

Abstract
Lipid mediators of inflammation play important roles in several diseases including skin cancer, the most prevalent type of cancer found in the industrialized world. Ultraviolet (UV) radiation is a complete carcinogen and is the primary cause of skin cancer. UV radiation is also a potent immunosuppressive agent, and UV-induced immunosuppression is a well-known risk factor for skin cancer induction. An essential mediator in this process is the glyercophosphocholine 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine commonly referred to as platelet-activating factor (PAF). PAF is produced by keratinocytes in response to diverse stimuli and exerts its biological effects by binding to a single specific G-protein-coupled receptor (PAF-R) expressed on a variety of cells. This review will attempt to describe how this lipid mediator is involved in transmitting the immunosuppressive signal from the skin to the immune system, starting from its production by keratinocytes, to its role in activating mast cell migration in vivo, and to the mechanisms involved that ultimately lead to immune suppression. Recent findings related to its role in regulating DNA repair and activating epigenetic mechanisms, further pinpoint the importance of this bioactive lipid, which may serve as a critical molecular mediator that links the environment (UVB radiation) to the immune system and the epigenome.
1. An introduction to PAF
Platelet-activating factor (PAF, 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine) is a glycerophospholipid that was first discovered in the early 1970s. As its name implies, it induced the aggregation of blood platelets following its release from immunoglobulin E-stimulated basophils [1]. Independently, around the same time another group reported on a lipid compound possessing potent hypotensive properties that was later shown to be PAF [2]. Since then, a broad and significant spectrum of pathophysiological effects and functions have been described for this biolipid, affecting many different cell types, unrelated to its platelet-activating activity, hence its name may be somewhat inappropriate but it has universally remained.
It is generally recognized that its primary role is to mediate intracellular processes through binding to a single highly specific seven-transmembrane G-protein-coupled receptor, which is expressed by many cells, including those of the innate immune system [3,4]. In fact, PAF was the first intact phospholipid known to have messenger functions by binding to a specific receptor on the cell membrane, and not simply via physicochemical effects on the plasma membrane of the target cell. The effects induced by PAF binding to its receptor can be non-inflammatory, such as its involvement in glycogen degradation, reproduction, brain function, blood circulation and its recently described role as an anti-obesity factor [5–9]. However, PAF is much better known for its role in pro-inflammatory and allergic processes and in regulating the immune response [10–12]. It may be regarded as both a friend, since it is presumed to have evolved as part of a protective mechanism in the innate host defense system, but also as a foe, because of its involvement in uncontrolled pathological conditions. When found in excess, it has been implicated in the pathogenesis of several diseases ranging from stroke, sepsis, myocardial infarction, colitis and multiple sclerosis. Therefore, its synthesis, distribution and degradation are all under strict control as would be predictable for such a potent molecule with such diverse actions.
As expected, a wide variety of reviews concerning the biosynthesis and catabolism of PAF, as well as the molecular and biochemical features of the PAF signaling cascade, and its known roles in health and disease have been published [13–24]. However, none have actually focused on the emerging role that this unique biolipid has on mediating sunlight-induced skin cancer induction and immune suppression, despite recent reviews on bioactive lipid mediators in skin inflammation and immunity [25]. UV-induced immunosuppression is a well-known risk factor for skin cancer induction, and each year there are more new cases of skin cancer reported than the combined incidence of cancers of the breast, prostate, lung and colon [26]. Therefore, it is important to understand how this ubiquitous environmental carcinogen transmits a signal from the skin to the immune system that promotes immune suppression and contributes to skin cancer induction. This review is intended to provide the reader with a summary of the new-found role that PAF specifically plays in this scenario, starting from the first report of its production by keratinocytes in 2000 and the progress made since then in understanding the connection between this lipid mediator of inflammation, immune suppression and skin cancer.
2. PAF structure and biosynthesis
PAF is an ether lipid characterized by an ether bond in sn-1 position bearing an alkyl group, usually the fatty alcohol, hexadecanol. Because of this ether linkage, it is an unusual lipid as such moieties are not common in animals, nor is it common to find the acetic acid esterified directly to glycerol at the sn-2 position (Fig. 1). In sn-3 it has a phosphocholine head group. These three structural features are all equally important requisites for PAF's optimal biological activity mediated by its stereo-specific binding to its specific receptor [17,27]. For instance, there are several compounds produced by a variety of cells that are similar to PAF, but bear a fatty acid instead of a fatty alcohol at sn-1 position. These moieties have less than 1% of the potency of PAF. Increasing the chain length beyond 3 carbons at the sn-2 position decreases its biological potency; likewise altering the polar group at sn-3 position decreases the potency of the molecule [28,29].
Fig. 1.
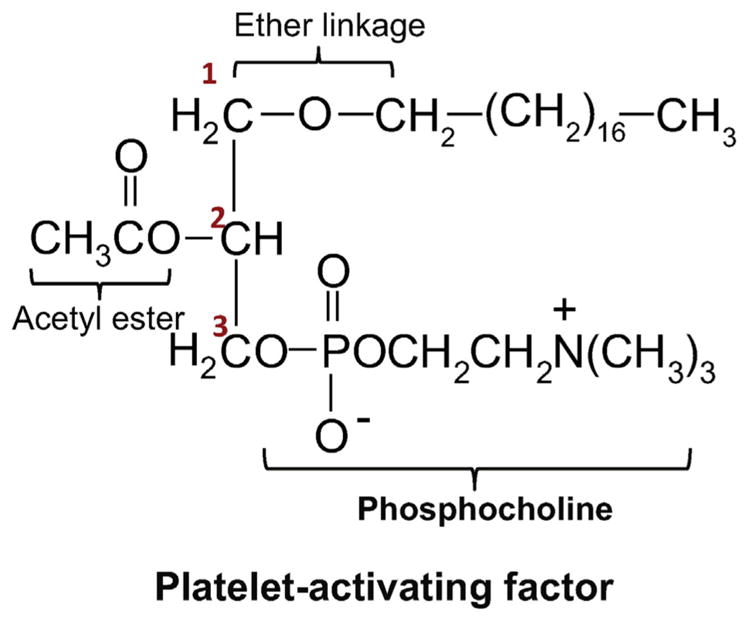
Structure of platelet activating factor. Structural features of PAF include: (1) an ether bond at the sn-1 position. (2) Acetic acid esterified to glycerol at sn-2. (3) Phosphocholine head group at sn-3.
PAF is synthesized by two distinct pathways. The first is the so-called remodeling pathway that involves remodeling of a membrane lipid constituent (a long-chain fatty acyl residue in sn-2 is replaced with an acetyl residue) and is the route utilized by inflammatory cells in response to cell-specific stimuli. The second is by de novo synthesis via a mechanism similar to the biosynthesis of phosphatidylcholine, in which a phosphocholine function is transferred to alkyl acetyl glycerol. This pathway is not activated by inflammatory stimuli, but rather is physiologically important in maintaining steady state levels of PAF in various tissues and blood. Both these pathways and the enzymes involved have been extensively reviewed in detail elsewhere [12,30], hence only a brief outline of the biosynthesis will be given here to familiarize the reader with how PAF is made in cells.
An essential first step for PAF biosynthesis through the remodeling pathway is the generation of lyso-PAF (1-alkyl-sn-glycero-3-phosphocholine). This can occur either directly by deacylation of alkylacylgly-cerophosphocholines via the action of phospholipase A2 (PLA2) or indirectly via a transacylation sequence involving PLA2 and a CoA-independent transacylase. This indirect path requires that exogenously or endogenously generated lyso-PAF must be present to trigger the transacylase reaction. Worthy of note is that those species containing arachidonate or other polyenoates at the sn-2 position are the membrane lipid precursors of lyso-PAF, since nutritional studies have shown that very little PAF is formed when arachidonate is deficient [31]. The enzyme PLA2 is phosphorylated by MAP kinases [32] in a Ca2+-dependent manner, and in addition to forming lyso-PAF, arachidonate is released for eicosanoid production. The second and final step in the synthesis of PAF is an actylation step performed by the action of a distinct membrane-bound acetyl-CoA-lyso PAF actyltransferase (LPCAT2), which catalyzes the transfer of an acetyl residue from acetyl-CoA to lyso-PAF to yield PAF with the simultaneous release of Coenzyme A (Fig. 2). This enzyme also appears to be regulated in a Ca2+ and phosphorylation-dependent fashion.
Fig. 2.

Essential steps in PAF biosynthesis: The remodeling pathway. Step 1: generation of Lyso-PAF. Step 2: Acetylation of Lyso-PAF. See Section 2 for details.
Two recent reports from Shimizu's laboratory have shed light on the mechanisms by which LPCAT2 is activated to rapidly produce PAF via the remodeling pathway. LPCAT2 is highly expressed in inflammatory cells and depending upon the inflammatory stimulus used to activate the cells, PAF is produced within seconds or within minutes to hours following stimulation. Morimoto and colleagues found that when lipopolysaccharide (LPS) was used to activate toll-like receptor 4 (TLR4) on macrophages, rapid (minutes to hours) production of PAF was noted. Activation of the TLR4 signaling cascade resulted in the activation of MAPK-activated protein kinase 2. This resulted in phosphorylation of LPCAT2 at the serine 34 residue, which enhanced enzymatic activity resulting in the rapid production of PAF [33].
PAF itself can act as an inflammatory signal and the binding of PAF to its receptor on inflammatory cells can promote the very rapid (within 30 s) production of PAF. Here again LPCAT2 enzymatic activity is involved, and phosphorylation at serine 34 is essential. However, in this situation PAF binding to its receptor activates phosphorylation of LPCAT2 within 10 s. Using siRNA, and selective inhibitors, Morimoto and colleagues demonstrated that PAF-induced protein kinase Cα-mediated phosphorylation of LPCAT2 enhanced enzymatic activity leading to the vary rapid production of PAF [34]. The data generated in these two reports provides important new information regarding the mechanisms by which PAF is rapidly produced by the remodeling pathway in response to inflammatory stimuli.
A second lyso-PAF actyltransferase (LPCAT1) is constitutively expressed in the lungs, where it produces PAF and dipalmitoyl-phosphatidylcholine of pulmonary surfactant, which is essential for respiration [35,36]. A number of recent reports have also indicated that LPCAT1 is expressed in a number of different cancers, including oral squamous cell carcinoma [37], prostate cancer [38], hepatocellular carcinoma [39], and colorectal carcinoma [40]. The biochemical characteristics of the different lysophospholipd actyltransferases have been recently reviewed [41].
The de novo pathway leading to PAF biosynthesis has been extensively reviewed by others [12,17,20], and will only be briefly summarized here. Three principal steps are involved, each catalyzed by three specific membrane-bound enzymes. The initial step is the acetylation of 1-alkyl-sn-glycero-3-phosphate, an important metabolic intermediate in the biosynthesis of ether-linked phospholipids. This step is catalyzed by an acetyl-CoA-alkyl-lysoglycero-P acetyltransferase. Subsequently, the acetylated product of this reaction is dephosphorylated by the action of an alkylacetylglycero-P phosphohydrolase. The third and final step involves the transfer of a phosphocholine moiety from CDP-choline to 1-alkyl-2-acetyl-sn-glycerol formed in the intermediate reaction and it is catalyzed by a distinct enzyme, CDP-choline alkylacetylglycerol cholinephosphotransferase. The availability of CDP-choline, which is formed by cytidylyltransferase, an important ancillary enzyme of this de novo pathway, appears to be rate limiting for PAF production. Hence any factor or condition that activates cytidylyl-transferase, such as fatty acids, or elevates the intracellular levels of CDP-choline can increase the biosynthesis of PAF by this pathway. These three steps are summarized in Fig. 3.
Fig. 3.

The de novo synthesis of PAF. Step 1: acetylation of 1-akyl-2-lyso-sn-glycero-3-P by acetyl-CoA-alkyl-lysoglycero-P acetyltransferase. Step 2: Dephosphorylation of 1-alkyl-2-sn-glycero-3-P to form 1-aklyl-2-lyso-sn-glycerol. Step 3: Transfer of phosphocholine from CDP-Choline to form PAF. The availability of CDP-choline appears to be the rate-limiting factor in the de novo synthesis of PAF.
The levels of PAF formed by either the remodeling route, or by de novo synthesis, are regulated not only by phosphorylation-dephosphophorylation, Ca2+, arachidonate, and CDP-choline levels, but also by a small important family of related phospholipase A2 catabolic enzymes known as PAF acetylhydrolases (PAF-AHs). Two subfamilies of PAF-AHs exist (group VII and group VIII phospholipases A2), which are both Ca2+-independent and they essentially inactivate PAF by removing the acetyl residue at sn-2 to generate lyso-PAF and acetate. The group VIII enzymes are completely specific for PAF and are found in brain and in erythrocytes, while the group VII enzymes are found associated with both circulating plasma LDL and HDL particles and function at the lipid-aqueous interface. These latter are less specific since they can also hydrolyze phospholipids structurally related to PAF, some of which contain oxidized functionalities at the sn-2 position. For more detail on the emerging role of PAF's catabolic enzymes, the reader is referred to the following articles [42,43].
