

Abstract
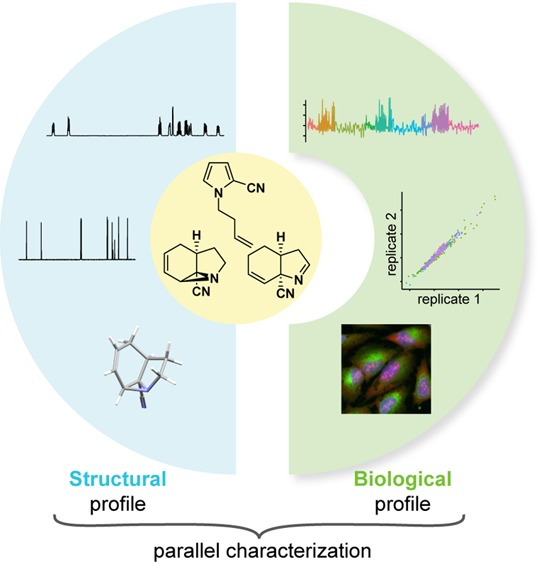
Organic chemists are able to synthesize molecules in greater number and chemical complexity than ever before. Yet, a majority of these compounds go untested in biological systems, and those that do are often tested long after the chemist can incorporate the results into synthetic planning. We propose the use of high-dimensional “multiplex” assays, which are capable of measuring thousands of cellular features in one experiment, to annotate rapidly and inexpensively the biological activities of newly synthesized compounds. This readily accessible and inexpensive “real-time” profiling method can be used in a prospective manner to facilitate, for example, the efficient construction of performance-diverse small-molecule libraries that are enriched in bioactives. Here, we demonstrate this concept by synthesizing ten triads of constitutionally isomeric compounds via complexity-generating photochemical and thermal rearrangements and measuring compound-induced changes in cellular morphology via an imaging-based “cell painting” assay. Our results indicate that real-time biological annotation can inform optimization efforts and library syntheses by illuminating trends relating to biological activity that would be difficult to predict if only chemical structure were considered. We anticipate that probe and drug discovery will benefit from the use of optimization efforts and libraries that implement this approach.
Introduction
Synthetic chemists routinely perform deep and comprehensive spectral characterization of compounds synthesized in the laboratory, often within 24 h of synthesis. However, it may take a year or more to learn if those compounds bind macromolecules or have unique biological activities in cells or animals.1 We recently reported that scientists at the Broad Institute used 130 cell-based assays to learn retrospectively about the performance of their synthetic compounds (ca. 100k prepared using diversity-oriented synthesis) four years after the syntheses were complete.1 If, instead, chemists could “annotate” their synthetic products in real time, within days of synthesis and using large numbers of multiplexed biological measurements, this critical delay in information feedback could be shortened from years to days. Despite the dramatically shortened time scale, this method would be capable of annotating each compound with thousands of measurements relating to cellular performance across multiple concentrations (Figure 1). While biological annotation may not be sufficiently granular to identify precise molecular targets consistently, it can reveal which molecules are bioactive. Furthermore, comparisons with reference compounds having known activities can inform whether “actives” function by known or novel mechanisms of action (MoA) and generate hypotheses for follow-up MoA studies.2,3 This real-time capability would provide a more immediate understanding of the consequences of specific chemical transformations on the biological performance of the resulting compounds.4 The resulting profiles would provide high-dimensional vector representations of the response of the cells, if any, to a given perturbagen, permitting rich comparisons of cellular responses using vector algebra.
Figure 1.
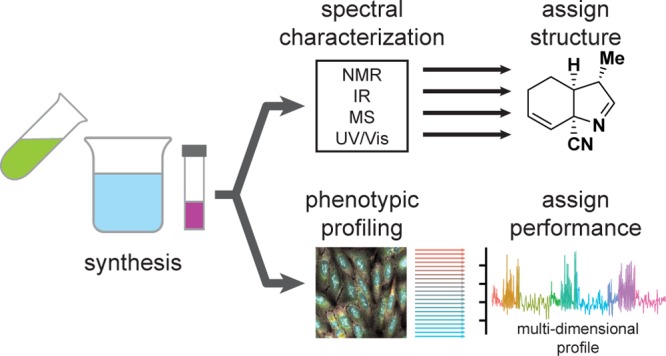
Schematic representation of concurrent structural assignment and biological annotation of compounds immediately following their synthesis.
Systematically coupling synthetic reactions with immediate biological annotation of reaction products could also facilitate the discovery of novel small-molecule drugs and probes. Whereas physicochemical descriptors are often inadequate predictors of biological performance, data from high-dimensional cell-based assays can provide a “snapshot” of a compound’s activity in multiple biological contexts. This capability would enrich optimization efforts that often focus on a few key metrics (e.g., potency or specificity) by determining whether underlying cellular mechanisms have unwittingly been changed. Only through routine biological annotation can collections of small molecules be curated such that individual members (or, ideally, small sets of compounds sharing similar biological profiles) have distinct MoA, whether they be known or novel. Thus, real-time biological annotation of synthetic and natural compounds is central to generating performance-diverse compound collections.1
Providing immediate feedback to chemists, having a better understanding of the molecular consequences of structural alterations during optimization, and delivering performance-diverse compound libraries all address another key challenge in modern therapeutics discovery. Human biology, especially the discovery of risk and protective gene variants and the determination of the altered activities of the corresponding variant proteins, affords a blueprint for the activities that drugs should confer on their targets to be safe and effective. Thus, prior to launching a drug-discovery project, we can know the consequences of modulating a target in the context of human physiology.5 But the insights gained thus far suggest that these modulations demand challenging mechanisms of action not yet seen in drug discovery. Novel MoA (nMoA) compounds are needed in order to translate insights from human biology into safe and effective therapeutic agents, and real-time annotation concurrent with synthesis may help to identify candidate nMoA compounds.
