

Abstract
Advances in computational and experimental methods in enzymology have aided comprehension of enzyme-catalyzed chemical reactions. The main difficulty in comparing computational findings to rate measurements is that the first examines a single energy barrier while the second frequently reflects a combination of many microscopic barriers. We present here intrinsic kinetic isotope effects and their temperature dependence as a useful experimental probe of a single chemical step in a complex kinetic cascade. Computational predictions are tested by this method for two model enzymes: dihydrofolate reductase (DHFR) and thymidylate synthase (TSase). The description highlights the significance of collaboration between experimentalists and theoreticians to develop a better understanding of enzyme-catalyzed chemical conversions.
Keywords: enzyme catalysis, dihydrofolate reductase, thymidylate synthase, kinetic isotope effect, simulations, kinetic complexity, commitment to catalysis
Introduction
Empirical studies are invaluable in understanding many kinetic, mechanistic, and structural features of enzyme catalysis. However, many microscopic aspects of catalysis, such as transition state (TS) structures, reactive intermediate constituents, and dynamic effects, should also be examined by computational methods, which complement experiments and can provide atomistic insights. One of the main challenges in comparing experimental studies to calculations is ensuring that the two approaches actually examine the same phenomenon. When the experiments involve enzyme kinetics, this task is not trivial: most kinetic measurements reflect a complex combination of microscopic events, while most computations address a single event, such as a single energy barrier crossing.
Enzymes such as dihydrofolate reductase (DHFR) and thymidylate synthase (TSase) catalyze the breaking and formation of carbon-hydrogen bonds; several computational methods, including molecular dynamics (MD) and hybrid quantum mechanics/molecular mechanics (QM/MM), have been used in the study of these catalytic events. A partial list of such studies includes (Dametto, Antoniou, & Schwartz, 2012; S. Hammes-Schiffer & Benkovic, 2006; Klinman & Kohen, 2013; H. Liu & Warshel, 2007a; Luk et al., 2013; Rod, Radkiewicz, & Brooks, 2003; Thorpe & Brooks, 2004; Z. Wang, Ferrer, Moliner, & Kohen, 2013; K. F. Wong, Selzer, Benkovic, & Hammes-Schiffer, 2005). Each of these computational studies examined barrier-crossing for a specific H-transfer reaction, and predicted the involvement of several protein residues in facilitating C-H bond cleavage. A means of experimental examination of those predictions is the focus of this chapter. We describe several experimental probes of specific reactions’ barriers including measurements of rate constants, of their kinetic isotope effects (KIE), and their temperature dependence. The comparison of experimental observations with computational predictions of individual catalytic events is also discussed.
A KIE is the ratio of reaction rates for two substrates that differ only in their isotopic composition (isotopologues). Please note that this chapter refers only to primary (1°) KIEs, i.e., isotopic effects in which the isotope under study is part of the bond being cleaved or formed. When only the hydrogen on the C-H bond under study is isotopically labeled, KIEs offer an important means of probing activation of that bond. Isotopic labeling of the C-H that is cleaved in the reaction has little or no effect on other physical and chemical events along the catalytic cycle. This assumption follows that the C-H bond is not polarized and thus contributes little to the binding of large biological molecules, such as DHFR’s and TSase’s reactants.
While experimental measurements always yield an observed KIE (KIEobs), the KIE for the bond cleavage per se is called the intrinsic KIE (KIEint). By this definition KIEint is free from kinetic complexities arising from the inclusion of other kinetic steps such as substrate binding, product release, or other chemical steps along the catalytic cascade that affect KIEobs. Consequently, experimental evaluation of KIEint (but not KIEobs) can parallel the computational method in the sense that each examines a single chemical step.
We find that it is is possible to use KIEint to examine predictions of high-level computations by exploiting the temperature dependence of KIEint. This can be used to probe the nature of C-H bond cleavage and sub-nanosecond protein motions coupled to the C-H bond cleavage (Klinman & Kohen, 2013; Kohen, 2015b; D. Roston, Islam, Z., Kohen, A., 2014). An empirical interpretation of the temperature dependence of KIEint is offered by “Activated Tunneling” models as described in the literature (Fan, Cembran, Ma, & Gao, 2013; Klinman, 2015; Klinman & Kohen, 2013; Kohen, 2015b; Layfield & Hammes-Schiffer, 2013; H. Liu & Warshel, 2007a, 2007b; Marcus, 2007; Nagel & Klinman, 2010; Jingzhi Pu, Gao, & Truhlar, 2006; Pudney et al., 2009; D. Roston, Islam, & Kohen, 2013). In the framework of these models small or no temperature dependence of KIE arise primarily from high frequency H-donor and H-acceptor distance (DAD) sampling, i.e., a narrow distribution of DADs. Temperature dependent KIE, on the other hand, stems from low-frequency DAD sampling (a broad distribution of DADs) at the transition state (TS). A high DAD sampling frequency indicates a well-organized TS, while a low DAD sampling frequency demonstrates a loose TS. A perturbation of the TS (for instance, through the mutation of a residue that affects the TS) alters the frequency of DAD sampling, and this manifests in an altered KIE temperature dependence. The temperature dependence of KIEint, in other words, reflects the nature of a single bond cleavage step (within the complex kinetic cascade of an enzymatic reaction), which allows experimental comparisons to relevant computations.
That said, extracting the value of KIEint from measured KIEobs is a considerable challenge. While the Northrop method described below can be used with any KIEobs (from single turnover, burst experiments, steady state parameters, etc.), we recommend measuring KIEobs on the second order rate constant V/K competitively. The main advantage here is that only trace labeling with radioactive tritium (T) is needed in competitive experiments in which two isotopically-labeled substrates compete for the enzyme at the same pot. Most other methods would require close to 100% tritium in the bond to be cleaved, and close to no tritium in any other positions on the substrate, which is experimentally most challenging.
In the sections below we describe the process of extracting KIEint from the observed value (KIEobs) using the Northrop method then turn to examples of computational studies carried out on DHFR and TSase, followed by tests of these computational predictions with studies of temperature dependence of KIEint. Finally, each case study includes the experimental procedure used to study a specific C-H bond activation.
Observed vs. Intrinsic KIE (KIEobs vs. KIEint)
Every experimental measurement of a KIE reflects a kinetically complex cascade rather than a single microscopic step. This kinetically complex chain of events may include binding and release of substrates and products, conformational changes associated with ligand binding or release, re-protonation of many enzymatic residues and the substrates, multiple chemical conversions within the reactive complex, etc. This problem is illustrated in Figure 1, and shows that steps other than the chemical step of interest (e.g., the C−H→C hydride transfer in the cases presented below) are partly rate-limiting for the different measurable kinetic parameters. This demonstrates how difficult it is to determine the rate constant or KIEint on the intended chemical step per se, either from steady-state kinetics (e.g., kcat or kcat/KM) or from pre-steady-state kinetics (e.g., single turnover or burst studies).
Figure 1.
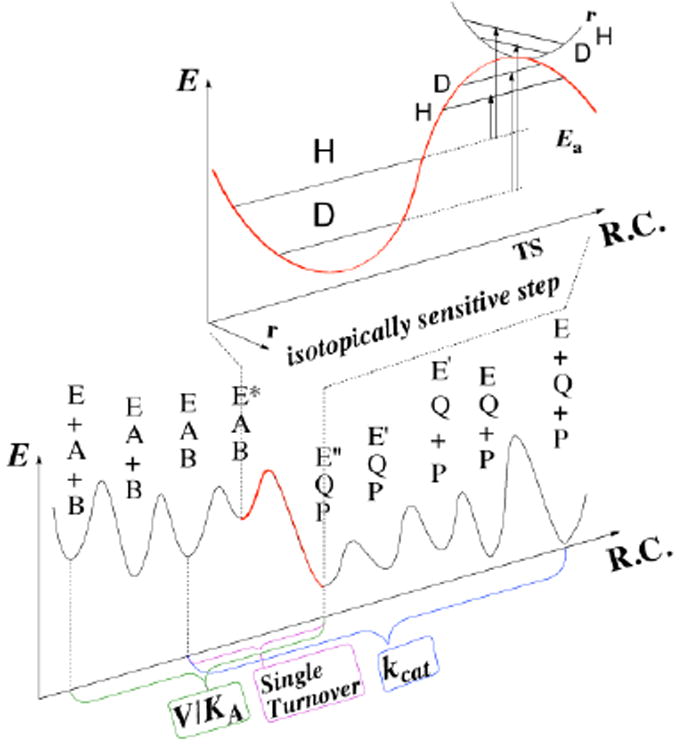
Relations between the chemical step (red) and measurable rate constants. Both steady-state parameters (e.g., kcat/KM (V/K) and kcat) and pre-steady-state rates (e.g., single-turnover rate) involve several microscopic rate constants. The KIEint reflects the nature of the chemical step, but assessing KIEint from observed values is quite challenging (see text). Reproduced from ref (Kohen, 2015a) with permission from Faculty 1000.
The KIEint in Figure 1 results from differences between the zero point energies (ZPE) of the ground state and the TS, and can be also affected by nuclear-quantum-mechanical tunneling (a phenomenon in which the atom is transferred under the classical energy barrier via its wave-like properties). These considerations are a matter of computational studies, but experimental information reflecting the cleavage of a single step is needed in order compare the two. The discrepancy between calculated and measured rates has led to some misunderstandings in the past: for example, the catalytic turnover rate, kcat, is the uni-molecular rate constant representing the reciprocal of the time for a single enzymatic turnover (Cook & Cleland, 2007). However, since most enzymes did not evolve under saturating substrate concentrations, kcat is often rate-limited by product release; thus comparing kcat’s rates or KIEs with calculations examining merely the chemical step is not always meaningful.