3. Skin and UV radiation
3.1. Structural/functional aspects of the skin
The skin is the largest organ of the body, and its specialized structure covers many essential functions that span from thermoregulation, sensation (touch, pain, temperature, pressure), prevention of water loss and the production of vitamin D. Importantly, it interfaces with the environment providing the first line of defense against several environmental factors such as microbial pathogens and ultraviolet radiation [44].
The structure of skin responsible for many of the above functions comprises three distinct layers — the epidermis, the dermis and the hypodermis (Fig. 4). The epidermis, which is the outermost layer, is a self-renewing tissue composed mainly (>90%) of keratinocytes. The basal keratinocytes are cuboidal in appearance and as they proliferate and differentiate in an ordered, tightly regulated sequence of events, they migrate to the skin surface where they become anucleate, keratin-packed and flattened to form the stratum corneum (the epidermal–environmental interface). The mature keratinocytes known as corneocytes are then shed from the stratum corneum (a process known as desquamation) every 12–14 days accounting for renewal of the epidermis. The keratinization of the upper layers of the epidermis provides barrier function, which protects the internal milieu from the external environment. Other cells present in the epidermis are melanocytes, located at the bottom layer of the epidermis and that give the skin its pigment, Merkel cells which are thought to function as touch receptors, and bone marrow derived Langerhans cells, which are antigen-presenting cells important for the immunologic function of skin [45].
Fig. 4.

Potential mechanisms that transmit the immune suppressive signal contained within UVB radiation to the immune system. The epidermis is the outermost layer of the skin comprised mostly of keratinocytes. Most of the UVB energy is absorbed here. Keratinocyte-derived PAF activates the up regulation of CXCR4 on dermal mast cells. PAF-induced histone acetylation of the CXCR4 promoter appears to be involved. This activates mast cell migration from the skin to the draining lymph nodes, where they secrete IL-10 and suppress CHS and antibody production. UV damaged keratinocytes also up-regulate RANK-L, which re-programs migrating Langerhans cells (LC) to activate T regulatory cells. This figure is modified from an image produced by The National Institutes of Health (Don Bliss artist, ID # 4606), which is in the public domain and can be freely reused.
Below the epidermis, and separated by a basement membrane, is the dermis which is composed primarily of fibroblasts and extracellular matrix proteins (elastin and collagen) providing structural support for the skin. Other components found here are hair follicles, sweat glands, small blood vessels and sensory nerves. Various immune cells, including macrophages, dermal dendritic cells, T cells, mast cells, eosinophils and neutrophils reside in the dermis and provide for routine immune surveillance. Inflammation activates a massive increase in the cutaneous immune cell population [46,47].
Beneath the dermis lies the hypodermis, which comprises a layer of subcutaneous tissue, and contains blood vessels and adipocytes. The main role of this layer is to provide storage of lipids for energy and as a reserve of fatty acids. Adipocytes also produce a range of bioactive lipid mediators and peptide hormones that influence other cutaneous cells and that regulate both local and systemic effects such as inflammation and appetite regulation [48,49]. Recently, this adipose tissue has been identified as a source of stem cells that have potent effects on keratinocytes and dermal fibroblasts [50]. A more detailed review of the anatomy and physiology of the skin can be found in reference [51].
3.2. UV radiation
In terms of skin health, the UV radiation present in sunlight is probably the most important ubiquitous and inescapable carcinogen in our environment. The UV radiation present in the sun's electromagnetic radiation is responsible for skin cancer and photoaging [52,53]. The UV radiation emitted by the sun is subdivided into three bands: UVC (100–290 nm), UVB (290–320 nm) and UVA (320–400 nm). UVC rays are mostly absorbed by atmospheric ozone and do not reach the earth's surface, hence its toxic effect in humans is of concern primarily to those exposed to artificial sources of sunlight (mercury arc lamps and germicidal lamps). Most of the UV radiation that does reach the earth's surface is UVA (≈ 95%) and only about 5% is UVB since these latter rays are mainly filtered by atmospheric ozone. Besides being more prevalent than UVB, UVA rays penetrate deeper into the skin reaching the dermal layers [54]. These rays are also present with relatively equal intensity during all daylight hours throughout the year, and can penetrate clouds and glass. On the other hand, the intensity of UVB rays varies by season, location, and time of day and UVB cannot penetrate beyond the upper epidermal layers of the skin.
3.3. Acute and chronic effects of UV radiation on the skin
Small amounts of UV radiation are essential for the production of vitamin D in people, yet prolonged exposure to UV radiation may result in acute and chronic health effects on the skin and immune system. The best-known acute effect of excessive UV exposure is sunburn (erythema), which is an acute inflammatory reaction of the skin associated with redness, pruritus, and pain [55]. The erythemal reaction is mainly induced by UVB, particularly at 299 nm, while UVA radiation is 1000-fold less potent and contributes to only 15% of total sun-induced erythema [56].
Long-term damage to skin by chronic sun exposure leads to premature skin aging known as photoaging, and to skin cancers [57,58]. Photoaged skin is characterized by numerous clinical signs that comprise fine and coarse wrinkling, laxity, leathery appearance, mottled pigmentation, fragility and telangiectasias [59]. Numerous studies have revealed that the major alterations in photoaged skin are found in the dermis, which is readily reached by the UVA rays. These rays are absorbed by skin chromophores triggering the generation of reactive oxygen species (ROS) in the resident dermal fibroblasts and in extracellular structures [60]. This contributes to oxidative damage, alterations in gene expression and DNA damage. One of the best-studied DNA lesions caused by UVA is 8-oxo-deoxy-guanosine (8-oxo-dG) resulting from oxidation of the guanine moiety [61]. This lesion is pre-mutagenic and is suspected to be involved in the photocarcinogenic process initiated by sunlight. On the other hand, UVB rays which reach the stratum corneum and upper layers of the epidermis, directly impact DNA by causing cyclobutane pyrimidine dimer (CPD) formation [62]. If this most common defect in UV-irradiated DNA is left unrepaired it leads to “UV signature mutations” (C → T or CC → TT transitions). UVA can also cause CPDs and the same mutations as UVB [63]. A large number of DNA repair enzymes exist in cells to correct these photolesions, but if cell division occurs before the damage has been successfully repaired, then the photolesions may lead to mutations being incorporated into daughter DNA and hence genetic instability. Pyrimidine dimer formation in the skin leads to erythema and immune suppression [64–66]. UV radiation also creates mutations to p53 tumor suppressor genes, some of which are involved in DNA repair and in apoptosis of cells with high genetic instability. Hence if p53 genes are mutated, the DNA repair process will be impaired resulting in loss of pro-apoptotic functions, expansion of mutated skin cells and initiation of skin cancer [67].
Skin cancer is the most common type of cancer in fair-skinned people around the world [68]. The most common ones associated with chronic sunlight exposure are the non-melanoma skin cancers, squamous cell carcinoma (SCC) and basal cell carcinoma (BCC) that affect around 2–3 million people worldwide each year. These cancers occur more frequently on the head, neck, arms and hands, which are the areas most frequently exposed to UV radiation. They are rarely fatal and are often surgically removed. Actinic keratoses (AK) which are pre-cancerous lesions, are also frequent in these body sites and about 5–20% of these lesions progress to SCC [69]. Melanoma instead is responsible for most of skin cancer related mortalities and approximately 130,000 malignant melanomas occur globally each year [26]. About 65–90% of all melanomas are attributable to UV exposure. Overall, a history of exposure to sunlight, particularly during childhood, is the most important behavioral risk factor for the development of both non-melanoma and melanoma skin cancers [70,71].
One of the principal risk factors by which transformed cells can develop into skin cancers is through UV-induced immune suppression. In both experimental animals and humans, the two most prevalent in vivo endpoints used to measure the effects of UV radiation on the immune response are suppression of contact hypersensitivity (CHS) or delayed type hypersensitivity (DTH). CHS is a T-cell mediated immune reaction to a contact allergen applied to the skin, analogous to contact dermatitis in humans. DTH is a T-cell mediated immune reaction to antigens injected into the skin, analogous to the tuberculin reaction in humans. A discussion of immunological mechanisms underlying CHS and DTH is well beyond the scope of this review, and they have been extensively reviewed recently [72,73]. Both UVB and UVA suppress CHS and DTH, albeit with some differences. For instance, UVB suppresses primary immune reactions (both CHS and DTH) while UVA suppresses the recall response (DTH) [74,75]. Also, the UV-dose response curve for UVA-induced suppression of CHS is bell-shaped, whereas that for UVB is linear [76,77]. In addition, studies with human volunteers suggest that doses of UVA that do not suppress CHS by themselves (i.e. the ends of the bell-shaped curve) can cooperate with UVB to enhance suppression of the CHS reaction [78]. A number of immunoregulatory factors are produced in the skin in response to UV that affect the immune response, including PAF, serotonin, histamines, prostaglandins such as PGE2, and cytokines including interleukin (IL)-10, IL-6 and tumor necrosis factor (TNF) [79,80]. The following section will specifically concentrate on the role played by PAF in UV-driven immune suppression, inflammation and skin cancer.
4. PAF and UV-induced immune suppression
4.1. Immune cells of the skin
The skin is an organ with immune function [81] and a special term (SALT: skin associated lymphoid tissue) was designated to describe the special relationship between the skin and elements of the immune system [82]. In fact, several immune cells reside within the skin: antigen-presenting dendritic cells, macrophages, T-cells, innate immune cells and mast cells. These cells are highly regulated by inflammatory mediators such as cytokines and bioactive lipids [25], and the resident skin cells themselves, such as keratinocytes and fibroblasts, are important sources of these mediators that regulate certain functions such as dendritic and mast cell migration [83,84].
Two resident antigen-presenting dendritic cells found in the skin are the Langerhans cells (LCs) present in the epidermis and the dermal dendritic cells. Once these cells have captured an antigen, they migrate from the skin to the draining lymph nodes (LNs) for antigen-presentation to T-cells. T-cells then differentiate and proliferate to give a population of activated, antigen specific T-cells, crucial in determining the type of immune response generated. LCs are now thought to be primarily involved in activating tolerance and immune regulation [85–87], while dermal dendritic cells act as the main antigen-presenting cells for a protective immune response [88,89]. Both LCs and dermal dendritic cells migrate to skin draining LNs under steady-state conditions, but their migration is accelerated by inflammatory environmental insults, such as UV radiation [90]. Recent data has shown that LCs are absolutely essential for UV-induced suppression of CHS in mice [90–92] and that their migration to the draining lymph nodes is induced by UV-induced up-regulation of Receptor Activator of NF-κβ-ligand (RANK-L) on keratinocytes [93].
Normal resting skin is also home to a population of resident T-cells, the majority of which are CD45RO+ memory cells deriving from T-helper (Th)-1 effector memory cells [94]. Present in the normal dermis are also CD4+ T-regulatory cells (Treg) and CD4+ T-helper cells that are distinguished into Th1, Th2 and Th17 cells, each of which secrete their own specific interleukins and other immune mediators [95]. For example, Th2 cells secrete IL-4 and IL-5, which can stimulate IgE production and eosinophil and mast cell activation. They also secrete IL-10 and IL-13, responsible for type 1 allergic or atopic inflammatory responses. Th17 cells secrete IL-17 and play a role in combating infection [96]. A population of long-lasting CD8+ tissue-resident memory cells is also found whose function is to protect the body from infection [97]. In murine skin, these cells have been found one-year post infection, indicating that a robust and long-lasting immune response to invading pathogens can be mounted by T-cells residing in the skin [98]. Other unique T-cells found in the skin are dermal cells that express the Vγ3+ receptor and that appear to play a role in the innate immune response and dendritic epidermal T cells that express the γδT-cell receptor and that appear to function in skin homeostasis and wound repair [99,100]. Skin homeostasis and wound repair are also mediated by resident macrophages since depleting these cells leads to a rapid reduction in skin wound repair [101]. Following UV-irradiation of the skin, macrophages migrate into the skin, where they suppress the generation of the local immune response, in part through the secretion of IL-10 [102].
The most important skin immune cell in the context of a review concerning the mechanism through which UV-induced PAF activates immune suppression is the dermal mast cell. The conventional view of mast cells initially limited their function to immune reactions associated with allergy and those controlling parasitic infections. This view has changed radically in the past few years, since mast cells have been shown to be strong immune regulators [103]. Mast cells in the skin reside in the dermis and their numbers increase after an inflammatory insult [104]. Hart and colleagues were among the very first to demonstrate that mast cells were essential for UV-induced immune suppression, and immune regulation in vivo. In their study, exposing mast-cell deficient mice to UV radiation did not suppress the CHS response, but when these mice were reconstituted with normal bone marrow-derived mast cells, UV-induced suppression of CHS was restored [105]. These observations have subsequently been confirmed by others [106,107] and mast cells have been shown to play an essential role in UVA-induced suppression of a secondary immune reaction (elicitation of the DTH response) as well [108]. It is not surprising then that mast cell density in the skin has been shown to correlate with the incidence of basal cell carcinoma and melanoma, suggesting that the immune regulatory function of mast cells may contribute to skin carcinogenesis [109,110]. A more in-depth role of mast cells in UV-induced immune suppression driven by PAF will be discussed below.
