

Abstract
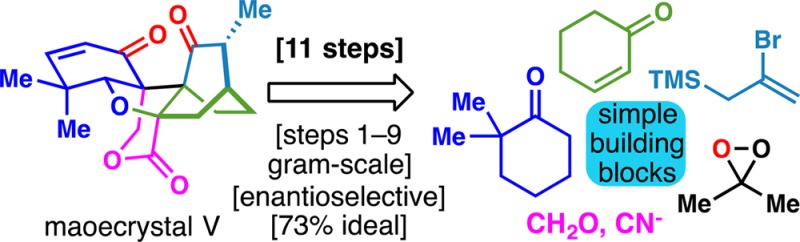
An expedient, practical, and enantioselective route to the highly congested ent-kaurane diterpene maoecrystal V is presented. This route, which has been several years in the making, is loosely modeled after a key pinacol shift in the proposed biosynthesis. Only 11 steps, many of which are strategic in that they build key skeletal bonds and incorporate critical functionalities, are required to access (−)-maoecrystal V. Several unique and unexpected maneuvers are featured in this potentially scalable pathway. Reevaluation of the biological activity calls into question the initial exuberance surrounding this natural product.
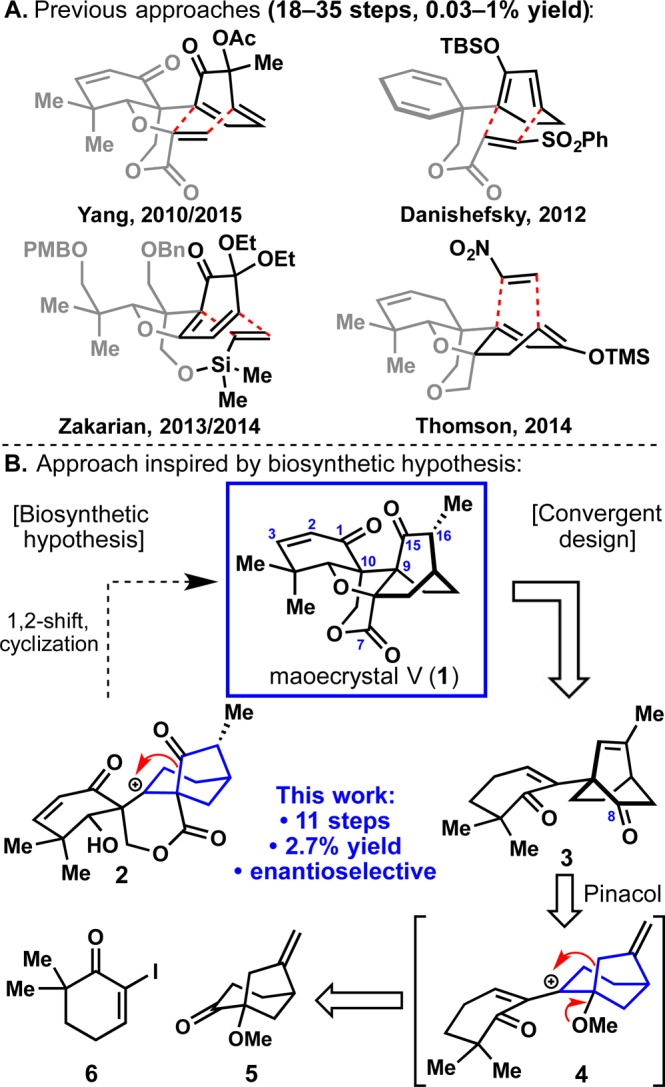
The combination of striking structural features and exciting bioactivity exhibited by the ent-kaurane diterpene maoecrystal V1 (1, Figure 1B) has attracted extensive interest within the organic community since 2004.2−4 As the most structurally complex member within this family of terpenes, it features a densely interlocked array of ring systems nestled within adjacent vicinal quaternary stereocenters (C-9/10, ent-kaurane numbering). The [2.2.2] bicyclic portion of the molecule is a clear signal for the use of the Diels–Alder transform, and thus all successful total syntheses4 to date have relied on this strategic maneuver (Figure 1A). In striking contrast, the biosynthetic route to this system is proposed to arise via an unusual pinacol-type shift of cation 2 (Figure 1B).5 It was reasoned that a route to 1 loosely patterned off of this theme might efficiently access the C-9 bridgehead quaternary center of the [2.2.2] bicycle and allow for a convergent fragment assembly at the C-9/10 juncture. Here, an 11-step enantioselective route to 1 is described based on the realization of this hypothesis. Using synthetic 1 it was also revealed that despite the original report, maoecrystal V exhibits virtually no cytotoxicity in any cancer cell line tested.
