

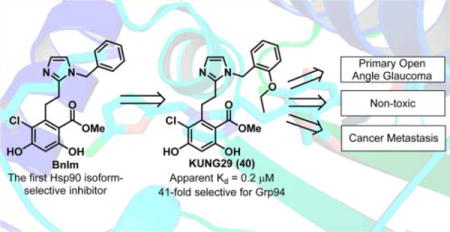
Abstract
Glucose regulated protein 94 (Grp94) is the endoplasmic reticulum resident of the heat shock protein 90 kDa (Hsp90) family of molecular chaperones. Grp94 associates with many proteins involved in cell adhesion and signaling, including integrins, Toll-like receptors, immunoglobulins, and mutant myocilin. Grp94 has been implicated as a target for several therapeutic areas including glaucoma, cancer metastasis, and multiple myeloma. While 85% identical to other Hsp90 isoforms, the N-terminal ATP-binding site of Grp94 possesses a unique hydrophobic pocket that was used to design isoform-selective inhibitors. Incorporation of a cis-amide bioisostere into the radamide scaffold led to development of the original Grp94-selective inhibitor, BnIm. Structure–activity relationship studies have now been performed on the aryl side chain of BnIm, which resulted in improved analogues that exhibit better potency and selectivity for Grp94. These analogues also manifest superior antimigratory activity in a metastasis model as well as enhanced mutant myocilin degradation in a glaucoma model compared to BnIm.
Graphical abstract
INTRODUCTION
Molecular chaperones are responsible for the conformational maturation of nascent polypeptides into their bioactive conformations.1 One family of chaperones is the 90 kDa heat shock proteins (Hsp90), which has emerged as a promising therapeutic target for the treatment of many diseases.2–7 The Hsp90 family is responsible for the maturation of more than 200 client proteins, many of which belong to signaling pathways that are commonly hijacked in cancer. In fact, Hsp90 clients are directly associated with all ten hallmarks of cancer and thus represents a unique opportunity to simultaneously target multiple oncogenic pathways.8,9 Hsp90 exists as a homodimer within cells and consists of three domains: an N-terminal domain, a middle domain, and a C-terminal dimerization domain. The N-terminal domain contains an ATP-binding pocket in which ATP hydrolysis provides the necessary energy for client protein maturation. The middle domain is responsible for client protein binding and interactions with cochaperones and partner proteins. The C-terminal domain contains a dimerization motif and is responsible for modulating client protein release.
The majority of Hsp90 research has focused on the development of N-terminal inhibitors, which compete with ATP within the binding site.10–12 N-terminal inhibitors have entered clinical studies for the treatment of various cancers.13–15 However, some concerns have been raised that must be addressed before new Hsp90 inhibitors are developed. A common factor among all clinical candidates is that they exhibit pan-Hsp90 inhibition, meaning they manifest similar affinities against all four Hsp90 isoforms: Hsp90α and Hsp90β are found in the cytosol, tumor necrosis factor receptor-associated protein 1 (Trap1) is localized to the mitochondria, and glucose regulated protein 94 (Grp94) resides in the endoplasmic reticulum (ER).
Many clients of the Hsp90-dependent protein folding process have been identified, however, clients with specific dependency on each isoform remain underinvestigated although some isoform-dependent substrates have been determined. For instance, maturation of the hERG channel and its trafficking to the cell surface was found to be solely dependent upon the Hsp90α isoform and suggests that inhibition of Hsp90α may contribute to some of the cardiotoxicity observed in clinical trials.16 It is also likely that other isoform-dependent client proteins contribute to other toxicities, which highlights the need to develop new strategies for Hsp90 inhibition. An alternative to pan-inhibition is the development of isoform-selective inhibitors. Such molecules would provide an opportunity to elucidate isoform-dependent client proteins associated with each isoform while simultaneously reducing/derisking potential liabilities associated with pan-inhibition. Unfortunately, the development of isoform-selective inhibitors is challenging due to the fact that 85% identity is shared within the N-terminal ATP-binding site of all four Hsp90 isoforms. In fact, Hsp90α and Hsp90β are 95% identical within their N-terminal ATP-binding pocket, while Grp94 is least similar to only 85% identity.17–19
Grp94 is responsible for the maturation of proteins associated with cell-to-cell signaling and cell adhesion. Client proteins dependent upon Grp94 include many integrins (α-2, α-4, α-L, and β-4), Toll-like receptors (TLR1, TLR2, TLR4, and TLR9), insulin-like growth factor-I and -II, immunoglobulins, and mutant myocilin.5,20–22 Grp94 is essential only during embryonic development and therefore appears to represent a nontoxic drug target. However, overexpression of Grp94 is associated with tumor aggressiveness and poor clinical prognosis for cancer patients.23 In fact, Grp94 is responsible for the maturation and trafficking of proteins required for metastasis and cell migration and thus represents a nontoxic target for the development of antimetastatic agents. More recently, mutant myocilin has also been identified as a Grp94-dependent substrate and studies have shown that inhibition of Grp94 leads to the disaggregation of mutant myocilin and consequently represents a novel target for the treatment of primary open-angle glaucoma (POAG).4,21,24 While Grp94 may represent an ideal target for cell migration and glaucoma, Grp94 has also been shown to be essential in multiple myeloma, which appears to possess increased ER stress. Inhibition of Grp94 has been shown to induce cell death in multiple myeloma, as disruption of interactions between Grp94 and LRP6 results in reduced cell-trafficking of LRP6 to the cell surface.3,5,25 LRP6 is a coreceptor of Frizzled in the Wnt pathway and reduction of these interactions leads to caspase 9 activation and apoptosis. Grp94 has also been shown to interact with Her2 and, consequently, disruption of these interactions leads to Her2 degradation via the lysosome and may represent an alternative mechanism to treat Her2 overexpressing breast cancers.26,27
Grp94 is least similar, in its N-terminal ATP-binding pocket, to other Hsp90 isoforms due to a five amino acid insertion (QEDGQ) into the primary sequence.22 This insertion results in the creation of a hydrophobic, secondary binding region within the N-terminal ATP-binding site of Grp94. As such, this unique pocket provides an opportunity to develop Grp94-selective inhibitors.28,29 Prior studies via a high throughput screening method identified NECA (Figure 1) as a Grp94-selective inhibitor that bound to this secondary pocket.22,30 Recently, a high throughput screen identified a series of Grp94-selective inhibitors based on the purine scaffold and subsequent optimization led to the development of 2.26 Co-crystallization of 3 (Figure 1) with Grp94 showed this inhibitor to induce a conformational change within Grp94 that revealed an extended binding pocket distinct from the secondary pocket observed with NECA and RDA (4).26,27,30–32 Co-crystallization of the resorcinol containing pan-Hsp90 inhibitor, radamide (RDA, Figure 1) with canine Grp94 (cGrp94), revealed that the amide bond existed in both the cis- and trans-amide conformations.33 The cis-amide conformation projected the quinone moiety into the secondary binding pocket, while the trans-amide did not. On the basis of the RDA co-crystal structure, it was clear that a cis-amide conformation was required for selective Grp94 inhibition. The incorporation of a cis-amide bioisostere (e.g., imidazole) into the RDA scaffold predisposes the aryl ring into the unique binding pocket found in Grp94 and ultimately led to the discovery of BnIm (Figure 1).28 BnIm demonstrates Grp94 selectivity in vitro as determined by inhibition of IGF-II secretion and the trafficking of Toll-like receptors at concentrations that did not result in cytosolic Hsp90-dependent client protein degradation. BnIm provided a lead compound for Grp94-selective inhibition, however, analysis of the unique binding pocket of Grp94 revealed further interactions could be realized improving the selectivity and affinity of this scaffold for Grp94, reported herein.