4.2. Generation of PAF, the immune suppressive signal, following UV radiation
The skin is endowed with a number of photoreceptors that are able to convert the energy contained within the UV wavelengths of sunlight into a biologically recognizable signal that induces immune suppression. The first to be identified were urocanic acid [111] and DNA [65] but then others followed, such as tryptophan and the arylhydrocarbon receptor [112], 7-dehydrocholesterol [113], elements in the complement pathway [114], and oxidized membrane lipids of which PAF is a prime example. Once these photoreceptors become activated, they generate an immune suppressive signal in the skin that must then be transmitted to the immune system. Here we will focus on how this is achieved through PAF.
Keratinocytes in the skin release bonafide PAF and PAF-R agonists following UVB exposure and they also respond to PAF since they express the PAF receptor on their surface [115–120]. Worth mentioning is also the production of significant amounts of PAF due to direct damage to keratinocytes by either heat or cold stimuli [121]. UV-irradiation of keratinocytes activates the generation of ROS under physiological conditions, and these promote the photoperoxidation of polyunsaturated cellular phospholipids. Oxidation of esterified fatty acyl residues introduces oxy functions, rearranges bonds and fragments carbon–carbon bonds by β-scission generating PAF and PAF-like lipids such as 1-alkyl-2-butanoyl and butenoyl-sn-glycero-3-phosphocholine in a non-enzymatic way [122,123]. The formation of PAF and PAF-agonists by UVB-mediated ROS has also been shown to be due in part to the activation of the epidermal growth factor receptor (EGF-R), since in vivo production of PAF/PAF agonists from UVB-irradiated mouse skin was blocked by both systemic administration of the antioxidant vitamin C and topical treatment with PD168393, which is a specific inhibitor of EGF-R [124]. The PAF analogs generated from phospholipid peroxidation have been reported to have only one-tenth of the potency of native PAF [125]. Interestingly, the time course of UVB-generated PAF/PAF agonists in human skin also resembles the time course of UVB-mediated ROS in keratinocytes [124] and UV-generated ROS have actually been shown to up-regulate PAF production [126]. Hence this has been suggested to be an important mechanism for initiating the immunosuppressive pathway. Data from experiments in which UV-irradiated cell-free phosphatidylcholine injected into mice suppressed the induction of DTH [127], coupled with the fact that PAF synthesis in cultured keratinocytes and human skin was fully suppressed by the antioxidants N-acetyl cysteine, 1,1,3,3-tetramethyl-2-thiourea, and vitamins C and E, are consistent with this idea [115,116]. Moreover, data in the literature indicating that UV-induced membrane effects activate gene expression provide further support for UV-induced lipid oxidation as an initial step in the process leading to UV-induced immune suppression [128,129]. PAF synthesis by keratinocytes in response to UVB radiation is also greatly enhanced by overexpression of the PAF receptor and is repressed by the PAF-R antagonists WEB 2086 and A-85783 [119]. Therefore, PAF and/or PAF-like molecules act as molecular sensors for cellular damage, such as UV-induced lipid peroxidation, triggering the immune suppressive cascade.
Once PAF is produced following UVB-irradiation, it signals through the epidermal PAF-R, which amplifies and sustains the biological signal by inducing further PAF synthesis via the remodeling pathway [119]. A variety of downstream effects are then generated. These include production of cytokines like TNF-α, IL-6, 8 and 10, cyclooxygenase (COX)-2 and prostaglandin PGE2 that mediate downstream systemic photo-immune suppression [80,130,131]. By using Affymetrix oligonucleotide arrays, Travers et al. showed that the epidermal PAF-R is implicated in the early expression of a wide variety of genes following UV exposure of the PAF-R-positive epithelial cell line (KBP) compared to KB cells transduced with a vector control (KBM) [132]. When total RNA was isolated from KBP cells 1 h after treatment with PAF or UVB radiation, over 200 genes, including those for cytokines and growth factors were altered. In particular, the pro-inflammatory cytokine TNF-α and the chemokine CCL20, which serve as part of the “alarm system” in skin, were selectively up-regulated [132]. The variety of chemokines and cytokines up-regulated by direct PAF-R activation suggests that PAF could play an important immunomodulatory role in keratinocyte function. These in vitro findings were confirmed in vivo where UVB-irradiation resulted in increased levels of epidermal TNF-α and CCL20 mRNA in wild-type (WT) but not in PAF-R-deficient mice [132]. However, one of the most important consequences of UV-induced PAF is its effect on mast cells.
4.3. PAF-derived effects on mast cells
Mast cells generally migrate to sites of inflammation. Therefore, as expected, mast cells have been shown to quickly accumulate in the skin within hours of UV exposure [104], a phenomenon later shown to be dependent upon UV-induced-keratinocyte derived IL-33 [133]. Surprisingly, 24 h post-irradiation, mast cell numbers in the skin return to normal while the mast cell density in the skin draining LNs increased significantly. Proof that the migrating cells came from the skin was provided by experiments in which skin from green fluorescent protein positive mice (GFP+) was grafted onto the backs of mast cell-deficient mice. The ‘green’ mast cells were shown to migrate into the B cell areas of the LNs of the UV-irradiated mice, thus confirming that the migrating mast cells came from the skin. Mast cells express the C–X–C chemokine receptor type 4 (CXCR4) on their surface [134], and UV-exposure up-regulates the expression of both CXCR4 on mast cells and its ligand, CXCL12 by lymph node B cells [104]. Treatment of UV-irradiated mice with the selective CXCR4 antagonist, AMD3100, blocked mast cell migration from the skin to the LNs and abrogated UV-induced suppression of the CHS response [104]. These data provide compelling evidence that the CXCR4–CXCL12 axis is essential for mast cell migration. Recently, Sarchio and colleagues confirmed that blocking mast cells migration with AMD3100 inhibits the suppression of the DTH reaction in UV-irradiated mice and further found that blocking mast cell migration suppresses tumor development [135]. Therefore, mast cells carry the UV-immune suppressive signal from the skin to the immune system in a CXCR4 dependent manner, representing a key early step in UV-induced immune suppression [104].
Data indicating that PAF is the signal that triggers mast cell migration from the skin to the draining LNs was presented in a series of experiments by Chacón-Salinas et al. [136]. The hypothesis that PAF may regulate mast cell migration in vivo was based on several lines of evidence. First, injecting PAF-receptor antagonists into mice prevented the UV-induced suppression of DTH, while injecting carbamyl-PAF (cPAF), a stable bioactive analog of PAF into mice, activated immune suppression [127]. Second, when PAF-receptor knockout mice were exposed to UV radiation, no suppression of DTH or CHS was observed [137, 138]. Third, PAF is known to regulate the migration of many cell types, including tumor cells, neutrophils and leukocytes as well as mast cells [139–144], and mast cells are known to express the PAF receptor and are able to respond to PAF [145]. Chacón-Salinas and colleagues confirmed that mast cell-deficient mice were resistant to the suppressive effect of UV-irradiation, and suppression of the CHS reaction was restored by reconstituting mast cell-deficient mice with normal bone marrow-derived mast cells. However, if the mast cells were derived from PAF-receptor deficient mice, UV exposure did not activate mast cell migration from the skin to the draining LNs, nor were the mice susceptible to the suppressive effects of UV radiation. Similarly, treating WT mice with cPAF activated mast cell migration, and injecting PAF-R antagonists into UV-irradiated WT mice interfered with migration of mast cells into the draining LNs. Moreover, they found increased accumulation of mast cells in the draining LNs of UV-irradiated WT mice but not in those of UV-irradiated PAF-R knockout mice. These findings indicate that PAF-R binding induces mast cells to migrate from the skin to the LNs, where they mediate UV radiation-induced suppression of the CHS reaction.
The other key important outcomes from this study were that PAF treatment up-regulates CXCR4 expression on mast cells and that PAF-treatment up-regulates the expression of CXCL12, the ligand of CXCR4, on draining LN cells. When bone marrow mast cells (BMMC) derived from normal C57BL/6 mice were cultured with cPAF, CXCR4 mRNA expression was increased. Similarly, surface expression of CXCR4 was increased following treatment with cPAF. Little expression of CXCR4 mRNA or protein surface expression was observed when BMMCs derived from PAF-R knockout mice were treated with cPAF. Furthermore, cPAF-treated C57BL/6 BMMC migrated in response to CXCL12, versus the minimal migration observed with PAF-treated BMMC isolated from PAF-R knockout mice. The effect of PAF on the expression of CXCL12 in LN cells was also investigated. Because others have shown that PGE2 up-regulates CXCL12 expression [146], this prostaglandin was used as a positive control in the experiments. The results showed that injecting an immunosuppressive dose of cPAF into C57BL/6 mice resulted in a significant increase in CXCL12 expression by LN cells whereas no CXCL12 expression was observed when cPAF was injected in PAF-R knockout mice [136]. In summary, PAF up-regulates the expression of CXCR4 on mast cells and expression of its ligand, CXCL12, on draining LNs. Similarly, incubating the human mast cell line HMC-1 with cPAF was recently found to increase both total and membrane-bound CXCR4 expression [147].
4.4. PAF and epigenetic modifications
The expression of CXCR4 has been linked to transcriptional regulation involving promoter methylation and histone modification in a wide variety of cancers, including cutaneous and uveal melanomas [148]. This raised the question as to whether PAF could be involved in activating epigenetic alterations that could be linked with the increased CXCR4 expression observed in mast cells. We recently addressed this question by demonstrating that cPAF suppressed the expression of the DNA methyltransferases (DNMTs) 1 and 3b, while increasing expression of histone acetyltransferase p300. This coincided with a decreased expression of the histone deacetylase enzyme HDAC2. Further support was provided by data demonstrating that cPAF caused an increase in acetylated histone H3, both in the transformed mast cell line HMC-1 and in freshly isolated, non-transformed human mast cells. Moreover, chromatin immunoprecipitation assays showed that cPAF treatment leads to the acetylation of the CXCR4 promoter. Finally, inhibiting histone acetylation with curcumin blocked p300 up-regulation and suppressed PAF-induced surface expression of CXCR4 [147]. Because histone acetylation is a well-recognized epigenetic mark that is generally associated with chromatin regions permissive for gene expression, these findings strongly support the hypothesis that cPAF participates in the up-regulation of CXCR4 expression through histone acetylation. Additional evidence supporting the role of PAF-induced transcriptional activation through acetylation comes from recent findings in human mast cells. The key cell cycle regulator p21 was shown to be up-regulated after cPAF treatment and its increased expression was linked to increased acetylated histone-H3, p300 and acetyl-CPB protein expression. We also noted acetylation of the promoter region of the p21 gene (Damiani, Puebla-Osorio and Ullrich; unpublished observations).
4.5. PAF and UV-induced immune suppression
UV exposure suppresses in vivo antibody formation to T-dependent antigens [149,150]. Chacón-Salinas and colleagues took advantage of this phenomenon when studying the effects of UV exposure on germinal center formation [151]. They showed that once PAF-driven mast cells arrive at the draining LNs they suppress T-dependent antibody formation by releasing IL-10, a well-known immune regulatory cytokine known to be essential for suppressing DTH and CHS in UV-irradiated mice [152,153]. They found that UV exposure suppressed antibody formation and germinal center formation in WT UV-irradiated mice and that the mechanism involved suppressing the function of T follicular helper cells, which are the unique T helper cells found in the LN essential for germinal center and antibody formation. No suppression of antibody production or germinal center formation was found in UV-irradiated mast cell deficient mice. As expected, reconstituting the mast cell deficient mice with normal bone marrow derived mast cells restored immune suppression. However, when the mice were reconstituted with mast cells grown from the bone marrow of IL-10-deficient animals, and then exposed to UV radiation, no immune suppression was noted: T follicular helper cell function, germinal center formation and antibody formation were not suppressed. This indicates that UV-activated mast cells are an important source of the IL-10 responsible for UV-induced suppression of antibody formation in vivo.