Here, we illustrate an experimental and analytical first step toward these challenging goals. A pilot study was performed using triads of constitutional isomers that, within triads, differ simply in the arrangement of their atoms and, across triads, differ in the appendages of three isomeric skeletons. Extending an earlier retrospective analysis,1,6 we used a multiplexed “cell painting” assay to measure changes in 1140 cell-morphology features induced by compound treatment and examined the effects of varying concentration on these measurements.7 Our goal is to use this analysis prospectively—to select performance-diverse compounds for use in future cell-based screens. The results, derived from real-time biological annotation of synthetic reaction products, reinforce the idea that rearranging the bond connectivity of constitutional isomers can dramatically alter their biological activity, and support the notion that similarity between chemical structures correlates poorly with their biological performance. In the process, we discovered a set of biologically active quaternary nitriles that induce striking changes in cell morphology. Adopting and extending the concept of real-time biological annotation to additional types of measurements (e.g., gene expression and in-cell protein binding) should advance chemical biology, facilitate the construction of performance-diverse compound libraries in the future, and enhance the ability of chemists to contribute to challenging problems posed by modern biology.8
Results
Synthesis of Skeletally Isomeric Triads
We designed our pilot compound collection (Table 1) to explore the effect of differing skeletons and appendages on compound activity in cells. Synthesizing and annotating triads of constitutionally isomeric compounds controls for variable atom composition within skeletons. These skeletons undergo significant reordering of atoms to afford triads comprising a monocyclic pyrrole, a tricyclic aziridine, and a bicyclic imine (or a structurally related secondary amine). We refer to the collections of all pyrroles, aziridines, or imines/amines, irrespective of triad, as distinct cohorts. The compounds that populate our collection contain several sites of appendage diversification (Table 1), which can be used to further categorize our compounds. Two such examples are the identity of the electron-withdrawing substituent (e.g., ketone, nitrile, ester, or amide) and the substitution pattern along the homoallyl arm of the pyrroles.
Table 1. Overall Synthetic Scheme and Enumeration of Ten Triads of Skeletally Isomeric Compoundsa.
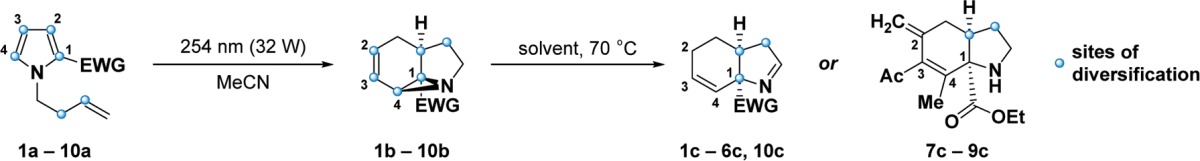

Sites of diversification are marked with a blue dot. Yields of transformations leading to skeletal isomers are indicated in parentheses below the corresponding structures.
We identified a photochemical rearrangement of pyrroles to (racemic) tricyclic aziridines9 as an isomerization that exhibits a large structural complexity differential (i.e., it is a complexity-generating reaction).10 This photoreaction converts relatively “flat” N-homoallyl-substituted pyrroles with an electron-withdrawing group at C2 into cup-shaped, tricyclic vinyl aziridines with three contiguous stereocenters. By a measure developed by the Böttcher group,11 the difference in complexity between the starting material and the product in this reaction is 96 units; comparatively, intermolecular aldol reactions score 41 units, and amide formation via the Schotten–Baumann reaction increases complexity by only 3 units.
While expanding the scope of this powerful transformation, we noted that these aziridines convert to constitutionally isomeric endocyclic imines via a type of retro-ene reaction upon exposure to elevated temperatures.12−14 While there is ample precedent for the thermal rearrangement of aziridines to azomethine ylides, transformations of this type have been less well-documented.15−17 This reagent-free rearrangement can be performed in a variety of organic solvents (Supporting Information, Table S1) and opens up several avenues for further synthetic exploration. Furthermore, the topography of the substrate allows this reaction to proceed under relatively mild conditions; the cup-shaped tricyclic skeleton orients the participating atoms in a conformation that is suitable for a [1,5]-hydrogen shift, and aziridine opening is accompanied by a significant relief of ring strain.12 A competing eliminative aziridine opening is observed with certain substrates, affording secondary amines that contain the same 5,6-fused ring system that characterizes the imines. Particularly important for our study, these two transformations dramatically reorganize the skeletal atoms of their substrates to afford compounds that are structurally distinct from those in the previous two cohorts.
Based on these two isomerizations, we envisioned a small library of compounds that were derived from a variety of N-substituted pyrroles. While it had previously been shown that a number of appendages on the pyrrole ring do not disrupt the photochemical rearrangement,9 we additionally discovered that various sites along the homoallylic alcohol-derived appendage could also be functionalized. Accordingly, we synthesized a cohort of ten N-substituted pyrroles to constitute the foundation of our library (Table 1). The generality of the subsequent photochemical rearrangement allowed us to incorporate substantial appendage diversity into our small set of pyrroles: analogs 1a–10a contain one of four electron-withdrawing groups at C2 and one of three homoallylic substituents at N1.