Figure 1.
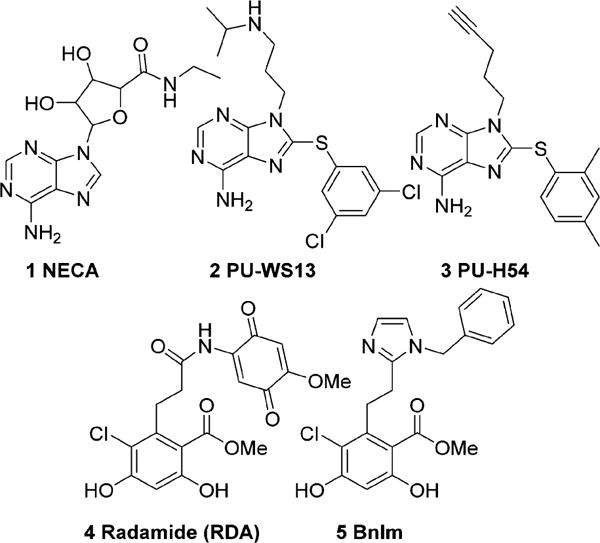
Purine-based Grp94-selective inhibitors (1, 2, and 3). Resorcinol-based pan-Hsp90 (4) and Grp94-selective (5) inhibitors.
GRP94-SELECTIVE INHIBITOR DESIGN, SYNTHESIS, AND BIOCHEMICAL SCREENING
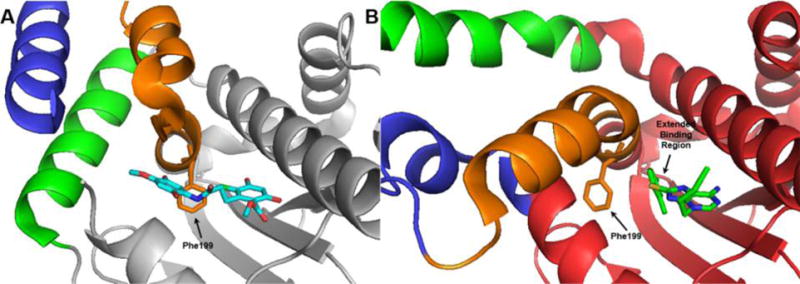
The co-crystal structure of RDA with cGrp94 revealed the cis-amide to project the aryl side chain into the secondary binding pocket, which is surrounded by hydrophobic amino acids (Val82, Ile166, Ala167, Phe195, Val197, Phe199, Tyr200, and Trp223), suggesting that affinity may be increased through additional hydrophobic interactions (Figure 2a).33 In contrast, Hsp90β contains the backbone carbonyl of Asn92 and the ɛ-NH2 of Lys98, which blocks access to these hydrophobic residues and can be used as a paradigm to predict Grp94-selective inhibition (Figure 2b). Docking of BnIm into the RDA co-crystal structure with Grp94 (PDB 2GFD) suggested that substitutions on the aryl side chain would provide additional interactions with the secondary pocket of Grp94, allowing for optimization of the aryl side chain for Grp94-selective inhibition. Nonpolar substitutions at the 4-position were proposed to extend further into the hydrophobic pocket and to produce increased affinity and selectivity. However, larger substitutions at this position would likely result in steric clash with Val82 and exhibit decreased affinity. Because of the close proximity of Tyr200 to the 3-position of the aryl side chain, substitutions at this position appeared detrimental to Grp94 affinity. Substitutions at the 2-position were likely to increase affinity through hydrophobic interactions with the secondary pocket. Furthermore, these substitutions have the potential to produce the conformational shift observed with the purine-based Grp94-selective inhibitor, 3 (see Figure 4), and project into the extended binding pocket.26 On the basis of these observations, the aryl side chain of BnIm was investigated to probe the unique pocket present in Grp94 and to elucidate structure–activity relationships for optimal affinity and selectivity.
Figure 2.
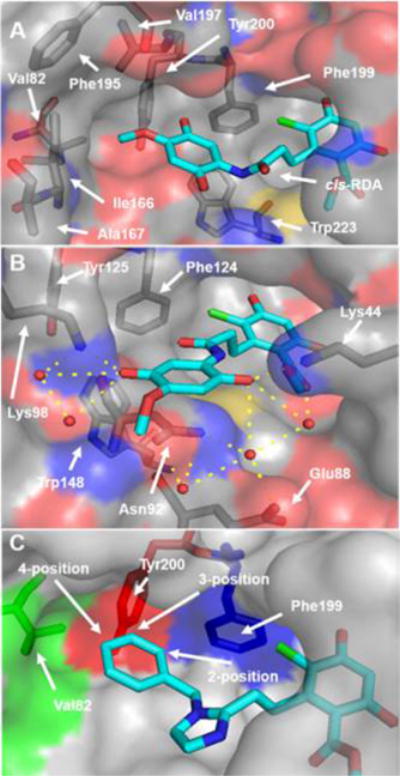
Co-crystal structures of RDA with Hsp90 isoforms. cGrp94 (A, PDB 2GFD) highlights the hydrophobic and π-rich nature of the unique secondary binding pocket of Grp94. Only cis-RDA shown for clarity. yHsp82 (B, PDB 2FXS) highlights the hydrogen bonding network present and restricted access to the aromatic residues. Residues are numbered as in their respective proteins. BnIm docked in to Gpr94 (C) highlighting substitution positions on the aryl side chain.
Figure 4.
