

Abstract
Golgi apparatus (GA) is a membrane-bound organelle that serves a multitude of critical cellular functions including protein secretion and sorting, and cellular polarity. Many Mendelian diseases are caused by mutations in genes encoding various components of GA. GOLGA2 encodes GM130, a necessary component for the assembly of GA as a single complex, and its deficiency has been found to result in severe cellular phenotypes. We describe the first human patient with a homozygous apparently loss of function mutation in GOLGA2. The phenotype is a neuromuscular disorder characterized by developmental delay, seizures, progressive microcephaly, and muscular dystrophy. Knockdown of golga2 in zebrafish resulted in severe skeletal muscle disorganization and microcephaly recapitulating loss of function human phenotype. Our data suggest an important developmental role of GM130 in humans and zebrafish.
Keywords: congenital disorder of glycosylation, microcephaly, muscular dystrophy, golgin
INTRODUCTION
Golgi apparatus (GA) is a membrane-bound cellular organelle composed of multiple stacks and cisterns that are interconnected to form a single complex (Lowe 2011). Classically, GA has been studied in the context of its role in the secretion and sorting of proteins as they move progressively from the endoplasmic reticulum to the cis- and medial Golgi cisternae to the trans-Golgi network (TGN) (anterograde transport). In this sense, GA functions as a major hub for posttranslational modification of proteins and their sorting to their final destination through vesicular transport, although some proteins are retained within specific GA cisternae and yet others are returned to the endoplasmic reticulum (retrograde transport) (Allan et al. 2002; Santiago-Tirado and Bretscher 2011). A more recently identified role of GA has been in relation to cytoskeleton and cellular polarity. Specifically, studies have shown that GA functions as a microtubule organizing center that nucleates microtubules, thereby revealing an essential role for GA in cell division, migration and ciliogenesis, and the molecular network that controls these processes has only recently been elucidated (Egea et al. 2015; Gurel et al. 2014; Wei et al. 2015; Yadav et al. 2009).
Many Mendelian diseases have been found to be caused by mutations in genes encoding various components of GA, and although the resulting phenotypes are highly variable and heterogeneous there is an overrepresentation of glycosylation disorders. These are multisystem disorders that result from perturbed posttranslational glycosylation of proteins, a process that is housed in GA (Hennet and Cabalzar 2015; Potelle et al. 2015). Interestingly, GA-related genes that are mutated in the above mentioned human disorders tend to encode enzymes and other proteins that are not essential for the assembly and maintenance of GA per se, presumably because the latter would not be compatible with life as demonstrated for TRIP11 (GMAP210) and USO1 (p115), deficiency of which results in embryonic lethality in humans and mouse, respectively (Kim et al. 2012; Smits et al. 2010).
GOLGA2 encodes GM130, a protein initially identified in 1995 in a screen for novel GA-associated proteins (Nakamura et al. 1995). Subsequent studies revealed that this protein, among other roles, provides the long sought molecular link between GA and the cytoskeleton such that its deficiency results in fragmentation of GA and failure of the cell to undergo normal mitosis (Barr et al. 1998; Lowe et al. 1998; Nakamura et al. 1997). No mutations have been reported in this gene and there are no available mouse models. In this study, we report the first human patient with a homozygous frameshift mutation in GOLGA2 and an associated neuromuscular disorder that is also recapitulated in the zebrafish morphants.
MATERIALS AND METHODS
Human Subject
Patient underwent full clinical evaluation for her multisystem disorder (see below). A standard clinical exome consent was used for the trio-whole genome sequencing analysis at a clinical lab. Prior to their enrollment in our IRB-approved research protocol (KFSHRC RAC#2080006), parents signed an informed consent form. Blood was then collected from index and parents in EDTA tubes for DNA extraction. Blood was also collected from index in a sodium heparin tube for the establishment of EBV-transformed lymphoblastoid cell line (LCL).
Characterization of GOLGA2 Mutation at the RNA and Protein Level
To check for the stability of GOLGA2 transcript, RT-PCR was carried out using RNA extracted from patient and control LCL, using RNeasy Qiagen kit as per the manufacturer's instructions. Primer sequence and PCR conditions are available upon request. Western blot was performed to check for the integrity of GOLGA2 protein, using protein extracted from patient and control LCL and GM130 antibody (Abcam Ep892Y).