The compounds in the aziridine cohort were then synthesized by irradiating the pyrroles with 254-nm light in a Rayonet photoreactor (32 W total power output). The benefits, particularly with regard to scale, of performing this reaction in a flow reactor have previously been demonstrated,18,19 but we achieved comparable yields on hundred-milligram scale using traditional batch chemistry in quartz round-bottom flasks. Notably, the methyl group in aziridines 2b, 5b, and 8b preferentially occupies the exo position, as confirmed by the X-ray crystal structure of derivative compounds (Supporting Information, Figure S1). With regard to the complexity-generating aspect of this reaction, pyrrole 1a has three sp3-hybridized carbons, no chiral centers, and one ring, whereas aziridine 1b contains seven sp3-hybridized carbons, three chiral centers, and three rings. A representation of this difference can be visualized by principal moment-of-inertia (PMI) analysis,20 which suggests that the pyrrole and aziridine cohorts occupy distinct areas of three-dimensional space (Figure 2; Supporting Information p S82, “Principal moment-of-inertia (PMI) calculations”).
Figure 2.
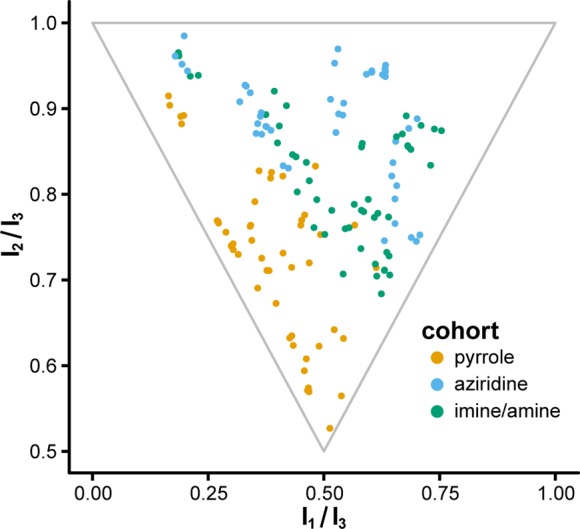
Principal moment-of-inertia analysis of newly synthesized compounds. Individual points indicate the location of a given conformer (up to five conformers per compound) in PMI space, as defined by the ratios of their computed principal moments of inertia (I1 < I2 < I3). Points near the top-left corner indicate one-dimensional “rod-like” character, points near the bottom indicate two-dimensional “disc-like” character, and points near the top-right corner indicate three-dimensional “sphere-like” character.
To finalize our pilot library, members of the aziridine cohort were heated to effect the strain-releasing aziridine-to-imine rearrangement. This transformation can take over 48 h to achieve complete conversion when performed at 70 °C, but further raising the temperature increases the rate of undesired side reactions and degradation pathways. Of note, rather than providing imines through the typical [1,5]-hydrogen shift, aziridines 7b, 8b, and 9b rearranged into secondary amines upon exposure to elevated temperatures via the competing eliminative transformation mentioned previously. 1H NMR spectra of compounds 7c, 8c, and 9c show the “loss” of a methyl group and the emergence of two exo-methylene peaks around 4.7 and 4.9 ppm. Weak spin–spin coupling can also be observed between the exo-methylene protons and the protons on the allylic methylene group.
As might be expected, PMI calculations indicate that members of the imine/amine cohort are more similar to aziridines than pyrroles with regard to three-dimensional shape. Yet, when taken together, our small library of 30 compounds covers a wide swath of topographic space (Figure 2). This outcome highlights the power of strategically designed chemical pathways and further emphasizes that topographically complex molecules need not be difficult to synthesize.21,22 Furthermore, the wide array of molecular architectures contained in our library allows us to begin to probe the relationship between three-dimensionality and biological activity in a systematic manner using cell painting.
Biological Profiling via Cell Painting
To perform the cell-painting assay, U-2 OS cells, which are derived from an osteosarcoma (bone cancer), were seeded in 384-well plates and treated with DMSO solutions of compounds in a concentration-dependent manner (six concentrations ranging from 3.125 μM to 100 μM). Treatments were performed in quadruplicate, and DMSO-alone conditions were used as negative controls. After 24 h, cells were exposed to six nonantibody-based dyes that stained seven organelles and cellular compartments (Supporting Information, Table S2).6 The cells were then imaged with an automated, high-throughput fluorescent microscope across five channels, acquiring images at nine sites per well. Once obtained, images of well sites were processed with CellProfiler software,23 which is capable of systematically extracting and quantifying cellular morphological features (Supporting Information p S66, “Image analysis and data processing”). For example, characteristics of nuclei and mitochondria, such as size, shape, and eccentricity, can be measured using nucleus-specific (Hoechst) and mitochondria-specific (MitoTracker Deep Red) stains, respectively, whereas characteristics of whole-cell architecture can be illuminated using a cytoskeleton-specific stain (actin-binding phalloidin). In this manner, 1140 quantitative features can be examined on a cell-by-cell basis. By comparing these features to those exhibited by DMSO-treated cells, we can construct profiles for each compound-treatment condition.