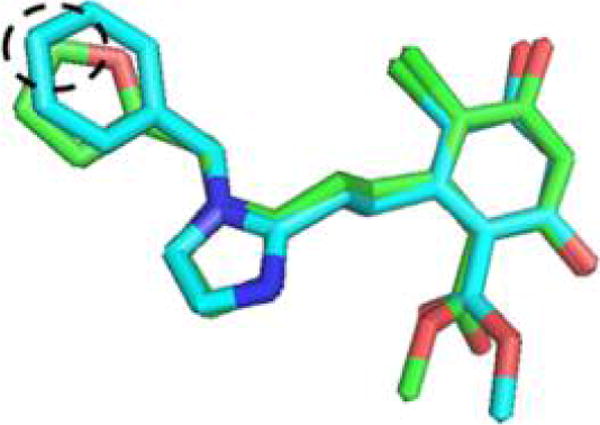
Overlay of the minimized structures of BnIm (cyan) and 46 (green) highlighting the overlapping 4- and 5-positions, respectively.
Because the cis-amide bioisostere, imidazole, could be prepared via a multicomponent cyclization reaction with an aromatic aldehyde, access to the desired analogues was achieved by varying the substitutions on the aryl amine component, which were either commercially available or readily accessible (Scheme 1). 4-Ethynylbenzylamine (6a) was synthesized from 4-iodobenzylamine following literature precedent.34 Aromatic nitriles were reduced using lithium aluminum hydride to produce the requisite amines (6b–e). The substituted benzoic acid was reduced to the corresponding benzyl alcohol, 8, using lithium aluminum hydride, and then converted to the corresponding azide, which was subsequently reduced to the desired amine, 6f, via Staudinger reduction. The basic amines were then cyclized with aldehyde 9 in the presence of ammonium bicarbonate and glyoxal, followed by desilylation with tetrabutylammonium fluoride to provide the desired analogues, 10–64 (Scheme 2).28,35,36
Scheme 1.
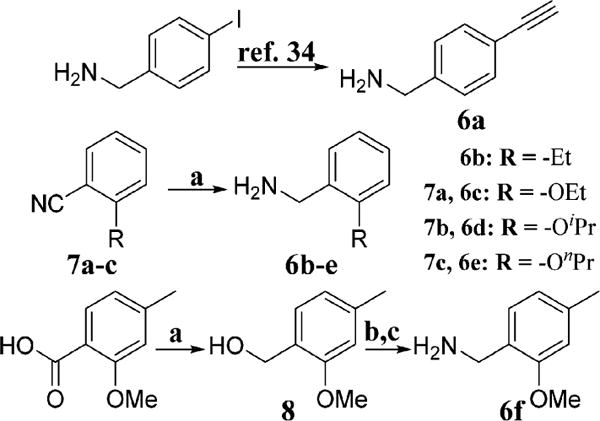
aConditions: (a) LiAlH4, THF, 0–25 °C, 12 h; (b) DBU, DPPA, toluene, 0–25 °C, 12 h; (c) PPh3, THF:H2O (10:1), 25 °C, 12 h.
Scheme 2.
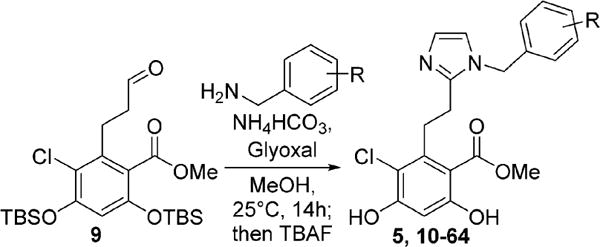
Once in hand, the analogues were screened via a competitive binding fluorescence polarization assay using Grp94 or Hsp90α and FITC-labeled geldanamycin (FITC-GDA). Geldanamcyin is a potent, natural product N-terminal pan-Hsp90 inhibitor that competes with Grp94 inhibitors for the N-terminal ATP-binding site.10 Analogues were originally screened at 25 μM to determine the percent of FITC-GDA (tracer) bound compared to vehicle control (0% tracer displaced, Table 1).
Table 1.
Apparent Kd Values with Grp94 and Hsp90α
| ||||||
|---|---|---|---|---|---|---|
| Entry | R | % Tracer Displaced Grp94a | % Tracer Displaced Hsp90αa | Apparent Kd Grp94 (μM) | Apparent Kd Hsp90α (μM) | Fold Selective for Grp94 |
| Bnlm |
|
98.9 ± 0.2 | 72.9 ± l.2 | 1.14 ± 0.l | 13.1 ± 1.1 | 12 |
| 10 |
|
90.3 ± 0.5 | 40.3 ± 3.2 | 3.2 ± 0.5 | 23.4 ± 1.2 | 7 |
| 11 |
|
99.1 ± 0.5 | 52.1 ± 2.1 | 0.81 ± 0.99 | 12.1 ± 1.6 | 15 |
| 12 |
|
99.0 ± 04 | 57.2 ± 3.7 | 0.96 ± 0.1 | 12.5 ± 1.8 | 13 |
| 13 |
|
43.6 ± 4.4 | 43.5 ± 2.5 | n.d. | n.d. | n.d. |
| 14 |
|
98.9 ± 0.8 | 33.5 ± 44 | 0.73 ± 0.1 | 25.2 ± 2.1 | 34 |
| 15 |
|
24.2 ± 8.7 | 17.3 ± 5.6 | n.d. | n.d. | n.d. |
| 16 |
|
23.9 ± 7.7 | 15.9 ± 6.3 | n.d. | n.d. | n.d. |
| 17 |
|
75.8 ± 4.1 | 49.7 ± 3.6 | 4.7 ± 0.7 | 18.2 ± 1.1 | 4 |
| 18 |
|
89.3 ± 3.2 | 43.9 ± 2.3 | 1.4 ± 0.1 | 15.8 ± 1.0 | 11 |
| 19 |
|
52.1 ± 5.5 | 10.0 ± 12.1 | n.d. | n.d. | n.d |
| 20 |
|
18.0 ± 8.5 | 12.8 ± 61 | n.d. | n.d. | n.d. |
| 21 |
|
78.7 ± 4.7 | 30.1 ± 5.3 | 6.9 ± 1.3 | >150 | >22 |
| 22 |
|
13.7 ± 7.2 | 17.1 ± 9.2 | n.d. | n.d. | n.d. |
| 23 |
|
29.5 ± 6.4 | 9.6 ± 10.3 | n.d | n.d. | n.d. |
| 24 |
|
34.0 ± 5.7 | 11.1 ± 9.7 | n.d. | n.d. | n.d. |
| 25 |
|
64.2 ± 3.9 | 35.5 ± 4.7 | n.d | n.d. | n.d. |
| 26 |
|
70.9 ± 4.6 | 57.0 ± 5.6 | 1.53 ± 0.1 | 9.63 ± 1.4 | 6 |
| 27 |
|
56.0 ± 7.1 | 25.8 ± 62 | n.d. | n.d. | n.d. |
| 28 |
|
35.6 ± 5.1 | 25.3 ± 6.7 | n.d. | n.d. | n.d. |
| 29 |
|
16.5 ± 8.5 | 14.9 ± 9.4 | n.d. | n.d. | n.d. |
| 30 |
|
27.0 ± 7.2 | 32.0 ± 6.4 | n.d. | n.d. | n.d. |
| 31 |
|
62.4 ± 4.0 | 39.0 ± 6.7 | n.d. | n.d. | n.d. |
| 32 |
|
67.3 ± 4.3 | 60.9 ± 4.5 | n.d. | n.d. | n.d. |
| 33 |
|
89.2 ± 3.7 | 59.8 ± 7.5 | 1.36 ± 0.1 | 11.4 ± 1.2 | 8 |
| 34 |
|
86.0 ± 4.2 | 60.4 ± 2.3 | 1.39 ± 0.1 | 8.47 ± 0.9 | 6 |
| 35 |
|
84.2 ± 2.1 | 45.5 ± 5.2 | 1.60 ± 0.3 | 21.l ± 3.7 | 13 |
| 36 |
|
79.9 ± 2.7 | 38.8 ± 6.6 | 3.0 ± 0.8 | 68.1 ± 8.7 | 23 |
| 37 |
|
30.8 ± 8.1 | 20.9 ± 8.3 | n.d. | n d. | n.d. |
| 38 |
|
93.5 ± 1.3 | 36.6 ± 5.8 | 1.3 ± 0.2 | 53.9 ± 7.6 | 41 |
| 39 |
|
99.5 ± 0.1 | 39.8 ± 7.3 | 0.81 ± 0.08 | 38.8 ± 5.3 | 48 |
| 40 |
|
99.6 ± 0.1 | 57.3 ± 5.8 | 0.20 ± 0.08 | 8.1 ± 1.8 | 41 |
| 41 |
|
99.7 ± 0.1 | 99.7 ± 0.22 | 0.09 ± 0.01 | 0.24 ± 0.03 | 3 |
| 42 |
|
99.6 ± 0.1 | 93.2 ± 1.2 | 0.25 ± 0.05 | 2.3 ± 0.2 | 9 |
| 43 |
|
