

Abstract
Summary: Commonly used multiplicity adjustments fail to control the error rate for reported findings in many expression quantitative trait loci (eQTL) studies. TreeQTL implements a hierarchical multiple testing procedure which allows control of appropriate error rates defined relative to a grouping of the eQTL hypotheses.
Availability and Implementation: The R package TreeQTL is available for download at http://bioinformatics.org/treeqtl.
Contact: sabatti@stanford.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The goal of eQTL analysis is to gain insight into the genetic regulation of gene expression. Typically, this is carried out by testing a vast collection of hypotheses Hvg probing association between the genotype at variant v and the measured expression for gene g, where , M is on the order of hundreds of thousands, and G of tens of thousands. Given the large number of hypotheses tested, the need to adjust for multiplicity is universally recognized and the false discovery rate (FDR) (Benjamini and Hochberg, 1995) is typically adopted as the target global error rate.
In an effort to improve interpretability, reporting of results is typically organized along more general findings such as the discovery of genes subject to local regulation (eGenes) (Göring et al., 2007) or regulatory SNPs (eSNPs) (Nica et al., 2010). The adopted strategy for multiplicity adjustment needs then to offer guarantees on these reported ‘global’ discoveries. For example, imagine testing the Hvg hypotheses using the BH rule (Benjamini and Hochberg, 1995) and defining as an eSNP those variants v for which Hvg is rejected for at least some gene g. While this would control the FDR among the Hvg rejections, it would not control any measure of global error for the discovery of eSNPs, as shown by the simulations in Peterson et al. (2016).
Researchers in the eQTL field have recognized this challenge, and have additionally noted that since local regulation is more common than distal (Albert and Kruglyak, 2015), hypotheses probing these two mechanisms should be tested separately. However, there is no single standard in the literature for error rates targeted or error-controlling strategies: for example, one finds the notion of per-gene error rates (Nica et al., 2010) or the application of Bonferroni across genes (Zeller et al., 2010) in local regulation, while for distal effects significance cut-offs vary from 5 × 10–8 (Grundberg et al., 2012) to 5.78 × 10–12 (Zeller et al., 2010). This makes comparison across studies and replicability challenging.
2 Approach
To overcome the confusion generated by the plurality of approaches and to provide guarantees relative to the discoveries reported, it is useful to recognize the structure among the hypotheses tested in an eQTL study and to introduce some terminology. We distinguish between hypotheses testing local (when the distance between variant v and gene g is less than a threshold) and distal regulation, indicating them with Lvg and Dvg, respectively.
Further, we recognize that we might be interested mainly in identifying which genes appear to have local (distal) regulation prior to obtaining a detailed list of the variants involved in this regulation; or we might want to pinpoint SNPs that appear to have local (distal) effects on multiple genes, even without committing to a comprehensive list of these genes. We use the term ‘local eGene’ to signify a gene whose expression is influenced by some local DNA variants and ‘local eSNP’ to designate a specific SNP associated to variability in expression for some local genes. With ‘local eAssociation’ we signify the local association between one specific SNP and one specific gene. For distal regulation, ‘distal eGene’, ‘distal eSNP’ and ‘distal eAssociation’ are similarly defined. These terms can be mapped back to the original collection of hypotheses noting, for example, that discovering a distal eSNP is equivalent to rejecting the intersection hypothesis , that is, the null stating that SNP v has no effect on any distal gene g.
To control global error rates defined in terms of the reported discoveries (local eSNPs, local eAssociations, etc.), we have implemented in TreeQTL a multi-resolution approach based on results in Benjamini and Bogomolov (2014) whose practical effectiveness in GWAS has been described in Peterson et al. (2016). Furthermore, TreeQTL has the potential to increase power: by focusing on the promising SNPs with possibly higher proportions of true eAssociations, one capitalizes on the adaptivity of FDR.
3 Methods
TreeQTL is a hierarchical testing procedure that distinguishes two levels of discoveries within each class of local or distal regulation. In Level 1, users can specify their primary interest as the identification of either eGenes or eSNPs. Given this choice, all the pair-wise association hypotheses are given a position in a tree similar to that shown in Figure 1: each eGene or eSNP hypothesis indexes the collection of simple hypotheses by whose intersection it is defined. Level 1 hypotheses are tested controlling for global errors within the two regulation classes. The more granular eAssociation hypotheses in Level 2 are tested only when they correspond to a global hypothesis rejected in Level 1.
Fig. 1.
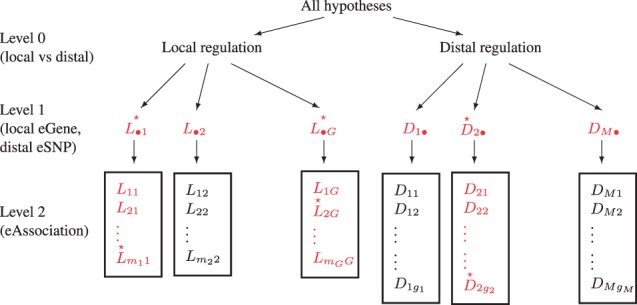
Organization of eQTL hypotheses in TreeQTL. Local regulation hypotheses have been grouped by gene and distal regulation hypotheses are grouped by SNP, so that Level 1 rejections will result in discoveries of local eGenes and distal eSNPs. Tested hypotheses are colored in red, and rejected hypotheses indicated with a star
TreeQTL takes as input the P-values for each hypothesis in Level 2: these may be computed via Matrix eQTL (Shabalin, 2012) and their validity is of crucial importance. Regardless of how the P-values were obtained, the input file for TreeQTL should follow the format used by Matrix eQTL, i.e. a tab-delimited file with columns SNP, gene, beta, t-stat, p-value and FDR (note that the fields beta, t-stat and FDR can be empty). The P-values for the Level 1 hypotheses are computed using Simes’ rule (Simes, 1986) on the families they index. This summary of the evidence for the global null hypotheses is relatively robust to dependence (Benjamini and Heller, 2008). Users can, however, input alternative P-values for Level 1, such as those obtained via permutation.