The relative ease with which this assay is performed and its low associated costs make it an attractive assay to annotate newly synthesized compounds in high throughput; the most significant costs are dyes and microscope time. Furthermore, the use of label-free compounds guarantees that entire classes of compounds are not precluded from screening because they lack a suitable derivatization point. It should also be noted that cell painting may be performed with a wide variety of cell lines derived from several tissue types, such as HeLa and A549 cells.6
Analysis of Biological Annotation Data
Having generated the profiles for all concentrations of the synthesized compounds (Supporting Information p S66, “Biological profiles”), we first determined which compounds could be considered “active” by identifying the compounds that exhibited profiles significantly different from that of DMSO alone (Figure 3). To arrive at an “activity score,” we needed a measure of the distance between two populations (DMSO and treatment) in a 1140-dimensional feature space. One such measure is Mahalanobis distance,24 which is analogous to the Euclidean distance between two points in a lower-dimensional space and also accounts for the spread of each population and the colinearity between features (to avoid “overcounting” redundant features).25 Using this metric, we calculated the activity scores for each compound treatment condition (e.g., 8b at 25 μM; Supporting Information p S82, “Compound activity calculation”). For the purposes of this analysis, compounds with an activity score greater than 8 were deemed “active.” Compounds that reduced cell count (perhaps by death, reduced proliferation, or surface detachment) tended to exhibit a high activity score; this result is unsurprising as these processes are often accompanied by drastic changes in cell morphology. By this assessment, 7 of the 30 compounds were found to be active at one or more concentrations (4b, 4c, 5b, 5c, 6c, 10a, and 10b). These active compounds revealed an enrichment of aziridines and imines compared to pyrroles. Separating compounds by cohort (pyrrole, aziridine, imine/amine) and taking the average signal from all six compound concentrations allows this trend to be seen more clearly (Figure 4). The six most active compounds (4b, 4c, 5b, 5c, 6c, 10b) belong to the aziridine or imine/amine cohort, a result that is consistent with the notion that sp3-rich molecules that are topographically complex exhibit more favorable protein-binding properties than flat, sp2-rich compounds.26 One potential confounding factor is that aziridines and imines are often considered to be reactive functional groups that might impart promiscuous activity. However, not all aziridines or imines are active in our assay, suggesting that a more nuanced and context-dependent mechanism is responsible for these results. We also note that pyrrole motifs can be found in a wide variety of bioactive molecules,27 suggesting that the pyrrole cohort’s relative lack of activity cannot be explained solely by the presence of a pyrrole ring.
Figure 3.
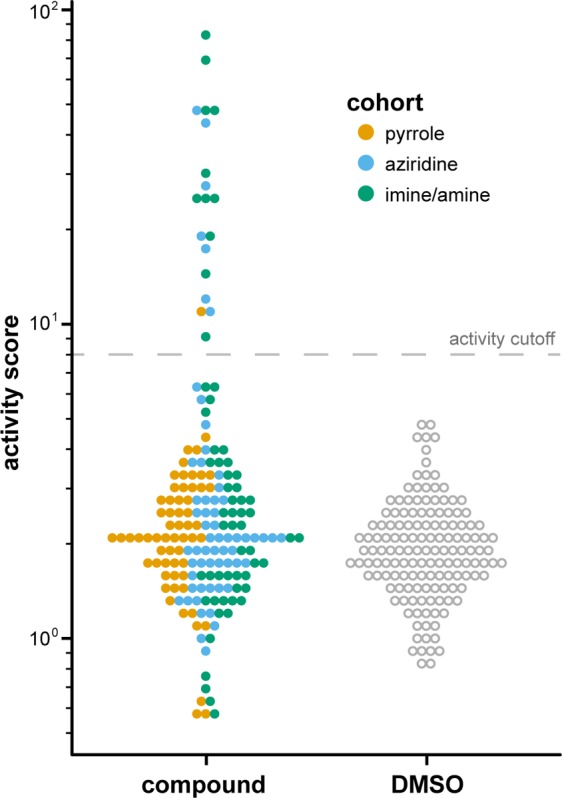
Dot plot depicting compound activities compared to DMSO alone. Activity scores describe the degree to which compound-induced morphological changes are distinct from those induced by DMSO alone and are calculated according to the distance metric described by Mahalanobis.24 Each point represents a single compound-treatment condition (compound A, concentration X), averaged across four replicates. The dashed line indicates the cutoff at which compound treatment conditions were considered “active”.
Figure 4.
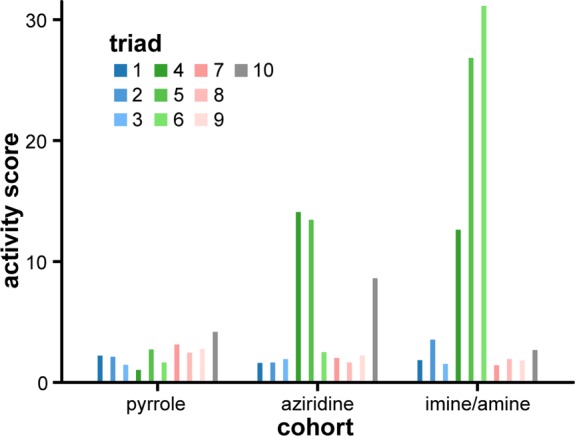
Bar graph of compound activities reveals contributions from both skeleton and appendage identities. Activity scores for each compound were averaged across replicates of all concentrations. Compounds are grouped by cohorts and colored by triads, with differing shades of a given color indicating a shared electron-withdrawing group (blue, acetyl; green, nitrile; salmon, ester; gray, amide). Representing the data in this format emphasizes the influence of both skeletons and appendages on biological activity. In particular, the combination of a nitrile appendage on an aziridine or imine/amine skeleton yields highly active compounds.
