

Abstract
Advanced stages of papillary and anaplastic thyroid cancer represent a highly aggressive subset, in which there are currently few effective therapies. We and others have recently demonstrated that c-Src is a key mediator of growth, invasion, and metastasis, and therefore represents a promising therapeutic target in thyroid cancer. However clinically, Src inhibitor efficacy has been limited, and therefore further insights are needed to define resistance mechanisms and determine rational combination therapies. We have generated four thyroid cancer cell lines with a greater than 30-fold increase in acquired resistance to the Src inhibitor, dasatinib. Upon acquisition of dasatinib-resistance, the two RAS-mutant cell lines acquired the c-Src gatekeeper mutation (T341M), whereas the two BRAF-mutant cell lines did not. Accordingly, Src signaling was refractory to dasatinib treatment in the RAS-mutant dasatinib-resistant cell lines. Interestingly, activation of the Mitogen Activated Protein (MAP) Kinase pathway was increased in all four of the dasatinib-resistant cell lines, likely due to B-Raf and c-Raf dimerization. Furthermore, MAP2K1/MAP2K2 (MEK1/2) inhibition restored sensitivity in all four of the dasatinib-resistant cell lines, and overcome acquired resistance to dasatinib in the RAS-mutant Cal62 cell line, in vivo. Together, these studies demonstrate that acquisition of the c-Src gatekeeper mutation and MAP Kinase pathway signaling play important roles in promoting resistance to the Src inhibitor, dasatinib. We further demonstrate that up-front combined inhibition with dasatinib and MEK1/2 or ERK1/2 inhibitors drives synergistic inhibition of growth and induction of apoptosis, indicating that combined inhibition may overcome mechanisms of survival in response to single agent inhibition.
Keywords: Src, thyroid cancer, dasatinib, drug resistance, MAPK
Introduction
The MAP Kinase pathway accounts for the majority of mutations in thyroid cancer with a high prevalence of BRAF and RAS mutations (1,2). While there has been great interest in targeting this pathway in thyroid cancer, clinically, thyroid cancers appear to exhibit primary resistance to MAP Kinase pathway inhibition, falling short of the responses seen in melanoma patients with similar activating mutations, but rather mimicking the lack of efficacy observed in colorectal cancer patients (3–5). Thus, with advanced stages of thyroid cancer continuing to maintain a dismal prognosis, it is clear that new therapeutic approaches are desperately needed (6,7).
To address the current dearth of therapies, our lab has focused on the role of Src due to its multiple pro-tumorigenic functions (8,9). We and others have previously demonstrated that inhibition of the Src signaling pathway with the Src inhibitors, dasatinib (BMS-354825) or saracatinib (AZD0530) effectively inhibits thyroid cancer growth and metastasis, both in vitro and in vivo (10–13). Despite dasatinib being a multi-kinase inhibitor, we have further shown that c-Src is a key mediator of these responses (10). Unfortunately however, clinical trials with Src inhibitors have not been as effective at this stage, likely due to resistance mechanisms in response to single agent therapy (14–18). Thus, it is important to define mechanisms of Src inhibitor resistance in order to develop new strategies to more effectively target this oncogenic pathway in the clinic (9).
Multiple mechanisms of resistance have been observed in response to single agent targeted therapies. Two major mechanisms include the activation of bypass pathways, and the disruption of drug binding due to targeted mutations (e.g. Gatekeeper mutations) (19). Mutation of the BCR-ABL gatekeeper residue has been reported in CML, whereas EGFR and ALK gatekeeper mutations have been reported in lung cancer (20–22). Bypass pathway mechanisms have also been frequently reported, with Met amplification and FGFR signaling promoting resistance to EGFR inhibition in lung cancer, and relief of feedback inhibition of the MAP Kinase pathway in response to vemurafenib (PLX4032) treatment in BRAFV600E-mutant melanoma and thyroid cancer (4,23–25). Additionally, mechanisms of reprogramming can allow for the survival of drug tolerant persisters, which then allow for more stable (typically genomic) mechanisms of resistance to be acquired (26).
To elucidate mechanisms of resistance and define strategies to more effectively target Src, we generated 2 BRAF-mutant (BCPAP and SW1736), and 2 RAS-mutant (C643 and Cal62) thyroid cancer cell lines with acquired resistance to the Src inhibitor, dasatinib. Interestingly, we observed acquisition of the c-Src gatekeeper mutation only in the RAS-mutant dasatinib-resistant (DasRes) cell lines, whereas reactivation of the MAP Kinase pathway was a conserved mechanism of resistance in response to dasatinib treatment in both the BRAF- and RAS-mutant DasRes cell lines. Consistent with an increased reliance on the MAP Kinase pathway upon acquisition of resistance, inhibition of the MAP Kinase pathway effectively inhibited growth both in vitro and in vivo. Additionally, combined Src and MEK1/2 inhibition resulted in synergistic inhibition of growth and increased apoptosis. Overall, these results indicate that inhibition of the MAP Kinase pathway represents a promising strategy to overcome resistance to the Src inhibitor, dasatinib, and that combined inhibition of Src and the MAP Kinase pathway may overcome early mechanisms of survival derived from either monotherapy.
Materials and Methods
Reagents
For the drug screening assays, selumetinib (AZD6244) and SCH772984 were purchased from SelleckChem, trametinib (GSK-1120212) was purchased from LC laboratories or SelleckChem, and dasatinib (BMS-354825) was generously provided by Bristol-Meyers Squibb. The drugs were dissolved in dimethyl sulfoxide. For in vivo studies, dasatinib was dissolved in 80mmol/L sodium citrate buffer, pH3.0 and trametinib (SelleckChem) was dissolved in 0.5% hydroxypropylenemethylcellulose (Sigma) and 0.2% Tween-80 in distilled water (pH 8.0).
Cell Culture
Human thyroid cancer cell lines C643, SW1736, BCPAP, and Cal62 were grown in RPMI (Invitrogen, Carlsbad, CA) supplemented with 5% FBS (HyClone Laboratories, Logan, UT), and the A375 cell line was grown in DMEM and supplemented with 10% FBS. All lines were maintained at 37°C in 5% CO2. All cell lines were validated using short tandem repeat profiling using the Applied Biosystems Identifiler kit (#4322288) in the Barbara Davis Center BioResources Core Facility, Molecular Biology Unit, at the University of Colorado, as previously described (27). The SW1736 and C643 cells were generously provided by Dr. K. Ain (University of Kentucky, Lexington, KY), with permission from Dr. N.E. Heldin (University Hospital, Uppsala, Sweden). The BCPAP and Cal62 cells were generously provided by Dr. M. Santoro (Medical School, University “Federico II” of Naples, Naples, Italy). All cell lines were routinely monitored for Mycoplasma contamination using the Lonza Mycoalert system (Lonza Walkersville, Inc., Walkersville, MD), according to the manufacturer's directions.
Generation of Dasatinib resistant cell lines
Cell lines were cultured in gradually increasing concentrations of dasatinib starting at 50nM, or in DMSO vehicle control alongside, for a period of nine months (20-45 passages). The dasatinib concentration was increased when cell confluency reached 70-80%. Dasatinib resistance was measured monthly by sulforhodamine B (SRB) growth assays, as previously described (10). Cells were then maintained as a heterogeneous population and in 2μM dasatinib once they reached a resistant state. DasRes cells were also authenticated by STR profiling, as described above.