Autozygome Mapping and Exome Analysis
Genomewide mapping of autozygous intervals (autozygome) was as described before (Alkuraya 2010, 2012). Briefly, runs of homozygosity of >2Mb were taken as surrogates of autozygosity and these were determined by AutoSNPa with the input file generated by genomewide SNP genotyping using the Axiom SNP Chip platform (Affymetrix) following the manufacturer's instructions. Exome capture was performed using TruSeq Exome Enrichment kit (Illumina) following the manufacturer's protocol. Samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using an Illumina HiSeq 2000 Sequencer. The reads were mapped against UCSC hg19 by BWA. The SNPs and Indels were detected by SAMTOOLS.
Variants from WES were filtered such that only novel, coding/splicing, homozygous variants that are within the autozygome of the index and are predicted to be pathogenic were considered as likely causal variants (Alazami et al. 2015; Alkuraya 2013). Frequency of variants was determined using publically available variant databases (1000 Genomes, Exome Variant Server and ExAC) as well as a database of 679 in-house ethnically matched exomes. Pathogenicity was likely if the mutation is loss-of-function (splicing/truncating) or, in the case of missense/in-frame indels, removes a highly conserved amino acid and is predicted to be pathogenic by the two in silico prediction modules PolyPhen and SIFT.
Expression Analysis of Golga2 in Mouse Tissues
To detect the expression of Golga2, RT-PCR reaction was performed using RNA extracted from different mouse tissue (kidney, heart liver spleen, lung and brain, and skeletal muscle), using RNeasy kit (Qiagen) as per the manufacturer's protocol.. Mouse RT-PCR primers covering Golga2 exons 2-9 were used for this experiment.
Fish and Embryo Maintenance
Fish were bred and maintained as described previously. Control embryos were obtained from the Oregon AB line and were staged by hours (hpf) or days (dpf) post fertilization at 28.5 °C. All animal work was performed with approval from the Boston Children's Hospital Animal Care and Use Committee.
Morpholino Knockdown, mRNA Rescue and Immunoblotting
Two antisense morpholinos (MOs), one targeting the translational start site (T-MO) and one targeting the exon13-intron13 splice site (e13i13-MO), were designed to knockdown the zebrafish golga2 transcript (ENSDART00000092087; GeneTools LLC, Philomath, OR, USA). The morpholino sequences were golga2 T-MO: 5’- GCGAACGACACCACGTCATCAGATC -3’ and golga2 e13i13-MO: 5’- ATGCTGATAAATGAAGAGACTCACT -3’. A morpholino against human β-globin, which is not homologous to any sequence in the zebrafish genome by BLAST search, was used as a negative control for all injections (5′-CCTCTTACCTCAGTTACAATTTATA-3′). Morpholinos were dissolved in 1X Danieau's buffer with 0.1% phenol red and 1–2 nL (1–3.5 ng) injected into the yolk of 1-cell stage wild-type embryos. For rescue experiments, full-length human GOLGA (NM_004486) cDNA was obtained from the DNA Resource Core at Harvard Medical School and cloned into a pCSDest destination vector (created by Nathan Lawson) using Gateway technology (Invitrogen, Carlsbad, CA, USA). mRNA was synthesized in vitro using mMessage mMachine SP6 kits (Ambion, Austin, TX, USA). mRNAs (100–200 pg) were injected into embryos at the 1-cell stage independently or in combination with morpholinos, and subsequent phenotypic analyses performed at 3 dpf.
For immunoblotting, zebrafish embryos at 3 dpf were homogenized in buffer containing Tris-Cl (20 mM, pH 7.6), NaCl (50 mM), EDTA (1 mM), NP-40 (0.1%) and complete protease inhibitor cocktail (Roche Applied Sciences, Indianapolis, IN, USA). Following centrifugation at 11000g at 4°C for 15 min, protein concentration in supernatants was determined by BCA protein assay (Pierce, Rockford, IL, USA). Proteins were separated by electrophoresis on 4–12% gradient Tris–glycine gels (Invitrogen) and transferred onto polyvinylidene difluoride membrane (Invitrogen). Membranes were blocked in PBS containing 5% casein and 0.1% Tween-20, then incubated with either rabbit polyclonal anti-GOLGA2 (1:100, ab30637, Abcam, Cambridge, MA, USA) or mouse monoclonal anti-β-actin (1:1000, A5441, Sigma, St. Louis, MO) primary antibodies. After washing, membranes were incubated with horseradish peroxidase-conjugated anti-mouse (1:5000, 170-6516) IgG secondary antibody (BioRad, Hercules, CA, USA). Proteins were detected using the SuperSignal chemiluminescent substrate kit (Pierce).