While it may be tempting to focus primarily on the compounds that elicit the strongest cellular responses, a brief survey of our inactive compounds shows that cell painting is capable of reliably distinguishing between constitutional isomers. Even when resorting to a binary active/inactive designation, we see that many triads contain both active and inactive compounds. This outcome may be unsurprising—many chiral compounds, for example, exhibit dramatically different activities from their enantiomers—but it provides further support that high-dimensional annotation is a valuable tool for compound characterization.
We further analyzed the compound profiles to determine if we could extract more nuanced insights into MoA. Pearson correlation coefficients were calculated between each pair of compounds, and the resulting pairwise correlations summarize the degree to which compounds’ patterns of induced changes in cell morphology correlate to one another (Figure 5; Supporting Information p S82, “Heat map generation and clustering”). Although we see a variety of relationships, the most striking feature of this analysis is the strong correlation between the six compounds in the shortest clade of the distance dendrogram (top-right corner of the heat map, as rendered), which correspond to the six compounds that we previously identified to be the most active (Figure 4). Strikingly, five out of these six compounds contain a nitrile connected to a quaternary sp3-hybridized carbon (only one of the nitrile-containing aziridines or imines did not score as active at any concentration). While it remains unclear how the nitrile motif may be influencing compound activity in this instance, previous studies have identified several classes of bioactive nitriles.28 Mechanistically, bioactive nitriles are capable of behaving as electrophiles or hydrogen-bond acceptors. It has also been recognized that nitriles lacking α-protons are appealing from the standpoint of their resistance to oxidative metabolism.29
Figure 5.

Heat map showing relationships of compounds (compound vs compound) based on the similarities of their biological profiles. Biological profiles are compared using pairwise Pearson correlation coefficients, calculated from the patterns of cell morphology changes (1140 total features) induced by compound treatment (average of six concentrations, each in quadruplicate). Structures of the six most highly correlated compounds (red clade in top dendrogram) are shown to the right of the heat map.
While these active compounds were found to exhibit distinct activities from the other members of our library, we attempted to resolve these activities from each other by exploiting the often nonlinear effects of varying compound concentration.30 For example, we imagine compounds having a single target showing linear changes in measurements relative to those having multiple targets with varying affinities. To this end, we performed a principal component (PC) analysis of the individual replicates of the active concentrations of six compounds (Figure 5); focusing on the first two principal components (Figure 6) reveals several noteworthy observations. First, the spread between replicates in this PC-space is minor compared to the distances between different treatment conditions. This high level of reproducibility allows us to distinguish between distinct compound-induced phenotypes, even for seemingly highly correlated compounds. Second, analyzing the activities of separate concentrations affords additional insights into biological activity. In particular, we note that three concentrations of 5c (25, 50, and 100 μM) form their own cluster, which indicates that 5c is likely acting via a distinct cellular MoA from the other five compounds. Compounds whose concentrations exhibit dissimilar profiles (e.g., 6c) could indicate a polypharmacological effect in which additional cellular targets are being engaged at higher concentrations.
Figure 6.
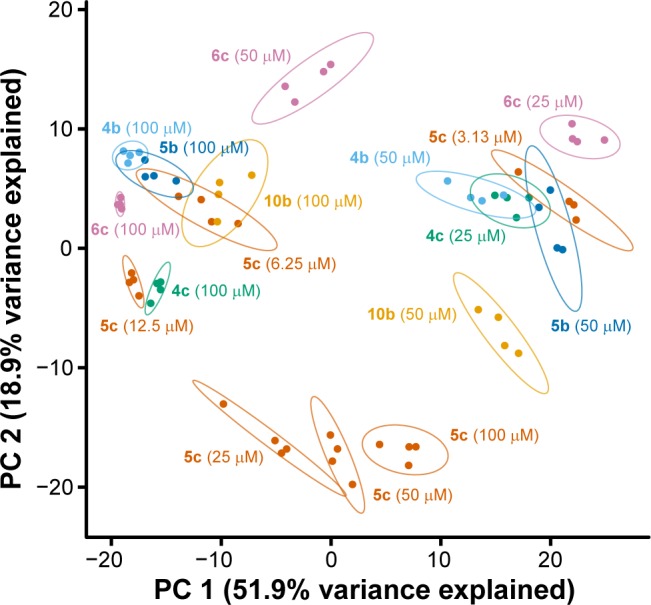
Principal component analysis of compound-induced changes in cell morphology illuminates differences in MoAs of active compounds. Tight clusters of treatment replicates, represented by individual points and encircled by ellipses, demonstrate the high reproducibility of compound-induced changes in cell morphology. Analyzing biological annotation data across various compound concentrations provides greater resolution with regard to compound MoA. Points and ellipses (drawn at 95% confidence intervals) are colored according to compound identities.
To illustrate representative cellular images, we have highlighted the visual differences between DMSO alone-, imine 5c-, and aziridine 10b-treated cells (Figure 7). The cell-morphological changes induced by 5c were particularly striking: cells exhibited filopodium-like protrusions31 that were observable in multiple channels, but most prominently in the Short Red channel (642 nm, cytoskeleton-stained). Another noticeable difference was the apparent “softening” of subcellular compartments near the nuclei, which can be best seen in the Short Red (642 nm, cytoskeleton-stained) and Long Red (692 nm, mitochondria-stained) channels. These images not only provide confirmation that 5c and 10b dramatically alter cell morphology (indicated by their high activity scores) but also do so in different ways, as suggested by PC analysis (Figure 6).
Figure 7.