Sanger Sequencing
Sequencing was performed on gDNA. gDNA was collected using the Quick-gDNA MiniPrep kit (Zymo Research) and amplified using exon 9 specific primers for the c-Src gatekeeper region. Exon 9 was sequenced using the forward primer c-Src 5′-CAGGAGGCCCAGGTCATG-3′ and reverse primer 5′-ATCTGAGCAGCCATGTCCAC-3′ at the University of Colorado, Department of Pathology DNA Sequencing core.
RNA Sequencing
mRNA sequencing was performed at the University of Colorado Cancer Center (UCCC) Genomics and Microarray Core on the HiSeq2000 (single read 100 cycles). On average, 70 million reads (53.5 - 93 million) per sample were obtained, with an average mapping of 98% (97.2-98.8%) to the hg19 reference genome using the tophat/cufflinks workflow as previously described (28). To determine the enriched pathways between the control and DasRes cell lines, fragments per kilobase per million mapped reads (FPKM) from each sample were estimated and analyzed using Gene Set Enrichment Analysis (GSEA). We used the pathways from the Kyoto Encyclopedia Genes and Genomes (KEGG) as the gene set and performed 1000 gene set permutations. As this is a discovery step, we considered pathways with nominal p-value < 0.15 as candidate hits for follow-up experiments.
Cellular Growth Assays
Cells (1500/well for BCPAP, SW1736, C643; 1000/ well for Cal62) were plated in triplicate in 96 well plates. Cells were treated with increasing concentrations of the indicated drugs and cell growth was measured by SRB assay after 3 days of drug treatment (10,29). Briefly, after 72 hours of treatment, cells were fixed with 10% trichloroacetic acid (TCA) at 4°C, stained with 0.057% SRB (Sigma), and unbound SRB was removed using 1% acetic acid. The remaining SRB bound to protein was dissociated using 10 mmol/L unbuffered Tris base, and the optical density of the solubilized SRB was measured at an absorbance wavelength of 570 nm using the SynergyH1 hybrid plate reader (BioTek). Cell growth was calculated by the intensity of the SRB staining in relation to a solvent control treated well, which was set to 100%. Synergy was calculated using the Calcusyn software, which is based upon Chou and Talalay statistics (30). Synergistic values represented as combination index values (CI) are indicated using varying shades of grey. CI values less than 0.7 are considered to be synergistic. Clonogenicity was measured by seeding cells at single cell densities (100-1000 cells) in a 6-well dish and treated with indicated inhibitors 24 hours later. Cells were maintained in the indicated inhibitors for a total of 6 days with media and inhibitors replaced on day 3. On day 6 the cells were washed and released from treatment for an additional 7 days. Wells were then rinsed with phosphate buffered saline, fixed with ice cold methanol, stained with 0.5% (wt/vol) crystal violet in 6.0% (vol/vol) gluteraldehyde solution (Fisher Scientific), and destained with distilled water. The plates were then imaged on the 700 channel and analyzed using the Odyssey CLx imager (Li-Cor). Signal Intensity was measured by generating an ellipse to best fit a well, and the ellipse was then copied throughout the experimental replicates.
Cellular Apoptotic Assay
Cells (7500/well for BCPAP, C643, and Cal62; 8000/well for SW1736) were plated in triplicate in 96 well plates and allowed to adhere overnight. Media was substituted with 0.1% FBS, and 6 hours later treated with indicated inhibitors for 24 hours. Cleaved caspase 3/7 luminescence was measured using the caspase-glo 3/7 assay (Promega) using the Synergy H1 hybrid plate reader (Biotek).
Immunoblotting and Immunoprecipitation
Cells were collected in NP-40 lysis buffer (containing 1% NP-40, 20 mmol/L Tris-HCl (pH 8.0), 137 mmol/L NaCl, and 10% glycerol) with 1× protease/phosphatase inhibitor cocktail (Thermo)). Protein concentration was determined using the DC protein assay (Bio-rad). Protein (30 μg) was separated using an 8% PAGE-SDS gel, and transferred to Immobilon-P membranes (Millipore). Membranes were incubated overnight at 4°C with the indicated antibodies: FAK (BD Biosciences), p-Y925-FAK, ppERK1/2, ERK1/2, c-Src, p-Y416-SFK, SFK (Cell Signaling), Raf-1 (C-20), Raf-B (F-7) (Santa Cruz), β-actin (Sigma), or α-Tubulin (Calbiochem). For ECL detection, blots were incubated with secondary goat anti-rabbit or goat anti-mouse horseradish peroxidase–conjugated antibodies (GE Healthcare) and detected by enhanced chemiluminescence (ECL) (Pierce). For Odyssey CLx imaging blots were incubated with secondary goat anti-rabbit (IRDye 800CW) or goat anti-mouse (IRDye 680RD) (Li-Cor).
For immunoprecipitation (IP) assays, lysates were rotated with pre-clearing matrix F beads for 30 minutes at 4°C. Protein (500 μg) in 500 μl of lysis buffer was then incubated for 1 hour at 4°C with the indicated antibody (5 μg Raf-1 (C-20) or rabbit IgG (cell signaling)) prior to being incubated with 40 μl of the IP/WB Optima F beads (Santa Cruz) overnight at 4°C.
Mouse Xenograft Study
The Cal62 parental, control, and DasRes cell lines were injected into the left and right flanks of athymic nude mice. Briefly, female Athymic Nude-Foxn1nu mice (Harlan Laboratories; 20-30g; 6-8 weeks old) were anesthetized with isoflourane. Thyroid cancer cells (Cal62 parental and DasRes) 5 × 106 in 100ul RPMI and 50% high concentration Matrigel (BD Biosciences) were injected into the left and right flanks of athymic nude mice. Tumor establishment and progression were monitored weekly through the use of caliper measurements. Mice were randomized 7-10 days post-injections and treated with either vehicle, (12.5mg/kg or 25mg/kg) dasatinib, or (0.5mg/kg or 1mg/kg) trametinib. Both dasatinib and trametinib were administered daily oral gavage (5 days/week). All animal studies were performed in accordance with the animal procedures approved by the Institutional Animal Care and Use Committee at the University of Colorado.
Statistical analysis
Experiments were performed with at least three separate replicates. Statistical analysis was performed using the GraphPad Prism software and the unpaired Student t test was used to compare two means. Error bars represent the standard error of the mean (SEM), unless otherwise noted in their respective figure legends.