Overall, the interaction between the bioactive lipid PAF and mast cells provides an excellent example by which the immune suppressive and carcinogenic potential present in UV light is conveyed from the upper layers of the skin to the immune system, thus activating systemic photo-immune suppression. The model proposed is summarized in Fig. 4. UV-irradiation of the skin induces epidermal keratinocytes to release PAF and oxidized phospholipids that bind to the PAF-R on mast cells. This triggers the surface expression of the chemokine CXCR4 on mast cells, in part by activating histone acetylation of the CXCR4 promoter, and the up-regulation of CXCL12 expression on LN cells. Since PAF is rapidly cleared from the circulation and has a limited half-life (3–13 min) due to its degradation by PAF-AH [12,154], it is unlikely that keratinocyte-derived PAF gets to the LNs. However, because PAF promotes production of prostaglandin PGE2 [104,136] it is reasonable to assume that PAF-induced PGE2 up-regulates CXCL12 expression in the lymph node, thus setting up the gradient needed for mast cell migration. This is supported in part, by the observation that injecting PGE2 into PAF-R knockout mice, over-rides the inability of PAF to up-regulate the expression of CXCL12 [136]. Mast cells then migrate from the skin to the draining LNs where they suppress the immune response by secreting IL-10. Moreover, since Walterscheid et al. previously demonstrated that PAF activates transcription of the IL-10 promoter [127], it is reasonable to assume that PAF is the signal that activates mast cell IL-10 secretion. Zhang et al. also confirmed the involvement of UV-induced PAF and PAF agonists acting on PAF-R-positive bone marrow-derived cells to up-regulate IL-10 through COX-2-generated prostaglandins [155]. Further involvement of PAF-mediated IL-10 production in photo-induced immune suppression has been reported by Rockel and colleagues during their investigations of the role of the osmolyte taurine (2-aminoethane sulfonic acid) and its receptor, taut in modulating UV-induced suppression of CHS [156]. Taut-deficient mice are more sensitive to the immuno-suppressive effects of UVB radiation. This increased sensitivity was associated with increased PAF production by keratinocytes in UVB-irradiated taut-knockout mice. They further showed that PAF induction correlated positively in a UVB dose-dependent manner with early IL-10 mRNA expression, suggesting possible involvement of PAF receptor signaling in photo-immune suppression in taut knockout mice. When PAF signaling was blocked by injecting the PAF-R antagonist PCA-4248, UVB-induced suppression of CHS was reversed in taut knockout mice. The immunosuppressive capacity of PAF was confirmed by injecting cPAF into taut knockout mice compared to WT mice. Hence, these results suggest a protective role for taurine against UVB-induced suppression of CHS, in particular by controlling the generation of signaling lipids like PAF, which induce immune regulatory IL-10.
The evidence reported here so far deals with PAF and its involvement in systemic photo-immune suppression (UV radiation applied to the skin, with the antigen applied or injected at a distant non-irradiated site). But what about its effects on local photo-immune suppression (UV radiation and the antigen both applied to the same site on the skin)? Sahu and colleagues addressed this issue by comparing local UVB irradiation on CHS responses in WT versus PAF-R knockout mice. They found that applying the contact allergen DNFB (2,4-dini-tro-1-fluorobenzene) onto UVB-exposed mouse skin resulted in a sig-nificant inhibition of CHS responses in both WT and PAF-R knockout mice. Furthermore, the expression of langerin, a marker for the presence of LC was reduced equally in the ears of both types of mice compared to their respective sham irradiated control groups [155,157]. These find-ings indicate that PAF does not mediate UVB-induced local immune suppression, suggesting that PAF accounts for some but not all of the effects of UV radiation on the immune system.
4.6. PAF and its effects on Langerhans cells
In addition to mast cells, migrating LCs also play an important role in transmitting the immune suppressive signal from the skin to the immune system following in vivo UV exposure [91]. UV irradiation of the skin promotes LCs to migrate to the LNs where they activate immune regulatory T cells (i.e. Tregs and Natural Killer T cells) that suppress DTH, CHS and tumor immunity [90,158,159]. UV exposure also up-regulates the expression of RANK-L in keratinocytes and this activates LCs migration to the LNs where they trigger the proliferation of Tregs [93]. PAF-induced PGE2 may be involved in up-regulating RANK-L on keratinocytes, since blocking the binding of PGE2 to its receptor (EP4) using selective receptor antagonists, blocks RANK-L up-regulation, impairs UV-induced Treg activation and blocks UV-induced suppression of CHS [160]. In addition, applying the hapten DNFB to wild type mice activates LCs migration, which was not observed when the same contact allergen was applied to the skin of PAF-R knockout mice [88]. Moreover, LC migration was suppressed in DNFB treated wild-type mice injected with the PAF-R antagonists, PCA4248 and CV3938. To determine whether the PAF-R on keratinocytes or LCs (or both) were more important for LCs migration, LC chimeric mice whose keratinocytes were PAF-R−/− and LC were PAF-R+/+, or whose LCs were PAF-R−/−and keratinocytes were PAF-R+/+ were produced. Fukunaga et al. observed that the PAF-R on keratinocytes, but not on LCs, plays an important role in hapten-induced LC migration [88]. These results suggest that UV-induced PAF binds to its receptor on keratinocytes and activates the keratinocytes to release cytokines (i.e., TNF-α, IL-1β) that drive LC migration and the subsequent activation of immune suppression.
4.7. PAF and skin cancer
UV-induced immunosuppression is a well-known risk factor for skin cancer induction. Since the PAF/PAF-R system is implicated in mediating this event, what are the consequences concerning skin cancer? Sreevidya et al. tested the hypothesis that blocking agents that contribute to immune suppression (i.e., PAF) could prevent skin cancer induction [161]. They used both PAF-R antagonists and serotonin receptor antagonists, the latter being the receptor for cis-urocanic acid [162], another important mediator of immune suppression produced in the skin [111]. They observed a significant and substantial decrease in skin cancer incidence and progression in hairless mice treated with these antagonists and then exposed to solar-simulated UV radiation. After a single UV exposure, they also observed inhibition of UV-induced skin damage, such as sunburn cell formation, hypertrophy, cytokine release and apo-ptosis [163].
UVB generated PAF/PAF-R agonists have also been shown to increase melanoma growth in an experimental murine melanoma model. When WT mice harboring the mouse melanoma B16F10 cell line were exposed to UVB, a significant increase in tumor growth was observed compared to what was seen with PAF-R knockout mice, which were resistant to UVB-mediated increase of melanoma tumor growth. This increase was inhibited with antioxidants, demonstrating the importance of UV-induced PAF and oxidized PAF agonists in skin cancer induction [164]. For further reading regarding the role of PAF and its receptor in melanoma growth and metastasis, we recommend the review by Melnikova and Bar-Eli [15].
PAF and PAF agonists not only appear to be involved in skin cancer induction but they have also recently been shown to hamper systemic chemotherapy against experimental melanoma. In fact, Sahu et al. reported that etopside-mediated suppression of melanoma tumor growth in syngeneic mice was blocked by the exogenous administration of cPAF [165]. Another study by the same group further illustrates the importance of the PAF system in interfering with skin cancer treatment. Pho-todynamic therapy (PDT) is a well-recognized procedure used for the treatment of pre-cancerous actinic keratosis as well as superficial skin cancers [166]. This type of therapy has been shown to cause immune suppression in both humans and mice, however, the mechanisms underlying these effects were not completely clear [167]. Therefore, the hypothesis that PAF-R activation mediates PDT-induced systemic im-munosuppression was tested. Ferracini et al. first demonstrated using KBP and KBM cells, that PDT generates PAF-R agonists that are enzymatically produced, and second, that PDT significantly inhibited CHS reactions in WT, but not in PAF-R deficient mice. Injecting cPAF had the same effect as PDT, inducing immune suppression only in WT mice [168].
Another hypothesis that was tested suggested that blocking mast cell migration, with the CXCR4 antagonist AMD3100, would prevent sunlight-induced skin cancer. When C57BL/6 mice were exposed to UVB prior to treatment with AMD3100, squamous cell carcinoma incidence and burden was significantly reduced compared to the control mice. A most striking result was the observation that AMD3100 treatment completely prevented the outgrowth of latent tumors that occurred once UV irradiation ceased and this was associated with reduced mast cell infiltration in the skin, draining LNs and the tumor itself [135].
4.8. PAF and cell cycle arrest, DNA damage response and apoptosis
The carcinogenic effect of UV radiation results from its ability to induce DNA mutations in tumor suppressor genes [169,170] and impair DNA repair. UV-induced PAF contributes to these effects as evidenced by the fact that blocking the binding of PAF to its receptor blocks the induction and progression of UV-induced skin cancer and DNA repair [161,163]. We recently provided data indicating that PAF arrests proliferation in human mast cells by reducing the expression of cyclin-B1 and cyclin-dependent kinase-1 and promoting the expression of a variety of genes that control the cell cycle, including cyclin-dependent kinase inhibitor p21CDKN1A [171]. p21 is a well-studied potent inhibitor that binds to, and inhibits the activity of several cyclins and cyclin-dependent kinase complexes [172]. It is a member of the CIP/KIP family of cell cycle regulators and plays an important role in regulating many fundamental biological processes [173]. It also serves as a regulator of cell cycle progression [174], and plays a pivotal role in regulating cell quiescence, senescence and differentiation [175–177]. In our study we noted cell-cycle arrest mainly at the G2-M checkpoint following PAF treatment [171]. p21 also participates in other regulatory roles such as in p53-dependent and/or independent apoptosis and in transcriptional regulation [178,179]. Additionally, p21 activity appears to be necessary for DNA repair in the presence of low levels of genotoxic stress, while it is degraded in the presence of extensive DNA damage to favor apoptotic cell death [180]. We found that PAF induced the expression of pro-apoptotic markers (Cleaved Parp-1 and Caspase-3) in treated cells [171]. We also found that PAF disrupted the expression of key components of the DNA repair machinery (γ-H2AX, ATR, ATR-interacting protein and Brit1/microcephalin) and the localization of these important DNA repair elements to the site of DNA damage. These findings are in agreement with data previously reported by Barber and colleagues showing that PAF activates apoptosis [119], and suggests that PAF's effect on the mitotic machinery may provide a reasonable explanation for this phenomena.
5. Conclusions and future directions
A quick review of the literature indicates that PAF plays an important role in a wide variety of biological processes, including glycogen degradation, reproduction, brain function, blood circulation and obesity [5–9]. It has also been implicated in a variety of disease states, ranging from myo-cardial infarction, autoimmunity and cancer, just to name a few [11,13–15]. Skin trauma activates PAF release by damaged keratinocytes. This affects basic biological processes in the skin, including DNA repair, pain perception and cutaneous inflammation [121,171,181].
In this review we focused on the role of PAF in activating systemic immune suppression, which has been shown to be a major risk factor for sunlight-induced skin cancer induction. One of the most intriguing aspects of the induction of systemic immune suppression by UVB radiation, and a subject of many research efforts over the years, revolved around the question as to how the immunosuppressive signal contained within UVB radiation, which is essentially absorbed in the very upper layers of the skin, is transmitted to the immune system. Here we summarize the data indicating that keratinocyte-derived PAF targets dermal mast cells and activates the up-regulation of CXCR4 on the surface of the mast cells, which is critical for the migration of the mast cells to the skin draining lymph nodes. Upon arrival of the mast cell to the lymph nodes, they secrete the immune regulatory cytokine IL-10 and suppress CHS, the activation of T follicular helper cells, germinal center formation and antibody production in vivo. Because PAF activates the transcription of the IL-10 gene in treated cells, we suspect that UV-induced keratinocyte-derived PAF is the signal that activates the immune suppressive program in mast cells.
Further, it is now apparent that PAF can activate an epigenetic program in mast cells that suppresses DNA methylation, increasing the expression of the histone acetylating enzyme, p300, which activates acetylation of histone H3, resulting in acetylation of the CXCR4 promoter [147]. These novel results identify a new molecular role for PAF by demonstrating that it can affect basic biological processes by activating the epigenetic machinery. These studies reveal a plausible connection between UV radiation in the external environment, keratinocyte-derived PAF and chromatin patterns in immune cells of the skin, suggesting that this bioactive lipid may serve as a molecular link between the environment and the epigenome. Whether it is possible to use PAF and or PAF-receptor-agonists as drugs to suppress DNA methylation, and activate histone H3 acetylation remains to be seen.
Apart from furthering our understanding of how this bioactive lipid modulates basic biological processes, recent findings also suggest that modulating PAF's functions in vivo may affect disease processes. For example, PAF-R antagonists have been shown to prevent skin cancer induction and progression in experimental animals [161,164]. Similarly, UV-irradiation of the skin results in DNA damage and inefficient DNA repair contributes to the mutations associated with cancer progression. Keratinocyte-derived PAF delays DNA repair [163,171]. Whether blocking the function of PAF in vivo (i.e., use of PAF-R antagonists or PAF-AH) is applicable to human skin cancer therapy remains to be seen. In addition PAF production is associated with PDT-induced immunosuppression [168]. Could the use of PAF-R antagonists, or PAF-AH divorce the immunosuppressive effects of PDT from its therapeutic activity?