Histology
Zebrafish (3 dpf) were fixed in 4% paraformaldahyde overnight at 4°C. Fish were subsequently washed with 1xPBS (Phosphate buffer saline) and incubated in 10% sucrose/1XPBS for 1 hour at room temperature followed by incubating in 20% sucrose/1xPBS solution overnight at 4°C. The fish embryos were embedded in OCT and 8um thin frozen section were cut. Slides were fixed in a formalin-ethyl alcohol-acetic acid solution for 30 seconds, washed with water (twice, 5 minutes each) and stained with DAPI solution (1:5000, D1306, Thermofisher Scientific). Slides were finally rinsed with water and visualized using a Perkin Elmer UltraVIEW VoX spinning disk confocal microscope.
Statistical analysis
Data were statistically analyzed by parametric Student t-test (two-tailed) and were considered significant when p < 0.01. All data analyses were performed using GraphPad Prism 6 software (GraphPad Software Inc., La Jolla, CA, USA), and are described as the mean ± standard deviation.
RESULTS
Clinical Report
Index is a 10.5m old infant girl born to double first cousin Saudi parents following uneventful full term pregnancy and spontaneous vaginal delivery. Parents have had two first trimester miscarriages. Birth growth parameters were as follows: weight 3.22 kg (37th percentile), length 49.5cm (53rd percentile) and head circumference 32.5 cm (7th percentile). Neonatal history is unremarkable. Microcephaly, hypotonia and failure to thrive were noticeable starting at 4m of age, around which time she was diagnosed with strabismus. At 6m of age, she developed a febrile illness that coincided with the onset of infantile spasms. The latter was fully controlled with vigabatrin therapy. The aforementioned clinical features persisted and were accompanied by developmental delay, mostly in the motor domain e.g. she was unable to sit at the time of her evaluation at 10.5m. Physical examination revealed poor weight gain (−2.7SD) and linear growth (−2.2SD), and progressive microcephaly (HC −4.2SD). Only subtle facial dysmorphism was noted in the form of tented upper lip, micrognathia, brachycephaly and strabismus. She also had central hypotonia.
Brain MRI revealed nonspecific cerebral volume loss with delayed myelination and thinning of corpus callosum. EEG revealed hypsarrhythmia that normalized after treatment. There was mild elevation of liver enzymes. Creatine Kinase (CK) was persistently elevated with a range of 959-1,368U/L. Isoelectric focusing of transferrin was normal but Apo CIII-0/Apo CIII-2 ratio was slightly elevated at 0.53 (normal is <0.48). A muscle biopsy revealed nonspecific mild muscle fiber atrophy. Clinical trio-exome sequencing revealed no likely pathogenic variants. Molecular karyotyping was largely normal except for a small maternally inherited duplication that was felt to be unrelated to the patient's illness.
Identification of a Likely Null GOLGA2 Mutation
We have previously shown that research-grade whole exome sequencing can uncover causal mutations that are missed by clinical exome sequencing (Ben-Omran et al. 2015; Shaheen et al. 2015; Shamseldin et al. 2015). Therefore, we enrolled this patient in a research protocol that entails combined autozygome/exome analysis. Our analysis revealed only three novel variants that met our filtering criteria (see Methods). These include a missense variant in FHOD3: NM_025135:c.926T>A:p.(Leu309Gln), a missense variant in SCFD2: NM_152540:c.806T>G:p.(Val269Gly), and a 4bp deletion in GOLGA2: NM_004486.4:c.1266_1269del:p.(Glu423Argfs*6). FHOD3 was deemed an unlikely candidate since knockout mice develop a primarily heart maturation defect and die in utero. A recent GWAS study has shown that SCFD2 locus is linked to cataract in Australian Shepherd. Therefore, the homozygous truncating candidate in GOLGA2 was an attractive candidate in view of its established role as a master regulator of GA (see below). In view of the truncating nature of the GOLGA2 mutation, we first sought to assess whether it led to NMD but found no evidence of that on RTPCR (Figure S1). However, immunoblotting using an anti-human GM130 antibody raised against a region of the protein that is both upstream and downstream to our mutation failed to identify the expected 130kDa band that was readily visible in the two controls used (Figure S1). Furthermore, we found no evidence of the predicted ~40kDa protein indicating that the mutation is likely null at the protein level. In view of these results and the multisystem nature of the phenotype observed in the patient, we sought to analyze the expression of the murine ortholog Golga2. In line with available expression data from the Expression Atlas (http://www.ebi.ac.uk/gxa), we found evidence for ubiquitous expression of Golga2 (Figure S2). To further validate our genetic findings, we proceeded with testing the effect of its deficiency in zebrafish as a model organism.