Results
Generation of a model of dasatinib resistance in thyroid cancer
To define mechanisms of resistance to the Src inhibitor, dasatinib, thyroid cancer cells expressing the BRAFV600E (BCPAP and SW1736) or KRASG12R/HRASG13R-mutation (Cal62 and C643) were cultured with gradually increasing concentrations of dasatinib (50nM - 2μM), or DMSO (control), over a period of nine months, until cells demonstrated resistance to dasatinib by SRB growth assays (IC50s > 2 μM). Upon acquired resistance, pooled populations of cells were maintained in 2μM dasatinib, and control cell lines were cultured in corresponding amounts of DMSO alongside. We and others have previously shown the IC50 values for the parental cell lines range between 35nM and 90nM (10,12), which is consistent with what we observed for the control cell lines (Figures 1A & Table S1). Upon acquisition of resistance, the DasRes cells exhibit a greater than 30-fold increase in relative resistance to dasatinib in comparison to their counterpart controls, with IC50 values ranging from 2 μM to >10 μM (Table S1). Additionally, all four DasRes cell lines exhibited cross-resistance to another Src inhibitor saracatinib, further confirming resistance to Src inhibition, and that the observed resistance is not a dasatinib-specific response (data not shown). To confirm cell line genetic identity, short tandem repeat (STR) profiling was performed upon acquisition of dasatinib resistance (Table S2) (27).
Figure 1.
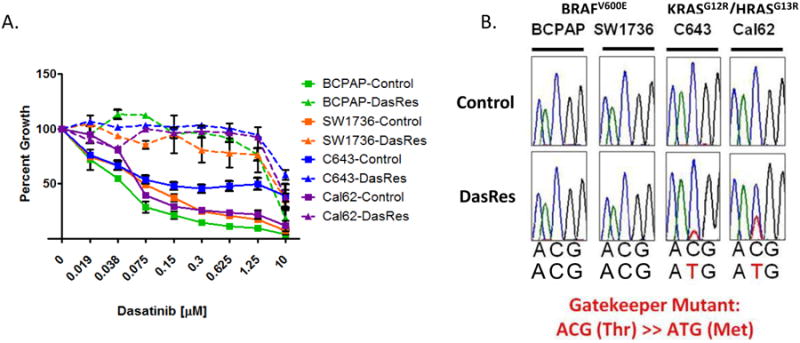
Generation of a model of Src Inhibitor resistance in two BRAF- and two RAS-mutant thyroid cancer cell lines. A.) Sulforhodamine B (SRB) growth analysis of the BCPAP, SW1736, C643, and Cal62, control and DasRes, cell lines in response to indicated concentrations of dasatinib (μM) after 72 hours of treatment. B.) Sanger Sequencing for exon 9 of c-Src in both control and dasatinib-resistant cell lines on gDNA.
Acquisition of the c-SRC gatekeeper mutation correlates with a stable mechanism of dasatinib resistance
We first hypothesized that a drug-resistant gatekeeper mutation in c-SRC may be driving resistance to dasatinib (31). Sequencing of exon 9 of c-SRC revealed the presence of the c-SRC gatekeeper mutation (c.C1022T; p. T341M) in both of the DasRes RAS-mutant cell lines (C643 and Cal62), but not in the DasRes BRAF-mutant cell lines (Fig. 1B). Acquisition of the drug-resistant c-SRC gatekeeper mutation in the RAS-mutant cell lines suggests these cells may exhibit a more permanent mechanism of resistance. We therefore released the BRAF- and RAS-mutant DasRes cell lines from dasatinib for 2 months (DasRes-2mo; ∼10 passages), and observed that the RAS-mutant DasRes-2mo cell lines maintained their resistance to dasatinib exhibiting 39- to 86-fold relative resistance to dasatinib, whereas the BRAF-mutant DasRes-2mo cell lines returned to a more sensitive state, exhibiting only 3-fold to 15-fold relative resistance compared to the control cell lines (Fig. 2A and Table S1). Of note, the BRAF-mutant DasRes-2mo cell lines did not regain full sensitivity to dasatinib, suggesting that mechanisms of transient and stable reprogramming may mediate resistance in the BRAF-mutant DasRes cell lines. Consistent with this, basal pY416Src levels in the BRAF-mutant DasRes-2mo cell line more closely resembled the basal pY416Src levels observed in the control cell line, suggesting that when released from dasatinib, the DasRes BRAF-mutant cells are able to reprogram back to being more dependent on Src (Fig. 2B; DasRes-2mo). As expected, in the BRAF- and RAS-mutant control and BRAF-mutant DasRes cell lines, pY416Src was inhibited by dasatinib (Fig. 2B & C). However in the RAS-mutant DasRes and DasRes-2mo cell lines, which acquired the c-Src gatekeeper mutation, pY416Src levels were not inhibited, as expected, and interestingly, exhibited a paradoxical increase when treated with 100nM dasatinib (Fig. 2C). Together, these data suggest that in the BRAF-mutant DasRes cell lines, transient mechanisms of cellular reprogramming are likely mediating resistance to dasatinib, whereas the primary driver of resistance in the RAS-mutant cell lines is likely the c-Src gatekeeper mutation.
Figure 2.

Analysis of Src inhibition in control versus dasatinib-resistant cell lines. A.) SRB growth analysis of the BCPAP, SW1736, C643, and Cal62 dasatinib-resistant cells released from dasatinib for 2 months (DasRes-Release) in response to indicated concentrations of dasatinib (μM) over 72 hours of treatment. DasRes-Release growth curve is overlaid onto the growth curves for both the control and dasatinib resistant cell lines from figure 1A. B & C.) Whole cell lysates were analyzed by Western blot analysis on the BRAF-mutant (panel B) BCPAP and SW1736 and the RAS-mutant (panel C) Cal62 and C643 control (Con), Dasatinib-Resistant (DR), and Dasatinib-Resistant released from dasatinib for 2 months (DR-2mo), which were treated with either DMSO or 100nM dasatinib for 24 hours. Proteins were detected by probing with antibodies against phospho-Src Family Kinase (SFK) Y416, SFK, and α-tubulin.
The MAP Kinase pathway is activated in dasatinib resistant thyroid cancer cell lines
To address alternative mechanisms of resistance and reprogramming, we next performed genome-wide RNA-sequencing on the control and DasRes cell lines. RNA-sequencing results further validated the presence of the c-SRCT341M gatekeeper mutation in the DasRes RAS-mutant cell lines; however RNA-sequencing was unable to detect any additional mutations that may provide evidence for genetic mechanisms of resistance in the BRAF-mutant DasRes cell lines (data not shown). Thus, we next performed gene set enrichment analysis (GSEA), and observed enrichment of the KEGG pathway Melanoma (hsa05218) across all four DasRes cell lines (Fig. S1). Consistent with the GSEA results, we observed a 1.5-3 fold increase in MAPK3/MAPK1 (ERK1/2) phosphorylation in all four DasRes cell lines in comparison to their respective counterpart controls (Fig. 3A and S2A). In addition, this increase in ERK1/2 phosphorylation appears to be a dasatinib specific event, as stable knockdown of c-Src does not promote increased ERK1/2 phosphorylation (Fig. S2B). Consistent with previous reports demonstrating that dasatinib can promote MAP Kinase pathway activation by promoting dimerization of B-Raf and c-Raf (32), increased B-Raf and c-Raf dimerization was observed in all four DasRes cell lines (Fig. 3B & S2C). Taken together, in addition to acquisition of the c-SRC gatekeeper mutation in the RAS-mutant DasRes cell lines; mechanisms of MAP Kinase pathway activation are conserved amongst all four DasRes cell lines.
Figure 3.