Finally, although our focus is on the deleterious effects of UV on the immune system and skin cancer induction, it has become apparent that the immune regulation activated by UV can also have advantageous effects. Multiple sclerosis and asthma are more prevalent at higher latitudes and some suspect that the lack of UV-induced immune suppression may play a role [182,183]. Although narrow band UV radiation has been used in phototherapy for decades, its carcinogenic potential is a considerable dangerous side effect. Could activating the PAF-pathway replace UV and induce immune regulation? We suggest that further efforts to understand the effect(s) of bioactive lipids, such as PAF, on the immune response may provide opportunities to develop new strategies to treat diseases such as cancer and autoimmunity in the future.
Abbreviations
- 8-oxo-dG
8-oxo-deoxy-guanosine
- AK
actinic keratosis
- BCC
basal cell carcinoma
- BMMC
bone marrow derived mast cells
- cPAF
carbamyl-PAF
- COX-2
cyclooxygenase-2
- CHS
contact hypersensitivity
- CPD
cyclobutane pyrimidine dimer
- CXCR4
C–X–C chemokine receptor type 4
- CXCL12
C–X–C chemokine ligand 12
- DNFB
dinitroflurobenzene
- DTH
delayed type hypersensitivity
- EGR-R
epidermal growth factor receptor
- IL
interleukin
- LC
Langerhans cells
- LN
lymph nodes
- LPCAT
lyso-PAF actyltransferase
- PAF
platelet activating factor
- PAF-R
platelet activating factor receptor
- PAF-AH
PAF-acetylhydrolase
- PGE
prostaglandin E2
- PLA2
phospholipase A2
- PDT
photodynamic therapy
- RANK
receptor activator of NF-κβ
- RANKL
receptor activator of NF-κβ ligand
- ROS
reactive oxygen species
- SCC
squamous cell carcinoma
- Th
T helper cell
- Treg
T regulatory cells
- TNF
tumor necrosis factor
- UV
ultraviolet
- UVA
UV (320–400 nm)
- UVB
UV (290–320 nm)
- UVC
UV (100–290 nm)
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Benveniste J, Henson PM, Cochrane CG. Leukocyte-dependent histamine release from rabbit platelets. The role of IgE, basophils, and a platelet-activating factor. J Exp Med. 1972;136:1356–1377. doi: 10.1084/jem.136.6.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muirhead EE. Antihypertensive functions of the kidney: Arthur C. Corcoran memorial lecture. Hypertension. 1980;2:444–464. doi: 10.1161/01.hyp.2.4.444. [DOI] [PubMed] [Google Scholar]
- 3.Ishii S, Nagase T, Shimizu T. Platelet-activating factor receptor. Prostaglandins Other Lipid Mediat. 2002;68–69:599–609. doi: 10.1016/s0090-6980(02)00058-8. [DOI] [PubMed] [Google Scholar]
- 4.Ishii S, Shimizu T. Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog Lipid Res. 2000;39:41–82. doi: 10.1016/s0163-7827(99)00016-8. [DOI] [PubMed] [Google Scholar]
- 5.Bellizzi MJ, Geathers JS, Allan KC, Gelbard HA. Platelet-activating factor receptors mediate excitatory postsynaptic hippocampal injury in experimental autoimmune encephalomyelitis. J Neurosci. 2016;36:1336–1346. doi: 10.1523/JNEUROSCI.1171-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang W, Zhang R, Sha L, Lv P, Shang E, Han D, et al. Platelet activating factor induces transient blood–brain barrier opening to facilitate edaravone penetration into the brain. J Neurochem. 2014;128:662–671. doi: 10.1111/jnc.12507. [DOI] [PubMed] [Google Scholar]
- 7.Kimura K, Moriyama M, Nishisako M, Kannan Y, Shiota M, Sakurada K, et al. Modulation of platelet activating factor-induced glycogenolysis in the perfused rat liver after administration of endotoxin in vivo. J Biochem. 1998;123:142–149. doi: 10.1093/oxfordjournals.jbchem.a021901. [DOI] [PubMed] [Google Scholar]
- 8.Silver RK, Caplan MS, Kelly AM. Amniotic fluid platelet-activating factor (PAF) is elevated in patients with tocolytic failure and preterm delivery. Prostaglandins. 1992;43:181–187. doi: 10.1016/0090-6980(92)90085-8. [DOI] [PubMed] [Google Scholar]
- 9.Sugatani J, Sadamitsu S, Yamaguchi M, Yamazaki Y, Higa R, Hattori Y, et al. Antiobese function of platelet-activating factor: increased adiposity in platelet-activating factor receptor-deficient mice with age. FASEB J. 2014;28:440–452. doi: 10.1096/fj.13-233262. [DOI] [PubMed] [Google Scholar]
- 10.Palgan K, Bartuzi Z. Platelet activating factor in allergies. Int J Immunopathol Pharmacol. 2015 doi: 10.1177/0394632015600598. [DOI] [PubMed] [Google Scholar]
- 11.Yost CC, Weyrich AS, Zimmerman GA. The platelet activating factor (PAF) signaling cascade in systemic inflammatory responses. Biochimie. 2010;92:692–697. doi: 10.1016/j.biochi.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Platelet-activating factor and related lipid mediators. Annu Rev Biochem. 2000;69:419–445. doi: 10.1146/annurev.biochem.69.1.419. [DOI] [PubMed] [Google Scholar]
- 13.Detopoulou P, Nomikos T, Fragopoulou E, Chrysohoou C, Antonopoulou S. Platelet activating factor in heart failure: potential role in disease progression and novel target for therapy. Curr Heart Fail Rep. 2013;10:122–129. doi: 10.1007/s11897-013-0131-2. [DOI] [PubMed] [Google Scholar]
- 14.Edwards LJ, Constantinescu CS. Platelet activating factor/platelet activating factor receptor pathway as a potential therapeutic target in autoimmune diseases. Inflamm Allergy Drug Targets. 2009;8:182–190. doi: 10.2174/187152809788681010. [DOI] [PubMed] [Google Scholar]
- 15.Melnikova V, Bar-Eli M. Inflammation and melanoma growth and metastasis: the role of platelet-activating factor (PAF) and its receptor. Cancer Metastasis Rev. 2007;26:359–371. doi: 10.1007/s10555-007-9092-9. [DOI] [PubMed] [Google Scholar]
- 16.Prescott SM, McIntyre TM, Zimmerman GA. The role of platelet-activating factor in endothelial cells. Thromb Haemost. 1990;64:99–103. [PubMed] [Google Scholar]
- 17.Prescott SM, Zimmerman GA, McIntyre TM. Platelet-activating factor. J Biol Chem. 1990;265:17381–17384. [PubMed] [Google Scholar]
- 18.Reznichenko A, Korstanje R. The role of platelet-activating factor in mesangial pathophysiology. Am J Pathol. 2015;185:888–896. doi: 10.1016/j.ajpath.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snyder F. Platelet-activating factor and its analogs: metabolic pathways and related intracellular processes. Biochim Biophys Acta. 1995;1254:231–249. doi: 10.1016/0005-2760(94)00192-2. [DOI] [PubMed] [Google Scholar]
- 20.Snyder F. Platelet-activating factor: the biosynthetic and catabolic enzymes. Biochem J. 1995;305(Pt 3):689–705. doi: 10.1042/bj3050689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stafforini DM, McIntyre TM, Zimmerman GA, Prescott SM. Platelet-activating factor, a pleiotrophic mediator of physiological and pathological processes. Crit Rev Clin Lab Sci. 2003;40:643–672. doi: 10.1080/714037693. [DOI] [PubMed] [Google Scholar]
- 22.Tiemann U. The role of platelet-activating factor in the mammalian female reproductive tract. Reprod Domest Anim. 2008;43:647–655. doi: 10.1111/j.1439-0531.2007.00959.x. [DOI] [PubMed] [Google Scholar]
- 23.Zimmerman GA, McIntyre TM, Prescott SM, Otsuka K. Brief review: molecular mechanisms of neutrophil binding to endothelium involving platelet-activating factor and cytokines. J Lipid Mediat. 1990;2(Suppl):S31–S43. [PubMed] [Google Scholar]
- 24.Gill P, Jindal NL, Jagdis A, Vadas P. Platelets in the immune response: revisiting platelet-activating factor in anaphylaxis. J Allergy Clin Immunol. 2015;135:1424–1432. doi: 10.1016/j.jaci.2015.04.019. [DOI] [PubMed] [Google Scholar]
- 25.Kendall AC, Nicolaou A. Bioactive lipid mediators in skin inflammation and immunity. Prog Lipid Res. 2013;52:141–164. doi: 10.1016/j.plipres.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 26.American Cancer Society. [Accessed January 9, 2016];Cancer Facts & Figures. 2015 http://www.cancer.org/acs/groups/content/@editorial/documents/document/acspc-044552.pdf.
- 27.Snyder F. Chemical and biochemical aspects of platelet activating factor: a novel class of acetylated ether-linked choline-phospholipids. Med Res Rev. 1985;5:107–140. doi: 10.1002/med.2610050105. [DOI] [PubMed] [Google Scholar]
- 28.Clay KL, Johnson C, Worthen GS. Biosynthesis of platelet activating factor and 1-O-acyl analogues by endothelial cells. Biochim Biophys Acta. 1991;1094:43–50. doi: 10.1016/0167-4889(91)90024-r. [DOI] [PubMed] [Google Scholar]
- 29.Tordai A, Franklin RA, Johnson C, Mazer BD, Clay KL, Gelfand EW. Autocrine stimulation of B lymphocytes by a platelet-activating factor receptor agonist, 1-palmitoyl-2-acetyl-sn-glycero-3-phosphocholine. J Immunol. 1994;152:566–573. [PubMed] [Google Scholar]
- 30.Hishikawa D, Hashidate T, Shimizu T, Shindou H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J Lipid Res. 2014;55:799–807. doi: 10.1194/jlr.R046094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramesha CS, Pickett WC. Platelet-activating factor and leukotriene biosynthesis is inhibited in polymorphonuclear leukocytes depleted of arachidonic acid. J Biol Chem. 1986;261:7592–7595. [PubMed] [Google Scholar]
- 32.Leslie CC. Properties and regulation of cytosolic phospholipase A2. J Biol Chem. 1997;272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- 33.Morimoto R, Shindou H, Oda Y, Shimizu T. Phosphorylation of lysophosphatidylcholine acyltransferase 2 at Ser34 enhances platelet-activating factor production in endotoxin-stimulated macrophages. J Biol Chem. 2010;285:29857–29862. doi: 10.1074/jbc.M110.147025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morimoto R, Shindou H, Tarui M, Shimizu T. Rapid production of platelet-activating factor is induced by protein kinase C alpha-mediated phosphorylation of lysophosphatidylcholine acyltransferase 2 protein. J Biol Chem. 2014;289:15566–15576. doi: 10.1074/jbc.M114.558874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakanishi H, Shindou H, Hishikawa D, Harayama T, Ogasawara R, Suwabe A, et al. Cloning and characterization of mouse lung-type acyl-CoA:lysophosphatidylcholine acyltransferase 1 (LPCAT1). Expression in alveolar type II cells and possible involvement in surfactant production. J Biol Chem. 2006;281:20140–20147. doi: 10.1074/jbc.M600225200. [DOI] [PubMed] [Google Scholar]
- 36.Chen X, Hyatt BA, Mucenski ML, Mason RJ, Shannon JM. Identification and characterization of a lysophosphatidylcholine acyltransferase in alveolar type II cells. Proc Natl Acad Sci U S A. 2006;103:11724–11729. doi: 10.1073/pnas.0604946103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shida-Sakazume T, Endo-Sakamoto Y, Unozawa M, Fukumoto C, Shimada K, Kasamatsu A, et al. Lysophosphatidylcholine acyltransferase1 overexpression promotes oral squamous cell carcinoma progression via enhanced biosynthesis of platelet-activating factor. PLoS One. 2015;10:e0120143. doi: 10.1371/journal.pone.0120143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou X, Lawrence TJ, He Z, Pound CR, Mao J, Bigler SA. The expression level of lysophosphatidylcholine acyltransferase 1 (LPCAT1) correlates to the progression of prostate cancer. Exp Mol Pathol. 2012;92:105–110. doi: 10.1016/j.yexmp.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morita Y, Sakaguchi T, Ikegami K, Goto-Inoue N, Hayasaka T, Hang VT, et al. Lysophosphatidylcholine acyltransferase 1 altered phospholipid composition and regulated hepatoma progression. J Hepatol. 2013;59:292–299. doi: 10.1016/j.jhep.2013.02.030. [DOI] [PubMed] [Google Scholar]
- 40.Mansilla F, da Costa KA, Wang S, Kruhoffer M, Lewin TM, Orntoft TF, et al. Lysophosphatidylcholine acyltransferase 1 (LPCAT1) overexpression in human co-lorectal cancer. J Mol Med (Berl) 2009;87:85–97. doi: 10.1007/s00109-008-0409-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shindou H, Hishikawa D, Harayama T, Eto M, Shimizu T. Generation of membrane diversity by lysophospholipid acyltransferases. J Biochem. 2013;154:21–28. doi: 10.1093/jb/mvt048. [DOI] [PubMed] [Google Scholar]