The relations between the KIEint of interest and the relevant measured KIEobs and computational studies can be calculated per system. The mathematical expression for this relation always has a similar general form, although the specific expressions vary depending on the details of each system. For example, consider the following simple kinetic cascade, with only one irreversible chemical step and a single substrate:
Here the chemical step is the conversion of ES to EP, represented by the rate constant k3. KIEint = k3 light / k3 heavy or, in the case of H/D KIE, Dk3 = k3H/k3D. As expressed in Eq. 1, the observed 1° KIE will be smaller than KIEint by an amount that depends on commitment to catalysis. The KIEobs on any steady-state rate constant is affected by commitments as follows (Cook & Cleland, 2007):
| (1) |
where EIE is the equilibrium isotope effect. The forward commitment (Cf) is defined as the ratio of the isotopically-sensitive rate constant (k3 in this case) in the forward direction to the net rate constant for the breakdown of the reactive ES complex into E and S (k2 in this case). The reverse commitment (Cr) is the ratio of the isotopically-sensitive rate constant in the reverse direction (from EP to ES, zero in the current example) to the isotopically-insensitive net rate constant of the decomposition of the the EP complex to E and P (k4). In cases where the chemistry is irreversible or cases where EIE = 1, Equation (1) simplifies to:
| (2) |
where C = Cf for irreversible reactions or C = Cf + Cr when EIE = 1. In cases where C is much smaller than KIEint, KIEobs is similar to KIEint. KIEint values can then be directly compared to a computed barrier crossing event, the assessment of which is the focus of this chapter.
The Northrop Method
There are several experimental methods that attempt to assess the nature of individual chemical steps: for instance, measurement of single-turnover rates as a function of pH (Fierke, Johnson, & Benkovic, 1987), or measurement of KIEobs as function of substrate concentration (Hong, Maley, & Kohen, 2007). Measuring single-turnover rates can dramatically reduce the kinetic complexity by reducing the number of microscopic steps involved in the measurement, but as the initiation of the reaction involves substrate binding, and there is still a C > 0 that results from protonation changes and the conformational steps that follow that substrate binding, and of course from the chemical steps other than the one under study, in systems catalyzing sequential chemical conversions (e.g., see TSase below).
The Northrop method eliminates C by making use of measurements of KIEobs for all three hydrogen isotopes (Northrop, 1991). The KIEobs acquired for this method can be measured for single turnover rates, steady state rate constants, or other kinetically well-defined parameters. For example, for the simplified kinetic scheme presented above the expression for the second order rate constant (kcat/KM, denoted as V/K) is given by:
| (3) |
where i represents a given isotopologue, i.e., a substrate with hydrogen (H), deuterium (D) or tritium (T). For H-transfer reactions, since there are only three possible isotopologues, every KIEobs has an isotope in common with one other, e.g., H/D KIE and H/T KIE share H as common isotope; H/D KIE and D/T KIE share D, and H/T KIE and D/T KIE share T as common isotope. All three combinations can be used, and each has different advantages and disadvantages as described in ref (Sen, Yahashiri, & Kohen, 2011). For each case, one must derive the Northrop equation so the commitment (C) for the shared isotope is moved to the denominator of the KIEobs expression, and is the C for the shared isotope in each case. For H/T KIE and D/T KIE, where T is the common isotope, C for T must be the only commitment in each expression. In this case, one derives the reciprocal of KIEobs on V/K for H/T, H(V/K)T,obs:
| (4) |
where (V/K)i is the rate for isotope i, and 1/KIEobs for H/T is H(V/K)T, obs.
| (5) |
Dividing the numerator and denominator by k3H k2 gives:
| (6) |
| (7) |
where CT = k3T/k2 is the commitment for tritium (k3T is the C-T bond cleavage rate in this simple example), H(k3)T = kT /kH is reciprocal of the intrinsic tritium KIE for isotopes H vs. T.
Similarly, the reciprocal of the KIEobs on V/K for D vs. T is given by:
| (8) |
where D(k3)T = kT /kD is the reciprocal of the KIEint for D/T. Subtracting 1 from both sides in Equations (7) and (8) gives:
| (9) |
| (10) |
Dividing Equation (9) by Equation (10) results in an expression with no commitment at all, no matter how complex the expression for C:
| (11) |
Equation 11 has two unknowns (the KIEint for H/T and D/T), but no commitment (C). The Swain-Schaad relationship for the pairs of hydrogen isotope (between H/T and D/T) is given by (Swain, Stivers, Reuwer, & Schaad, 1958):
| (12) |
where the value at the exponent (3.3) can be assessed by QM/MM calculations (like in the case of ecDHFR presented below), or calculated from basic principals as discussed below. Using the reciprocal of this relationship, the H(k3)T can be numerically determined from a pair of observed KIEs:
| (13) |
Further rearrangement of Eq. 13 leads to:
| (14) |
where T (V /K)H,obs, T (V /K)D,obs, and T(k3)H are observed H/T , observed D/T, and intrinsic H/T KIEs respectively. This equation only has one unknown, which is the KIEint that represents KIE on only the bond cleavage event (k3).
Thus KIEint is extracted from its respective observed values using a numerical solution of Eq. 14. The equations were numerically solved for using a program that is freely available on our web site under tools: https://chem.uiowa.edu/kohen-research-group/calculation-intrinsic-isotope-effects.
Like any other experimental method, the Northrop method makes assumptions, and its associated assumptions have to be carefully tested for each system. Three assumptions are embedded in the Northrop method: 1) KIEs on binding or other kinetic steps (except the one under study) are insignificant; 2) Cr in Eq. 1 is close to zero or EIE is close to unity; and 3) the two KIEint in Eq. 12 are related to each other, for example, via the Swain-Schaad exponent (SSE). The SSE can be calculated using QM/MM or assessed from (Swain et al., 1958), where μC-i is the reduced mass for isotope i and carbon 12. At the high temperature limit (i.e., above 273 K), 1° SSE falls between 3.34 and 3.26, as the carbon’s mass in the C-H/D/T bond can vary between 12 and infinity, respectively (depending on its coupling to the rest of the heavy atoms). While at temperatures below 270 K SSE can vary, SSE is mostly temperature-independent in the range in which most biochemical experiments are conducted (Shelton, Hrovat, & Borden, 2007; Smedarchina & Siebrand, 2005). The value of SSE for specific systems can be calculated from QM/MM calculations with a tunneling correction (J. Pu, Ma, Gao, & Truhlar, 2005; J. Pu, Ma, Garcia-Viloca, et al., 2005), e.g., 3.3 for the enzyme DHFR.
A recent suggestion that there is another, “hidden” assumption in the Northrop method resulted from an false procedure in which CT is calculated indirectly from T(V/K)H and T(V/K)D (Z. Wang, Antoniou, Schwartz, & Schramm, 2016), rather than from their reciprocals as used in the Northrop method (Eq. 14). The correct procedure described above (Eqs. 3-14) can be used for the complex kinetic scheme used in ref (Z. Wang et al., 2016) to calculate CT directly, without the use of any further assumptions.
Case study 1 – Thymidylate Synthase (TSase)
Thymidylate synthase (TSase) catalyzes the last committed step of the de novo synthesis of the DNA building block thymidylate (2’-deoxythymidine-5’-monophosphate, dTMP). It plays an essential role in regulating the balance of nucleotide pools and the replication of DNA, and thus is a major target for chemotherapeutic drugs (Carosati et al., 2012; Wilson, Danenberg, Johnston, Lenz, & Ladner, 2014). In the TSase-catalyzed production of thymidylate, the cofactor methylene tetrahydrofolate (CH2H4folate) sequentially provides a methylene and a hydride to the substrate dUMP (2’-deoxyuridine-5’-monophosphate). Following the formation of the ternary complex of TSase and its substrates, the catalyzed chemistry involves a series of bond cleavages and formations, including two C-H activations: a proton abstraction and a hydride transfer. In the conventionally proposed mechanism of E. coli TSase (Carreras & Santi, 1995; Finer-Moore, Santi, & Stroud, 2003; McMurry & Begley, 2016), a highly-conserved cysteine (C146) initiates the reaction by Michael addition at C6 of dUMP (step 1 in Scheme 1), forming an enolate. This enolate forms a methylene bridge with the pre-activated CH2H4folate iminium cation (step 2 and 3). Two H-transfers follow: (i) a proton abstraction from the C5 of dUMP (step 4), leading to the elimination of tetrahydrofolate (H4folate) and the formation of an exocyclic methylene intermediate (step 4); and (ii) a hydride transfer from the C6 of the H4folate to the C7 of the exocyclic methylene intermediate to produce the product dTMP (step 5). The traditionally proposed mechanism (left column in Scheme 1) was constructed from numerous experimental studies including X-ray crystallography (Stroud & Finer-Moore, 2003), kinetic isotope effects (Agrawal, Hong, Mihai, & Kohen, 2004; Spencer, Villafranca, & Appleman, 1997), steady-state kinetics, and mutagenesis (Carreras & Santi, 1995; Finer-Moore et al., 2003).
Scheme 1.
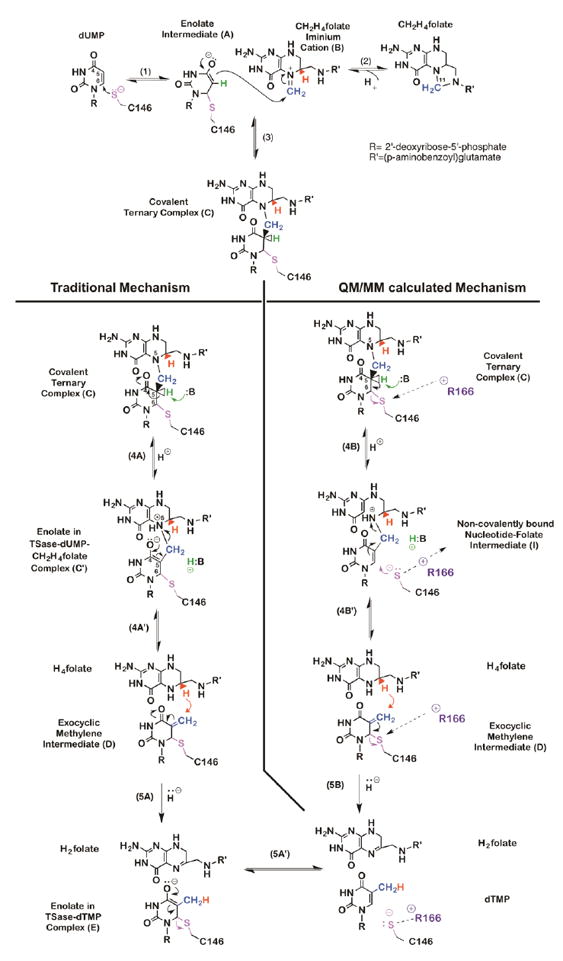
Chemical mechanism of TSase. Traditional (left panel) and QM/MM calculated (right panel) mechanisms for proton abstraction and hydride transfer.