Increased MAPK pathway activation upon acquisition of dasatinib resistance. A.) Whole cell lysates were analyzed by Western blot analysis by probing for phospho-ERK1/2 (ppERK1/2), ERK2, and β-actin in the BCPAP, SW1736, C643, and Cal62 control (C) and dasatinib-resistant (DR) cell lines. B.) Immunoprecipitation of c-Raf and Western blot analysis for co-immunoprecipitation of B-Raf and c-Raf as well as whole cell lysate (WCL) immunoblots for B-Raf, c-Raf, and β-actin in the BCPAP, SW1736, C643, and Cal62 control (C) and dasatinib-resistant (DR) cell lines. C.) Western blot for phospho-ERK (ppERK1/2), ERK2, pFAK Y925, FAK, and α-Tubulin following treatment of cells with 100nM dasatinib for 0,2,4,8,24, 48, or 46 hours plus a 2 hour challenge with 100nM dasatinib (48+2). D.) Immunoprecipitation for c-Raf and western blot for B-Raf and c-Raf and western blot for B-Raf and c-Raf in whole cell lysates (WCL) following treatment of cells with 100nM dasatinib for 0,2,48, or 46 hours plus a 2 hour challenge with 100nM dasatinib (48+2).
MAP Kinase pathway activation occurs as an early response to dasatinib therapy
We next examined the response of the MAP Kinase pathway to dasatinib treatment at early time points. Specifically, parental cell lines were treated with 100 nM dasatinib for increasing periods of time (0, 2, 4, 8, 24, and 48 hours). Interestingly, dasatinib treatment resulted in an initial reduction in ERK1/2 phosphorylation, however a recovery was observed between 4-48 hours (Fig. 3C & S2D). The recovery in ERK1/2 phosphorylation was not a result of dasatinib metabolism, because inhibition of the Src dependent phosphorylation site, tyrosine Y925 of Focal Adhesion Kinase (FAK/PTK2) was maintained for 48 hours (Fig. 3C). To further confirm a functional recovery in ERK1/2 phosphorylation, we transfected the BCPAP cell line with a c-fos serum response element luciferase reporter construct (c-Fos SRE-luc), which has previously been demonstrated to be responsive to ERK1/2 (33). Consistent with ERK1/2 phosphorylation data, luciferase activity was decreased after 2-4 hrs of treatment and exhibited a recovery in activity between 8-48 hrs of treatment (Fig. S2E). Additionally, we challenged the 48 hour timepoint with an additional 100 nM dasatinib, 2 hours prior to harvest, and the recovery in ERK1/2 phosphorylation was maintained in both the BRAF- (BCPAP) and RAS- (Cal62) mutant cell lines (Fig. 3C, S2C-E). We therefore evaluated B-Raf and c-Raf dimerization in relation to the early recovery in ERK1/2 phosphorylation. Accordingly, we observed an increase in dimerization in the RAS- (Cal62) mutant cell line after treatment with dasatinib for 48 hours, which was maintained when challenged with dasatinib for the last 2 hours (Fig. 3D). Interestingly, RAF dimerization was more variable in the BRAF-mutant BCPAP cell line at the 48-hour time point in response to 100 nM dasatinib (Fig. 3D). In contrast, when we tested a higher dose of dasatinib (5 μM) at a shorter 3-hour time point, we observed a consistent increase in RAF dimerization in both the BRAF-mutant BCPAP and RAS-mutant Cal62 cell line (Fig S3A-C). To better define the role of B-Raf and c-Raf dimerization in BRAF-mutant thyroid cancer cells, we compared responses between the BRAF-mutant melanoma cell line A375 and the thyroid cancer cell line BCPAP. Consistent with a previous report, we see very little dimerization in the A375 cell line, however we observe a larger increase in the BCPAP cell line (Fig S3A & B) (32). Additionally, these data are also consistent with previous reports demonstrating that RAF dimerization is more robust in RAS-mutant versus BRAF-mutant cells, as we see a larger increase in dimerization in the RAS-mutant Cal62 cell line compared to the BRAF-mutant BCPAP cell line (24,32). Finally, B-Raf and c-Raf dimerization is an off-target effect that does not appear to be conserved amongst all Src inhibitors, as the Src inhibitor, saracatinib, does not strongly induce B-Raf and c-Raf dimerization in either the BRAF-mutant (BCPAP) or RAS-mutant (Cal62) cell lines (Fig. S3C), which is consistent with lack of phospho-ERK1/2 induction in response to c-Src knockdown (Fig. S2B). Overall, a recovery in ERK1/2 phosphorylation suggests that the MAP Kinase pathway may promote drug tolerant persistence, which is sustained and allows for additional resistance mechanisms to develop.
MEK1/2 inhibition restores sensitivity in dasatinib-resistant cell lines
We next tested whether reactivation of the MAP Kinase pathway in dasatinib-resistant cells results in an increased dependence on the MAP Kinase pathway for growth. We therefore performed growth assays in the control and DasRes cell lines in the presence of increasing concentrations of the MEK1/2 inhibitors, trametinib (GSK-1120202) or selumetinib (AZD6244) (Fig. 4A & Fig. S4). MEK1/2 inhibition with trametinib effectively inhibited growth in all four control and DasRes cell lines, with IC50s between 0.01 and 1 μM, with the RAS-mutant, C643, cell line exhibiting enhanced sensitivity in the DasRes line over the control (Fig. 4A and Table S3). Interestingly, the BRAF-mutant (BCPAP and SW1736) and RAS-mutant (C643) control and DasRes cell lines showed differential sensitivity to the MEK1/2 inhibitor, selumetinib, with control IC50 values ranging from 6 μM to >10 μM (Fig. S4 & Table S3; control), and enhanced sensitivity in the DasRes cell lines with IC50 values ranging from 1.6 μM to 4 μM (Fig. S4 and Table S3; DasRes). Despite these differential responses, ERK1/2 phosphorylation was completely abrogated in the BRAF- and RAS-mutant control and DasRes cell lines (Fig. 4B & 4C). As expected, inhibition of the downstream target of Src, pY925FAK, was observed in the BRAF-mutant DasRes cell lines, due to maintenance of these cells in 2 μM dasatinib (Fig. 4B). Consistent with acquisition of the c-Src gatekeeper mutation in the RAS-mutant DasRes cells, pY925FAK levels were not inhibited in these cells (Fig. 4C). Overall, these results indicate that the DasRes cell lines have an increased dependence on the MAP Kinase pathway upon acquisition of dasatinib resistance.
Figure 4.

Increased trametinib sensitivity when DasRes cell lines are maintained in 2μM dasatinib. A.) SRB growth analysis of the BCPAP, SW1736, C643, and Cal62 control and DasRes cell lines in response to indicated concentrations of trametinib (μM) for 72 hours. B. & C.) Western blot analysis on BRAF-mutant (B.) BCPAP, SW1736, and RAS-mutant (C.) C643, Cal62 control (C), dasatinib-resistant (DR) treated with either DMSO or 100 trametinib 24 hours. Cell lines were probed for ppERK1/2, ERK, pFAK Y925, FAK, ppERK1/2, ERK, and α-tubulin.