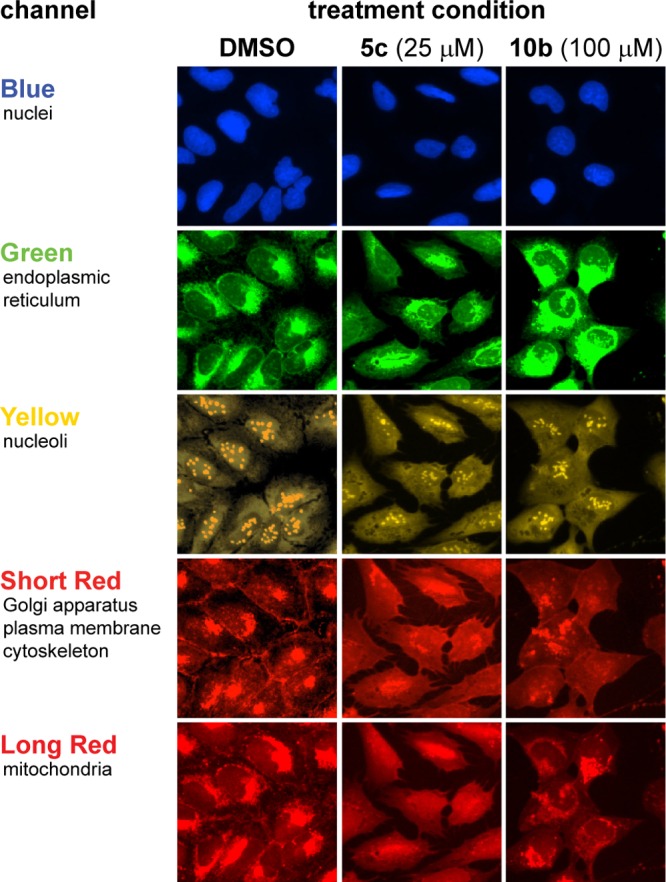
Actual images of vehicle-, compound 5c- (25 μM), and compound 10b-treated (100 μM) cells observed across five channels. The images reinforce that the two highly active compounds depicted have distinct effects on cell morphology, despite their membership in a distinct clade when compared to all other compounds (cf., Figure 5). The discrete morphological features best visualized in each channel are listed below the channel names.
Follow-up experiments that incorporate additional types of multiplexed measurements may be performed to gain deeper insights into this peculiar phenotype. One next step will be to compare the cell-painting profile of 5c to the profiles of known MoA compounds that have been annotated in this fashion.1 By analogy to our earlier study, this process should allow us to distinguish known mechanism-of-action compounds from ones having novel mechanisms of action (kMoA vs nMoA). These insights are key to creating performance-diverse compound collections for cellular or organismal phenotype-based screens.
Of course, another potential direction is to drill deeper into the newly uncovered activities of compounds with potential for having novel mechanisms of action. Additional analogs of 5c may be synthesized and annotated to generate more robust structure–activity relationships, establishing a valuable feedback loop between synthetic chemistry and biological performance. This type of analysis may also be performed with any compound known to elicit a particular phenotype of interest, potentially expediting the challenging task of identifying the target(s) of a bioactive small molecule.
Discussion
Small-molecule collections used in screening and selection experiments often facilitate the discovery of new chemical probes and therapeutics. Since some screens of compound libraries provide unsatisfying results, including yielding no starting points for further investigation, there is a widespread view that larger libraries are better than smaller ones. But this premise may be false if the primary effect of increasing the size of a library is to increase performance redundancy—adding compounds that either have no effects or have actions that mirror those of existing library members. We have advocated for a different approach: creating compound libraries in which each member (ideally, small subsets of molecules) functions by a distinct MoA. This strategy allows libraries, regardless of size, to achieve vastly greater levels of performance diversity and functionality, so long as unique MoA compounds can be discovered efficiently.1 Our work here represents an advance toward this goal, among others.
Because of the often-substantial temporal disconnection between chemical synthesis and biological testing, most modern library generation has been guided by chemical structures and their associated physical properties. The hope has been that structural diversity will yield performance diversity. However, it has been demonstrated that these structural descriptors are often insufficient predictors of biological performance (in our recent study, no better than random chance)1 and that data from high-dimensional cellular assays may better inform a compound’s activity in biological contexts.32 Applying readily accessible and inexpensive assays in real time could greatly facilitate cellular mechanism-informed optimization of compounds and the design and synthesis of performance-diverse screening collections by providing rapid feedback on compound activity. Just as synthetic chemists annotate each compound with an ensemble of spectroscopic techniques to establish its structure, broad arrays of phenotypic measurements could provide critical insights into its biological function in a matter of days. Conversely, it would take several years and hundreds of experiments to reach a similar level of understanding using the current model for HTS library synthesis and evaluation.
We have demonstrated a pilot-scale version of this concept by describing the synthesis of a small collection of compounds and their real-time biological annotation via cell painting. We are able to distinguish compounds having little or no effect on cells from those engendering either known or novel phenotypes. Furthermore, we see small clusters of compounds whose actions on cells appear to be related, despite their structural differences. Importantly, our analysis was able to distinguish between not only constitutional isomers but also compounds that may appear to be more structurally similar, even to the trained eye. Looking through a broader lens, our focus on isomeric triads allowed our results to support the notion that structural complexity is a valuable feature of molecules that populate HTS compound libraries. Real-time analysis provides chemists with powerful insights into the relationship between their chemical intuition and imagination and the consequences of perturbing cells in novel ways, in a manner that would be impossible otherwise. Despite the small size of the compound collection, we were able to identify a particularly interesting compound that elicits an unusual phenotype in U-2 OS cells.