The mechanism of TSase has also been investigated by hybrid quantum mechanics/molecular mechanics (QM/MM) calculations, which paint a slightly different picture (Kanaan et al., 2011; Kanaan, Marti, Moliner, & Kohen, 2007, 2009; Kanaan, Roca, Tunon, Marti, & Moliner, 2010; Z. Wang, Ferrer, et al., 2013) (right column in Scheme 1). One of the main differences between the two proposed mechanisms is in the hydride transfer step (step 5 in Scheme 1), which the traditional view (left colum) presents as a step-wise mechanism in which the hydride transfer precedes the elimination of enzymatic nucleophile C146 (step 5A and 5A’). The QM/MM calculations (right column), on the other hand, suggest a concerted process in which the transfer of hydride and the cleavage of the thio-ether bond happen together (step 5B). The difference in hydride transfer mechanisms is significant: the concerted mechanism involves the formation of a charged species (compound E in Scheme 1) while the other seems to bypass it, an important consideration for designing a transition state analogue. Key to the concerted mechanism’s favorability, according to calculations, is a highly conserved, positively charged residue R166 that moves closer to the thioether bond and promotes its cleavage at the transition state of the hydride transfer.
The QM/MM calculations also examined the mechanism of the proton abstraction from the C5 of dUMP in the ternary complex (compound C in Scheme 1) (Kanaan et al., 2007, 2009; Z. Wang, Ferrer, et al., 2013). The calculations suggest the formation of an intermediate that had not been proposed in the traditional mechanism. It was calculated that the removal of the C5 proton from intermediate C causes dissociation of the C146, leaving an intermediate in which the folate and the nucleotide are bound via a methylene bridge, but are not covalently bound to the enzyme (intermediate in Scheme 1)! If confirmed experimentally, this previously unrecognized intermediate might serve as a new target for antibiotic and chemotherapeutic drugs. In contrast to current drugs, which are either derivatives of pyrimidine or folate, this hypothetical new class of inhibitors could comprise part of the nucleotide and part of the folate.
These theoretical predictions were examined experimentally: The calculations predicted that as in the hydride transfer step R166 also plays a vital role in polarizing and cleaving the thio-ether bond during proton abstraction (Z. Wang, Ferrer, et al., 2013). A mutation at R166 should have altered the temperature dependence of KIEint only if it was involved in TSs of H-transfers. KIEobss for H/T and D/T were measured over a range of temperatures for the both the hydride transfer and the proton abstraction in WT and in a variant of R166 (R166K, the only active mutant of R166) (Agrawal, Hong, et al., 2004; Islam, Strutzenberg, Ghosh, & Kohen, 2015; Islam, Strutzenberg, Gurevic, & Kohen, 2014; Z. Wang & Kohen, 2010). KIEints were extracted from the KIEobss using the Northrop method described above and plotted against the inverse of temperature (Figure 2).
Figure 2.
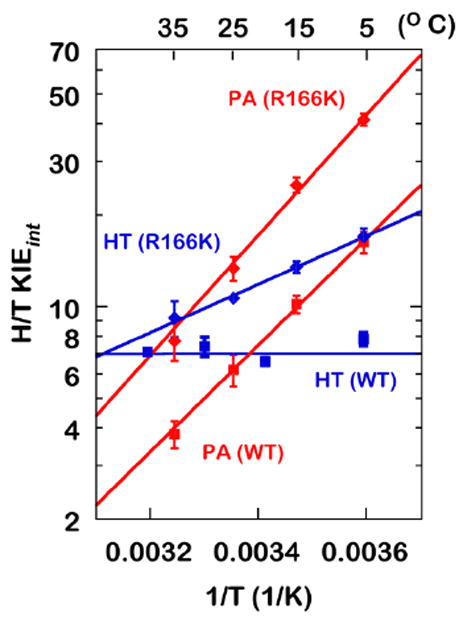
KIEints on the hydride transfer (blue) and the proton abstraction (red) for the WT (cubes) and R166K (diamonds). HT: Hydride Transfer, PA: Proton Abstraction. Reproduced from ref (Islam et al., 2015) with permission from ACS.
As is apparent in Figure 2, the mutation caused an increase in temperature dependence of KIEint for both H-transfers. The KIEint for the hydride transfer, which had been temperature-independent in the WT, became temperature-dependent in R166K, while the temperature dependence of the proton abstraction KIE increased in R166K (relative to WT). In addition, the mutation also increased the absolute magnitude of KIEint for both H-transfers. Seen in the light of the activated tunneling model, the larger magnitudes and the steeper temperature dependence of KIEs in R166K suggest lower-frequency DAD sampling and a higher average DAD at the TS. Figure 3 illustrates the DAD sampling potential along the DAD coordinate.
Figure 3.
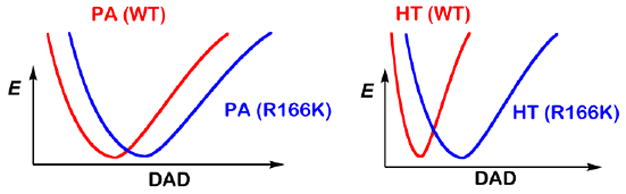
Schematic representations of PESs for the H-transfers along the DAD coordinate. Reproduced from ref (Islam et al., 2015) with permission from ACS.
A stiff potential energy surface (PES) represents a high DAD sampling frequency, suggesting a well-coordinated TS -- as is seen in the hydride transfer in WT ecTSase -- while a broad PES indicates a low DAD sampling frequency, demonstrating a loose TS, as can be seen in the proton abstraction in the wild type. The mutation at R166 caused the PES for the DAD to become wider for the both H-transfers, implying perturbation of the TSs. The mutation also caused a shift in the PESs’ minima, yielding larger DADs for both H-transfers.
These effects were attributed to R166’s being an indispensable part of the reaction’s TS for both the hydride transfer and the proton abstraction, in good agreement with the QM/MM calculations. Additionally, an observed normal secondary (2°) KIE on C6 of dUMP supported the concerted mechanism for the hydride transfer (Islam et al., 2014). As a control, mutation to several other residues closer to the reaction center were found to produce no or very minor changes in the temperature dependence of KIEint (Abeysinghe & Kohen, 2015; Agrawal, Hong, et al., 2004; Z. Wang, Abeysinghe, Finer-Moore, Stroud, & Kohen, 2012).
In summary, examining chemical steps such as C-H bond activations in enzyme-catalyzed reactions is of contemporary interest. While computational approaches have advantages and the virtue of examining a single chemical step, experimental tools for such examinations are few. The temperature dependence of KIEint probes the TS of a given chemical transfer, thereby offering a test of computational predictions. As an example, TSase has conventionally been thought to catalyze two sequential H-transfers in a complex cascade of physical and chemical steps; QM/MM calculations predicted new mechanisms for the both hydride transfer and the proton abstraction. In both cases the experimental findings, using KIE temperature dependence as a probe, offer strong support for the predictions made by QM/MM calculations. Not only was the role of a particular residue, R166, confirmed in both steps, but secondary KIEs (Islam et al., 2014) confirmed the concerted nature of the hydride transfer step proposed by QM/MM calculations.
The existence of the newly predicted intermediate (Z. Wang, Ferrer, et al., 2013) has not yet been directly demonstrated because the involvement of R166 in steps before and after that proposed intermediate is quite indirect. More experiments and computational studies are needed to further examine these proposals, such as QM/MM calculations and temperature dependence KIEint measurements with TSase from other organisms, for isotopically labeled enzymes, and other studies that would further expose the nature of TSase catalysis.
Experimental section for TSase
[2-14C]dUMP (specific radioactivity of 52 Ci/mol) was purchased from Moravek Biochemicals. [5-3H]dUMP (15-30 Ci/mmol) and [3H]NaBH4 (15Ci/mmol) were purchased from American Radiolabeled Chemicals. [2-3H]isopropanol was synthesized by reducing acetone using [3H]NaBH4. . Isopropanol-d8 was purchased from Sigma-Aldrich.
Measurements of KIEs on the hydride transfer step
The upper panel in Scheme 2 shows the isotopic labeling pattern for the hydride-transfer KIE measurements, where the hydrogen at C6 of CH2H4folate (to be transferred at that step) was labeled as deuterium or tritium. The labeled (R)-[6-xH]CH2H4folate ((xH= D or T) was synthesized through a chemoenzymatic process described elsewhere (Agrawal, Mihai, & Kohen, 2004). The KIE measurements were performed in 100 mM Tris buffer (pH= 7.5), 2 mM TCEP, 1 mM EDTA and 7 mM HCHO. Both H/T and D/T KIE were measured competitively, yielding a KIEobs on the second order rate constant (V/K)(Cook & Cleland, 2007).
Scheme 2.
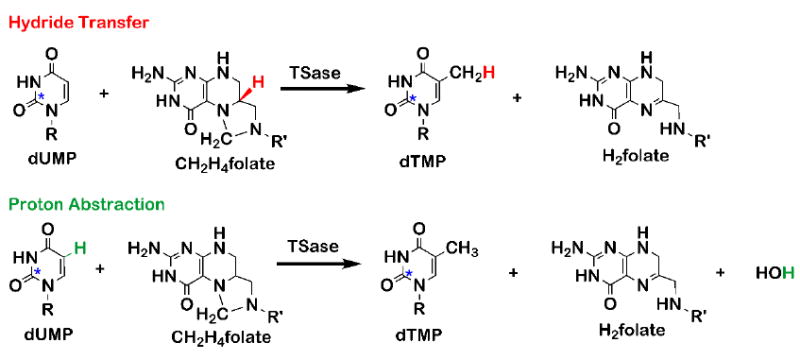
Isotopic labeling for measuring KIEs on the hydride transfer (top) and the proton abstraction (bottom). The hydride (red) and the proton (green) are the sites of isotopic labeling. The asterisks on dUMP represent remote 14C labeling to trace lighter isotopes.