Figure 1.
A unique approach to maoecrystal V (1).
The key strategic disconnections made on 1 (Figure 1B) involved: (1) removal of the unsaturation at C-2/3; (2) masking of the C-15 ketone as an olefin to avoid epimerization at C-16; (3) nitrile-assisted delivery of C-7; and (4) extrusion of formaldehyde from C-10 to furnish 3. Whereas the first and second tasks were anticipated to be straightforward, the remaining were not. For example, there was no clear way to predict the stereoselectivity of cyanide addition (or any other small nucleophile) to C-8. In addition, the challenge of installing the hydroxymethyl group onto a ketone such as 3 was even reported to be insurmountable.3a A pinacol shift and olefin isomerization of 4 traces back to two simple ketones (5 and 6). The simplicity of this approach beckoned to be experimentally probed despite the clear warning signs present at the outset.
The synthesis of 1 commenced (Scheme 1) with a highly enantioselective conjugate addition of an allyl silane to cyclohexenone to deliver 7 in 80% isolated yield (99% ee). Among the many ligands explored, the TADDOL-derived phosphine-phosphite L1 designed by Schmalz was singularly successful.6 The use of CuI·0.75DMS was also critical to minimize dimerization of the Grignard reagent.7 A profound solvent effect was also observed with a mixture of PhMe/MeTHF being essential to obtain consistently high yield and enantioselectivity on 20 g scale.
Scheme 1. Total Synthesis of (−)-Maoecrystal V (1).

Reagents and conditions: (1) CuI·0.75DMS (0.60 mol %), L1 (0.80 mol %), TMSCH2C(MgBr)CH2 (2.5 equiv), PhMe/MeTHF, −78 °C (80%, 99% ee); (2) LiTMP (1.02 equiv), THF, −78 °C; then Davis oxaziridine (1.3 equiv), THF, DMPU, −78 °C; then Ac2O (1.2 equiv), −78 to 0 °C (64%); (3) EtAlCl2 (2.0 equiv), PhMe, 0 °C (77%); (4) NaH (1.2 equiv), Bu4NI (1.0 equiv), Me2SO4 (3.0 equiv), DMF, 23 °C; then aq. LiOH (8.5 equiv), 23 °C; (5) Py·SO3 (3.5 equiv), Et3N (8.5 equiv), DMSO, DCM, 0 to 23 °C (81%, 2 steps); (6) i-PrMgCl·LiCl (1.5 equiv), 6 (1.5 equiv), PhMe, −78 to 0 °C; then aq. TsOH (6.5 equiv), 0 to 85 °C (45%); (7) NaHMDS (1.3 equiv), LaCl3·2LiCl (1.0 equiv), THF, DMPU, CH2O(g) (10 equiv), −45 °C (84%, 2:1 dr); (8) TFA (0.50 equiv), HC(OMe)3, MeOH, 60 °C; then Zn(OTf)2 (2.0 equiv), LiBH4 (8.5 equiv), DCM, 23 °C (83%, 3:1 dr); (9) TsOH (5.0 mol %), H2O (2.0 equiv), THF, 23 °C; then DNBCl (3.5 equiv), DMAP (0.20 equiv), Et3N (5.0 equiv), 23 °C (86%); (10) MsOH (5 × 0.50 equiv), HC(OMe)3, MeOH, 65 °C; then ZnI2 (0.30 equiv), TMSCN (5.0 equiv), 23 °C; then aq. LiOH (12 equiv); then aq. HCl (20 equiv), 65 °C (82%); (11) DMDO (2 × 3.0 equiv), acetone, 23 °C; then InI3 (5.0 mol %), MgI2 (1.2 equiv), MeCN, 23 °C; then DMP (3.0 equiv), 23 °C; then aq. Oxone (10 equiv), Bu4NHSO4 (0.10 equiv), pH = 7.4 buffer, 23 °C (76%); DMS = dimethyl sulfide, MeTHF = 2-methyltetrahydrofuran, LiTMP = lithium 2,2,6,6-tetramethylpiperidide, DMPU = N,N′-dimethylpropyleneurea, DMSO = dimethyl sulfoxide, DCM = dichloromethane, TsOH = p-toluenesulfonic acid, NaHMDS = sodium bis(trimethylsilyl)amide, TFA = trifluoroacetic acid, DNBCl = 3,5-dinitrobenzoyl chloride, MsOH = methanesulfonic acid, DMDO = dimethyldioxirane, DMP = Dess–Martin periodinane.