We suggest that real-time annotation of synthetic compounds should become a routine aspect of synthetic organic chemistry in the future as a means to increase the potential of chemistry to impact biology and medicine. The robustness and granularity of our results highlight the promise of readily accessible and inexpensive cell painting as a potential profiling method in the context of real-time biological annotation of compounds. Similarly, additional annotation methodologies (such as cellular thermal proteome profiling and bar-coded gene-expression analysis by multiplexed RNA-Seq)33,34 and cell lines representing a diverse array of cell states35 may be incorporated to paint a more comprehensive portrait of a given compound’s biological activity. Not only would performance-diverse compound collections assembled in this way increase the chance of discovering compounds with a wide range of desired activities, they may also assist MoA determination for any individual compound by incorporating multi-feature annotations such as those described in this work.36,37 Lastly, real-time annotation of compounds empowers chemists to discover the consequences of their chemical reactions well in advance of the traditional route of waiting and hoping for a chance observation, most often by a biologist, years later.
Acknowledgments
We thank Kate Hartland for assistance with the cell-painting assay; Dr. Stephen Johnston for analytical support; Dr. Marshall Morningstar for providing critical feedback on the manuscript; Drs. Jochen Zoller, Sina Haftchenary, and Matthias Leiendecker for valuable comments; and the compound-management team for curating the synthesized compounds, all at the Broad Institute. We thank Dr. Peter Müller, Massachusetts Institute of Technology, for X-ray crystallographic analysis. The National Institutes of General Medical Sciences (GM038627 awarded to S.L.S.) and the Engineering and Physical Sciences Research Council (EP/K006053/1 and EP/L003325/1 awarded to K.I.B.-M.) supported this research. C.J.G. is supported in part by an Eli Lilly Graduate Fellowship in Chemistry. S.D.N. Jr. is funded in part by Harvard University’s Graduate Prize Fellowship. O.V. is a recipient of postdoctoral funding from the Wenner–Gren Foundation and the Swedish Chemical Society (Stiftelsen Bengt Lundqvists Minne). S.L.S. is an Investigator and Z.V.B. is a Research Associate with the Howard Hughes Medical Institute.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b04614.
The authors declare no competing financial interest.
Supplementary Material
References
- Wawer M. J.; Li K.; Gustafsdottir S. M.; Ljosa V.; Bodycombe N. E.; Marton M. A.; Sokolnicki K. L.; Bray M.-A.; Kemp M. M.; Winchester E.; Taylor B.; Grant G. B.; Hon C. S.-Y.; Duvall J. R.; Wilson J. A.; Bittker J. A.; Dančík V.; Narayan R.; Subramanian A.; Winckler W.; Golub T. R.; Carpenter A. E.; Shamji A. F.; Schreiber S. L.; Clemons P. A. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 10911. 10.1073/pnas.1410933111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner B. K.; Clemons P. A. Curr. Opin. Chem. Biol. 2009, 13, 539. 10.1016/j.cbpa.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J.; Crawford E. D.; Peck D.; Modell J. W.; Blat I. C.; Wrobel M. J.; Lerner J.; Brunet J.-P.; Subramanian A.; Ross K. N.; Reich M.; Hieronymus H.; Wei G.; Armstrong S. A.; Haggarty S. J.; Clemons P. A.; Wei R.; Carr S. A.; Lander E. S.; Golub T. R. Science 2006, 313, 1929. 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Scannell J. W.; Bosley J. PLoS One 2016, 11, e0147215. 10.1371/journal.pone.0147215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge R. M.; Scolnick E. M.; Altshuler D. Nat. Rev. Drug Discovery 2013, 12, 581. 10.1038/nrd4051. [DOI] [PubMed] [Google Scholar]
- Gustafsdottir S. M.; Ljosa V.; Sokolnicki K. L.; Wilson J. A.; Walpita D.; Kemp M. M.; Seiler K. P.; Carrel H. A.; Golub T. R.; Schreiber S. L.; Clemons P. A.; Carpenter A. E.; Shamji A. F. PLoS One 2013, 8, e80999. 10.1371/journal.pone.0080999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray M.-A.; Singh S.; Han H.; Davis C. T.; Borgeson B.; Hartland C.; Kost-Alimova M.; Gustafsdottir S. M.; Gibson C. C.; Carpenter A. E.. Nat. Protoc., DOI: http://dx.doi.org/10.1101/049817, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent F.; Loria P.; Pregel M.; Stanton R.; Kitching L.; Nocka K.; Doyonnas R.; Steppan C.; Gilbert A.; Schroeter T.; Peakman M.-C. Sci. Transl. Med. 2015, 7, 293ps15. 10.1126/scitranslmed.aab1201. [DOI] [PubMed] [Google Scholar]