82.6 ± 3.2 | 54.8 ± 3.6 | 1.53 ± 0.23 | 19.3 ± 2.8 | 13 |
| 44 |
|
94.9 ± 1.6 | 31.0 ± 6.7 | 2.35 ± 0.31 | 72.5 ± 8.4 | 30 |
% Tracer bound determined when incubated with 25 μM of inhibitors. Data are the average of at least two experiments ± SEM. n.d. = not determined.
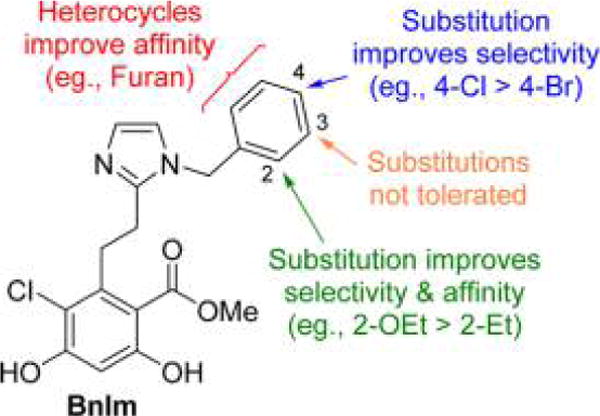
The 25 μM screen provided insight into the structural requirements for binding of the aryl side chain to the Grp94 secondary pocket. Substitutions at the 4-position typically maintained similar affinity as BnIm, but provided increased selectivity in several cases. As hypothesized, polar substitutions (17 and 21) manifested lower affinity due to the hydrophobic nature of the secondary binding pocket (Figure 2). Incorporation of a bicyclic system was not tolerated as the naphthyl (23 and 24) and quinoline (25) derivatives manifested decreased affinity for both Grp94 and Hsp90α. Substitutions at the 3-position were not beneficial, as all of the analogues resulted in decreased affinity for both isoforms compared to BnIm. Alternatively, substitutions at the 2-position typically increased Grp94 affinity and improved selectivity versus Hsp90α.
Analogues that displaced ≥70% of the tracer when incubated with Grp94 at 25 μM were subsequently evaluated to determine their apparent Kd against Grp94 and Hsp90α. BnIm manifests an apparent Kd of 1.1 μM for Grp94 and 13.1 μM for Hsp90α. Substitutions at the 4-position were designed to extend further into the hydrophobic pocket present in Grp94 and to increase selectivity over other isoforms. Modest increases in selectivity were observed with these compounds (11, 12, and 18), while increases in affinity were observed with analogues containing halogen substitutions. Incorporation of a larger methyl group (14) resulted in a substantial increase in selectivity for Grp94, as well as increased affinity. Extended alkyl chains (15 and 16) were too large for accommodation into the secondary pocket of Grp94 and resulted in decreased affinity.
As observed in the 25 μM screen, substitutions at the 3-position did not manifest improved affinity nor selectivity for Grp94 compared to BnIm (26–32). Alternatively, substitutions at the 2-position provided additional insights into the modes of binding for these analogues. Small substitutions at the 2-position were tolerated, however, such substitutions did not increase selectivity or affinity. Larger substitutions produced increased selectivity for Grp94 versus other Hsp90 isoforms, as 38, 39, and 40 manifested >40-fold selectivity for Grp94 over Hsp90α, which is likely due to induction of a conformational shift in the tertiary structure of Grp94. Patel and colleagues previously reported the co-crystallization of 3 with Grp94 (PDB 3O2F), in which a conformational shift was observed that resulted in an extended binding region within the Grp94 ATP-binding site.26,31 This pocket is distinct from the secondary pocket observed with NECA and RDA (Figure 3), as this region opens due to the migration of Phe199, which allows additional access to the binding pocket as observed with 3. Substitutions at the 2-position may also induce a similar conformational change in Grp94, which allows access to this region and increases selectivity for Grp94. In the case of 40 (Grp94 apparent Kd = 0.2 μM; 41-fold selective), the increased selectivity was accompanied by a ~6-fold increase in affinity for Grp94 compared to BnIm. Extension or branching of the alkyl ether (41 and 42) improved affinity for Grp94, although a loss in selectivity was observed. Larger alkyl ethers were not accommodated within the secondary binding pocket of Grp94 and likely push the benzyl side chain toward the solvent exposed region to form hydrogen bonds with polar amino acids. Such polar residues are present in both Grp94 and Hsp90 and provides an explanation for the loss of selectivity and an increase in affinity. 44 was synthesized in an attempt to combine beneficial substitutions observed with the 2- and 4-positions, however, affinity was compromised as compared to the parent compounds (14 and 38), suggesting the effects of these substitutions are not additive (Table 1).