The seemingly simple α-acetoxylation of ketone 7 turned out to be remarkably difficult to execute in a scalable way. Rubottom oxidation was unsuccessful due to competitive oxidation of electron-rich allyl silane moiety. Oxidants such as MoOPH, Pb(OAc)4 and Mn(OAc)3 gave complex mixtures, while Tomkinson’s reagent8 and similar enamine chemistry suffered from poor regioselectivity. Gratifyingly, deprotonation of 7 with LiTMP followed by sequential treatment with Davis oxaziridine9 and Ac2O in a mixture of THF/DMPU gave the desired α-acetoxylated product 8 in 64% yield as an inconsequential mixture of diastereomers (2:1) on a 30 g scale.
The ensuing Sakurai reaction10 initially suffered from extensive protodesilylation, possibly due to the fact that such a cyclization is disfavored by Baldwin rules, being analogous to a 5-enolendo-exo-trig in terms of orbital alignment.11 However, in an extensive evaluation of >50 Lewis acids, EtAlCl2 was found to be singularly successful, providing the desired [3.2.1] bicycle 9 in 77% yield. It is worth noting that in addition to the use of EtAlCl2, the α-acetoxy group was essential as silyl or benzyl ethers and the free alcohol all failed to react productively. Methylation with Me2SO4 and in situ hydrolysis, followed by Parikh–Doering oxidation afforded ketone 5 in 81% over 2 steps (>7 g were prepared in a single pass).
With ample quantities of 5 and 6(12) in hand, the stage was set to investigate the key 1,2-addition/pinacol rearrangement. Initially, attempts to add organometallic species generated from protected versions of 6 (such as ketal or silyl enol ether) resulted in no observed addition to ketone 5, presumably due to steric hindrance. Interestingly, it was found that treating unprotected 6 with i-PrMgCl·LiCl13 in PhMe resulted in Mg/I exchange and the obtained Grignard reagent added smoothly to ketone 5 to afford 10 (not isolated).14 Addition of aq. TsOH to the reaction mixture and heating to 85 °C resulted in gradual pinacol rearrangement and olefin isomerization to give key intermediate 3 in 45% isolated yield (7 g scale). The remaining mass balance primarily consisted of an undesired isomer (22% yield) formed during the pinacol shift (see SI for details).
Not surprisingly, the most challenging step in this synthesis was the enolate-based installation of the hydroxymethyl group at C-10 onto 3 to forge the final quaternary center. Aside from obvious difficulties posed by the steric hindrance of this position, there were unique chemo- and regioselectivity issues that needed to be overcome (six possible monohydroxymethylated products could be formed). In the first instance, there was the hurdle of engaging the more hindered C-5/10 enolate in the presence of the more accessible one at C-8/14. Even if that could be accomplished, it would be extremely difficult to achieve regioselective hydroxymethylation at the desired C-10 position versus the less sterically hindered and undesired C-2 position. To address the chemoselectivity challenge, the C-8 ketone was initially protected as an ethylene glycol ketal or a cyanohydrin; these modifications completely shut down the reactivity despite the multiple conditions evaluated. All ensuing attempts enlisted 3 in the hydroxymethylation event. Roughly 1000 experiments were conducted changing every conceivable variable from the base used to deprotonate, the solvent employed, additives, and the electrophile. Emerging from this exhaustive study was the remarkable finding that the addition of LaCl3·2LiCl15 to the extended sodium enolate of 3, followed by quenching with freshly prepared formaldehyde gas led to the desired adduct 11 in 84% yield as a 2:1 diastereomeric mixture favoring 11 (3 g scale). Complete chemoselectivity was achieved because the more accessible C-8 ketone did not enolize under these conditions presumably due to (1) increasing angle strain introduced onto the bicyclic system upon C-8/14 enolate generation and (2) destabilizing non-bonding interactions between π-systems (see inset graphic and SI). To the best of our knowledge, this represents the first use of LaCl3·2LiCl to control the course of an aldol reaction of an extented enolate and might be of general utility in similarly challenging contexts.