In competitive KIE measurements, both the light and the heavy isotopologue are present in the same reaction mixture. Here, the 14C-labeled substrate ([2- 14C]dUMP) was used to trace the light isotope (H or D). For each reaction, trace (R)-[6-3H]CH2H4folate (about 2 million disintegrations per minute, Mdpm) in protiated or deuterated CH2H4folate for H/T or D/T KIE experiments, respectively, was mixed with 14C-labeled substrate [2- 14C]dUMP (~ 0.3 Mdpm) in a ratio of 6/1 (3H/14C) or higher. Since tritium radiation is weaker than 14C radiation, this ratio contributed to higher accuracies in the Liquid Scintillation Counting (LSC) analyses. Using unlabeled dUMP, the concentration of [2- 14C]dUMP was adjusted to 20% in excess over the total CH2H4folate in order to enable measurement of fractional conversion of dUMP to dTMP (f). Since high CH2H4folate concentration (~500 μM) was previously found to be inhibitory (Agrawal, Hong, et al., 2004; Z. Wang, Sapienza, et al., 2013), the chosen total concentration of dUMP was in the range of 100-400 μM so that with 20% in excess, CH2H4folate stayed below the inhibitory concentration. Aliquots of the reaction mixture were equilibrated to 5, 15, 25, and 35 °C and adjusted to pH 7.5. Prior to initiating the reaction by adding enzyme, two aliquots (t0) that served as controls were also taken and quenched (for consistency). At each temperature, the reaction was initiated with the addition of TSase, and six to eight aliquots of reaction mixture were removed and quenched at different time points (t) leading to fraction conversion between 20 and 80%. All aliquots were quenched with at least 5 times molar excess of 5-fluoro-dUMP, a nanomolar competitive inhibitor of TSase, over the substrate dUMP, and stored at -80 °C for further HPLC analysis. A solution of concentrated enzyme was added to the remaining reaction mixture and incubated further to reach the completion of the reaction, then divided into three aliquots that were quenched (for consistency) and served as infinite time points (t∞). All of the aliquots (t0, t and t∞) were subjected to RP HPLC analyses in which reactants and products were separated from each other and their radioactivity measured by LSC. The observed KIE for each time point was calculated using Equation 15, as mentioned in an earlier section:
| (15) |
where f, Rt and R∞ are the fractional conversion, the ratio of 3H/14C in the product, and the ratio of 3H/14C in the product at the infinity time points, respectively. R∞ was averaged over the infinite time points. The fractional conversion f for all time points was calculated using the following equation:
| (16) |
The exact excess of [2- 14C]dUMP over CH2H4folate was calculated from the infinite time points using following equation:
| (17) |
Figure 4 shows a typical plot of KIEobs versus fraction conversion (f). The fact that the observed KIEs are independent of fraction conversion serves as an important quality control, because most artifacts would lead to f-dependent KIEobs. Each of the observed KIEs was averaged over at least five time points at each temperature. The KIEints were calculated using Eq 14.
Figure 4.
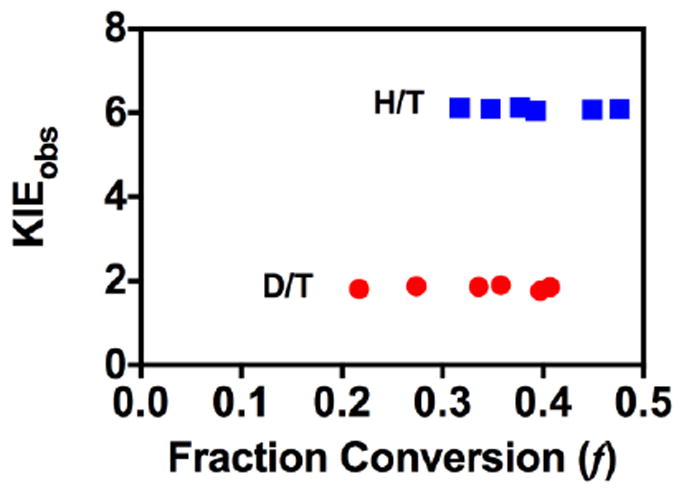
Observed H/T and D/T KIEs on hydride transfer for R166K at 35 °C.
Measurements of KIEs on the proton abstraction
The lower panel of Scheme 2 shows the isotopic labeling pattern for the proton-abstraction KIE measurements. Apart from different isotopic labeling, the experimental processes for the proton-abstraction KIEs are mostly identical to those of the hydride-transfer KIE measurements. Therefore, in this section, only the experimental procedures unique to hydride transfer are highlighted.
[2- 14C]dUMP and[5- 3H]dUMP were purchased from Moravek Biochemicals and American Radiolabeled Chemicals, respectively. [2- 14C, 5-2H]dUMP was synthesized following a published procedure (Wataya & Hayatsu, 1972).
To measure the isotope effects in the proton abstraction, trace [5- 3H]dUMP was mixed with [2- 14C]dUMP or [2- 14C, 5-2H]dUMP for H/T or D/T KIE measurements, respectively. The concentration of CH2H4folate was taken in excess to total dUMP concentration in order to obtain complete conversion of the labeled substrates.
It should be noted that the KIEobs for proton abstraction is dependent on the concentration of CH2H4folate, as can be seen in Eqs. 2 and 18 (Ghosh, Islam, Krueger, Abeysinghe, & Kohen, 2015; Hong et al., 2007; Z. Wang & Kohen, 2010):
| (18) |
where Cf is the forward commitment and k9 denotes the proton abstraction step (Scheme 3).
Scheme 3.
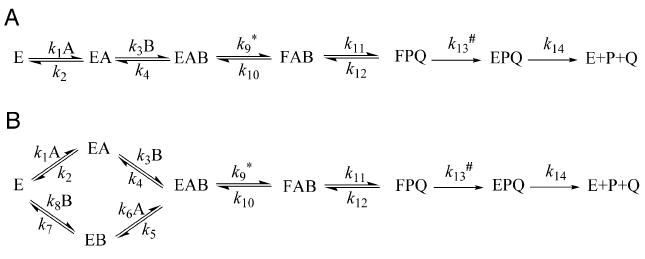
Ordered (A) and random (B) binding mechanisms for substrates dUMP (A) and CH2H4folate (B) for TSase. Asterisk (*) and number sign (#) represent the proton abstraction (k9) and the hydride transfer (k13), respectively.
In some cases, at high concentration of CH2H4folate KIEobs goes to unity, which indicates a strictly-ordered binding mechanism with dUMP binding first (Panel A in Scheme 3), as can be seen in Eq. 19 (Ghosh et al., 2015; Hong et al., 2007; Z. Wang & Kohen, 2010):
| (19) |
In Eq. 19 Cf goes to infinity at high concentrations of CH2H4folate, so KIEobs goes to one (Eq. 1). For WT ecTSase an optimal concentration of CH2H4folate (3 μM) was chosen to yield large H/T and D/T KIEobs with low errors (Z. Wang & Kohen, 2010). However, for many mutants (Abeysinghe & Kohen, 2015) including R166K (Islam et al., 2015) that follow a random binding mechanism, high concentrations of CH2H4folate yield large KIEobs. The reaction mixture for the proton-abstraction KIE measurements contains 50 mM MgCl2, which was added to reduce the commitment factor on this step (Z. Wang & Kohen) (Islam et al.). Note that the presence of MgCl2 does not affect the intrinsic KIEs for the hydride transfer (Z. Wang, Sapienza, et al., 2013). The observed KIE was calculated using Eq. 15 while the fraction conversion was determined using the following equation:
| (20) |
As in the hydride transfer, the absence of any trends in observed KIEs with fraction conversion (Figure 5) indicates that no significant artifact such as protium contamination distorts? the D/T KIEs. Though the proton abstraction is reversible, Eq. 14 for extracting intrinsic KIEs is still valid since the EIE was found to be close to unity for this step (Hong et al., 2007). Furthermore, the reverse commitment on the tritium is essentially zero since once released, the tritium is diluted into 110 M protiated water. Given all these assumptions, quality control (QC) is most important in this case, and our experience is that the most sensitive QC is f-independent KIEint (Figure 5). Almost every deviation from the above assumptions leads to an f-dependent KIEint.
Figure 5.
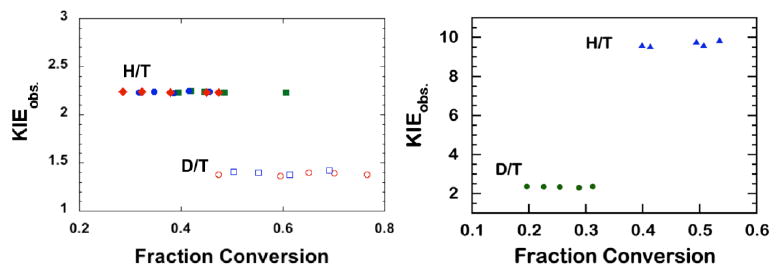
Examples of KIEobs for the proton abstraction step for the WT ecTSase (left) (Z. Wang & Kohen, 2010) and its R166K mutant (right) (Islam et al., 2015). Reproduced from ref (Islam et al., 2015) with permission from ACS.
