMed] [Google Scholar]
- 16. Sandwall E., O'Callaghan P., Zhang X., Lindahl U., Lannfelt L., and Li J. P. (2010) Heparan sulfate mediates amyloid-β internalization and cytotoxicity. Glycobiology 20, 533–541 [DOI] [PubMed] [Google Scholar]
- 17. Iannuzzi C., Irace G., and Sirangelo I. (2015) The effect of glycosaminoglycans (GAGs) on amyloid aggregation and toxicity. Molecules 20, 2510–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fraser P. E., Darabie A. A., and McLaurin J. A. (2001) Amyloid-β interactions with chondroitin sulfate-derived monosaccharides and disaccharides. Implications for drug development. J. Biol. Chem. 276, 6412–6419 [DOI] [PubMed] [Google Scholar]
- 19. Saridaki T., Zampagni M., Mannini B., Evangelisti E., Taddei N., Cecchi C., and Chiti F. (2012) Glycosaminoglycans (GAGs) suppress the toxicity of HypF-N prefibrillar aggregates. J. Mol. Biol. 421, 616–630 [DOI] [PubMed] [Google Scholar]
- 20. Maji S. K., Perrin M. H., Sawaya M. R., Jessberger S., Vadodaria K., Rissman R. A., Singru P. S., Nilsson K. P., Simon R., Schubert D., Eisenberg D., Rivier J., Sawchenko P., Vale W., and Riek R. (2009) Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 325, 328–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dannies P. S. (2012) Prolactin and growth hormone aggregates in secretory granules: the need to understand the structure of the aggregate. Endocr. Rev. 33, 254–270 [DOI] [PubMed] [Google Scholar]
- 22. Mellman I., Fuchs R., and Helenius A. (1986) Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 55, 663–700 [DOI] [PubMed] [Google Scholar]
- 23. Madine J., Clayton J. C., Yates E. A., and Middleton D. A. (2009) Exploiting a 13C-labelled heparin analogue for in situ solid-state NMR investigations of peptide-glycan interactions within amyloid fibrils. Org. Biomol. Chem. 7, 2414–2420 [DOI] [PubMed] [Google Scholar]
- 24. Madine J., Pandya M. J., Hicks M. R., Rodger A., Yates E. A., Radford S. E., and Middleton D. A. (2012) Site-specific identification of an Aβ fibril-heparin interaction site by using solid-state NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 51, 13140–13143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chesnut C. H. 3rd, Azria M., Silverman S., Engelhardt M., Olson M., and Mindeholm L. (2008) Salmon calcitonin: a review of current and future therapeutic indications. Osteoporos. Int. 19, 479–491 [DOI] [PubMed] [Google Scholar]
- 26. Galante L., Horton R., Joplin G. F., Woodhouse N. J., and MacIntyre I. (1971) Comparison of human, porcine and salmon synthetic calcitonins in man and in the rat. Clin. Sci. 40, 9P–10P [DOI] [PubMed] [Google Scholar]
- 27. Cudd A., Arvinte T., Das R. E., Chinni C., and MacIntyre I. (1995) Enhanced potency of human calcitonin when fibrillation is avoided. J. Pharm. Sci. 84, 717–719 [DOI] [PubMed] [Google Scholar]
- 28. Diociaiuti M., Macchia G., Paradisi S., Frank C., Camerini S., Chistolini P., Gaudiano M. C., Petrucci T. C., and Malchiodi-Albedi F. (2014) Native metastable prefibrillar oligomers are the most neurotoxic species among amyloid aggregates. Biochim. Biophys. Acta 1842, 1622–1629 [DOI] [PubMed] [Google Scholar]
- 29. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., and Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 30. Diociaiuti M., Polzi L. Z., Valvo L., Malchiodi-Albedi F., Bombelli C., and Gaudiano M. C. (2006) Calcitonin forms oligomeric pore-like structures in lipid membranes. Biophys. J. 91, 2275–2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Andreotti G., and Motta A. (2004) Modulating calcitonin fibrillogenesis: an antiparallel α-helical dimer inhibits fibrillation of salmon calcitonin. J. Biol. Chem. 279, 6364–6370 [DOI] [PubMed] [Google Scholar]