The next step required the reduction of the more sterically encumbered C-5 ketone in the presence of the C-8 ketone. Since reagent-based strategies failed, an internal protection strategy was devised by selective in situ formation of ketal 12 (TFA, CH(OMe)3), followed by reduction with LiBH4 in the presence of Zn(OTf)2 to furnish alcohol 13 in 83%. As with the hydroxymethylation step, this combination of reagents has no precedent (freshly prepared Zn(BH4)2 showed no reactivity and the Zn salt must be added before the borohydride). Of the >100 reducing conditions brought to bear on this step, this was the only one capable of providing the desired diastereomer as the major product (for a more extensive list, see SI).
The final carbon atom was appended onto C-8 of 13 in a relatively straightforward fashion. Thus, rupture of the ketal and concomitant masking of the primary alcohol with a 3,5-dinitrobenzoyl (DNB) group delivered 14 in 86% yield (steps 7–9 were all carried out on a gram-scale). It was essential to transform 13 to 14 without isolation of any intermediates, as the free alcohol equilibrates to a mixture of hemiketals that are difficult to manipulate. The DNB group was fortuitously chosen in order to render intermediates crystalline for stereochemical assignment but was later realized to be essential (use of a Bz ester was unsuccessful). Formation of the strained THF ring of 1 could be accomplished using a 6:1 ratio of CH(OMe)3:MeOH with methanesulfonic acid. The intermediate ketal 15 was directly exposed to ZnI2 and TMSCN, smoothly incorporating the C-7 carbon which after saponification in the same flask delivered 16, the complete carbon skeleton of 1.
The synthesis of 1 was completed in a single step from 16 via the net installation of two oxygen atoms (C-15, C-1), olefin isomerization (C-1/2 to C-2/3), and tertiary stereogenic center formation (C-16). The cascade of seven reactions in a single flask initiated upon exposure to DMDO to generate diepoxide 17 (not isolated). When subjected to InI3 and MgI2 the C-1/2 epoxide is ruptured to the corresponding iodohydrin and a stereospecific 1,2-hydride shift is elicited to install the C-16 stereochemistry. Subsequent addition of Dess–Martin periodinane oxidized the C-1 hydroxyl group to produce α-iodoketone 18 that, upon exposure to aqueous Oxone, oxidized the iodine atom to a putative α-iodoso species16 which spontaneously eliminated to deliver enantiopure (−)-1 in 76% isolated yield (verified by X-ray crystallography, [α]D23 = −98.2° (c 1.00, MeOH)). The dehydroiodination of 18 could not be accomplished under basic conditions as the C–I bond appears to adopt an unfavorable conformation for E2 elimination.
With a robust route to the natural product in hand, it was screened against 32 different cancer cell lines (including HeLa) across four different laboratories and showed little to no activity in any of them, suggesting that the originally exciting findings were either due to a flawed assay or impure 1.17 The route described herein was used to prepare >80 mg of (−)-1 to date, but it could easily be adapted to procure gram quantities had the biological data been more compelling.
The primary goal of this study was to demonstrate that the notorious difficulties surrounding the exotic architecture of 1 could be solved by total synthesis in a practical way. This was eventually2b,3b accomplished by employing a strategy not wedded to Diels–Alder type disconnections18 but rather a desire to maximize convergency and minimize concession steps (steps 4, 5, and 9).19 Some of the memorable lessons from this concise synthesis (73% ideal) include: (1) short access to 5 via a highly enantioselective conjugate addition and anti-Baldwin cyclization; (2) convergent coupling of fragments 5 and 6 with concomitant pinacol shift and olefin isomerization to establish the [2.2.2] bicycle; (3) the first use of a lanthanide Lewis acid to control the regio- and stereochemical course of an aldol reaction with an extended enolate; (4) Zn(OTf)2-assisted reversal of the stereoselectivity of a bis-neopentyl ketone reduction; (5) an efficient cascade sequence to install key oxidations and unsaturation.