Case study 2 – Dihydrofolate Reductase (DHFR)
DHFR catalyzes the NADPH-dependent reduction of 7,8-dihydrofolate (H2F) to 5,6,7,8-tetrahydrofolate (H4F) through a stereospecific transfer of the pro-R hydrogen from NADPH to C6 of the si face of the pterin ring (Scheme 4) (S. Hammes-Schiffer & Benkovic, 2006). DHFR from E. coli (ecDHFR) is a common model system for various studies due to its small size, flexibility and the simple chemical transformation it catalyzes. The enzyme has served as a platform for many calculations and experimental studies, and the relationship between protein dynamics and its function has been examined by both theoreticians and experimentalists. A partial list of references includes (Arora & Brooks III, 2009; Bystroff & Kraut, 1991; Dametto et al., 2012; Fan et al., 2013; Fierke et al., 1987; Klinman & Kohen, 2013; H. Liu & Warshel, 2007b; Loveridge, Behiry, Guo, & Allemann, 2012; McElheny, Schnell, Lansing, Dyson, & Wright, 2005; Pauling, 1948; Singh, Abeysinghe, & Kohen, 2015; Singh, Francis, & Kohen, 2015; Singh, Morris, Tivanski, & Kohen, 2015a, 2015b; Z. Wang, Singh, Czekster, Kohen, & Schramm, 2014; Kim F. Wong, Watney, & Hammes-Schiffer, 2004). The focus of this section is to demonstrate a combination of different approaches to studying the DHFR-catalyzed reaction emphasizing the hydride transfer step, primarily through KIE experiments and hybrid quantum mechanical/molecular mechanics/molecular dynamics (QM/MM/MD) simulations. The current section emphasizes the interplay between experimental and computational approaches.
Scheme 4.
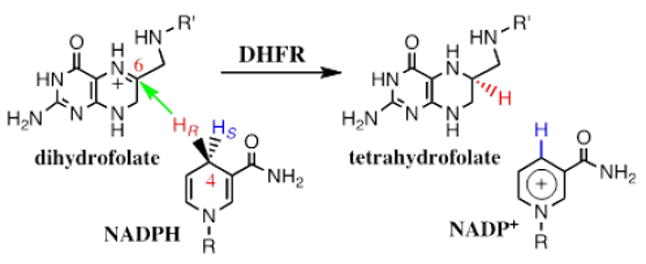
DHFR catalyzes the stereospecific transfer of the pro-R hydride of C4 of NADPH to C6 of protonated N5-DHF, producing THF and the oxidized cofactor NADP+. It is important for both measurements and computations that the hydride transfer (green arrow) takes place after DHF is already protonated (C. T. Liu, Francis, K., Layfield , J., Huang, X., Hammes-Schiffer, S., Kohen, A., Benkovic, S.J., 2014).
Computational vs. experimental studies
Network of coupled motions in DHFR
The concept of remote residues influencing events at the active site has been debated for several enzymes (Fraser et al., 2009; Ghanem, Li, Wing, & Schramm, 2008; Henzler-Wildman & Kern, 2007; Loria, Berlow, & Watt, 2008; Meadows, Tsang, & Klinman, 2014). Calculations by Brooks and Hammes-Schiffer independently predicted that a “network of dynamically coupled motions” was part of the reaction coordinate of the C-H→C hydride transfer in ecDHFR (S. Hammes-Schiffer & Benkovic, 2006; Radkiewicz & Brooks, 2000; Rod et al., 2003; K. F. Wong et al., 2005). These coupled motions denote thermal equilibrium; i.e., thermally-averaged conformational changes along the collective reaction-coordinate, producing configurations conducive to the hydride transfer reaction with the pre-protonated DHF (Scheme 4). A portion of this network of coupled motions in DHFR is illustrated in Figure 6.
Figure 6.
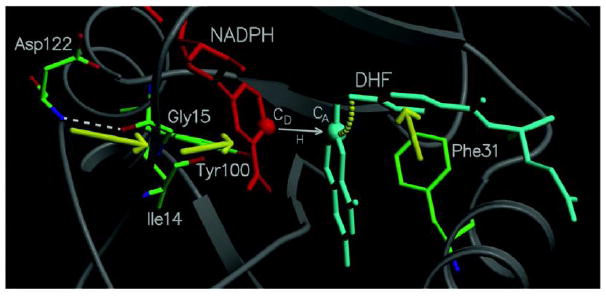
Diagram of a portion of the network of coupled promoting motions in ecDHFR. The yellow arrows and arc indicate the coupled promoting motions. From ref (Agarwal, Billeter, Rajagopalan, Benkovic, & Hammes-Schiffer, 2002) with permission from Proc. Natl. Acad. Sci.
The equilibrium motions produce configurations that promote hydride transfer through short DADs, a proper electrostatic environment for charge transfer, and correct orientation of the substrate and cofactor. Further insights into this network of coupled motion have been provided by calculations performed on mutants of DHFR. For example, G121V, one of the most intensively studied DHFR mutants, results in a 40-fold decrease in NADPH binding affinity and a 200-fold decrease in the single turnover rates, despite the fact that the mutation is located ~15 Å away from the active site (Boehr et al., 2013; Fan et al., 2013; H. Liu & Warshel, 2007a, 2007b; Mauldin, Sapienza, Petit, & Lee, 2012; Ohmae, Iriyama, Ichihara, & Gekko, 1998; Rajagopalan, Stefan, & Benkovic, 2002; Rod et al., 2003; Singh, Abeysinghe, et al., 2015; Singh, Sen, Francis, & Kohen, 2014; Swanwick, Shrimpton, & Allemann, 2004; Thorpe & Brooks, 2003, 2004; L. Wang, Tharp, Selzer, Benkovic, & Kohen, 2006). Simulations of G121V have resulted in a rate decrease that is consistent with experimental rate measurements and suggested that mutations may alter the network of coupled motions through nonlocal structural perturbations, raising the free energy barrier and reducing the reaction rate (Watney, Agarwal, & Hammes-Schiffer, 2003). These calculations have suggested that remote mutations far from the active site could bring subtle structural perturbations that affect the catalytic rate by altering the conformational sampling of the entire enzyme, leading to alterations in the network of coupled motions of the wild type enzyme. This concept provides an explanation for the experimentally observed non-additive rates (Rajagopalan et al., 2002): the introduction of a mutation modifies the thermal motions of the entire enzyme because the remote regions of the enzyme are coupled to each other through long-range electrostatics and extended hydrogen bonding networks (Kim F. Wong et al., 2004); in turn, altering the thermal motions of the enzyme affects the probability of the occurrence of sampling conformations conducive to the catalyzed chemical reaction, thereby influencing the free energy barrier and the rate. Other theoretical studies by Moliner, Allemann and co-workers on WT-DHFR and G121V also imply that mutation causes nonlocal structural effects that may lead to perturbation of the network of coupled motions (Luk et al., 2013).
The QM/MM/MD calculations predicted that distal residues M42, G121, and F125, as well as active-site residue I14, are part of the network in question (S. Hammes-Schiffer & Benkovic, 2006; Sharon Hammes-Schiffer & Watney, 2006; Rod et al., 2003; K. F. Wong et al., 2005; Kim F. Wong et al., 2004). Single turnover rates examined by Benkovic and co-workers on G121, M42, and their double mutants provided some support for these predictions (Rajagopalan et al., 2002). Intrinsic KIEs and their temperature dependence were examined experimentally to test for the presence of the predicted network and effects of mutants on that network. As is true of most wild-type enzymes, WT ecDHFR has a KIEint that is temperature independent (Sikorski et al., 2004), indicating a narrow DAD distribution and an active site that is perfect for hydride transfer. The temperature dependence of KIEint was measured for I14A and M42W, G121V, F125M, and W133F, as well as for their double mutants (Singh et al., 2014; L. Wang, Goodey, Benkovic, & Kohen, 2006a, 2006b; L. Wang, Tharp, et al., 2006). Synergy between single mutations was used to indicate that the tested residues “work together”, i.e., are part of a network of motions coupled or correlated with the hydride transfer step. “Synergy” in this context means that the sum of changes in single turnover rates or temperature dependence of KIEint caused by single mutants is smaller than that caused by the relevant double mutant.
Figure 7 summarizes the temperature dependences of the intrinsic KIEs for remote single mutants, their double mutants, the single active site mutant, and a remote-active site double mutant. The synergistic effect of double-mutant KIEs is apparent in the non-additive values of the temperature dependence, i.e., ΔEa, which is the difference between the energy of activations (Ea) of the two isotopes used per study. (Please note that for KIEs ΔEa = ΔH‡ because Ea = H‡ - RT, and RT is the same for both isotopes.) Figure 8 summarizes the isotope effect on activation energy (ΔEa) and the Arrhenius pre-exponential factors (Alight/Aheavy) for WT-ecDHFR and its mutants. Figure 8 also reveals that the effect of single distal mutation is similar to the effect of active site mutations I14V and I14A, while the effect of double distal mutation is similar to the effect of active site mutation I14G, suggesting similarities in the way those mutants affect DAD distribution at the active site.
Figure 7.
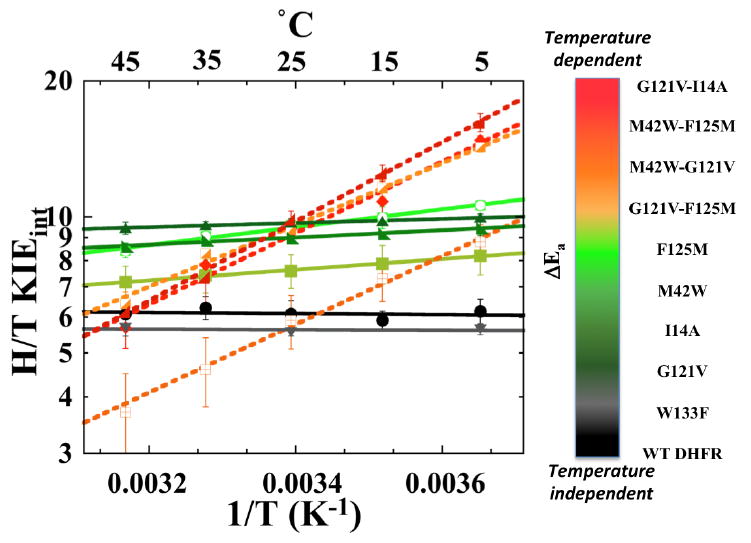
Comparison of Arrhenius plots of intrinsic H/T KIEs of WT DHFR and its mutants. The lines represent the non-linear regression to the Arrhenius equation and the error bars represent standard deviation.
Figure 8.