Testing proceeds from Level 1, where three options are available: controlling FDR at level q1 using BH or BY (Benjamini and Yekutieli, 2001) or controlling the family-wise error rate via Bonferroni.
In Level 2, significance is established using a modified BH within each set of eAssociation hypotheses corresponding to an eSNP or eGene identified at Level 1, using a more stringent target FDR to account for selection. The expected average proportion of false discoveries across the selected Level 2 families is controlled to the user-specified target level q2. (See Supplementary Material).
4 Example application
Purely to demonstrate feasibility, we applied TreeQTL to whole-blood data from the pilot phase of the GTEx project (Ardlie et al., 2015): genotype data at 6 820 472 SNPs and expression levels for 30 115 genes are available across 156 subjects. Local associations correspond to SNP-gene pairs where the SNP is within 1 Mb of the transcription start site (TSS) of the gene; all other SNP-gene associations are considered distal. This definition results in approximately 142 million local tests (reflecting an average of 21 genes in the local region for each SNP) and 205 billion distal tests. Following the steps in Ardlie et al. (2015), P-values were obtained by applying Matrix eQTL to normalized gene expression, adjusting for both known and unknown technical covariates by the inclusion of gender, 3 genotype principal components and 15 PEER factors. In applying TreeQTL, we identified eSNPs as the discovery of interest in Level 1, choose the BH procedure in level 1 and set q1 = q2 = 0.01. This led to the discovery of 136 609 local eSNPs (with 229 821 local eAssociations) and 164 860 distal eSNPs (with 216 933 distal eAssociations). This analysis required around 2 h in R version 3.1.0 to complete. Note that because of linkage disequilibrium, the number of eSNPs (and eAssociation) discoveries is likely much larger than the number of true causal variants. A discussion of this point as well as an indicative comparison with the analysis of these data published in Ardlie et al. (2015) is included in the Supplementary Material.
5 Conclusion
By analyzing local and distal regulation separately, TreeQTL has less stringent cut-offs for tests probing local effects, where it is expected that a larger number of hypotheses will be non-null. By grouping hypotheses relative to the same SNP or the same gene, TreeQTL capitalizes on the inherent heterogeneity of the problem. For example, while few SNPs will act as distal regulators of expression and might influence multiple genes, the vast majority of SNPs will not have such an effect: testing all the eAssociation hypotheses relative to one SNP together, separately from those concerning other loci, increases the discovery of eSNPs with many associations, which may play a true regulatory role, and reduces the discovery of SNPs with few associations, which are more likely to correspond to false positives. Finally, the hierarchical structure of TreeQTL assures control of the FDR for eSNP and eGene discoveries. While the current version of TreeQTL implements methodology relative to studies involving only one tissue, future releases will incorporate approaches for the more complex structure of multi-tissue investigations.
Supplementary Material
Acknowledgements
The pilot release of the GTEx data (accession no. phs000424.v3.p1) is available through dbGaP (http://www.ncbi.nlm.nih.gov/gap).
Funding
This work was supported by the National Institutes of Health [MH101782 to C.P. and C.S., HG006695 to C.S. and Y.B.]; and the Israel Science Foundation [1112/14 to M.B.].
Conflict of Interest: none declared.
References
- Albert F., Kruglyak L. (2015) The role of regulatory variation in complex traits and disease. Nat. Rev. Genet., 16, 197–212. [DOI] [PubMed] [Google Scholar]
- Ardlie K.G. et al. (2015) The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science, 8, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y., Bogomolov M. (2014) Selective inference on multiple families of hypotheses. JRSS B, 76, 297–318. [Google Scholar]
- Benjamini Y., Heller R. (2008) Screening for partial conjunction hypotheses. Biometrics, 64, 1215–1222. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. JRRS B, 57, 289–300. [Google Scholar]
- Benjamini Y., Yekutieli D. (2001) The control of the false discovery rate in multiple testing under dependency. Ann. Statist., 29, 1165–1188. [Google Scholar]
- Göring H.H. et al. (2007) Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat. Genet., 39, 1208–1216. [DOI] [PubMed] [Google Scholar]
- Grundberg E. et al. (2012) Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat. Genet., 44, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica A.C. et al. (2010) Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet., 6, e1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson C.B. et al. (2016) Many phenotypes without many false discoveries: error controlling strategies for multi-trait association studies. Genet. Epidemiol., 40, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin A. (2012) Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics, 28, 1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simes R. (1986) An improved Bonferroni procedure for multiple tests of significance. Biometrika, 73, 751–754. [Google Scholar]
- Zeller T. et al. (2010) Genetics and beyond – the transcriptome of human monocytes and disease susceptibility. PloS One, 5, e10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.