MEK1/2 inhibition overcomes dasatinib-resistance in vivo
To further define the dependence of the dasatinib-resistant cells on the MAP Kinase pathway, we evaluated the ability of MEK1/2 inhibition to overcome resistance to dasatinib in vivo. For these studies, we injected the Cal62 parental and DasRes cell lines into the flanks of athymic nude mice, and monitored tumor volume weekly (Fig. 5A). Therapies were initiated 7 days after the cells were injected and when tumor volumes were approximately 100mm3 (parental = 126mm3; DasRes = 104mm3). Consistent with our in vitro data, the DasRes tumors remained resistant to 25mg/kg dasatinib in vivo after 4 weeks of treatment (Fig. 5A, left). Additionally, dasatinib treatment resulted in a 30% inhibition of tumor growth in comparison to vehicle treatment in the Cal62 parental tumors, similar to previous reports (Fig. 5A, right) (12). Consistent with the importance of the MAP kinase pathway in dasatinib resistance, MEK1/2 inhibition with trametinib (1 mg/kg) resulted in significant inhibition of tumor growth in the Cal62 parental tumors (5.26 fold-inhibition; p-value = 0.0031) and DasRes tumors (20.4 fold-inhibition; p-value = 0.0007) in comparison to vehicle controls (Fig. 5A & B). Notably, MEK1/2 inhibition with trametinib also resulted in 3-fold smaller tumor weights in the DasRes cells compared to trametinib-treated parental tumors (Fig S5; p < 0.0001). Furthermore, the parental tumors started to become resistant to trametinib after only 4 weeks of treatment, as demonstrated by increased tumor volume (Fig. 5A, right). To further define the differential MEK1/2 inhibitor sensitivities between the parental and DasRes tumors, we continued to monitor additional tumors treated trametinib (0.5mg/kg) for 9 weeks (Fig. 5B). Interestingly, the DasRes tumors had a 5.5-fold greater inhibition of tumor volume in response to trametinib (p = 0.0009) with two of the DasRes tumors appearing to be completely eradicated, in comparison to the parental tumors (Fig. 5C). Taken together our in vivo and in vitro data indicates that the MAP Kinase pathway plays an important role in early and late resistance to Src inhibition, and that inhibition of MEK1/2 with trametinib is an effective secondary therapy to overcome resistance to Src inhibition with dasatinib.
Figure 5.

Trametinib overcomes dasatinib resistance in a flank model of thyroid tumorigenesis. A.) Cal62 DasRes (left) and Parental (right) cell lines were injected into the left and right flanks of Athymic Nude-Foxn1nu mice and treated with either vehicle, 25mg/kg dasatinib, or 1mg/kg trametinib and tumors were measured by caliper weekly. Day 0 represents treatment iniatiation (7 days post cell injection) B.) Comparison of the Cal62 Parental and DasRes growth curves in the presence of 0.5mg/kg trametinib. C.) Comparison of the Cal62 Parental and DasRes Final tumor volumes after 63 days of treatment with 0.5mg/kg trametinib. Data as means +/- SEM (n=6-8; student t-test; **, P < 0.005)
Increased sensitivity and cell death in response to combined Src and MAP Kinase pathway inhibition
To further test the hypothesis that reactivation of the MAP Kinase pathway drives resistance to dasatinib, we tested whether combined inhibition would result in enhanced anti-growth responses. Thus, we treated all four parental cell lines with increasing concentrations of dasatinib (0.019 – 1.25 μM) in combination with the MEK1/2 inhibitors, trametinib (0.001-0.1 μM) or selumetinib (0.05 - 0.8 μM), as well as the BCPAP and Cal62 cell lines with the ERK1/2 inhibitor, SCH772984 (0.005-0.5 μM). In support of the MAP Kinase pathway mediating resistance to dasatinib, all three MAP Kinase pathway inhibitors resulted in synergistic inhibition of growth when combined with dasatinib across all four cell lines (Figure 6A, S6A, & S6B) (30), with combination index (CI) values < 0.7, indicated by varying shades of grey along the curve. Additionally, combined Src and MEK1/2 inhibition also resulted in decreased clonogenic growth in both the BRAF- (BCPAP) and RAS- (Cal62) mutant cell lines in comparison to single agent treatments (Fig. S7A & B). To better understand if the combination therapy results in greater elimination of cancer cells, we analyzed cleavage of the downstream effectors of the apoptosis pathway, caspase 3/7. Consistent with a potential role for apoptosis driven drug synergy, we observed enhanced induction of caspase 3/7 activity in response to combination therapy compared to single-agent (≥2-fold; Fig. 6B). Lastly, we tested the combined treatment with the Src inhibitor, dasatinib, and the MEK1/2 inhibitor, trametinib in the RAS-mutant parental Cal62 tumors in vivo (Fig. 6C). Therapies were initiated 10 days after the cells were injected and when tumor volumes were approximately 100 mm3. After 42 days of treatment both the vehicle control and dasatinib treated groups reached the criteria for euthanasia. At this timepoint, a subset of the trametinib and combination treated tumors were harvested alongside for comparison of tumor volumes. Both the single agent trametinib and combination treatment groups exhibited significantly smaller tumor volumes (>3-fold; Fig. S7C). In addition, a subset of the trametinib and combination treated mice remained on therapy, however consistent with data from Fig. 5B, the single agent trametinib treatment group started to develop resistance to therapy after approximately 50 days of treatment resulting in a significant 1.89-fold increase in tumor volume compared to the combination group at day 67 (Fig. 6C; p = 0.0182). The prolonged inhibition of tumor growth in the combination therapy group also resulted in enhanced survival (110 vs. 81 days) in comparison to the single agent trametinib group (Fig. 6D; p = 0.0837). Thus, this data indicates that combined treatment with the Src inhibitor, dasatinib, and the MEK1/2 inhibitor, trametinib can overcome resistance to either single agent therapy, in vivo. Taken together, these data demonstrate that co-targeting the Src and MAP Kinase signaling pathways more effectively eliminates cancer cells than inhibition of either pathway alone, and represents a promising therapeutic strategy for advanced thyroid cancer patients for which few effective therapies are available.
Figure 6.