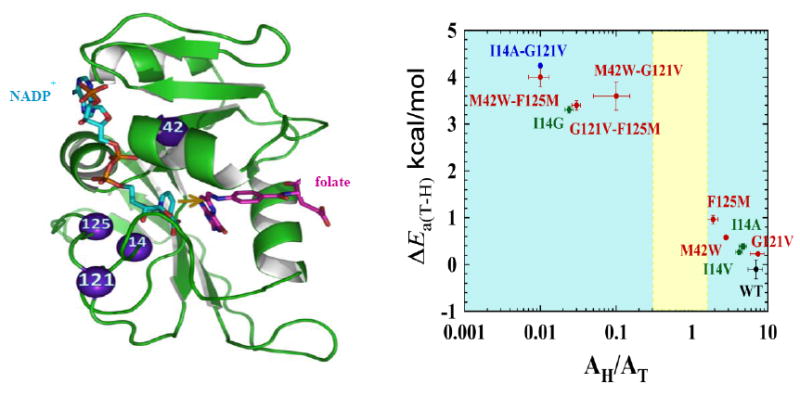
Roles of active site and distal residues on the DHFR-catalyzed reaction. Left panel: Structure of WT-ecDHFR (PDB Code 1RX2), with folate in magenta and NADP+ in light blue. A yellow arrow marks the hydride’s path from C4 of the nicotinamide to C6 of the folate, and the residues studied in ref (Singh et al., 2014) are marked as purple spheres. Right panel: Presentation of the temperature-dependence parameters of KIEint for WT (black), distal (red), and active site I14 (green) mutants of DHFR, where error bars represent standard deviation. For each point, the ordinate is the isotope effect on activation energy (ΔEa) and the abscissa is the isotope effect on the Arrhenius pre-exponential factor (AH/AT), both of which were determined from a non-linear regression of KIEint to the Arrhenius equation. The yellow block represents the semi-classical range of the Arrhenius pre-exponential factor (0.3-1.7) (Kohen, 2006). Reprinted from ref. (Singh, Francis, et al., 2015) with permission from the American Chemical Society.
In summary, the influence of remote residues participating in a network of coupled motions on the catalyzed chemistry was validated for all the experimentally verified residues proposed by QM/MM simulations. Furthermore, W133F, the residue that had not been predicted by these calculations to be part of that network, did not seem to have an effect on hydride transfer. Because both the calculations and KIEint examined the same single chemical step, the experimental findings can be compared directly to the computations that predicted that network. The findings thus validate calculations proposing that both remote and active-site residues constitute a network of coupled promoting motions correlated to the bond activation step (Singh, Francis, et al., 2015).
After the experimental findings for G121V and M42W were published (L. Wang, Goodey, et al., 2006a), Warshel and coworkers calculated the temperature dependences of KIEint for these mutants and for the wild type using the empirical valence bond-quantum classical path method (EVB-QCP) (H. Liu & Warshel, 2007b). Those studies specifically explored the relationship between DAD and KIEint at different temperatures. In good agreement with the empirical activated tunneling models mentioned above, Warshel’s calculations found a broader DAD distribution for steeper temperature dependence of KIEs (ΔEa).
A very different computational study of ecDHFR used transition-path sampling (TPS) and examined the role of protein promoting vibrations (PPV) in ecDHFR catalysis (Dametto et al., 2012). This study found no such PPVs, which could be taken as contradictory to all the computational and experimental studies discussed above. However, the “network of coupled motions” along the reaction coordinate simulated by Hammes-Schiffer and others includes no non-statistical elements. The calculated PPVs, on the other hand, are non-statistical dynamic motions, so the apparent contradictions between the two methods likely reflect the fact that they examine very different phenomena. It seems that the experimental studies using KIEint agree well with the first but did not test the second theoretical approach. Additionally, TPS calculatins of the human DHFR (hsDHFR) predicted significant PPVs, but KIEint for both light and heavy hsDHFR where identical throughout the temperature range (Francis, Sapienza, Lee, & Kohen, 2016), falling to support the theoretical prediction. Similarly, in a different TPS study of liver alcohol dehydrogenase, it was predicted that the valine residues at position 203 and 207 facilitate enzyme catalysis as part of the PPV (Caratzoulas, Mincer, & Schwartz, 2002). However, no experimental evidence was found to support a role of V207 in the dynamics of catalysis (Yahashiri, Rubach, & Plapp, 2014).
Experimental section for DHFR
The cofactors [Ad-14C]-NADPH (50 mCi/mmol), (R)-[Ad-14C, 4-2H]-NADPH (50 mCi/mmol) and (R)-[4-3H]-NADPH (680 mCi/mmol) were synthesized according to previously published procedures (N. Agrawal & Kohen, 2003; Markham, Sikorski, & Kohen, 2004a, 2004b; McCracken, Wang, & Kohen, 2003; Sen et al., 2011; McCracken et al., 2003).
Competitive KIE measurements
For H/T 1° KIE, (R)-[4-3H]-NADPH and [Ad-14C]-NADPH were combined in a radioactivity ratio close to 5:1. Likewise, for D/T 1° KIE, (R)-[4-3H]-NADPH and (R)-[Ad-14C, 4-2H]-NADPH were combined in the same manner. Each mixture was co-purified on an analytical reverse phase HPLC column. The purified material was divided into aliquots containing 300,000 dpm of 14C and stored at -80 °C for short-term storage.
All measurements were done in MTEN (50 mM MES, 25 mM Tris, 25 mM ethanolamine and 100 mM NaCl) buffer at pH=9.0 over the temperature range of 5-45 °C. NADPH was added to the reaction mixture so that the final concentration was 4 μM, while dihydrofolate was added to a final concentration of 0.85 mM (~200-fold excess over NADPH). A pH electrode was calibrated at each temperature using standard buffers at the respective temperatures and used to check the pH of all reaction mixtures. For DHFR, the largest KIEobs can be measured at pH 9.0, though KIEobs at pH 8.0 and 7.0 have also been measured with good outcomes (C. T. Liu, Francis, K., Layfield , J., Huang, X., Hammes-Schiffer, S., Kohen, A., Benkovic, S.J., 2014). The reaction was initiated by addition of DHFR, and allowed to proceed until a fraction conversion of between 20-80% was reached, followed by quenching by adding an excess of methotrexate to a final concentration of 1.7 mM before storing in dry ice. Fraction conversions of 20-80% have been shown to produce minimal error when determining KIEobs (Northrop, 1991). Samples were thawed and bubbled with oxygen before HPLC-LSC analysis to ensure complete oxidation of the product tetrahydrofolate. The samples were then separated by reverse phase HPLC and analyzed on a liquid scintillation counter (LSC). The KIEobs was calculated using the following equation:
| (15) |
where the fraction conversion (f) was determined from the ratio of 14C in the product and reactant, and Rt and R∞ are the ratios of 3H/14C at a particular time point and at infinite time, respectively. Subsequently, KIEint was extracted from its respective observed values using the Northrop method as explained in an earlier section. All quality controls were performed for DHFR as for TSase, as described under the experimental section for TSase.
Summary
In this chapter, we underscore the importance of examining the same chemical step, or microscopic event, in both calculations and experiments, and illustrate the necessity of such precise, parallel computational and experimental studies of DHFR and Tsase. The utility of intrinsic KIE as a probe for TS structures and their distribution is described. In the two case studies discussed above, the objective of the calculations is not only to explain experimental results but also to create experimentally testable predictions that permit the examination of the theory’s underlying rationale. The objective of the experimental work is to conduct measurements that afford the examination of the same phenomenon studied by the relevant calculations. In that context, we suggest that assessment of intrinsic KIE (KIEint) from measured observed KIE (KIEobs) is an important tool, and that in some cases the Northrop method can assist in that task. The two examples presented above started from predictions made by QQM/MM calculations (a newly proposed intermediate in TSase and a network of coupled motions in ecDHFR), and demonstrated how measurement of the temperature dependence of KIEint supported theses predictions, then suggested further need for calculations and experimental studies.
We suggest that a collaborative blend of theoretical and experimental approaches is beneficial to resolving quandaries of theoretical models and experimental results. Collaborative approaches are likely to deepen our knowledge of enzyme mechanisms in general, as well as our knowledge of the role of protein motions in catalysis, allowing the development of more detailed and comprehensive models of enzyme catalysis. The expected gain is most significant: experimental findings that agree with computations lend reassurance to both methods; and exposing “hidden” features in enzyme mechanisms may lead to the identification of new targets for inhibitors and potential drugs.
Acknowledgments
This work was supported by NIH (R01GM65368) and NSF (CHE-1149023) and the Iowa Center of Biocatalysis and Bioprocessing associated with NIH T32 GM008365 to ZI.