Figure 3.
(A) Tertiary structure of RDA bound to Grp94 (PDB 2GFD) showing the open conformation of the N-terminal domain of Grp94. (B) Tertiary structure of 3 bound to Grp94 (PDB 3O2F) showing the conformational shift (blue, green, and orange helices) of the N-terminal domain induced by the inhibitor binding to Grp94 revealing the extended binding pocket.
Because of the high density of aromatic amino acids within the unique secondary binding pocket of Grp94, the incorporation of heterocycles was proposed to improve both π–π interactions and affinity for Grp94.33 The requisite heterocyclic amines (6g–l) were synthesized from the corresponding aldehydes through conversion to the oximes (66a–e) followed by reduction via lithium aluminum hydride (Scheme 3). Chlorination of thiophen-2-ylmethanamine via sulfuryl chloride provided 6m (Scheme 4). Radical bromination of 5-methylisoxazole followed by conversion to the azide and subsequent reduction resulted in 6n. The aromatic carboxylic acid 68 was reduced to the corresponding alcohol using lithium aluminum hydride followed by conversion to the azide and then Staudinger reduction to yield 6o. Deprotonation of 3-chlorothiophene with n-butyllithium followed by the addition of CO2(g) provided a mixture of carboxylate regioisomers which were separable via flash chromatography after reduction to the corresponding alcohols (69b and 69c) with lithium aluminum hydride. These isomers were then converted to the corresponding azides, followed by Staudinger reduction to provide the requisite basic amines, 6p and 6q (Scheme 4). The desired analogues were obtained through the multicomponent reaction detailed in Scheme 2 to produce analogues 45–64.
Scheme 3. Synthesis of Amines from Aromatic Aldehydesa.
aConditions: (a) HNO3, H2SO4, Ac2O, 0 °C, 1 h; (b) 50% H2SO4 (aq), 110 °C, 5 min; (c) conc HCl, 40 °C, 12 h; (d) NH2OH·HCl, NaOAc, MeOH, 25 °C, 8 h; (e) LiAlH4, THF, 0–25 °C, 12 h; (f) potassium vinyltrifluoroborate, Pd(dppf)Cl2, iPr2EtN, toluene, 110 °C, 14 h; (g) H2, 10% Pd/C, EtOH, 25 °C, 6 h.
Scheme 4.
aConditions: (a) SO2Cl2, 15 °C, AcOH:Et2O (9:1), 1 h; (b) NBS, AIBN, CCl4, 80 °C, 4 h; (c) NaN3, MeOH:H2O (10:1), 25 °C, 12 h; (d) PPh3, THF:H2O (10:1), 25 °C, 12 h; (e) LiOH, THF:MeOH:-H2O (9:1:1), 25 °C, 10 h; (f) LDA, MeI, THF, −40 °C, 3 h; (g) LiAlH4, THF, 0–25 °C, 12 h; (h) DBU, DPPA, toluene, 25 °C, 12 h; (i) n-BuLi, CO2(g), THF, −78 °C, 2 h.
As can be seen in Table 2, the incorporation of an electron poor (compared to benzene) pyridine ring (61–64) resulted in decreased selectivity and affinity for Grp94. In contrast, the incorporation of electron rich five-membered heterocycles proved beneficial for Grp94 affinity. Converting the phenyl ring of BnIm to the bioisosteric replacement, thiophene (50), resulted in a significant increase in Grp94 affinity compared to BnIm. The furan and thiophene (46 and 50, respectively) analogues exhibited both increased affinity and selectivity and consequently, substitutions about these rings were explored. The energy minimized structures of BnIm and 46 were overlaid and revealed the 5-position of the heterocycle to align similar to the 4-position of the BnIm benzyl side chain (Figure 4). Therefore, substitutions at the 5-position of both the furan and thiophene rings were sought to achieve increased selectivity and affinity.
Table 2.
Apparent Kd of Heterocyclic Analogues with Grp94 and Hsp90α
| ||||||
|---|---|---|---|---|---|---|
| Entry | R | % Tracer Bound Grp94a | %Tracer Bound Hsp90αa | Apparent Kd Grp94 (μM) | Apparent Kd Hsp90α (μM) | Fold Selective for Grp94 |
| Bnlm |
|
98.9 ± 0.2 | 72.9 ± 1.2 | 1.14 ± 0.1 | 13.1 ± 1.1 | 12 |
| 45 |
|
97.4 ± 0.8 | 78.3 ± 4.7 | 1.50 ± 0.12 | 3.77 ± 0.41 | 3 |
| 46 |
|
99.2 ± 0.1 | 75.0 ± 2.3 | 0.55 ± 0.06 | 5.91 ± 0.88 | 11 |
| 47 |
|
99.1 ± 0.07 | 73.4 ± 2.3 | 0.65 ± 0.10 | 26.6 ± 2.3 | 41 |
| 48 |
|
99.3 ± 0.09 | 91.7 ± 1.2 | 0.44 ± 0.05 | 8.3 ± 1.2 | 19 |
| 49 |
|
99.3 ± 0.1 | 65.1 ± 2.4 | 0.35 ± 0.06 | 9.2 ± 1.6 | 26 |
| 50 |
|
99.4 ± 0.08 | 76.7 ± 2.1 | 0.47 ± 0.07 | 3.92 ± 0.51 | 8 |
| 51 |
|
87.9 ± 1.9 | 64.5 ± 3.8 | 2.14 ± 0.22 | 6.54 ± 0.86 | 3 |
| 52 |
|
99.0 ± 0.2 | 49.8 ± 6.1 | 0.68 ± 0.08 | 5.13 ± 0.42 | 8 |
| 53 |
|
99.0 ± 0.2 | 59.2 ± 3.6 | 0.93 ± 009 | 34.7 ± 2.6 | 37 |
| 54 |
|
99.1 ± 0.1 | 97.6 ± 1.1 | 0.67 ± 0.08 | 1.8 ± 0.17 | 3 |
| 55 |
|
99.1 ± 0.08 | 97.2 ± 1.2 | 0.76 ± 0.07 | 3.16 ± 0.41 | 4 |
| 56 |
|
99.3 ± 0.06 | 99.1 ± 0.08 | 0.40 ± 0.02 | 0.81 ± 0.07 | 2 |
| 57 |
|
91.7 ± 2.1 | 62.5 ± 4.0 | 2.5 ± 0.23 | 16.0 ± 1.8 | 6 |
| 58 |
|
99.1 ± 0.2 | 46.2 ± 7.6 | 0.75 ± 0.12 | 36.7 ± 3.2 | 49 |
| 59 |
|
94.8 ± 1.3 | 59.9 ± 5.0 | 2.36 ± 0.17 | 7.5 ± 1.2 | 3 |
| 60 |
|
99.1 ± 0.1 | 63.1 ± 3.6 | 0.72 ± 0.08 | 6.94 ± 0.94 | 10 |
| 61 |
|
82.9 ± 3.3 | 56.3 ± 5.7 | 4.94 ± 0.46 | 9.92 ± 1.5 | 2 |
| 62 |
|
93.3 ± 1.5 | 73.3 ± 4.9 | 4.04 ± 0.39 | 5.41 ± 0.67 | 1 |
| 63 |
|
62.1 ± 2.7 | 49.7 ± 4.2 | n.d | n.d. | n.d. |
| 64 |
|
73.9 ± 3.9 | 59.2 ± 5.0 | 9.18 ± 1.1 | 9.92 ± 1.6 | 1 |
% Tracer bound determined when incubated with 25 μM of inhibitors. Data are the average of at least two experiments ± SEM. n.d. = not determined.