Acknowledgments
Financial support for this work was provided by NIH (GM-118176) and a graduate fellowship from Bristol-Myers Squibb (to A.C.). We thank Calibr, Stemcentrx, and Sirenas for assistance with evaluating the bioactivity of 1, Dr. D.-H. Huang and Dr. L. Pasternack for NMR spectroscopic assistance, and Prof. A. L. Rheingold and Dr. C. E. Moore for X-ray crystallographic analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b06623.
Experimental procedures, analytical data (1H and 13C NMR, MS) for all new compounds as well as summaries of unsuccessful approaches (PDF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Crystallographic data (CIF)
Author Present Address
† LEO Pharma, Industriparken 55, 2750 Ballerup, Denmark
Author Contributions
§ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Li S.-H.; Wang J.; Niu X.-M.; Shen Y.-H.; Zhang H.-J.; Sun H.-D.; Li M. L.; Tian Q.-E.; Lu Y.; Cao P.; Zheng Q.-T. Org. Lett. 2004, 6, 4327. 10.1021/ol0481535. [DOI] [PubMed] [Google Scholar]
- For studies toward maoecrystal V, see:; a Gong J.; Lin G.; Li C. C.; Yang Z. Org. Lett. 2009, 11, 4770. 10.1021/ol9014392. [DOI] [PubMed] [Google Scholar]; b Krawczuk P. J.; Schöne N.; Baran P. S. Org. Lett. 2009, 11, 4774. 10.1021/ol901963v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nicolaou K. C.; Dong L.; Deng L.; Talbot A. C.; Chen D. Y.-K. Chem. Commun. 2010, 46, 70. 10.1039/B917045F. [DOI] [PubMed] [Google Scholar]; d Peng F.; Yu M.; Danishefsky S. J. Tetrahedron Lett. 2009, 50, 6586. 10.1016/j.tetlet.2009.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Singh V.; Bhalerao P.; Movin S. M. Tetrahedron Lett. 2010, 51, 3337. 10.1016/j.tetlet.2010.04.082. [DOI] [Google Scholar]; f Lazarski K. E.; Hu D. X.; Stern C. L.; Thomson R. J. Org. Lett. 2010, 12, 3010. 10.1021/ol101025r. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Baitinger I.; Mayer P.; Trauner D. Org. Lett. 2010, 12, 5656. 10.1021/ol102446u. [DOI] [PubMed] [Google Scholar]; h Peng F.; Danishefsky S. J. Tetrahedron Lett. 2011, 52, 2104. 10.1016/j.tetlet.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Gu Z.; Zakarian A. Org. Lett. 2011, 13, 1080. 10.1021/ol1031238. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Dong L.; Deng L.; Lim Y. H.; Leung G. Y. C.; Chen D. Y.-K. Chem. - Eur. J. 2011, 17, 5778. 10.1002/chem.201100232. [DOI] [PubMed] [Google Scholar]; k Lazarski K. E.; Akpinar B.; Thomson R. J. Tetrahedron Lett. 2013, 54, 635. 10.1016/j.tetlet.2012.11.135. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Carberry P.; Viernes D. R.; Choi L. B.; Fegley M. W.; Chisholm J. D. Tetrahedron Lett. 2013, 54, 1734. 10.1016/j.tetlet.2013.01.075. [DOI] [Google Scholar]; m Jansone-Popova S.; May J. A. Tetrahedron 2016, 72, 3734. 10.1016/j.tet.2016.03.101. [DOI] [Google Scholar]
- For Ph.D. theses on maoecrystal V, see:; a Smith M.Efforts Toward the Synthesis of Maoecrystal V and Atropurpuran with the Discovery of Novel Skeletal Rearrangements. Ph.D. Dissertation, Princeton University, 2013. [Google Scholar]; b Krawczuk P. J.Studies Toward the Total Synthesis of Maoecrystal V. Ph.D. Dissertation, The Scripps Research Institute, 2011. [Google Scholar]