References
- Abeysinghe T, Kohen A. Role of Long-Range Protein Dynamics in Different Thymidylate Synthase Catalyzed Reactions. Int J Mol Sci. 2015;16:7304–7319. doi: 10.3390/ijms16047304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc Natl Acad Sci U S A. 2002;99:2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal N, Hong B, Mihai C, Kohen A. Vibrationally Enhanced Hydrogen Tunneling in the Escherichia coli Thymidylate Synthase Catalyzed Reaction. Biochemistry. 2004;43:1998–2006. doi: 10.1021/bi036124g. [DOI] [PubMed] [Google Scholar]
- Agrawal N, Mihai C, Kohen A. Microscale synthesis of isotopically labeled R-[6-xH]N5,N10-methylene-5,6,7,8-tetrahydrofolate as a cofactor for thymidylate synthase. Anal Biochem. 2004;328:44–50. doi: 10.1016/j.ab.2004.01.029. [DOI] [PubMed] [Google Scholar]
- Arora K, Brooks CL., III Functionally Important Conformations of the Met20 Loop in Dihydrofolate Reductase are Populated by Rapid Thermal Fluctuations. J Am Chem Soc. 2009;131:5642–5647. doi: 10.1021/ja9000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr DD, Schnell JR, McElheny D, Bae S-H, Duggan BM, Benkovic SJ, Wright PE, et al. A Distal Mutation Perturbs Dynamic Amino Acid Networks in Dihydrofolate Reductase. Biochemistry. 2013;52:4605–4619. doi: 10.1021/bi400563c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystroff C, Kraut J. Crystal structure of unliganded Escherichia coli dihydrofolate reductase. Ligand-induced conformational changes and cooperativity in binding. Biochemistry. 1991;30:2227–2239. doi: 10.1021/bi00222a028. [DOI] [PubMed] [Google Scholar]
- Caratzoulas S, Mincer JS, Schwartz SD. Identification of a Protein-Promoting Vibration in the Reaction Catalyzed by Horse Liver Alcohol Dehydrogenase. J Am Chem Soc. 2002;124:3270–3276. doi: 10.1021/ja017146y. [DOI] [PubMed] [Google Scholar]
- Carosati E, Tochowicz A, Marverti G, Guaitoli G, Benedetti P, Ferrari S, Costi MP, et al. Inhibitor of Ovarian Cancer Cells Growth by Virtual Screening: A New Thiazole Derivative Targeting Human Thymidylate Synthase. J Med Chem. 2012;55:10272–10276. doi: 10.1021/jm300850v. [DOI] [PubMed] [Google Scholar]
- Carreras CW, Santi DV. The catalytic mechanism and structure of thymidylate synthase. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. New York, NY: Taylor and Francis Group LLC; 2007. pp. 253–324. [Google Scholar]
- Dametto M, Antoniou D, Schwartz SD. Barrier Crossing in Dihydrofolate Reductasedoes not involve a rate-promoting vibration. Mol Phys. 2012;110:531–536. doi: 10.1080/00268976.2012.655337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Cembran A, Ma S, Gao J. Connecting Protein Conformational Dynamics with Catalytic Function As Illustrated in Dihydrofolate Reductase. Biochemistry. 2013;52:2036–2049. doi: 10.1021/bi301559q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- Finer-Moore JS, Santi DV, Stroud RM. Lessons and Conclusions from Dissecting the Mechanism of a Bisubstrate Enzyme: Thymidylate Synthase Mutagenesis, Function, and Structure. Biochemistry. 2003;42:248–256. doi: 10.1021/bi020599a. [DOI] [PubMed] [Google Scholar]
- Francis K, Sapienza PJ, Lee AL, Kohen A. The Effect of Protein Mass Modulation on Human Dihydrofolate Reductase. Biochemistry, Accepted for publication. 2016 doi: 10.1021/acs.biochem.5b00945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462:669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem M, Li L, Wing C, Schramm VL. Altered Thermodynamics from Remote Mutations Altering Human toward Bovine Purine Nucleoside Phosphorylase†. Biochemistry. 2008;47:2559–2564. doi: 10.1021/bi702132e. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Islam Z, Krueger J, Abeysinghe T, Kohen A. The general base in the thymidylate synthase catalyzed proton abstraction. Phys Chem Chem Phys. 2015;17:30867–30875. doi: 10.1039/c5cp01246e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S, Watney JB. Hydride transfer catalysed by Escherichia coli and Bacillus subtilis dihydrofolate reductase: coupled motions and distal mutations. Philos Trans R Soc B. 2006;361:1365–1373. doi: 10.1098/rstb.2006.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Hong B, Maley F, Kohen A. Role of Y94 in Proton and Hydride Transfers Catalyzed by Thymidylate Synthase. Biochemistry. 2007;46:14188–14197. doi: 10.1021/bi701363s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam Z, Strutzenberg TS, Ghosh AK, Kohen A. Activation of Two Sequential H Transfers in the Thymidylate Synthase Catalyzed Reaction. ACS Catal. 2015;5:6061–6068. doi: 10.1021/acscatal.5b01332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam Z, Strutzenberg TS, Gurevic I, Kohen A. Concerted versus Stepwise Mechanism in Thymidylate Synthase. J Am Chem Soc. 2014;136:9850–9853. doi: 10.1021/ja504341g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan N, Ferrer S, Marti S, Garcia-Viloca M, Kohen A, Moliner V. Temperature Dependence of the Kinetic Isotope Effects in Thymidylate Synthase. A Theoretical Study. J Am Chem Soc. 2011;133:6692–6702. doi: 10.1021/ja1114369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan N, Marti S, Moliner V, Kohen A. A Quantum Mechanics/Molecular Mechanics Study of the Catalytic Mechanism of the Thymidylate Synthase. Biochemistry. 2007;46:3704–3713. doi: 10.1021/bi061953y. [DOI] [PubMed] [Google Scholar]
- Kanaan N, Marti S, Moliner V, Kohen A. QM/MM Study of Thymidylate Synthase: Enzymatic Motions and the Temperature Dependence of the Rate Limiting Step. J Phys Chem A. 2009;113:2176–2182. doi: 10.1021/jp810548d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan N, Roca M, Tunon I, Marti S, Moliner V. Theoretical study of the temperature dependence of dynamic effects in thymidylate synthase. Phys Chem Chem Phys. 2010;12:11657–11664. doi: 10.1039/c003799k. [DOI] [PubMed] [Google Scholar]
- Klinman JP. Dynamically Achieved Active Site Precision in Enzyme Catalysis. Acc Chem Res. 2015;48:449–456. doi: 10.1021/ar5003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman JP, Kohen A. Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu Rev Biochem. 2013;82:471–496. doi: 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohen A. Isotopes Effects in Chemistry and Biology. Boca Raton, Florida: Taylor and Francis; 2006. pp. 743–764. [Google Scholar]
- Kohen A. Dihydrofolate Reductase as a Model for Studies of Enzyme Dynamics and Catalysis. F1000Research. 2015a;4:1464. doi: 10.12688/f1000research.6968.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohen A. Role of Dynamics in Enzyme Catalysis: Substantial versus Semantic Controversies. Acc Chem Res. 2015b;48:466–473. doi: 10.1021/ar500322s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layfield JP, Hammes-Schiffer S. Hydrogen Tunneling in Enzymes and Biomimetic Models. Chem Rev. 2013;114:3466–3494. doi: 10.1021/cr400400p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CT, Francis K, Layfield J, Huang X, Hammes-Schiffer S, Kohen A, Benkovic SJ. The Escherichia coli Dihydrofolate Reductase Catalyzed Proton and Hydride Transfers: Order and the Roles of Asp27 and Tyr100. Proc Nat, Acad Sci USA. 2014;111:18231–18236. doi: 10.1073/pnas.1415940111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Warshel A. The Catalytic Effect of Dihydrofolate Reductase and Its Mutants Is Determined by Reorganization Energies†. Biochemistry. 2007a;46:6011–6025. doi: 10.1021/bi700201w. [DOI] [PubMed] [Google Scholar]
- Liu H, Warshel A. Origin of the Temperature Dependence of Isotope Effects in Enzymatic Reactions: The Case of Dihydrofolate Reductase. J Phys Chem B. 2007b;111:7852–7861. doi: 10.1021/jp070938f. [DOI] [PubMed] [Google Scholar]
- Loria JP, Berlow RB, Watt ED. Characterization of Enzyme Motions by Solution NMR Relaxation Dispersion. Acc Chem Res. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- Loveridge EJ, Behiry EM, Guo J, Allemann RK. Evidence that a ‘dynamic knockout’ in Escherichia coli dihydrofolate reductase does not affect the chemical step of catalysis. Nat Chem. 2012;4:292–297. doi: 10.1038/nchem.1296. [DOI] [PubMed] [Google Scholar]
- Luk LYP, Javier Ruiz-Pernía J, Dawson WM, Roca M, Loveridge EJ, Glowacki DR, Allemann RK, et al. Unraveling the role of protein dynamics in dihydrofolate reductase catalysis. Proc Natl Acad Sci U S A. 2013;110:16344–16349. doi: 10.1073/pnas.1312437110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus RA. H and Other Transfers in Enzymes and in Solution: Theory and Computations, a Unified View. 2. Applications to Experiment and Computations. J Phys Chem B. 2007;111:6643–6654. doi: 10.1021/jp071589s. [DOI] [PubMed] [Google Scholar]
- Mauldin RV, Sapienza PJ, Petit CM, Lee AL. Structure and Dynamics of the G121V Dihydrofolate Reductase Mutant: Lessons from a Transition-State Inhibitor Complex. PLoS ONE. 2012;7:e33252. doi: 10.1371/journal.pone.0033252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElheny D, Schnell JR, Lansing JC, Dyson HJ, Wright PE. Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. Proc Natl Acad Sci U S A. 2005;102:5032–5037. doi: 10.1073/pnas.0500699102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurry JE, Begley TP. The Organic Chemistry of Biological Pathways. Greenwood Village, CO: Roberts and Company; 2016. Nucleotide Metabolism; p. 333. [Google Scholar]
- Meadows CW, Tsang JE, Klinman JP. Picosecond-Resolved Fluorescence Studies of Substrate and Cofactor-Binding Domain Mutants in a Thermophilic Alcohol Dehydrogenase Uncover an Extended Network of Communication. J Am Chem Soc. 2014;136:14821–14833. doi: 10.1021/ja506667k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel ZD, Klinman JP. Update 1 of: Tunneling and Dynamics in Enzymatic Hydride Transfer. Chem Rev. 2010;110:PR41–PR67. doi: 10.1021/cr1001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrop DB. Enzyme Mechanism from Isotope Effects. Boca Raton, FL: CRC Press; 1991. pp. 181–202. [Google Scholar]