53 was shown to manifest improved selectivity for Grp94 compared to the 3- and 4-chloro substituted thiophene analogues (55 and 55), supporting the hypothesis that 5-substituents bind similar to the 4-substituted phenyl ring. Incorporation of a 5-chloro substitution on the smaller furan ring (48) resulted in increased affinity for Grp94 while maintaining selectivity. However, incorporation of an ethyl substitution at the 3-position of the thiophene to mimic the 2-position of the benzene ring resulted in both reduced affinity and selectivity.
CO-CRYSTALLIZATION OF GRP94 WITH A GRP94-SELECTIVE INHIBITOR
The co-crystal structure of the N-terminal domain of Grp94 lacking the acidic linker (NΔ41, see Experimental Section) in complex with 48 (referred to as VC3 in the PBD file) was solved to 2.6 Å resolution to support the proposed docking interactions (Figure 5a, Supporting Information, Table 1). The structure reveals 48 in the ATP-binding site is stabilized by hydrogen bonding interactions with Asp149 (Figure 5b,c), similar to other resorcinol-based pan-Hsp90 inhibitors. There are few other direct interactions between 48 and the NΔ41 ATP-binding site, which are mediated through conserved water molecules (Figure 5c). In contrast to RDA, 48 is bound to Grp94 in a single orientation, illustrating the effectiveness of the cis-amide bioisostere in reducing binding heterogeneity.
Figure 5.
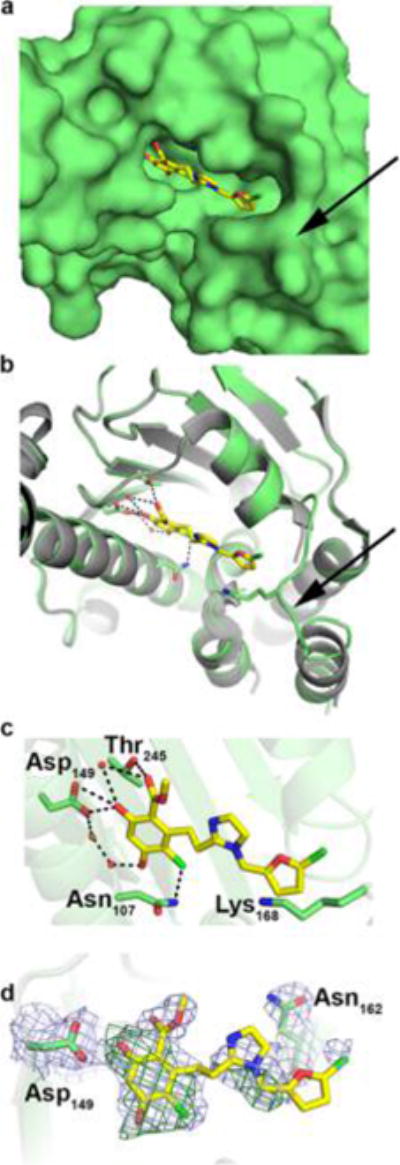
Crystal structure of 48 bound to Grp94. (a) Surface representation of ATP-binding pocket with 48 as ball and stick. Arrow points to a well-ordered loop at the mouth of the active site. (b) Cartoon representation of the structure as in (a) highlighting H-bonding interactions (≤3.5 Å) and loop configuration. (c) Zoomed view of 48 in active site with H-bonding interactions as in (b). (d) Final 2Fo – Fc electron density contoured at 1 σ (gray mesh) superimposed with initial Fo – Fc density contoured at 3 σ (green) after molecular replacement (see Experimental Section).
The electron density for 48 was largely continuous for the length of the molecule, with the notable exception of the chlorinated furan moiety (Figure 5d), which apparently samples multiple conformations within the ATP-binding site. It is possible that the chlorinated furan dwells in the extended hydrophobic region to increase selectivity (as predicted through modeling studies); however, the final binding mode modeled, based on optimal fit to 2Fo − Fc (Figure 5d) and Fo − Fc difference density (not shown), is one in which the chloride substituent is nestled in an ordered loop (residues 165–170) at the entry to the ATP-binding site (arrow, Figure 5a,b). Notably, this loop is disordered in the structure complexed with RDA. The furan moiety also appears to be involved in a cation–π interaction with Lys168 to stabilize this loop. In general, phenyl rings form stronger cation–π interactions due to a larger quadrapole moment compared to furan rings. However, modeling studies suggest that the phenyl ring of BnIm cannot orient in a manner that allows this interaction (data not shown) and therefore accounts for the increased affinity manifested by the smaller heterocycles (45–60). Taken together, 48, and by analogy other analogues described within this series, bind to the ATP-binding site of Grp94 in a mode that manifests increased selectivity over the other Hsp90 isoforms.
GRP94-SELECTIVE INHIBITION IN CANCER
Grp94 is responsible for the maturation and trafficking of several proteins associated with cell signaling and adhesion. One such client of Grp94 are the integrins, which are essential for cell adhesion and migration through promoting interactions between the intracellular actin cytoskeleton and the extracellular matrix.37–39 Integrins are dependent upon Grp94 for not only their maturation but also their transport to the cell surface. Therefore, inhibition of Grp94 leads to decreased trafficking of integrins to the cell surface and results in decreased integrin expression at the cell surface. As a result, decreased cell migration is observed and provides a new opportunity for the development of antimetastatic agents.29,40,41 For example, selective inhibition of Grp94 results in decreased migration of MDA-MB-231 cells, an aggressive form of metastatic breast cancer. In a wound-healing scratch assay, Grp94-selective inhibitors, 40 and 48, produced decreased wound closing at 24 h compared to BnIm and vehicle control (70% and 73% closed at 500 nM, respectively, Figure 6). In fact, these analogues manifested superior antimigratory activity compared to BnIm at 10-fold lower concentrations. Furthermore, these analogues were evaluated for antiproliferative activity against the same cell line and were found to manifest no antiproliferative activity up to 100 μM (78% and 107% viable at 100 μM, respectively, Figure 6), confirming that the antimigratory activity is not linked to cell viability. These data are in agreement with the nonessential nature of Grp94 and provides a large therapeutic window for the development of Grp94-selective inhibitors as a new class of antimetastatic agents.