- Maskill K. G.; Knowles J. P.; Elliott L. D.; Alder R. W.; Booker-Milburn K. I. Angew. Chem., Int. Ed. 2013, 52, 1499. 10.1002/anie.201208892. [DOI] [PubMed] [Google Scholar]
- Bertz S. H.; Sommer T. J. Chem. Commun. 1997, 2409. 10.1039/a706192g. [DOI] [Google Scholar]
- Böttcher T. J. Chem. Inf. Model. 2016, 56, 462. 10.1021/acs.jcim.5b00723. [DOI] [PubMed] [Google Scholar]
- Knowles J. P.; Booker-Milburn K. I.. Chem. - A Eur. J., DOI: 10.1002/chem.201600479, in press. [DOI] [Google Scholar]
- Åhman J.; Somfai P.; Tanner D. J. Chem. Soc., Chem. Commun. 1994, 2785. 10.1039/C39940002785. [DOI] [Google Scholar]
- Åhman J.; Somfai P. Tetrahedron 1999, 55, 11595. 10.1016/S0040-4020(99)00658-4. [DOI] [Google Scholar]
- Huisgen R.; Scheer W.; Huber H. J. Am. Chem. Soc. 1967, 89, 1753. 10.1021/ja00983a052. [DOI] [Google Scholar]
- Dauban P.; Malik G. Angew. Chem., Int. Ed. 2009, 48, 9026. 10.1002/anie.200904941. [DOI] [PubMed] [Google Scholar]
- Hugel G.; Levy J. J. Org. Chem. 1984, 49, 3275. 10.1021/jo00192a005. [DOI] [Google Scholar]
- Knowles J. P.; Elliott L. D.; Booker-Milburn K. I. Beilstein J. Org. Chem. 2012, 8, 2025. 10.3762/bjoc.8.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker-Milburn K. Nat. Chem. 2012, 4, 433. 10.1038/nchem.1356. [DOI] [PubMed] [Google Scholar]
- Sauer W. H. B.; Schwarz M. K. J. Chem. Inf. Comput. Sci. 2003, 43, 987. 10.1021/ci025599w. [DOI] [PubMed] [Google Scholar]
- Uchida T.; Rodriquez M.; Schreiber S. L. Org. Lett. 2009, 11, 1559. 10.1021/ol900173t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.; Sello J. K.; Schreiber S. L. Org. Lett. 2000, 2, 709. 10.1021/ol005574n. [DOI] [PubMed] [Google Scholar]
- Carpenter A. E.; Jones T. R.; Lamprecht M. R.; Clarke C.; Kang I. H.; Friman O.; Guertin D. A.; Chang J. H.; Lindquist R. A.; Moffat J.; Golland P.; Sabatini D. M. Genome Biol. 2006, 7, R100. 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalanobis P. C. Proc. Natl. Inst. Sci. Calcutta 1936, 2, 49. [Google Scholar]
- Hutz J. E.; Nelson T.; Wu H.; McAllister G.; Moutsatsos I.; Jaeger S. A.; Bandyopadhyay S.; Nigsch F.; Cornett B.; Jenkins J. L.; Selinger D. W. J. Biomol. Screening 2013, 18, 367. 10.1177/1087057112469257. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. J. Med. Chem. 2009, 52, 6752. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Domagala A.; Jarosz T.; Lapkowski M. Eur. J. Med. Chem. 2015, 100, 176. 10.1016/j.ejmech.2015.06.009. [DOI] [PubMed] [Google Scholar]
- Fleming F. F.; Yao L.; Ravikumar P. C.; Funk L.; Shook B. C. J. Med. Chem. 2010, 53, 7902. 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saillenfait A. M.; Sabaté J. P. Toxicol. Appl. Pharmacol. 2000, 163, 149. 10.1006/taap.1999.8839. [DOI] [PubMed] [Google Scholar]
- Perlman Z. E.; Slack M. D.; Feng Y.; Mitchison T. J.; Wu L. F.; Altschuler S. J. Science 2004, 306, 1194. 10.1126/science.1100709. [DOI] [PubMed] [Google Scholar]
- Mattila P. K.; Lappalainen P. Nat. Rev. Mol. Cell Biol. 2008, 9, 446. 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- Petrone P. M.; Wassermann A. M.; Lounkine E.; Kutchukian P.; Simms B.; Jenkins J.; Selzer P.; Glick M. Drug Discovery Today 2013, 18, 674. 10.1016/j.drudis.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Savitski M. M.; Reinhard F. B. M.; Franken H.; Werner T.; Savitski M. F.; Eberhard D.; Martinez Molina D.; Jafari R.; Dovega R. B.; Klaeger S.; Kuster B.; Nordlund P.; Bantscheff M.; Drewes G. Science 2014, 346, 1255784. 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- Macosko E. Z.; Basu A.; Satija R.; Nemesh J.; Shekhar K.; Goldman M.; Tirosh I.; Bialas A. R.; Kamitaki N.; Martersteck E. M.; Trombetta J. J.; Weitz D. A.; Sanes J. R.; Shalek A. K.; Regev A.; McCarroll S. A. Cell 2015, 161, 1202. 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees M. G.; Seashore-Ludlow B.; Cheah J. H.; Adams D. J.; Price E. V.; Gill S.; Javaid S.; Coletti M. E.; Jones V. L.; Bodycombe N. E.; Soule C. K.; Alexander B.; Li A.; Montgomery P.; Kotz J. D.; Hon C. S.-Y.; Munoz B.; Liefeld T.; Dančík V.; Haber D. A.; Clish C. B.; Bittker J. A.; Palmer M.; Wagner B. K.; Clemons P. A.; Shamji A. F.; Schreiber S. L. Nat. Chem. Biol. 2016, 12, 109. 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futamura Y.; Kawatani M.; Kazami S.; Tanaka K.; Muroi M.; Shimizu T.; Tomita K.; Watanabe N.; Osada H. Chem. Biol. 2012, 19, 1620. 10.1016/j.chembiol.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Duan Q.; Flynn C.; Niepel M.; Hafner M.; Muhlich J. L.; Fernandez N. F.; Rouillard A. D.; Tan C. M.; Chen E. Y.; Golub T. R.; Sorger P. K.; Subramanian A.; Ma’ayan A. Nucleic Acids Res. 2014, 42, W449. 10.1093/nar/gku476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.