golga2 Deficiency in Zebrafish Recapitulates Human Muscle and Brain Abnormalities
The effect of golga2 deficiency in zebrafish was investigated by knocking down the golga2 gene with antisense morpholinos. Two independent morpholinos targeting an exon-intron splice site and translational start site were used. Knockdown with either splice site or translational morpholinos resulted in highly similar phenotypes in golga2 morphants: a dorsal curvature in skeletal muscle indicative of myopathy and smaller brains (Figures 2 and 3). The development of other organs such as eyes and heart were not affected in morphant fish suggesting that Golga2 deficiency primarily affects skeletal muscle and brain. The skeletal muscles of these morphants were analyzed by polarized light that showed a reduced birefringence in axial skeletal muscles suggesting disorganized skeletal muscles (Figure 2). The skeletal muscle disorganization was also associated with impaired mobility in golga2 morphants. Behavioral characterization of 3 dpf morphant fish, knocked down for golga2, using the touch evoked response assay showed significantly diminished motility in comparison to control fish (WT fish: 4.32 ± 0.66 cm/0.1 sec; golga (e13-i13-MO): 2.13 ± 0.69 cm/0.1 sec; golga2 (T-MO): 1.84 ± .64 cm/0.1 sec) suggesting overall muscle weakness. Immunoblotting confirmed the knockdown in golga2 morphants and a corresponding decrease in protein levels (Figure 2). Brain was phenotypically smaller in morphant fish in comparison to controls (Figure 3, top). Quantification of cross-section area in brain showed a significant decrease in morphant brains (26 ± 5.6%) as compared to controls suggesting microcephaly. golga2 morphant fish die between 3-4 dpf suggesting a crucial requirement of GOLGA2 function. Overexpression of human GOLGA2 mRNA in morphants resulted in a significant decrease in fish exhibiting muscle and brain phenotypes suggesting the specificity of morpholino injections and demonstrating the ability of human ortholog to complement zebrafish gene function. These studies highlight the crucial requirement of GOLGA2 function in skeletal muscles in vertebrates.
Figure 2. golga2 gene knockdown results in skeletal muscle defects in zebrafish.
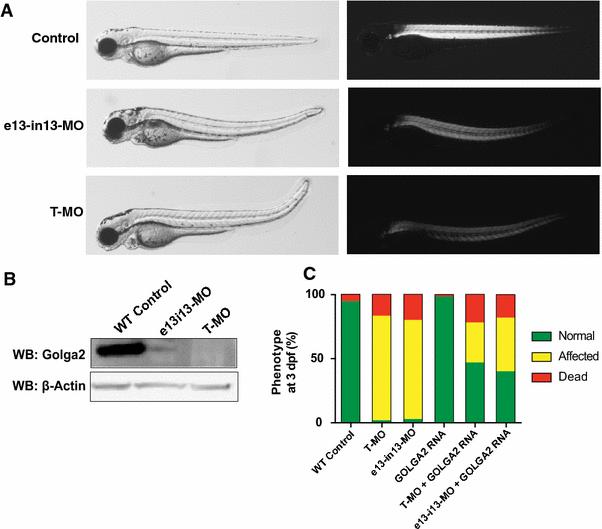
(A) Embryos injected with 2.5 ng of golga2 translation- (T-MO) and splice-blocking (e13i13-MO) morpholinos both show dorsally curved phenotype at 3 dpf in comparison to WT-controls (left panel). Visualization of embryos underpolarized light showed an overall reduction in birefringence indicative of disorganized sarcomeric structure (B) Western blot demonstrates that T- and e13i13-MO-mediated knockdowns of golga2 result in significantly decreased levels of zebrafish golga2 protein by 3 dpf. (C) Overexpression of human GOLGA2 mRNA in morphants resulted in recue of skeletal muscle and brain phenotypes. Green indicates normal embryos, yellow indicates embryos displaying muscle and brain abnormalities, and red indicates dead embryos a 3 dpf. Two independent experiments were performed for all rescue studies, with at least 100 embryos injected for each group. Statistical significance relative to the MO-only experimental group was determined by a Student's t-test, P < 0.01.