Figure 6.
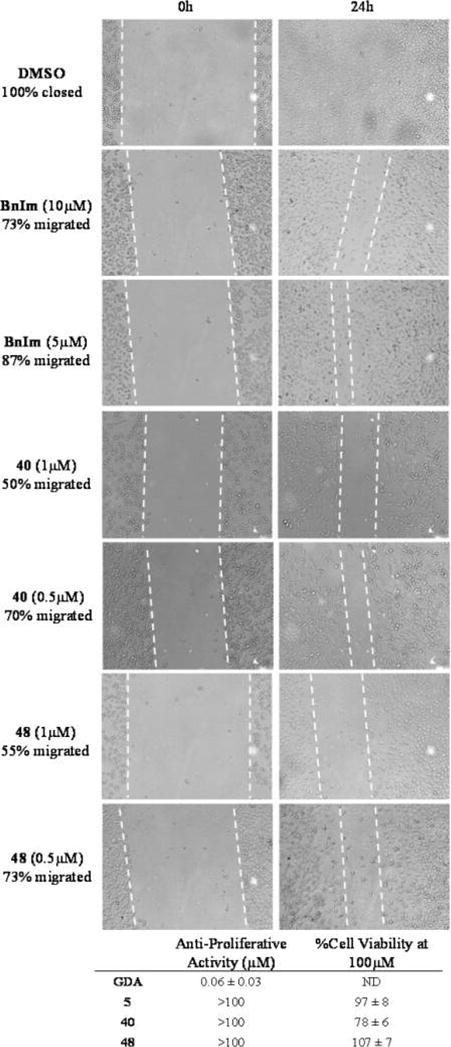
Wound-healing scratch assay results after 24 h treatment with BnIm, 40, 48, or vehicle. Antiproliferative activity given is relative to vehicle control (n = 4). ND = not determined.
Recently, integrin α2 was determined to be dependent upon Grp94 for its maturation and trafficking to the cell surface through proteomic and Western blot analyses of Grp94 knockdown cells.42 Similarly, selective inhibition of Grp94 resulted in the degradation of integrin α2 (Figure 7A,B). Integrin α2 forms a heterodimer with integrin β1 on the cell surface,38 which is responsible for binding to collagen in the extracellular matrix to promote metastasis and invasion. 40 and 48 both induced the degradation of integrin α2, providing evidence that the decrease in cell migration results from Grp94-selective inhibition. Furthermore, 40 and 48 do not result in the degradation of Akt (Figure 7C), a well-known client protein that is dependent upon the cytosolic isoforms of Hsp90. It should be noted that a slight induction of the pro-survival heat shock response was observed with 40 and 48 (Figure 7D). However, this occurred at concentrations 10–50-fold higher than antimigratory activity was demonstrated.
Figure 7.
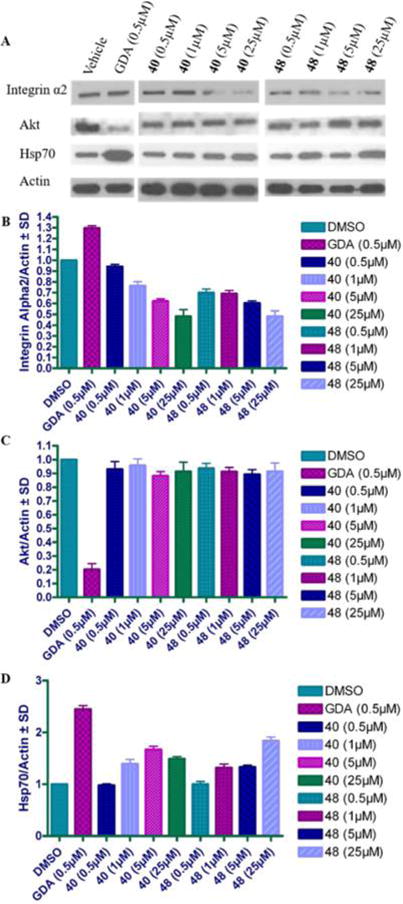
(A) Western blot analysis of MDA-MB-231 cells treated with Grp94-selective inhibitors, geldanamycin (GDA, a pan-Hsp90 inhibitor), or vehicle control (DMSO, 0.25% final concentration). Ratio of Integrin α2 (B), Akt (C), and Hsp70 (D) normalized to actin for each compound concentration.
PRIMARY OPEN ANGLE GLAUCOMA AND GRP94-SELECTIVE INHIBITION
Although POAG is characterized clinically by irreversible optic nerve damage and retinal ganglion cell death that leads to vision loss, the risk factor of elevated intraocular pressure typically results from decreased outflow of the aqueous humor through Schlemm’s canal of the trabecular meshwork (TM) extracellular matrix in the anterior segment of the eye. A contributor to TM dysfunction is TM cell death,24,43 which can be brought about by the aggregation of mutant myocilin.44 Nonsynonymous mutations in myocilin, localized to its olfactomedin domain, result in non-native tertiary structures, which promote facile aggregation and leads to TM cell death.43,45 Recently, it was demonstrated that Grp94 associates with amyloid-like aggregates of mutant myocilin but cannot triage these aggregates for degradation through the ER-associated degradation (ERAD) pathway.4 Our prior studies showed that inhibition of Grp94 allows mutant myocilin degradation through autophagy, which decreases intracellular levels of myocilin and, ultimately, reduces toxicity.21 Therefore, these newly designed Gpr94-selective inhibitors were evaluated for their ability to promote mutant myocilin clearance in an inducible HEK model. Treatment with 48 resulted in a substantial decrease of mutant myocilin levels at 1 μM (Figure 8A,C), however, treatment with 40 only resulted in a slight decrease in myocilin levels. The difference in activity between 40 and 48 is not readily clear and is currently being investigated in our laboratory. As expected, 40 and 48 did not induce the degradation of the cytosolic Hsp90 isoform-dependent client, Akt (Figure 8B,D), nor was there any measurable induction of the pro-survival heat shock response, as monitored by Hsp70 induction (Figure 8E). This is in contrast to Western blot analysis of the MDA-MB-231 cell line above (Figure 7) and is due to the Hsp90 isoforms in cancer cells having a higher affinity for inhibitors (and ATP) compared to the Hsp90 isoforms in nontransformed cells.46
Figure 8.