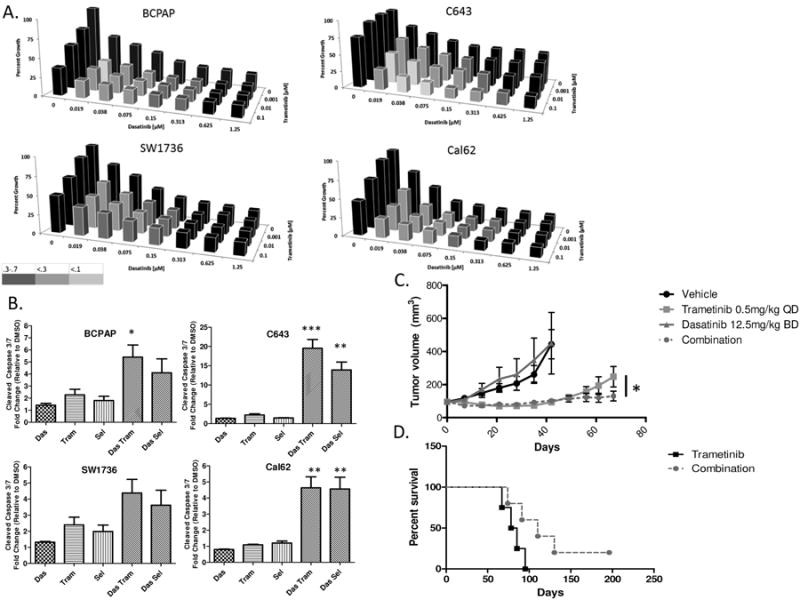
Increased sensitivity of Thyroid Cancer cells to Dasatinib when combined with MEK1/2 Inhibition. A.) Cell lines, C643, Cal62, BCPAP, and SW1736, were treated with increasing doses of dasatinib ranging from (0.019 μM to 1.25 μM) for 72 hours, in combination with increasing doses of trametinib (0.001 μM to 0.1 μM). Cell growth was measured using the Sulforhodamine B assay. Synergy was measured by determining the combination index using the Calcusyn software. Combinations that elicited a synergistic response are depicted by their corresponding shade of grey. (0.3-0.7, Synergism; 0.1-0.3, Strong Synergism; <0.1, Very Strong Synergism). B.) Cleaved caspase 3/7 was measured after a 24 hour incubation with indicated inhibitors in the BCPAP, SW1736, C643, and Cal62 cell lines. Data as means +/- SEM (n=4-5; student t-test; *, P < 0.05, **, P < 0.005, ***, P < 0.0005). Combination treatments were compared to respective single agent MEK1/2 inhibitors. C.) The Cal62 Parental cell line was injected into the left and right flanks of Athymic Nude-Foxn1nu mice and treated with either vehicle, 12.5mg/kg BD dasatinib, 0.5mg/kg QD trametinib, or the combination and tumors were measured by caliper weekly. Day 0 represents treatment iniatiation (10 days post cell injection). Data as means +/- SEM (n=8-10; student t-test; *, P < 0.05). D.) Overall survival analysis comparing the single agent trametinib treated tumors to the combination treated tumors. (Log-Rank (Mantel-Cox) Test; p = 0.0837)
Discussion
The development of Gleevac (imatinib) shed light on our ability to effectively inhibit cancer progression through the targeting of oncogenic drivers (34). Likewise, targeted therapies including vemurafenib and gefitinib, have exhibited strong therapeutic efficacy against specific onogenic drivers in other cancers including BRAF-mutant melanoma and EGFR-mutant non-small cell lung cancer, respectively. Unfortunately, this is not the case for all cancer types defined by a common oncogenic driver, as both BRAF-mutant thyroid and colorectal cancers do not exhibit similar sensitivities to MAP Kinase pathway inhibition, in comparison to BRAF-mutant melanoma (4,5). Due to the limited efficacy of targeted pathway inhibition in thyroid cancer, we have focused on the role of Src as an alternative, clinically relevant target, and have demonstrated that Src inhibition with dasatinib or saracatinib has strong therapeutic efficacy against multiple thyroid cancer cell lines in vitro and in vivo, and that c-Src is a key mediator of these responses (10,11). However, despite promising preclinical results, the efficacy of Src inhibitors has also been limited in the clinic (14–18). Thus, it is important to understand resistance mechanisms in order to effectively target this pathway in the clinic.
To further understand mechanisms of resistance to Src inhibition, we engineered four thyroid cancer cell lines resistant to the Src inhibitor, dasatinib. Interestingly, we observed acquisition of the c-Src gatekeeper mutation in the RAS-mutant cell lines, but not in the BRAF-mutant cell lines (Fig 1C). Reports of gatekeeper mutation acquisition have been previously observed in response to targeted therapies directed against mutated oncogenic drivers (BCR-ABL-imatinib (CML); EGFR-gefitinib (Lung); DDR2-dasatinib (lung) (35–37), however in contrast, our study discovered c-Src gatekeeper mutation acquisition specifically in RAS-mutant thyroid cancer cell lines. To the best of our knowledge, this is the first demonstration of differential mechanisms of gatekeeper acquisition in the context of different oncogenic mutations (BRAF versus RAS). As acquisition of the gatekeeper mutation is dependent on a cytosine to thymine transition, we hypothesize that two different possibilities may be occurring to promote the transition, which include increased cytosine deamination or malfunctions in DNA repair. Interestingly, a recent study highlighted a role for wild type RAS in mediating the DNA damage response in RAS-mutant cancers, and in conjunction, studies in thyroid cancer have demonstrated that oncogenic Ras promotes genome instability (38,39). Therefore a differential DNA damage response, between BRAF- and RAS-mutant cancers, may drive differential acquisition of the c-SRC gatekeeper mutation. Thus, further studies are needed to define the mechanism(s) of gatekeeper acquisition in order to enhance our ability to predict and combat resistance mechanisms to dasatinib.
In addition, increased levels of ERK1/2 phosphorylation were observed in all four DasRes cell lines, which correlated with increased B-Raf and c-Raf dimerization (Fig. 3A & 3B). While our data indicates B-Raf and c-Raf dimerization is likely a key mechanism promoting MAP Kinase pathway reactivation in response to dasatinib, we have also observed downregulation of negative regulators of the MAP Kinase pathway, including DUSP1, DUSP4, and SPRY1 (data not shown), as well as increased B-Raf protein levels in the BRAF-mutant, BCPAP, cell line in the dasatinib-resistant cells (Fig. 3B). These results are similar to previous reports showing that loss of negative regulators of the MAP Kinase pathway and BRAF gene amplification can promote resistance to targeted therapies (40–42), and will be evaluated in more detail in future studies. We further determined an early role for the MAP Kinase pathway in mediating dasatinib resistance, as treatment of parental cells with dasatinib resulted in an initial inhibition of ERK1/2 phosphorylation, and a recovery in ERK1/2 phosphorylation within 4-48 hours after dasatinib treatment (Fig. 3C).
Interestingly, whereas we observed increased dimerization in our BRAF-mutant cell lines upon dasatinib treatment, a previous report did not observe dimerization in two BRAF-mutant melanoma cell lines upon treatment with dasatinib and concluded that B-Raf and c-Raf dimerization occurs in a Ras-dependent manner (32). To better understand the observed dimerization in the thyroid cancer BRAF-mutant cell lines, we compared B-Raf and c-Raf dimerization in our thyroid cancer cell lines to the melanoma cell line A375 upon treatment with dasatinib. Consistent with dasatinib having limited effects on dimerization in the melanoma cell lines, we observed larger increases in dimerization in the thyroid cancer cell lines in comparison to the melanoma cell line A375 with the largest levels being observed in the RAS-mutant Cal62 cell line. In support of this, recent evidence demonstrates that BRAF-mutant thyroid cancer cells may be more primed to increase RAF dimers upon MAP KINASE pathway inhibition in comparison to melanoma (4,5). Therefore, future studies will aim to explore in more detail the discrepancies between dasatinib-mediated dimerization in thyroid versus melanoma cancer cell lines.