- 42.Marathe GK, Pandit C, Lakshmikanth CL, Chaithra VH, Jacob SP, D'Souza CJ. To hydrolyze or not to hydrolyze: the dilemma of platelet-activating factor acetylhydrolase. J Lipid Res. 2014;55:1847–1854. doi: 10.1194/jlr.R045492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McIntyre TM, Prescott SM, Stafforini DM. The emerging roles of PAF acetylhydrolase. J Lipid Res. 2009;50(Suppl):S255–S259. doi: 10.1194/jlr.R800024-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Proksch E, Brandner JM, Jensen JM. The skin: an indispensable barrier. Exp Dermatol. 2008;17:1063–1072. doi: 10.1111/j.1600-0625.2008.00786.x. [DOI] [PubMed] [Google Scholar]
- 45.Clausen BE, Kel JM. Langerhans cells: critical regulators of skin immunity? Immunol Cell Biol. 2010;88:351–360. doi: 10.1038/icb.2010.40. [DOI] [PubMed] [Google Scholar]
- 46.Friedmann PS, Strickland I, Memon AA, Johnson PM. Early time course of recruitment of immune surveillance in human skin after chemical provocation. Clin Exp Immunol. 1993;91:351–356. doi: 10.1111/j.1365-2249.1993.tb05908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kish DD, Li X, Fairchild RL. CD8 T cells producing IL-17 and IFN-gamma initiate the innate immune response required for responses to antigen skin challenge. J Immunol. 2009;182:5949–5959. doi: 10.4049/jimmunol.0802830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Falcao-Pires I, Castro-Chaves P, Miranda-Silva D, Lourenco AP, Leite-Moreira AF. Physiological, pathological and potential therapeutic roles of adipokines. Drug Discov Today. 2012;17:880–889. doi: 10.1016/j.drudis.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 49.Iyer A, Fairlie DP, Prins JB, Hammock BD, Brown L. Inflammatory lipid mediators in adipocyte function and obesity. Nat Rev Endocrinol. 2010;6:71–82. doi: 10.1038/nrendo.2009.264. [DOI] [PubMed] [Google Scholar]
- 50.Lee SH, Jin SY, Song JS, Seo KK, Cho KH. Paracrine effects of adipose-derived stem cells on keratinocytes and dermal fibroblasts. Ann Dermatol. 2012;24:136–143. doi: 10.5021/ad.2012.24.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Monteiro-Riviere NA. Structure and function of skin. In: Monteiro-Riviere NA, editor. Toxicology of the Skin. New York, NY: Informa Healthcare; 2010. pp. 1–18. [Google Scholar]
- 52.Hussein MR. Ultraviolet radiation and skin cancer: molecular mechanisms. J Cutan Pathol. 2005;32:191–205. doi: 10.1111/j.0303-6987.2005.00281.x. [DOI] [PubMed] [Google Scholar]
- 53.Nishigori C. Cellular aspects of photocarcinogenesis. Photochem Photobiol Sci. 2006;5:208–214. doi: 10.1039/b507471a. [DOI] [PubMed] [Google Scholar]
- 54.Madronich S, McKenzie RL, Bjorn LO, Caldwell MM. Changes in biologically active ultraviolet radiation reaching the Earth's surface. J Photochem Photobiol B. 1998;46:5–19. doi: 10.1016/s1011-1344(98)00182-1. [DOI] [PubMed] [Google Scholar]
- 55.Farr PM, Diffey BL. The erythemal response of human skin to ultraviolet radiation. Br J Dermatol. 1985;113:65–76. doi: 10.1111/j.1365-2133.1985.tb02045.x. [DOI] [PubMed] [Google Scholar]
- 56.Halliday GM, Byrne SN, Damian DL. Ultraviolet A radiation: its role in immunosuppression and carcinogenesis. Semin Cutan Med Surg. 2011;30:214–221. doi: 10.1016/j.sder.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 57.Soehnge H, Ouhtit A, Ananthaswamy ON. Mechanisms of induction of skin cancer by UV radiation. Front Biosci. 1997;2:d538–d551. doi: 10.2741/a211. [DOI] [PubMed] [Google Scholar]
- 58.Narayanan DL, Saladi RN, Fox JL. Ultraviolet radiation and skin cancer. Int J Dermatol. 2010;49:978–986. doi: 10.1111/j.1365-4632.2010.04474.x. [DOI] [PubMed] [Google Scholar]
- 59.Tsoureli-Nikita E, Watson RE, Griffiths CE. Photoageing: the darker side of the sun. Photochem Photobiol Sci. 2006;5:160–164. doi: 10.1039/b507492d. [DOI] [PubMed] [Google Scholar]
- 60.Wondrak GT, Jacobson MK, Jacobson EL. Endogenous UVA-photosensitizers: mediators of skin photodamage and novel targets for skin photoprotection. Photochem Photobiol Sci. 2006;5:215–237. doi: 10.1039/b504573h. [DOI] [PubMed] [Google Scholar]
- 61.Kvam E, Tyrrell RM. Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis. 1997;18:2379–2384. doi: 10.1093/carcin/18.12.2379. [DOI] [PubMed] [Google Scholar]
- 62.Setlow RB. Shedding light on proteins, nucleic acids, cells, humans and fish. Mutat Res. 2002;511:1–14. doi: 10.1016/s1383-5742(02)00004-2. [DOI] [PubMed] [Google Scholar]
- 63.Mouret S, Baudouin C, Charveron M, Favier A, Cadet J, Douki T. Cyclobutane pyrim-idine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci U S A. 2006;103:13765–13770. doi: 10.1073/pnas.0604213103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mouret S, Leccia MT, Bourrain JL, Douki T, Beani JC. Individual photosensitivity of human skin and UVA-induced pyrimidine dimers in DNA. J Invest Dermatol. 2011;131:1539–1546. doi: 10.1038/jid.2011.47. [DOI] [PubMed] [Google Scholar]
- 65.Kripke ML, Cox PA, Alas LG, Yarosh DB. Pyrimidine dimers in DNA initiate systemic immunosuppression in UV-irradiated mice. Proc Natl Acad Sci U S A. 1992;89:7516–7520. doi: 10.1073/pnas.89.16.7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuchel JM, Barnetson RS, Halliday GM. Cyclobutane pyrimidine dimer formation is a molecular trigger for solar-simulated ultraviolet radiation-induced suppression of memory immunity in humans. Photochem Photobiol Sci. 2005;4:577–582. doi: 10.1039/b504068j. [DOI] [PubMed] [Google Scholar]
- 67.Benjamin CL, Ananthaswamy HN. p53 and the pathogenesis of skin cancer. Toxicol Appl Pharmacol. 2007;224:241–248. doi: 10.1016/j.taap.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Breitbart EW, Greinert R, Volkmer B. Effectiveness of information campaigns. Prog Biophys Mol Biol. 2006;92:167–172. doi: 10.1016/j.pbiomolbio.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 69.Preston DS, Stern RS. Nonmelanoma cancers of the skin. N Engl J Med. 1992;327:1649–1662. doi: 10.1056/NEJM199212033272307. [DOI] [PubMed] [Google Scholar]
- 70.Kricker A, Armstrong BK, English DR. Sun exposure and non-melanocytic skin cancer. Cancer Causes Control. 1994;5:367–392. doi: 10.1007/BF01804988. [DOI] [PubMed] [Google Scholar]
- 71.Westerdahl J, Olsson H, Ingvar C. At what age do sunburn episodes play a crucial role for the development of malignant melanoma. Eur J Cancer. 1994;30A:1647–1654. doi: 10.1016/0959-8049(94)00337-5. [DOI] [PubMed] [Google Scholar]
- 72.Honda T, Egawa G, Grabbe S, Kabashima K. Update of immune events in the murine contact hypersensitivity model: toward the understanding of allergic contact dermatitis. J Invest Dermatol. 2013;133:303–315. doi: 10.1038/jid.2012.284. [DOI] [PubMed] [Google Scholar]
- 73.O'Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MPR. The immune response in tuberculosis. Annu Rev Immunol. 2013;31:475–527. doi: 10.1146/annurev-immunol-032712-095939. [DOI] [PubMed] [Google Scholar]
- 74.Nghiem DX, Kazimi N, Clydesdale G, Ananthaswamy HN, Kripke ML, Ullrich SE. Ultraviolet a radiation suppresses an established immune response: implications for sunscreen design. J Invest Dermatol. 2001;117:1193–1199. doi: 10.1046/j.0022-202x.2001.01503.x. [DOI] [PubMed] [Google Scholar]
- 75.Moyal DD, Fourtanier AM. Broad-spectrum sunscreens provide better protection from the suppression of the elicitation phase of delayed-type hypersensitivity response in humans. J Invest Dermatol. 2001;117:1186–1192. doi: 10.1046/j.0022-202x.2001.01545.x. [DOI] [PubMed] [Google Scholar]
- 76.Byrne SN, Spinks N, Halliday GM. Ultraviolet a irradiation of C57BL/6 mice suppresses systemic contact hypersensitivity or enhances secondary immunity depending on dose. J Invest Dermatol. 2002;119:858–864. doi: 10.1046/j.1523-1747.2002.00261.x. [DOI] [PubMed] [Google Scholar]
- 77.Matthews YJ, Halliday GM, Phan TA, Damian DL. Wavelength dependency for UVA-induced suppression of recall immunity in humans. J Dermatol Sci. 2010;59:192–197. doi: 10.1016/j.jdermsci.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 78.Poon TS, Barnetson RS, Halliday GM. Sunlight-induced immunosuppression in humans is initially because of UVB, then UVA, followed by interactive effects. J Invest Dermatol. 2005;125:840–846. doi: 10.1111/j.0022-202X.2005.23894.x. [DOI] [PubMed] [Google Scholar]
- 79.Shreedhar V, Giese T, Sung VW, Ullrich SE. A cytokine cascade including prostaglandin E2, IL-4, and IL-10 is responsible for UV-induced systemic immune suppression. J Immunol. 1998;160:3783–3789. [PubMed] [Google Scholar]
- 80.Ullrich SE. Mechanisms underlying UV-induced immune suppression. Mutat Res. 2005;571:185–205. doi: 10.1016/j.mrfmmm.2004.06.059. [DOI] [PubMed] [Google Scholar]
- 81.Bangert C, Brunner PM, Stingl G. Immune functions of the skin. Clin Dermatol. 2011;29:360–376. doi: 10.1016/j.clindermatol.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 82.Streilein JW. Skin-associated lymphoid tissue: the next generation. In: Bos D, editor. Skin Immune System. Boca Raton, Fl: CRC Press; 1989. pp. 25–48. [Google Scholar]
- 83.Homey B, Steinhoff M, Ruzicka T, Leung DY. Cytokines and chemokines orchestrate atopic skin inflammation. J Allergy Clin Immunol. 2006;118:178–189. doi: 10.1016/j.jaci.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 84.Saalbach A, Klein C, Schirmer C, Briest W, Anderegg U, Simon JC. Dermal fibroblasts promote the migration of dendritic cells. J Invest Dermatol. 2010;130:444–454. doi: 10.1038/jid.2009.253. [DOI] [PubMed] [Google Scholar]
- 85.Gomez de Aguero M, Vocanson M, Hacini-Rachinel F, Taillardet M, Sparwasser T, Kissenpfennig A, et al. Langerhans cells protect from allergic contact dermatitis in mice by tolerizing CD8+ T cells and activating Foxp3+ regulatory T cells. J Clin Invest. 2012;122:1700–1711. doi: 10.1172/JCI59725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Seneschal J, Clark RA, Gehad A, Baecher-Allan CM, Kupper TS. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity. 2012;36:873–884. doi: 10.1016/j.immuni.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shklovskaya E, O'Sullivan BJ, Ng LG, Roediger B, Thomas R, Weninger W, et al. Langerhans cells are precommitted to immune tolerance induction. Proc Natl Acad Sci U S A. 2011;108:18049–18054. doi: 10.1073/pnas.1110076108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fukunaga A, Khaskhely NM, Sreevidya CS, Byrne SN, Ullrich SE. Dermal dendritic cells, and not Langerhans cells, play an essential role in inducing an immune response. J Immunol. 2008;180:3057–3064. doi: 10.4049/jimmunol.180.5.3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kimber I, Cumberbatch M, Dearman RJ. Langerhans cell migration: not necessarily always at the center of the skin sensitization universe. J Invest Dermatol. 2009;129:1852–1853. doi: 10.1038/jid.2009.54. [DOI] [PubMed] [Google Scholar]
- 90.Fukunaga A, Khaskhely NM, Ma Y, Sreevidya CS, Taguchi K, Nishigori C, et al. Langerhans cells serve as immunoregulatory cells by activating NKT cells. J Immunol. 2010;185:4633–4640. doi: 10.4049/jimmunol.1000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schwarz A, Noordegraaf M, Maeda A, Torii K, Clausen BE, Schwarz T. Langerhans cells are required for UVR-induced immunosuppression. J Invest Dermatol. 2010;130:1419–1427. doi: 10.1038/jid.2009.429. [DOI] [PubMed] [Google Scholar]
- 92.Yoshiki R, Kabashima K, Sakabe J, Sugita K, Bito T, Nakamura M, et al. The mandatory role of IL-10-producing and OX40 ligand-expressing mature Langerhans cells in local UVB-induced immunosuppression. J Immunol. 2010;184:5670–5677. doi: 10.4049/jimmunol.0903254. [DOI] [PubMed] [Google Scholar]