- 32. Kamihira M., Naito A., Tuzi S., Nosaka A. Y., and Saitô H. (2000) Conformational transitions and fibrillation mechanism of human calcitonin as studied by high-resolution solid-state 13C NMR. Protein Sci. 9, 867–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Andreotti G., Vitale R. M., Avidan-Shpalter C., Amodeo P., Gazit E., and Motta A. (2011) Converting the highly amyloidogenic human calcitonin into a powerful fibril inhibitor by three-dimensional structure homology with a non-amyloidogenic analogue. J. Biol. Chem. 286, 2707–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Micsonai A., Wien F., Kernya L., Lee Y. H., Goto Y., Réfrégiers M., and Kardos J. (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 112, E3095–E3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nielsen J. T., Bjerring M., Jeppesen M. D., Pedersen R. O., Pedersen J. M., Hein K. L., Vosegaard T., Skrydstrup T., Otzen D. E., and Nielsen N. C. (2009) Unique identification of supramolecular structures in amyloid fibrils by solid-state NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 48, 2118–2121 [DOI] [PubMed] [Google Scholar]
- 36. Tamiola K., Acar B., and Mulder F. A. (2010) Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Chem. Soc. 132, 18000–18003 [DOI] [PubMed] [Google Scholar]
- 37. Brunden K. R., Richter-Cook N. J., Chaturvedi N., and Frederickson R. C. (1993) pH-dependent binding of synthetic β-amyloid peptides to glycosaminoglycans. J. Neurochem. 61, 2147–2154 [DOI] [PubMed] [Google Scholar]
- 38. Andreasen M., Skeby K. K., Zhang S., Nielsen E. H., Klausen L. H., Frahm H., Christiansen G., Skrydstrup T., Dong M., Schiøtt B., and Otzen D. (2014) The importance of being capped: terminal capping of an amyloidogenic peptide affects fibrillation propensity and fibril morphology. Biochemistry 53, 6968–6980 [DOI] [PubMed] [Google Scholar]
- 39. Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ø., Riekel C., and Eisenberg D. (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457 [DOI] [PubMed] [Google Scholar]
- 40. Giannattasio G., Zanini A., Rosa P., Meldolesi J., Margolis R. K., and margolis R. U. (1980) Molecular organization of prolactin granules. III. Intracellular transport of sulfated glycosaminoglycans and glycoproteins of the bovine prolactin granule matrix. J. Cell Biol. 86, 273–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chan C. W. (2000) Fmoc Solid Phase Peptide Synthesis, Oxford University Press, Oxford, UK [Google Scholar]
- 42. Shi T., and Rabenstein D. L. (2002) Convenient synthesis of human calcitonin and its methionine sulfoxide derivative. Bioorg. Med. Chem. Lett. 12, 2237–2240 [DOI] [PubMed] [Google Scholar]
- 43. Schägger H. (2006) Tricine-SDS-PAGE. Nat. Protoc. 1, 16–22 [DOI] [PubMed] [Google Scholar]
- 44. Dyrlund T. F., Poulsen E. T., Scavenius C., Sanggaard K. W., and Enghild J. J. (2012) MS Data Miner: a web-based software tool to analyze, compare, and share mass spectrometry protein identifications. Proteomics 12, 2792–2796 [DOI] [PubMed] [Google Scholar]
- 45. Nagasawa K., and Uchiyama H. (1978) Preparation and properties of biologically active fluorescent heparins. Biochim. Biophys. Acta 544, 430–440 [DOI] [PubMed] [Google Scholar]
- 46. Pedersen J. S. (2004) A flux- and background-optimized version of the NanoSTAR small-angle x-ray scattering camera for solution scattering. J. Appl. Crystallogr. 37, 369–380 [Google Scholar]
- 47. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 48. Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., and Svergun D. I. (2007) Structural characterization of flexible proteins using small-angle x-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 [DOI] [PubMed] [Google Scholar]
- 49. Takegoshi K., Nakamura S., and Terao T. (2001) C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 344, 631–637 [DOI] [PubMed] [Google Scholar]
- 50. Fung B. M., Khitrin A. K., and Ermolaev K. (2000) An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 142, 97–101 [DOI] [PubMed] [Google Scholar]
- 51. Gallagher J. T., and Lyon M. (2000) Molecular structure of heparan sulfate and interactions with growth factors and morphogens, in Proteoglycans: Structure, Biology and Molecular Interactions (Iozzo R. V., ed) pp. 27–59, Marcel Dekker, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.