Having demonstrated that the MAP KINASE pathway is primed for activation upon dasatinib treatment in thyroid cancer cells, we next analyzed MEK1/2 inhibition in the control and DasRes cell lines, and demonstrate the MEK1/2 effectively inhibits growth with a trend towards greater MAP Kinase pathway dependence in the DasRes cell lines (Fig. 4A & S4). Interestingly, despite having acquired the c-Src gatekeeper mutation, the RAS-mutant DasRes cell lines still remained sensitive to MEK1/2 inhibition. A potential explanation for maintained MEK1/2 inhibitor sensitivity is through an increased in Src-mediated phosphorylation of Y925FAK in the DasRes RAS-mutant cell lines (Fig 4C). Previous studies have shown that phosphorylation of Y925FAK generates a Grb2 binding site, and therefore activation of the MAP Kinase pathway, representing another potential mechanism by which gatekeeper mutant Src signaling contributes to MAP Kinase pathway dependence (43).
Next, we further defined the role of MAP kinase signaling in dasatinib resistance through evaluation of MEK1/2 inhibition in overcoming dasatinib-resistance in vivo. Using the RAS-mutant Cal62 cell line as a model, we observed sensitivity to MEK1/2 inhibition in both the parental and DasRes tumors. Intriguingly, we observed a significant increase in MEK1/2 inhibitor sensitivity in the Cal62 DasRes tumors in vivo even though we observed similar responses in vitro (Fig. 5A, 5B & S5), which was further exemplified by two complete tumor responses in the DasRes tumors (Fig. 5C). The in vivo data therefore further supports a role for increased MAP Kinase pathway dependence in the DasRes tumors.
Herein, we report increased dimerization of B-Raf and c-Raf, as a potential mechanism of resistance to dasatinib, as recently reported by Packer et al (Fig. 3B & 4B) (32). Increased B-Raf and c-Raf dimerization is important from a therapeutic perspective, as this is a key mechanism of resistance to B-Raf inhibitors in BRAF-mutant melanoma (24,44,45), and this mechanism may be more primed in BRAF-mutant thyroid cancer cells (4,5). In order to combat this mechanism of resistance, current therapies are focused on combined BRAF and MEK1/2 inhibition. Unfortunately, however, a potential caveat may derive from this strategy, as a recent study from Moriceau et al suggests that inhibition of multiple nodes in the same pathway (e.g. BRAF and MEK1/2) primes and amplifies resistance mechanisms that enhance the activation of resistant signaling pathways (46). Thus, the inhibition of distinct pathways (e.g. Src and MEK1/2) may be more effective. Consistent with this hypothesis, here we demonstrated that MEK1/2 inhibition was able to effectively overcome resistance to dasatinib both in vitro and in vivo (Figs. 4 and 5). Additionally, we and others have shown enhanced anti-growth and pro-apoptotic responses over single agent therapy when Src and MAP Kinase pathway inhibitors are used in combination in vitro (13,47) (Fig. 6). Furthermore, we show for the first time that combined Src inhibition with dasatinib and MEK1/2 inhibition with trametinib results in enhanced anti-tumor responses and increased survival (Fig. 6), similar to a previous study that evaluated BRAF inhibition with vemurafenib in combination with dasatinib (13). In support of this, recent data with a dual dimerization-breaking RAF inhibitor that also targets Src appears promising, and may be an effective new strategy to prevent or delay resistance to single agent therapy (48). Finally, recent data has demonstrated that MEK1/2 inhibition can improve standard of care radioiodine for patients with advanced thyroid cancer (49). Therefore further analysis of the potential for combined Src and MAP KINASE pathway inhibition enhance radioiodine uptake represents a promising therapeutic direction, and an important area for continued investigation.
In summary, we have discovered that thyroid cancer cell lines acquire the c-Src gatekeeper mutation, and mechanisms of MAP Kinase pathway activation upon acquisition of resistance to the Src inhibitor, dasatinib. Importantly dasatinib resistant cell lines exhibit increased sensitivity to MAP Kinase pathway inhibition both in vitro and in vivo. Taken together, MAP Kinase pathway inhibition is a promising strategy to overcome or prevent resistance to the Src inhibitor, dasatinib, in thyroid cancer.
Supplementary Material
Acknowledgments
We would like to thank Dr. Christopher Korch, UCCC, Department of Pathology, and Randall Wong at the B. Davis Center BioResources Core Facility, Molecular Biology Unit, for STR profiling of the cell lines. We also thank Drs. Lynn Heasley and Arthur Gutierrez-Hartmann for critical review of our manuscript. We also thank Bristol-Myers Squibb for generously providing dasatinib for these studies.
Grant support: This work was supported by NCI grant K12-CA086913, ACS RSG-13-060-01-TBE (RE Schweppe), NIH NRSA T32CA174648-01, NIH NRSA 1F31CA192805-01 (TC Beadnell), NIH P50CA058187, P30CA046934, and the Cancer League of Colorado (AC Tan). The UCCC DNA Sequencing is supported by NCI Cancer Center, grant P30 CA046934. The contents of this study are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Financial support: This work was supported by National Cancer Institute (NCI) grant K12-CA086913 (RES), American Cancer Society IRG #57-001-50 (RES), American Cancer Society RSG-13-060-01-TBE (RES), NIH NRSA T32CA174648-01 (TCB), NIH NRSA 1F31CA192805-01 (TCB), NIH P50CA058187, NIH NCI Cancer Center grant P30CA046934, and the Cancer League of Colorado (ACT).