Figure 3. Knockdown of golga2 results in microcephaly in fish.
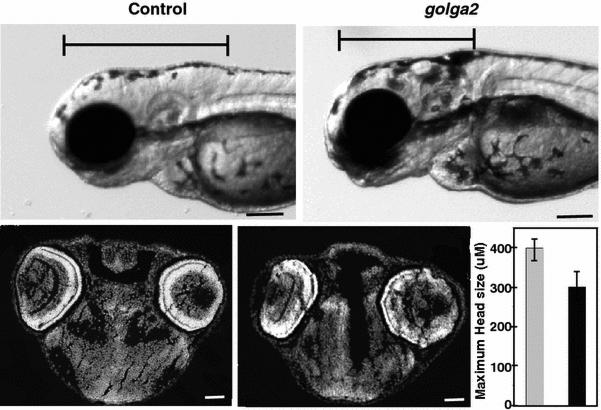
Morphant fish (e13-i13) exhibited smaller heads in comparison to wild-type controls. The length of brain was measured from forebrain to the beginning of first somite in both control and morphants (top panel). Cross-sections of brain (stained with DAPI) and quantification of brain diameter showed ~25% smaller brains in morphants (bottom panel).
DISCUSSION
Consanguinity provides a rich source of homozygosity for even very rare alleles that are statistically highly improbable to be rendered homozygous otherwise in an outbred population (Alkuraya 2010, 2014; Alsalem et al. 2013). When these alleles effect apparent loss of function i.e. truncations, individuals who harbor them are in essence naturally occurring “knockouts”, and phenotyping them represents an opportunity to study the physiological role of the involved genes in a clinically relevant context (Alkuraya 2015a; Alsalem et al. 2013). The apparently loss of function allele we identified in GOLGA2 is yet another example of a very rare allele that was rendered homozygous in the index by virtue of autozygosity owing to the double consanguinity loops connecting her parents.
Available data on GM130 domain mapping suggest that amino acids 690-986 are required for its binding to cis-golgi, which is the predominant localization of GM130, and this domain is presumably lost as a result of our mutation (Nakamura et al. 1997). A truncated protein that retains the first 436 amino acids can still mediate multiple well characterized interactions of GM130. Specifically, serine 25 (Ser25) is a key regulatory amino acid for the mitotic action of GM130 (Lowe et al. 2000; Lowe et al. 1998). CDK2 phosphorylates Ser25, which leads to liberation of GM130 from its binding partner p115. In a model proposed by Wei et al, the dissociation of GM130 from p115 allows the former to bind alpha α-importin, in turn liberating TPX2 from the α-importin-TPX2 complex (Wei et al. 2015). TPX2 is a potent spindle assembly factor in its free form such that it enucleates microtubules in a process that is further facilitated by GM130 itself. Importantly, the nuclear localization signal (aa 14,15,16,18, 33, 34, 35, 36, 37 and 39) and the binding domain to alpha importin (aa 1-74), both required for the efficient binding to α-importin are also upstream of our mutation (Wei et al. 2015). However, the results we obtained on immunoblotting suggest a truncated protein is either not made or very unstable. Therefore, we suggest that our mutation is a null and that the above listed functions of GM130 are likely lost as a result.
The pathogenesis of the neuromuscular disorder we describe is unclear. Loss of the mitotic function of GM130 is likely to have contributed to the microcephaly and growth deficiency as shown previously for many genes involved in microcephalic primordial dwarfism (Alkuraya 2015b; Shaheen et al. 2014). Our patient's phenotype may have been ameliorated by potential redundancy with other GM proteins. One may also speculate that the patient's clinical manifestations are the result of abnormal sorting or posttranslational modification of one or more proteins due to loss of GM130 localization to the cis-golgi. In this regard, it is worth highlighting that a combination of neurological (infantile spasm, progressive microcephaly, thin corpus callosum) and muscular (elevated CK and muscle fiber atrophy) clinical signs are common features of congenital disorders of glycosylation and this may hint at a possible mild perturbation of glycosylation as suggested by the slightly elevated Apo CIII-2/Apo CIII-0 ratio. Unfortunately, lack of patient fibroblasts precluded a more comprehensive analysis of her glycosylation profile.