- Ohmae E, Iriyama K, Ichihara S, Gekko K. Nonadditive Effects of Double Mutations at the Flexible Loops, Glycine-67 and Glycine-121, of Escherichia coli Dihydrofolate Reductase on Its Stability and Function. J Biochem. 1998;123:33–41. doi: 10.1093/oxfordjournals.jbchem.a021913. [DOI] [PubMed] [Google Scholar]
- Pauling L. Chemical Achievement and Hope for the Future. Am Sci. 1948;36:51–58. [PubMed] [Google Scholar]
- Pu J, Gao J, Truhlar DG. Multidimensional Tunneling, Recrossing, and the Transmission Coefficient for Enzymatic Reactions. Chem Rev. 2006;106:3140–3169. doi: 10.1021/cr050308e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J, Ma S, Gao J, Truhlar DG. Small temperature dependence of the kinetic isotope effect for the hydride transfer reaction catalyzed by Escherichia coli dihydrofolate reductase. J Phys Chem B. 2005;109:8551–8556. doi: 10.1021/jp051184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J, Ma S, Garcia-Viloca M, Gao J, Truhlar DJ, Kohen A. Nonperfect synchronization of reaction center rehybridization in the transition state of the hydride transfer catalyzed by dihydrofolate reductase. J Am Chem Soc. 2005;127:14879–14886. doi: 10.1021/ja054170t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pudney CR, Hay S, Levy C, Pang J, Sutcliffe MJ, Leys D, Scrutton NS. Evidence to Support the Hypothesis that Promoting Vibrations Enhance the Rate of an Enzyme Catalyzed H-Tunneling Reaction. J Am Chem Soc. 2009;131:17072–17073. doi: 10.1021/ja908469m. [DOI] [PubMed] [Google Scholar]
- Radkiewicz JL, Brooks CL. Protein Dynamics in Enzymatic Catalysis: Exploration of Dihydrofolate Reductase. J Am Chem Soc. 2000;122:255–231. [Google Scholar]
- Rajagopalan PTR, Stefan L, Benkovic SJ. Coupling Interactions of Distal Residues Enhance Dihydrofolate Reductase Catalysis: Mutaional Effects on Hydride Transfer Rates. Biochemistry. 2002;41:12618–12628. doi: 10.1021/bi026369d. [DOI] [PubMed] [Google Scholar]
- Rod TH, Radkiewicz JL, Brooks CL. Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc Natl Acad Sci U S A. 2003;100:6980–6985. doi: 10.1073/pnas.1230801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roston D, Islam Z, Kohen A. Isotope Effects as Probes for Enzyme Catalyzed Hydrogen-Transfer Reactions. Molecules. 2013;18:5543–5567. doi: 10.3390/molecules18055543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roston D, Islam Z, Kohen A. Kinetic Isotope Effects as a Probe of Hydrogen Transfers to and from Common Enzymatic Cofactors. Arch Biochem Biophys. 2014;544:96–104. doi: 10.1016/j.abb.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen A, Yahashiri A, Kohen A. Triple isotopic labeling and kinetic isotope effects: exposing H-transfer steps in enzymatic systems. Biochemistry. 2011;50:6462–6468. doi: 10.1021/bi2003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton GR, Hrovat DA, Borden WT. Calculations of the Effect of Tunneling on the Swain Schaad Exponents (SSEs) for the 1,5-Hydrogen Shift in 5-Methyl-1,3-cyclopentadiene. Can SSEs Be Used to Diagnose the Occurrence of Tunneling? J Am Chem Soc. 2007;129:16115–16118. doi: 10.1021/ja076132a. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Wang L, Markham KA, Rajagopalan PT, Benkovic SJ, Kohen A. Tunneling and coupled motion in the Escherichia coli dihydrofolate reductase catalysis. J Am Chem Soc. 2004;126:4778–4779. doi: 10.1021/ja031683w. [DOI] [PubMed] [Google Scholar]
- Singh P, Abeysinghe T, Kohen A. Linking Protein Motion to Enzyme Catalysis. Molecules. 2015;20:1192. doi: 10.3390/molecules20011192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Francis K, Kohen A. Network of Remote and Local Protein Dynamics in Dihydrofolate Reductase Catalysis. ACS Catalysis. 2015;5:3067–3073. doi: 10.1021/acscatal.5b00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Morris H, Tivanski AV, Kohen A. A calibration curve for immobilized dihydrofolate reductase activity assay. Data in Brief. 2015a;4:19–21. doi: 10.1016/j.dib.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Morris H, Tivanski AV, Kohen A. Determination of concentration and activity of immobilized enzymes. Anal Biochem. 2015b;484:169–172. doi: 10.1016/j.ab.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Sen A, Francis K, Kohen A. Extension and Limits of the Network of Coupled Motions Correlated to Hydride Transfer in Dihydrofolate Reductase. J Am Chem Soc. 2014;136:2575–2582. doi: 10.1021/ja411998h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedarchina Z, Siebrand W. Generalized Swain Schaad relations including tunneling and temperature dependence. Chem Phys Letters. 2005;410:370–376. [Google Scholar]
- Spencer HT, Villafranca JE, Appleman JR. Kinetic scheme for thymidylate synthase from Escherichia coli: determination from measurements of ligand binding, primary and secondary isotope effects, and pre-steady-state catalysis. Biochemistry. 1997;36:4212–4222. doi: 10.1021/bi961794q. [DOI] [PubMed] [Google Scholar]
- Stroud RM, Finer-Moore JS. Conformational Dynamics along an Enzymatic Reaction Pathway: Thymidylate Synthase, “the Movie”. Biochemistry. 2003;42:239–247. doi: 10.1021/bi020598i. [DOI] [PubMed] [Google Scholar]
- Swain CG, Stivers EC, Reuwer JF, Schaad LJ. Use of Hydrogen Isotope Effects to Identify the Attacking Nucleophile in the Enolization of Ketones Catalyzed by Acetic Acid1-3. J Am Chem Soc. 1958;80:5885–5893. [Google Scholar]
- Swanwick RS, Shrimpton PJ, Allemann RK. Pivotal Role of Gly 121 in Dihydrofolate Reductase from Escherichia coli: The Altered Structure of a Mutant Enzyme May Form the Basis of Its Diminished Catalytic Performance†. Biochemistry. 2004;43:4119–4127. doi: 10.1021/bi036164k. [DOI] [PubMed] [Google Scholar]
- Thorpe IF, Brooks CL. Barriers to Hydride Transfer in Wild Type and Mutant Dihydrofolate Reductase from E. coli. J Phys Chem B. 2003;107:14042–14051. [Google Scholar]
- Thorpe IF, Brooks CL. The coupling of structural fluctuations to hydride transfer in dihydrofolate reductase. Proteins: Structure, Function, and Bioinformatics. 2004;57:444–457. doi: 10.1002/prot.20219. [DOI] [PubMed] [Google Scholar]
- Wang L, Goodey NM, Benkovic SJ, Kohen A. Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase. Proc Natl Acad Sci U S A. 2006a;103:15753–15758. doi: 10.1073/pnas.0606976103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Goodey NM, Benkovic SJ, Kohen A. The role of enzyme dynamics and tunnelling in catalysing hydride transfer: studies of distal mutants of dihydrofolate reductase. Philos Trans R Soc B. 2006b;361:1307–1315. doi: 10.1098/rstb.2006.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tharp S, Selzer T, Benkovic SJ, Kohen A. Effects of a distal mutation on active site chemistry. Biochemistry. 2006;45:1383–1392. doi: 10.1021/bi0518242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Abeysinghe T, Finer-Moore JS, Stroud RM, Kohen A. A Remote Mutation Affects the Hydride Transfer by Disrupting Concerted Protein Motions in Thymidylate Synthase. J Am Chem Soc. 2012;134:17722–17730. doi: 10.1021/ja307859m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Antoniou D, Schwartz SD, Schramm VL. Hydride Transfer in DHFR by Transition Path Sampling, Kinetic Isotope Effects, and Heavy Enzyme Studies. Biochemistry. 2016;55:157–166. doi: 10.1021/acs.biochem.5b01241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ferrer S, Moliner V, Kohen A. QM/MM calculations suggest a novel intermediate following the proton abstraction catalyzed by thymidylate synthase. Biochemistry. 2013;52:2348–2358. doi: 10.1021/bi400267q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kohen A. Thymidylate Synthase Catalyzed H-Transfers: Two Chapters in One Tale. J Am Chem Soc. 2010;132:9820–9825. doi: 10.1021/ja103010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Sapienza PJ, Abeysinghe T, Luzum C, Lee AL, Finer-Moore JS, Kohen A, et al. Mg2+ Binds to the Surface of Thymidylate Synthase and Affects Hydride Transfer at the Interior Active Site. Journal of the American Chemical Society. 2013;135:7583–7592. doi: 10.1021/ja400761x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Singh P, Czekster CM, Kohen A, Schramm VL. Protein Mass-Modulated Effects in the Catalytic Mechanism of Dihydrofolate Reductase: Beyond Promoting Vibrations. J Am Chem Soc. 2014;136:8333–8341. doi: 10.1021/ja501936d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wataya Y, Hayatsu H. Cysteine-catalyzed hydrogen isotope exchange at the 5 position of uridylic acid. J Am Chem Soc. 1972;94:8927–8928. doi: 10.1021/ja00780a059. [DOI] [PubMed] [Google Scholar]
- Watney JB, Agarwal PK, Hammes-Schiffer S. Effect of Mutation on Enzyme Motion in Dihydrofolate Reductase. J Am Chem Soc. 2003;125:3745–3750. doi: 10.1021/ja028487u. [DOI] [PubMed] [Google Scholar]
- Wilson PM, Danenberg PV, Johnston PG, Lenz HJ, Ladner RD. Standing the test of time: targeting thymidylate biosynthesis in cancer therapy. Nat Rev Clin Oncol. 2014;11:282–298. doi: 10.1038/nrclinonc.2014.51. [DOI] [PubMed] [Google Scholar]
- Wong KF, Selzer T, Benkovic SJ, Hammes-Schiffer S. Impact of distal mutations on the network of coupled motions correlated to hydride transfer in dihydrofolate reductase. Proc Natl Acad Sci U S A. 2005;102:6807–6812. doi: 10.1073/pnas.0408343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KF, Watney JB, Hammes-Schiffer S. Analysis of Electrostatics and Correlated Motions for Hydride Transfer in Dihydrofolate Reductase. J Phys Chem B. 2004;108:12231–12241. [Google Scholar]
- Yahashiri A, Rubach JK, Plapp BV. Effects of Cavities at the Nicotinamide Binding Site of Liver Alcohol Dehydrogenase on Structure, Dynamics and Catalysis. Biochemistry. 2014;53:881–894. doi: 10.1021/bi401583f. [DOI] [PMC free article] [PubMed] [Google Scholar]