Footnotes
Conflicts of interest: None
References
- 1.Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13:184–99. doi: 10.1038/nrc3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfister DG, Fagin Ja. Refractory thyroid cancer: a paradigm shift in treatment is not far off. J Clin Oncol. 2008;26:4701–4. doi: 10.1200/JCO.2008.17.3682. [DOI] [PubMed] [Google Scholar]
- 3.Hayes DN, Lucas AS, Tanvetyanon T, Krzyzanowska MK, Chung CH, Murphy Ba, et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res. 2012;18:2056–65. doi: 10.1158/1078-0432.CCR-11-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf Ja, Viale A, Sherman EJ, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF mutant thyroid carcinomas. Cancer Discov. 2013;3:520–33. doi: 10.1158/2159-8290.CD-12-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sos ML, Levin RS, Gordan JD, Oses-Prieto JA, Webber JT, Salt M, et al. Oncogene Mimicry as a Mechanism of Primary Resistance to BRAF Inhibitors. Cell Rep. 2014;6:1–12. doi: 10.1016/j.celrep.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995. Cancer. 1998;83:2638–48. doi: 10.1002/(sici)1097-0142(19981215)83:12<2638::aid-cncr31>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 7.Bernet V, Smallridge R. New therapeutic options for advanced forms of thyroid cancer. Expert Opin Emerg Drugs. 2014:1–17. doi: 10.1517/14728214.2014.894017. [DOI] [PubMed] [Google Scholar]
- 8.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–9. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- 9.Sen B, Johnson FM. Regulation of SRC family kinases in human cancers. J Signal Transduct. 2011;2011:1–14. doi: 10.1155/2011/865819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan CM, Jing X, Pike La, Zhou Q, Lim DJ, Sams SB, et al. Targeted inhibition of SRC kinase with dasatinib blocks thyroid cancer growth and metastasis. Clin Cancer Res. 2012;18:3580–91. doi: 10.1158/1078-0432.CCR-11-3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schweppe RE, Kerege Aq, French JD, Sharma V, Grzywa RL, Haugen BR. Inhibition of Src with AZD0530 reveals the Src-Focal Adhesion kinase complex as a novel therapeutic target in papillary and anaplastic thyroid cancer. J Clin Endocrinol Metab. 2009;94:2199–203. doi: 10.1210/jc.2008-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan D, Tyner JW, Chng WJ, Bi C, Okamoto R, Said J. Effect of dasatinib against thyroid cancer cell lines in vitro and a xenograft model in vivo. 2012:807–15. doi: 10.3892/ol.2012.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borre P, Vanden Gunda V, Mcfadden DG, Sadow PM, Varmeh S, Bernasconi M, et al. Combined BRAFV600E- and SRC-inhibition induces apoptosis, evokes an immune response and reduces tumor growth in an immunocompetent orthotopic mouse model of anaplastic thyroid cancer. 2014;5:3996–4010. doi: 10.18632/oncotarget.2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma M, Wroblewski K, Polite B. Dasatinib in previously treated metastatic colorectal cancer: a phase II trial of the University of Chicago Phase II Consortium. Invest New Drugs. 2012;30:1211–5. doi: 10.1007/s10637-011-9681-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson FM, Bekele BN, Feng L, Wistuba I, Tang XM, Tran HT, et al. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:4609–15. doi: 10.1200/JCO.2010.30.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayer EL, Baurain JF, Sparano J, Strauss L, Campone M, Fumoleau P, et al. A phase 2 trial of dasatinib in patients with advanced HER2-positive and/or hormone receptor-positive breast cancer. Clin Cancer Res. 2011;17:6897–904. doi: 10.1158/1078-0432.CCR-11-0070. [DOI] [PubMed] [Google Scholar]
- 17.Herold CI, Chadaram V, Peterson BL, Marcom PK, Hopkins J, Kimmick GG, et al. Phase II trial of dasatinib in patients with metastatic breast cancer using real-time pharmacodynamic tissue biomarkers of Src inhibition to escalate dosing. Clin Cancer Res. 2011;17:6061–70. doi: 10.1158/1078-0432.CCR-11-1071. [DOI] [PubMed] [Google Scholar]
- 18.Brooks H, Glisson B, Bekele BN, Ginsber L, El-Naggar A, Culotta K, et al. Phase II study of dasatinib in the treatment of head and neck squamous cell carcinoma (HNSCC) Cancer. 2011;117:2112–9. doi: 10.1002/cncr.25769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glickman MS, Sawyers CL. Converting cancer therapies into cures: lessons from infectious diseases. Cell. 2012;148:1089–98. doi: 10.1016/j.cell.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J, Maheswaran S, et al. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther. 2008;7:874–9. doi: 10.1158/1535-7163.MCT-07-2387. [DOI] [PubMed] [Google Scholar]
- 21.Gibbons DL, Pricl S, Kantarjian H, Cortes J, Quintás-Cardama A. The rise and fall of gatekeeper mutations? The BCR-ABL1 T315I paradigm. Cancer. 2012;118:293–9. doi: 10.1002/cncr.26225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–82. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engelman Ja, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 24.Lito P, Pratilas Ca, Joseph EW, Tadi M, Halilovic E, Zubrowski M, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–82. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marek L, Ware KE, Fritzsche A, Hercule P, Helton WR, Smith JE, et al. Fibroblast Growth Factor (FGF) and FGF Receptor-Mediated Autocrine Signaling in Non – Small-Cell Lung Cancer Cells. Mol Pharmacol. 2009;75:196–207. doi: 10.1124/mol.108.049544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell Elsevier Ltd. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schweppe RE, Klopper JP, Korch C, Pugazhenthi U, Benezra M, Knauf Ja, et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab. 2008;93:4331–41. doi: 10.1210/jc.2008-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–6. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 30.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 31.Azam M, Seeliger Ma, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15:1109–18. doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Packer LM, Rana S, Hayward R, O'Hare T, Eide Ca, Rebocho A, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011;20:715–27. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato-Stankiewicz J, Hakimi I, Zhi G, Zhang J, Serebriiskii I, Guo L, et al. Inhibitors of Ras/Raf-1 interaction identified by two-hybrid screening revert Ras-dependent transformation phenotypes in human cancer cells. Proc Natl Acad Sci U S A. 2002;99:14398–403. doi: 10.1073/pnas.222222699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001 doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Yuan H, Roth M, Stark JM, Bhatia R, Chen WY. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene. 2013;32:589–98. doi: 10.1038/onc.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beauchamp EM, Woods Ba, Dulak AM, Tan L, Xu C, Gray NS, et al. Acquired Resistance to Dasatinib in Lung Cancer Cell Lines Conferred by DDR2 Gatekeeper Mutation and NF1 Loss. Mol Cancer Ther. 2014;13:475–82. doi: 10.1158/1535-7163.MCT-13-0817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chong CR, Jänne P. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grabocka E, Pylayeva-Gupta Y, Jones MJK, Lubkov V, Yemanaberhan E, Taylor L, et al. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. Elsevier Inc. 2014;25:243–56. doi: 10.1016/j.ccr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saavedra HI, Knauf Ja, Shirokawa JM, Wang J, Ouyang B, Elisei R, et al. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene. 2000;19:3948–54. doi: 10.1038/sj.onc.1203723. [DOI] [PubMed] [Google Scholar]
- 40.Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, et al. Disruption of CRAF-Mediated MEK Activation Is Required for Effective MEK Inhibition in KRAS Mutant Tumors. Cancer Cell. 2014:1–14. doi: 10.1016/j.ccr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ercan D, Xu C, Yanagita M, Monast C. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2:934–47. doi: 10.1158/2159-8290.CD-12-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate aJ, Settleman J, Engelman Ja. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitra SK, Mikolon D, Molina JE, Hsia Da, Hanson Da, Chi a, et al. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006;25:5969–84. doi: 10.1038/sj.onc.1209588. [DOI] [PubMed] [Google Scholar]
- 44.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–9. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 45.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moriceau G, Hugo W, Hong A, Shi H, Kong X, Yu CC, et al. Tunable-Combinatorial Mechanisms of Acquired Resistance Limit the Efficacy of BRAF/MEK Cotargeting but Result in Melanoma Drug Addiction. Cancer Cell. 2015;27:240–56. doi: 10.1016/j.ccell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henderson YC, Serra RT, Chen Y, Ryu J, Frederick MJ, Zhou G, et al. Src inhibitors in suppression of papillary thyroid carcinoma growth. Head Neck. 2013:1–10. doi: 10.1002/hed.23316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Girotti MR, Lopes F, Preece N, Niculescu-Duvaz D, Zambon A, Davies L, et al. Paradox-Breaking RAF Inhibitors that Also Target SRC Are Effective in Drug-Resistant BRAF Mutant Melanoma. Cancer Cell. 2014;27:85–96. doi: 10.1016/j.ccell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368:623–32. doi: 10.1056/NEJMoa1209288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.