- 93.Loser K, Mehling A, Loeser S, Apelt J, Kuhn A, Grabbe S, et al. Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat Med. 2007;12:1372–1379. doi: 10.1038/nm1518. [DOI] [PubMed] [Google Scholar]
- 94.Bos JD, Zonneveld I, Das PK, Krieg SR, van der Loos CM, Kapsenberg ML. The skin immune system (SIS): distribution and immunophenotype of lymphocyte subpopulations in normal human skin. J Invest Dermatol. 1987;88:569–573. doi: 10.1111/1523-1747.ep12470172. [DOI] [PubMed] [Google Scholar]
- 95.Tada T, Takemori T, Okumura K, Nonaka M, Tokuhisa T. Two distinct types of helper T cells involved in the secondary antibody response: independent and synergistic effects of Ia− and Ia+ helper T cells. J Exp Med. 1978;147:446–458. doi: 10.1084/jem.147.2.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee GR, Flavell RA. Transgenic mice which overproduce Th2 cytokines develop spontaneous atopic dermatitis and asthma. Int Immunol. 2004;16:1155–1160. doi: 10.1093/intimm/dxh117. [DOI] [PubMed] [Google Scholar]
- 97.Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature. 2012;483:227–231. doi: 10.1038/nature10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci U S A. 2014;111:5307–5312. doi: 10.1073/pnas.1322292111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jameson J, Ugarte K, Chen N, Yachi P, Fuchs E, Boismenu R, et al. A role for skin gammadelta T cells in wound repair. Science. 2002;296:747–749. doi: 10.1126/science.1069639. [DOI] [PubMed] [Google Scholar]
- 100.Sumaria N, Roediger B, Ng LG, Qin J, Pinto R, Cavanagh LL, et al. Cutaneous immunosurveillance by self-renewing dermal gammadelta T cells. J Exp Med. 2011;208:505–518. doi: 10.1084/jem.20101824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Muller W, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184:3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 102.Toichi E, Lu KQ, Swick AR, McCormick TS, Cooper KD. Skin-infiltrating monocytes/ macrophages migrate to draining lymph nodes and produce IL-10 after contact sensitizer exposure to UV-irradiated skin. J Invest Dermatol. 2008;128:2705–2715. doi: 10.1038/jid.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kalesnikoff J, Galli SJ. New developments in mast cell biology. Nat Immunol. 2008;9:1215–1223. doi: 10.1038/ni.f.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Byrne SN, Limón-Flores AY, Ullrich SE. Mast cell migration from the skin to the draining lymph nodes upon ultraviolet irradiation represents a key step in the induction of immune suppression. J Immunol. 2008;180:4648–4655. doi: 10.4049/jimmunol.180.7.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hart PH, Grimbaldeston MA, Swift GJ, Jaksic A, Noonan FP, Finlay-Jones JJ. Dermal mast cells determine susceptibility to ultraviolet B-induced systemic suppression of contact hypersensitivity responses in mice. J Exp Med. 1998;187:2045–2053. doi: 10.1084/jem.187.12.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alard P, Niizeki H, Hanninen L, Streilein JW. Local ultraviolet B irradiation impairs contact hypersensitivity induction by triggering release of tumor necrosis factor-alpha from mast cells. Involvement of mast cells and Langerhans cells in susceptibility to ultraviolet B. J Invest Dermatol. 1999;113:983–990. doi: 10.1046/j.1523-1747.1999.00772.x. [DOI] [PubMed] [Google Scholar]
- 107.Alard P, Kurimoto I, Niizeki H, Doherty JM, Streilein JW. Hapten-specific tolerance induced by acute, low-dose ultraviolet B radiation of skin requires mast cell de-granulation. Eur J Immunol. 2001;31:1736–1746. doi: 10.1002/1521-4141(200106)31:6<1736::aid-immu1736>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 108.Ullrich SE, Nghiem DX, Khaskina P. Suppression of an established immune response by UVA-a critical role for mast cells. Photochem Photobiol. 2007;83:1095–1100. doi: 10.1111/j.1751-1097.2007.00184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Grimbaldeston MA, Pearce AL, Robertson BO, Coventry BJ, Marshman G, Finlay-Jones JJ, et al. Association between melanoma and dermal mast cell prevalence in sun-unexposed skin. Br J Dermatol. 2004;150:895–903. doi: 10.1111/j.1365-2133.2004.05966.x. [DOI] [PubMed] [Google Scholar]
- 110.Sarchio SN, Kok LF, O'Sullivan C, Halliday GM, Byrne SN. Dermal mast cells affect the development of sunlight-induced skin tumours. Exp Dermatol. 2012;21:241–248. doi: 10.1111/j.1600-0625.2012.01438.x. [DOI] [PubMed] [Google Scholar]
- 111.De Fabo EC, Noonan FP. Mechanism of immune suppression by ultraviolet irradiation in vivo. I. Evidence for the existence of a unique photoreceptor in skin and its role in photoimmunology. J Exp Med. 1983;158:84–98. doi: 10.1084/jem.158.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Esser C, Bargen I, Weighardt H, Haarmann-Stemmann T, Krutmann J. Functions of the aryl hydrocarbon receptor in the skin. Semin Immunopathol. 2013;35:677–691. doi: 10.1007/s00281-013-0394-4. [DOI] [PubMed] [Google Scholar]
- 113.Hart PH, Gorman S, Finlay-Jones JJ. Modulation of the immune system by UV radiation: more than just the effects of vitamin D? Nat Rev Immunol. 2011;11:584–596. doi: 10.1038/nri3045. [DOI] [PubMed] [Google Scholar]
- 114.Stapelberg MP, Williams RB, Byrne SN, Halliday GM. The alternative complement pathway seems to be a UVA sensor that leads to systemic immunosuppression. J Invest Dermatol. 2009;129:2694–2701. doi: 10.1038/jid.2009.128. [DOI] [PubMed] [Google Scholar]
- 115.Calignano A, Cirino G, Meli R, Persico P. Isolation and identification of platelet-activating factor in UV-irradiated guinea pig skin. J Pharmacol Methods. 1988;19:89–91. doi: 10.1016/0160-5402(88)90049-6. [DOI] [PubMed] [Google Scholar]
- 116.Sheng Y, Birkle DL. Release of platelet activating factor (PAF) and eicosanoids in UVC-irradiated corneal stromal cells. Curr Eye Res. 1995;14:341–347. doi: 10.3109/02713689508999931. [DOI] [PubMed] [Google Scholar]
- 117.Travers JB, Harrison KA, Johnson CA, Clay KL, Morelli JG. Platelet-activating factor biosynthesis induced by various stimuli in human HaCaT keratinocytes. J Invest Dermatol. 1996;107:88–94. doi: 10.1111/1523-1747.ep12298295. [DOI] [PubMed] [Google Scholar]
- 118.Pei Y, Barber LA, Murphy RC, Johnson CA, Kelley SW, Dy LC, et al. Activation of the epidermal platelet-activating factor receptor results in cytokine and cyclooxygenase-2 biosynthesis. J Immunol. 1998;161:1954–1961. [PubMed] [Google Scholar]
- 119.Barber LA, Spandau DF, Rathman SC, Murphy RC, Johnson CA, Kelley SW, et al. Expression of the platelet-activating factor receptor results in enhanced ultraviolet B radiation-induced apoptosis in a human epidermal cell line. J Biol Chem. 1998;273:18891–18897. doi: 10.1074/jbc.273.30.18891. [DOI] [PubMed] [Google Scholar]
- 120.Travers JB, Berry D, Yao Y, Yi Q, Konger RL. Ultraviolet B radiation of human skin generates platelet-activating factor receptor agonists. Photochem Photobiol. 2010;86:949–954. doi: 10.1111/j.1751-1097.2010.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Alappatt C, Johnson CA, Clay KL, Travers JB. Acute keratinocyte damage stimulates platelet-activating factor production. Arch Dermatol Res. 2000;292:256–259. doi: 10.1007/s004030050483. [DOI] [PubMed] [Google Scholar]
- 122.Marathe GK, Johnson C, Billings SD, Southall MD, Pei Y, Spandau D, et al. Ultraviolet B radiation generates platelet-activating factor-like phospholipids underlying cutaneous damage. J Biol Chem. 2005;280:35448–35457. doi: 10.1074/jbc.M503811200. [DOI] [PubMed] [Google Scholar]
- 123.Konger RL, Marathe GK, Yao Y, Zhang Q, Travers JB. Oxidized glycerophosphocholines as biologically active mediators for ultraviolet radiation-mediated effects. Prostaglandins Other Lipid Mediat. 2008;87:1–8. doi: 10.1016/j.prostaglandins.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yao Y, Wolverton JE, Zhang Q, Marathe GK, Al-Hassani M, Konger RL, et al. Ultraviolet B radiation generated platelet-activating factor receptor agonist formation involves EGF-R-mediated reactive oxygen species. J Immunol. 2009;182:2842–2848. doi: 10.4049/jimmunol.0802689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Heery JM, Kozak M, Stafforini DM, Jones DA, Zimmerman GA, McIntyre TM, et al. Oxidatively modified LDL contains phospholipids with platelet-activating factor-like activity and stimulates the growth of smooth muscle cells. J Clin Invest. 1995;96:2322–2330. doi: 10.1172/JCI118288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lewis MS, Whatley RE, Cain P, McIntyre TM, Prescott SM, Zimmerman GA. Hydrogen peroxide stimulates the synthesis of platelet-activating factor by endothelium and induces endothelial cell-dependent neutrophil adhesion. J Clin Invest. 1988;82:2045–2055. doi: 10.1172/JCI113825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Walterscheid JP, Ullrich SE, Nghiem DX. Platelet-activating factor, a molecular sensor for cellular damage, activates systemic immune suppression. J Exp Med. 2002;195:171–179. doi: 10.1084/jem.20011450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Devary Y, Gottlieb RA, Smeal T, Karin M. The mammalian ultraviolet response is triggered by activation of Src tyrosine kinases. Cell. 1992;71:1081–1091. doi: 10.1016/s0092-8674(05)80058-3. [DOI] [PubMed] [Google Scholar]
- 129.Simon MM, Aragane Y, Schwarz A, Luger TA, Schwarz T. UVB light induces a nuclear factor kB (NFkB) activity independently from chromosomal DNA damage in cell-free cytosolic extracts. J Invest Dermatol. 1994;102:422–427. doi: 10.1111/1523-1747.ep12372194. [DOI] [PubMed] [Google Scholar]
- 130.Ullrich SE. Ultraviolet carcinogenesis and immune suppression. In: Weber RS, Moore BA, editors. Cutaneous Malignancy of the Head and Neck: A Multidisciplinary Approach. San Diego, CA: Plural Publishing; 2011. pp. 57–78. [Google Scholar]
- 131.Ullrich SE, Byrne SN. The immunologic revolution: photoimmunology. J Invest Dermatol. 2012;132:896–905. doi: 10.1038/jid.2011.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Travers JB, Edenberg HJ, Zhang Q, Al-Hassani M, Yi Q, Baskaran S, et al. Augmentation of UVB radiation-mediated early gene expression by the epidermal platelet-activating factor receptor. J Invest Dermatol. 2008;128:455–460. doi: 10.1038/sj.jid.5701083. [DOI] [PubMed] [Google Scholar]
- 133.Byrne SN, Beaugie C, O'Sullivan C, Leighton S, Halliday GM. The immune-modulating cytokine and endogenous Alarmin interleukin-33 is upregulated in skin exposed to inflammatory UVB radiation. Am J Pathol. 2011;179:211–222. doi: 10.1016/j.ajpath.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Juremalm M, Hjertson M, Olsson N, Harvima I, Nilsson K, Nilsson G. The chemokine receptor CXCR4 is expressed within the mast cell lineage and its ligand stromal cell-derived factor-1alpha acts as a mast cell chemotaxin. Eur J Immunol. 2000;30:3614–3622. doi: 10.1002/1521-4141(200012)30:12<3614::AID-IMMU3614>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 135.Sarchio SN, Scolyer RA, Beaugie C, McDonald D, Marsh-Wakefield F, Halliday GM, et al. Pharmacologically antagonizing the CXCR4–CXCL12 chemokine pathway with AMD3100 inhibits sunlight-induced skin cancer. J Invest Dermatol. 2014;134:1091–1100. doi: 10.1038/jid.2013.424. [DOI] [PubMed] [Google Scholar]