In summary, we report the first human case of biallelic truncation of the GOLGA2 and suggest that the resulting phenotype includes progressive microcephaly, infantile spasms, hypotonia, strabismus, thin corpus callosum and muscular dystrophy. Deficiency of golga2 in zebrafish also results in muscle dysfunction associated with microcephaly. Future reports of GOLGA2 mutations are needed to elucidate the full GM130-related phenotype in humans.
Supplementary Material
Figure 1. Identification of a patient with a homozygous truncating mutation in GOLGA2.
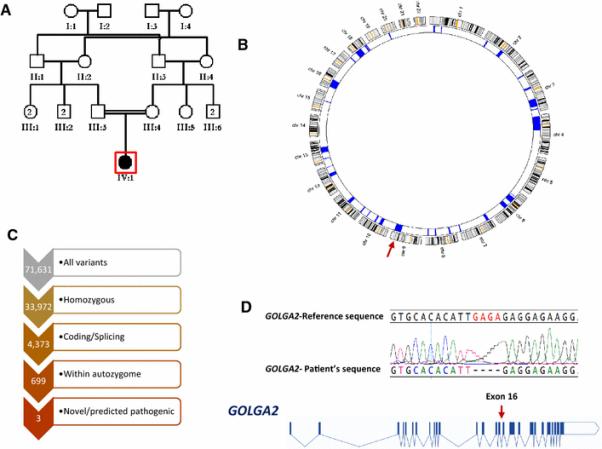
A) Pedigree of study family showing the double consanguineous loops between the parents. B) Autozygome analysis showing multiple block of autozygosity throughout the genome indicated by navy blocks on the corresponding chromosome ideograms. A red arrow indicates the autozygosity block that harbors GOLGA2. C) Filtering steps of the exome variants. D) Cartoon of GOLGA2 with the identified mutation indicated by a red arrow. The sequence chromatogram of the mutation is show on top.
ACKNOWLEDGEMENTS
We thank the family for their enthusiastic participation. This work was supported in part by King Salman Center for Disability Research (FSA). This work was supported by King Abdulaziz City for Science and Technology (13-BIO1113-20 to FSA), the National Institutes of Health, the National Institute of Arthritis and Musculoskeletal and Skin Diseases [K01 AR062601] as well as the Charles H. Hood Foundation Child Health Research Grant to VAG.
Footnotes
Authors declare no conflict of interest
REFERENCES
- Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, Hijazi H, Alshammari M, Aldahmesh MA, Salih MA. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell reports. 2015;10:148–161. doi: 10.1016/j.celrep.2014.12.015. [DOI] [PubMed] [Google Scholar]
- Alkuraya FS. Autozygome decoded. Genetics in Medicine. 2010;12:765–771. doi: 10.1097/GIM.0b013e3181fbfcc4. [DOI] [PubMed] [Google Scholar]
- Alkuraya FS. Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Current protocols in human genetics. 2012;6.12:1–6. 12, 13. doi: 10.1002/0471142905.hg0612s75. [DOI] [PubMed] [Google Scholar]
- Alkuraya FS. The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Human genetics. 2013;132:1197–1211. doi: 10.1007/s00439-013-1344-x. [DOI] [PubMed] [Google Scholar]
- Alkuraya FS. Genetics and genomic medicine in Saudi Arabia. Molecular genetics & genomic medicine. 2014;2:369–378. doi: 10.1002/mgg3.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkuraya FS. Human knockout research: new horizons and opportunities. Trends in Genetics. 2015a;31:108–115. doi: 10.1016/j.tig.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Alkuraya FS. Primordial dwarfism: an update. Current Opinion in Endocrinology, Diabetes and Obesity. 2015b;22:55–64. doi: 10.1097/MED.0000000000000121. [DOI] [PubMed] [Google Scholar]
- Allan VJ, Thompson HM, McNiven MA. Motoring around the Golgi. Nature cell biology. 2002;4:E236–E242. doi: 10.1038/ncb1002-e236. [DOI] [PubMed] [Google Scholar]
- Alsalem AB, Halees AS, Anazi S, Alshamekh S, Alkuraya FS. Autozygome sequencing expands the horizon of human knockout research and provides novel insights into human phenotypic variation. PLoS genetics. 2013;9:e1004030. doi: 10.1371/journal.pgen.1004030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr FA, Nakamura N, Warren G. Mapping the interaction between GRASP65 and GM130, components of a protein complex involved in the stacking of Golgi cisternae. The EMBO journal. 1998;17:3258–3268. doi: 10.1093/emboj/17.12.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Omran T, Alsulaiman R, Kamel H, Shaheen R, Alkuraya FS. Intrafamilial clinical heterogeneity of CSPP1-related ciliopathy. American Journal of Medical Genetics Part A. 2015 doi: 10.1002/ajmg.a.37175. [DOI] [PubMed] [Google Scholar]