- For completed total syntheses of maoecrystal V, see:; a Gong J.; Lin G.; Sun W.; Li C.-C.; Yang Z. J. Am. Chem. Soc. 2010, 132, 16745. 10.1021/ja108907x. [DOI] [PubMed] [Google Scholar]; b Peng F.; Danishefsky S. J. J. Am. Chem. Soc. 2012, 134, 18860. 10.1021/ja309905j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lu P.; Gu Z.; Zakarian A. J. Am. Chem. Soc. 2013, 135, 14552. 10.1021/ja408231t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lu P.; Mailyan A.; Gu Z.; Guptill D. M.; Wang H.; Davies H. M. L.; Zakarian A. J. Am. Chem. Soc. 2014, 136, 17738. 10.1021/ja510573v. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zheng C.; Dubovyk I.; Lazarski K. E.; Thomson R. J. J. Am. Chem. Soc. 2014, 136, 17750. 10.1021/ja5109694. [DOI] [PubMed] [Google Scholar]; f Zhang W.-B.; Shao W.-B.; Li F.-Z.; Gong J.-X.; Yang Z. Chem. - Asian J. 2015, 10, 1874. 10.1002/asia.201500564. [DOI] [PubMed] [Google Scholar]
- Han Q.-B.; Cheung S.; Tai J.; Qiao C.-F.; Song J.-Z.; Tso T.-F.; Sun H.-D.; Xu H.-X. Org. Lett. 2006, 8, 4727. 10.1021/ol061757j. [DOI] [PubMed] [Google Scholar]
- Robert T.; Velder J.; Schmalz H.-G. Angew. Chem., Int. Ed. 2008, 47, 7718. 10.1002/anie.200803247. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Shimizu M. J. Am. Chem. Soc. 1982, 104, 4299. 10.1021/ja00379a066. [DOI] [Google Scholar]
- Beshara C. S.; Hall A.; Jenkins R. L.; Jones K. L.; Jones T. C.; Killeen N. M.; Taylor P. H.; Thomas S. P.; Tomkinson N. C. O. Org. Lett. 2005, 7, 5729. 10.1021/ol052474e. [DOI] [PubMed] [Google Scholar]
- Davis F. A.; Vishwakarma L. C.; Billmers J. M.; Finn J. J. Org. Chem. 1984, 49, 3241. 10.1021/jo00191a048. [DOI] [Google Scholar]
- Hosomi A.; Sakurai H. Tetrahedron Lett. 1976, 17, 1295. 10.1016/S0040-4039(00)78044-0. [DOI] [Google Scholar]
- Baldwin J. E.; Lusch M. J. Tetrahedron 1982, 38, 2939. 10.1016/0040-4020(82)85023-0. [DOI] [Google Scholar]
- Enone 6 was made via a modified literature procedure in four steps, no column chromatography, see SI.
- Ren H.; Krasovskiy A.; Knochel P. Org. Lett. 2004, 6, 4215. 10.1021/ol048363h. [DOI] [PubMed] [Google Scholar]
- When THF was used as a solvent, the organomagnesium species generated from 6 reacted with itself upon formation, thus 4–5 equiv of 6 were required to achieve full conversion. In PhMe, this side reaction was not observed perhaps due to greater aggregation in the non-coordinating solvent.
- a Krasovskiy A.; Kopp F.; Knochel P. Angew. Chem., Int. Ed. 2006, 45, 497. 10.1002/anie.200502485. [DOI] [PubMed] [Google Scholar]; b Hall L. P.; Gilchrist J. H.; Collum D. B. J. Am. Chem. Soc. 1991, 113, 9571. 10.1021/ja00025a023. [DOI] [Google Scholar]
- Horiuchi C. A.; Ji S.-J.; Matsushita M.; Chai W. Synthesis 2004, 2, 202. 10.1055/s-2003-44371. [DOI] [Google Scholar]
- For another recent example of irreproducible biological assays for a natural product, see:Villaume M. T.; Sella E.; Saul G.; Borzilleri R. M.; Fargnoli J.; Johnston K. A.; Zhang H.; Fereshteh M. P.; Dhar M. T. G.; Baran P. S. ACS Cent. Sci. 2016, 2, 27. 10.1021/acscentsci.5b00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou K. C.; Snyder S. A.; Montagnon T.; Vassilikogiannakis G. Angew. Chem., Int. Ed. 2002, 41, 1668.. [DOI] [PubMed] [Google Scholar]
- Gaich T.; Baran P. S. J. Org. Chem. 2010, 75, 4657. 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.