- 136.Chacón-Salinas R, Chen L, Chávez-Blanco AD, Limón-Flores AY, Ma Y, Ullrich SE. An essential role for platelet-activating factor in activating mast cell migration following ultraviolet irradiation. J Leukoc Biol. 2014;95:139–148. doi: 10.1189/jlb.0811409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wolf P, Nghiem DX, Walterscheid JP, Byrne S, Matsumura Y, Matsumura Y, et al. Platelet-activating factor is crucial in psoralen and ultraviolet A-induced immune suppression, inflammation, and apoptosis. Am J Pathol. 2006;169:795–805. doi: 10.2353/ajpath.2006.060079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang Q, Yao Y, Konger RL, Sinn AL, Cai S, Pollok KE, et al. UVB radiation-mediated inhibition of contact hypersensitivity reactions is dependent on the platelet-activating factor system. J Invest Dermatol. 2008;128:1780–1787. doi: 10.1038/sj.jid.5701251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bussolati B, Biancone L, Cassoni P, Russo S, Rola-Pleszczynski M, Montrucchio G, et al. PAF produced by human breast cancer cells promotes migration and proliferation of tumor cells and neo-angiogenesis. Am J Pathol. 2000;157:1713–1725. doi: 10.1016/S0002-9440(10)64808-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lefebvre JS, Marleau S, Milot V, Levesque T, Picard S, Flamand N, et al. Toll-like receptor ligands induce polymorphonuclear leukocyte migration: key roles for leukotriene B4 and platelet-activating factor. FASEB J. 2010;24:637–647. doi: 10.1096/fj.09-135624. [DOI] [PubMed] [Google Scholar]
- 141.Melnikova VO, Mourad-Zeidan AA, Lev DC, Bar-Eli M. Platelet-activating factor mediates MMP-2 expression and activation via phosphorylation of cAMP-response element-binding protein and contributes to melanoma metastasis. J Biol Chem. 2006;281:2911–2922. doi: 10.1074/jbc.M508683200. [DOI] [PubMed] [Google Scholar]
- 142.Moreno SE, Alves-Filho JC, Rios-Santos F, Silva JS, Ferreira SH, Cunha FQ, et al. Signaling via platelet-activating factor receptors accounts for the impairment of neutrophil migration in polymicrobial sepsis. J Immunol. 2006;177:1264–1271. doi: 10.4049/jimmunol.177.2.1264. [DOI] [PubMed] [Google Scholar]
- 143.Nilsson G, Metcalfe DD, Taub DD. Demonstration that platelet-activating factor is capable of activating mast cells and inducing a chemotactic response. Immunology. 2000;99:314–319. doi: 10.1046/j.1365-2567.2000.00972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rastogi P, White MC, Rickard A, McHowat J. Potential mechanism for recruitment and migration of CD133 positive cells to areas of vascular inflammation. Thromb Res. 2008;123:258–266. doi: 10.1016/j.thromres.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kajiwara N, Sasaki T, Bradding P, Cruse G, Sagara H, Ohmori K, et al. Activation of human mast cells through the platelet-activating factor receptor. J Allergy Clin Immunol. 2010;125:1137–1145. e6. doi: 10.1016/j.jaci.2010.01.056. [DOI] [PubMed] [Google Scholar]
- 146.Katoh H, Hosono K, Ito Y, Suzuki T, Ogawa Y, Kubo H, et al. COX-2 and prostaglandin EP3/EP4 signaling regulate the tumor stromal proangiogenic microenvironment via CXCL12–CXCR4 chemokine systems. Am J Pathol. 2010;176:1469–1483. doi: 10.2353/ajpath.2010.090607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Damiani E, Puebla-Osorio N, Gorbea E, Ullrich SE. Platelet-activating factor induces epigenetic modifications in human mast cells. J Invest Dermatol. 2015;135:3034–3040. doi: 10.1038/jid.2015.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EGE, et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219–230. doi: 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 149.Brown EL, Rivas JM, Ullrich SE, Young CR, Norris SJ, Kripke ML. Modulation of immunity to Borrelia burgdorferi by ultraviolet irradiation: differential effect on Th1 and Th2 immune responses. Eur J Immunol. 1995;25:3017–3022. doi: 10.1002/eji.1830251105. [DOI] [PubMed] [Google Scholar]
- 150.Ullrich SE. The effect of ultraviolet radiation-induced suppressor cells on T cell activity. Immunology. 1987;60:353–360. [PMC free article] [PubMed] [Google Scholar]
- 151.Chacón-Salinas R, Limón-Flores AY, Chávez-Blanco AD, Gonzalez-Estrada A, Ullrich SE. Mast cell-derived IL-10 suppresses germinal center formation by affecting T follicular helper cell function. J Immunol. 2011;186:25–31. doi: 10.4049/jimmunol.1001657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rivas JM, Ullrich SE. Systemic suppression of delayed-type hypersensitivity by supernatants from UV-irradiated keratinocytes. An essential role for keratinocyte-derived IL-10. J Immunol. 1992;149:3865–3871. [PubMed] [Google Scholar]
- 153.Beissert S, Hosoi J, Kühn R, Rajewsky K, Müller W, Granstein RD. Impaired immunosuppressive response to ultraviolet radiation in interleukin-10-deficient mice. J Invest Dermatol. 1996;107:553–557. doi: 10.1111/1523-1747.ep12582809. [DOI] [PubMed] [Google Scholar]
- 154.Liu J, Chen R, Marathe GK, Febbraio M, Zou W, McIntyre TM. Circulating platelet-activating factor is primarily cleared by transport, not intravascular hydrolysis, by lipoprotein-associated phospholipase A2/PAF acetylhydrolase. Circ Res. 2011;108:469–477. doi: 10.1161/CIRCRESAHA.110.228742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhang Q, Mousdicas N, Yi Q, Al-Hassani M, Billings SD, Perkins SM, et al. Staphylococcal lipoteichoic acid inhibits delayed-type hypersensitivity reactions via the platelet-activating factor receptor. J Clin Invest. 2005;115:2855–2861. doi: 10.1172/JCI25429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rockel N, Esser C, Grether-Beck S, Warskulat U, Flogel U, Schwarz A, et al. The osmolyte taurine protects against ultraviolet B radiation-induced immunosuppression. J Immunol. 2007;179:3604–3612. doi: 10.4049/jimmunol.179.6.3604. [DOI] [PubMed] [Google Scholar]
- 157.Sahu RP, Yao Y, Konger RL, Travers JB. Platelet-activating factor does not mediate UVB-induced local immune suppression. Photochem Photobiol. 2012;88:490–493. doi: 10.1111/j.1751-1097.2011.01071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Moodycliffe AM, Nghiem D, Clydesdale G, Ullrich SE. Immune suppression and skin cancer development: regulation by NKT cells. Nat Immunol. 2000;1:521–525. doi: 10.1038/82782. [DOI] [PubMed] [Google Scholar]
- 159.Schwarz T. 25 years of UV-induced immunosuppression mediated by T cells-from disregarded T suppressor cells to highly respected regulatory T cells. Photochem Photobiol. 2008;84:10–18. doi: 10.1111/j.1751-1097.2007.00223.x. [DOI] [PubMed] [Google Scholar]
- 160.Soontrapa K, Honda T, Sakata D, Yao C, Hirata T, Hori S, et al. Prostaglandin E2-prostoglandin E receptor subtype 4 (EP4) signaling mediates UV irradiation-induced systemic immunosuppression. Proc Natl Acad Sci U S A. 2011;108:6668–6673. doi: 10.1073/pnas.1018625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sreevidya CS, Khaskhely NM, Fukunaga A, Khaskina P, Ullrich SE. Inhibition of photocarcinogenesis by platelet-activating factor or serotonin receptor antagonists. Cancer Res. 2008;68:3978–3984. doi: 10.1158/0008-5472.CAN-07-6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Walterscheid JP, Nghiem DX, Kazimi N, Nutt LK, McConkey DJ, Norval M, et al. cis-Urocanic acid, a sunlight-induced immunosuppressive factor, activates immune suppression via the 5-HT2A receptor. Proc Natl Acad Sci U S A. 2006;103:17420–17425. doi: 10.1073/pnas.0603119103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sreevidya CS, Fukunaga A, Khaskhely NM, Masaki T, Ono R, Nishigori C, et al. Agents that reverse UV-induced immune suppression and photocarcinogenesis affect DNA repair. J Invest Dermatol. 2010;130:1428–1437. doi: 10.1038/jid.2009.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sahu RP, Turner MJ, DaSilva SC, Rashid BM, Ocana JA, Perkins SM, et al. The environmental stressor ultraviolet B radiation inhibits murine antitumor immunity through its ability to generate platelet-activating factor agonists. Carcinogenesis. 2012;33:1360–1367. doi: 10.1093/carcin/bgs152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sahu RP, Ferracini M, Travers JB. Systemic chemotherapy is modulated by platelet-activating factor-receptor agonists. Mediat Inflamm. 2015;2015:820543. doi: 10.1155/2015/820543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Morton CA, McKenna KE, Rhodes LE. British Association of Dermatologists Therapy G, Audit S, the British Photodermatology G. Guidelines for topical photodynamic therapy: update. Br J Dermatol. 2008;159:1245–1266. doi: 10.1111/j.1365-2133.2008.08882.x. [DOI] [PubMed] [Google Scholar]
- 167.Mroz P, Hamblin MR. The immunosuppressive side of PDT. Photochem Photobiol Sci. 2011;10:751–758. doi: 10.1039/c0pp00345j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ferracini M, Sahu RP, Harrison KA, Waeiss RA, Murphy RC, Jancar S, et al. Topical photodynamic therapy induces systemic immunosuppression via generation of platelet-activating factor receptor ligands. J Invest Dermatol. 2015;135:321–323. doi: 10.1038/jid.2014.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Brash DE, Rudolf JA, Simon JA, Lin A, McKenna GJ, Baden HP, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci U S A. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang Y, Digiovanna JJ, Stern JB, Hornyak TJ, Raffeld M, Khan SG, et al. Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci U S A. 2009;106:6279–6284. doi: 10.1073/pnas.0812401106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Puebla-Osorio N, Damiani E, Bover L, Ullrich SE. Platelet-activating factor induces cell cycle arrest and disrupts the DNA damage response in mast cells. Cell Death Dis. 2015;6:e1745. doi: 10.1038/cddis.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 173.Elledge SJ, Winston J, Harper JW. A question of balance: the role of cyclin-kinase inhibitors in development and tumorigenesis. Trends Cell Biol. 1996;6:388–392. doi: 10.1016/0962-8924(96)10030-1. [DOI] [PubMed] [Google Scholar]
- 174.Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 175.Gartel AL, Serfas MS, Gartel M, Goufman E, Wu GS, el-Deiry WS, et al. p21 (WAF1/ CIP1) expression is induced in newly nondividing cells in diverse epithelia and during differentiation of the Caco-2 intestinal cell line. Exp Cell Res. 1996;227:171–81. doi: 10.1006/excr.1996.0264. [DOI] [PubMed] [Google Scholar]
- 176.Herbig U, Sedivy JM. Regulation of growth arrest in senescence: telomere damage is not the end of the story. Mech Ageing Dev. 2006;127:16–24. doi: 10.1016/j.mad.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 177.Perucca P, Cazzalini O, Madine M, Savio M, Laskey RA, Vannini V, et al. Loss of p21 CDKN1A impairs entry to quiescence and activates a DNA damage response in normal fibroblasts induced to quiescence. Cell Cycle. 2009;8:105–114. doi: 10.4161/cc.8.1.7507. [DOI] [PubMed] [Google Scholar]
- 178.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- 179.Perkins ND. Not just a CDK inhibitor: regulation of transcription by p21(WAF1/ CIP1/SDI1) Cell Cycle. 2002;1:39–41. [PubMed] [Google Scholar]
- 180.Cazzalini O, Scovassi AI, Savio M, Stivala LA, Prosperi E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat Res. 2010;704:12–20. doi: 10.1016/j.mrrev.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 181.Zhang Q, Sitzman LA, Al-Hassani M, Cai S, Pollok KE, Travers JB, et al. Involvement of platelet-activating factor in ultraviolet B-induced hyperalgesia. J Invest Dermatol. 2009;129:167–174. doi: 10.1038/jid.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Simpson S, Jr, Blizzard L, Otahal P, Van der Mei I, Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82:1132–1141. doi: 10.1136/jnnp.2011.240432. [DOI] [PubMed] [Google Scholar]
- 183.Staples JA, Ponsonby AL, Lim LL, McMichael AJ. Ecologic analysis of some immune-related disorders, including type 1 diabetes, in Australia: latitude, regional ultraviolet radiation, and disease prevalence. Environ Health Perspect. 2003;111:518–523. doi: 10.1289/ehp.5941. [DOI] [PMC free article] [PubMed] [Google Scholar]