- Egea G, Serra-Peinado C, Gavilan MP, Rios RM. Cytoskeleton and Golgi-apparatus interactions: a two-way road of function and structure. Cell Health & Cytoskeleton. 2015;7 [Google Scholar]
- Gurel PS, Hatch AL, Higgs HN. Connecting the cytoskeleton to the endoplasmic reticulum and Golgi. Current Biology. 2014;24:R660–R672. doi: 10.1016/j.cub.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennet T, Cabalzar J. Congenital disorders of glycosylation: a concise chart of glycocalyx dysfunction. Trends in biochemical sciences. 2015 doi: 10.1016/j.tibs.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Kim S, Hill A, Warman ML, Smits P. Golgi disruption and early embryonic lethality in mice lacking USO1. 2012 doi: 10.1371/journal.pone.0050530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M. Structural organization of the Golgi apparatus. Current opinion in cell biology. 2011;23:85–93. doi: 10.1016/j.ceb.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Lowe M, Gonatas NK, Warren G. The mitotic phosphorylation cycle of the cis-Golgi matrix protein GM130. The journal of cell biology. 2000;149:341–356. doi: 10.1083/jcb.149.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M, Rabouille C, Nakamura N, Watson R, Jackman M, Jämsä E, Rahman D, Pappin DJ, Warren G. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell. 1998;94:783–793. doi: 10.1016/s0092-8674(00)81737-7. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Lowe M, Levine TP, Rabouille C, Warren G. The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell. 1997;89:445–455. doi: 10.1016/s0092-8674(00)80225-1. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. The Journal of cell biology. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potelle S, Klein A, Foulquier F. Golgi post-translational modifications and associated diseases. Journal of inherited metabolic disease. 2015:1–11. doi: 10.1007/s10545-015-9851-7. [DOI] [PubMed] [Google Scholar]
- Santiago-Tirado FH, Bretscher A. Membrane-trafficking sorting hubs: cooperation between PI4P and small GTPases at the trans-Golgi network. Trends in cell biology. 2011;21:515–525. doi: 10.1016/j.tcb.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Faqeih E, Ansari S, Abdel-Salam G, Al-Hassnan ZN, Al-Shidi T, Alomar R, Sogaty S, Alkuraya FS. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome research. 2014;24:291–299. doi: 10.1101/gr.160572.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen R, Patel N, Shamseldin H, Alzahrani F, Al-Yamany R, ALMoisheer A, Ewida N, Anazi S, Alnemer M, Elsheikh M. Accelerating matchmaking of novel dysmorphology syndromes through clinical and genomic characterization of a large cohort. Genetics in Medicine. 2015 doi: 10.1038/gim.2015.147. [DOI] [PubMed] [Google Scholar]
- Shamseldin H, Alazami AM, Manning M, Hashem A, Caluseiu O, Tabarki B, Esplin E, Schelley S, Innes AM, Parboosingh JS. RTTN Mutations Cause Primary Microcephaly and Primordial Dwarfism in Humans. The American Journal of Human Genetics. 2015;97:862–868. doi: 10.1016/j.ajhg.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits P, Bolton AD, Funari V, Hong M, Boyden ED, Lu L, Manning DK, Dwyer ND, Moran JL, Prysak M. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. New England Journal of Medicine. 2010;362:206–216. doi: 10.1056/NEJMoa0900158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J-H, Zhang ZC, Wynn RM, Seemann J. GM130 Regulates Golgi-Derived Spindle Assembly by Activating TPX2 and Capturing Microtubules. Cell. 2015;162:287–299. doi: 10.1016/j.cell.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S, Puri S, Linstedt AD. A primary role for Golgi positioning in directed secretion, cell polarity, and wound healing. Molecular biology of the cell. 2009;20:1728–1736. doi: 10.1091/mbc.E08-10-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.