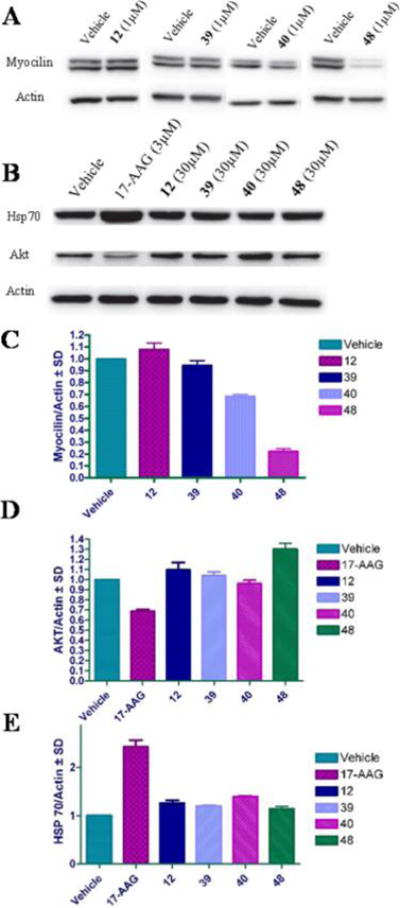
Western blot analysis of HEK cells overexpressing the I477N myocilin mutant and treated with 1 μM of indicated Grp94-selective inhibitors illustrating (A) myocilin degradation and (B) lack of AKT degradation and Hsp70 induction. Ratio of myocilin (C), AKT (D), and Hsp70 (E) normalized to actin for each compound. Vehicle = DMSO and 17-AAG = 17-(allylamino)-17-demethoxygeldanamycin (pan-Hsp90 N-terminal inhibitor).
CONCLUSION
Structure–activity relationship studies were performed on the aryl side chain of BnIm, which interacts with the unique secondary pocket of Grp94 (Figure 9). Substitutions at the 2-and 4-positions were tolerated within this binding region. Incorporation of an ethoxy group at the 2-position manifested a significant increase in affinity, as well as selectivity. 40 manifests nearly a 10-fold increase in affinity for Grp94 compared to BnIm and 40-fold selectivity for Grp94 over the cytosolic Hsp90 isoforms. Replacing the phenyl ring of the side chain with five-membered heterocycles also resulted in increased affinity for Grp94 as observed with 48. A crystal structure of the N-terminal domain of Grp94 was solved revealing a cation–π interaction between the furan ring and Lys168, which accounts for the increased affinity observed with the five-membered heterocycles. Grp94-selective inhibition reduced cell migration of aggressive breast cancer cells without manifesting toxicity and thus provided a large therapeutic index. Additionally, Grp94 inhibition resulted in the degradation of myocilin aggregates and provides a nontoxic approach to the treatment of POAG. These data presented herein provide a strong foundation for the further development of rationally designed Grp94-selective inhibitors for the treatment of metastasis and POAG.
Figure 9.
Summary of structure–activity relationships for the BnIm series of Grp94-selective inhibitors.
EXPERIMENTAL SECTION
Chemistry General
1H NMR were recorded at 400 (Bruker AVIIIHD 400 MHz NMR with a broadband X-channel detect gradient probe) or 500 MHz (Avance AVIII 500 MHz spectrometer with a dual carbon/proton cryoprobe), and 13C were recorded at 125 MHz (Bruker AVIII spectrometer equipped with a cryogenically cooled carbon observe probe); chemical shifts are reported in δ (ppm) relative to the internal standard (CDCl3, 7.26 ppm or MeOD, 3.31 ppm). HRMS spectra were recorded with a LCT Premier (Waters Cor., Milford, MA). The purity of compounds was determined by HPLC (Agilent 1100 series quaternary pump; 60% MeCN/40% water; Agilent C-18 column, 4.6 mm × 150 mm, 5 μM) with UV detection. All biologically tested compounds were determined to be >95% pure. TLC analysis was performed on glass backed silica gel plates and visualized by UV light. All solvents were reagent grade and used without further purification.
General Procedure for Multicomponent Cyclization Reaction
Basic amines (e.g., 6a–6q, 0.26 mmol) were added to a stirred solution of 9 (125 mg, 0.26 mmol) in wet MeOH (2 mL) and stirring continued for 30 min at rt before the addition of NH4HCO3 (0.26 mmol) and glyoxal (0.26 mmol). After stirring for 12 h, tetrabutylammonium fluoride (0.52 mL of 1 M solution in THF, 0.52 mmol) was added and then stirred for 30 min before the reaction was quenched with saturated aqueous NH4Cl (10 mL) and extracted with EtOAc (3 × 10 mL). The organic layers were combined, dried (Na2SO4), filtered, and concentrated. The residue was purified via flash chromatography (SiO2, 1:49 MeOH:DCM) to afford the desired product as amorphous solids.
Methyl 2-(2-(1-Benzyl-1H-imidazol-2-yl)ethyl)-3-chloro-4,6-dihydroxybenzoate (5)
Yield 54 mg (54%), white amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.28−7.23 (m, 3H), 7.01−6.97 (m, 2H), 6.95 (d, J = 1.5 Hz, 1H), 6.81 (d, J = 1.5 Hz, 1H), 6.40 (s, 1H), 5.02 (s, 2H), 3.79 (s, 3H), 3.48−3.41 (m, 2H), 2.89−2.83 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.81, 162.06, 158.06, 147.65, 141.73, 136.17, 128.93, 127.99, 126.91, 126.53, 120.10, 114.76, 105.88, 102.46, 52.45, 49.38, 30.77, 26.08. HRMS (ESI) m/z [M –H]− C20H19ClN2O4 385.0955, found 385.0953. tR = 5.062 min, 99.0%.
(4-Ethynylphenyl)methanamine (6a)
6a was synthesized from 4-iodobenzylamine hydrochloride following procedures detailed in ref 34. Yield 93 mg (78%) as a dark-yellow oil. 1H NMR (500 MHz, MeOD) δ 7.54 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 8.3 Hz, 2H), 4.13 (s, 2H), 3.60 (s, 1H). 13C NMR (126 MHz, MeOD) δ 133.56, 132.27 (2C), 128.71 (2C), 123.34, 82.10, 78.58, 42.51. HRMS (ESI) m/z [M + H]+ for C9H10N 132.0813, found 132.0809.
General Procedure for Reduction of Aromatic Nitriles
Benzonitriles (e.g., 7a–7c, 0.7 M in THF, 1 equiv) were added dropwise to a stirred solution of LiAlH4 (0.5 M in dry THF, 5 equiv) at 0 °C. The reaction mixture warmed to rt and stirred for 12 h. H2O (1 mL/g of LiAlH4) was added dropwise to quench the excess hydride, followed by 4 M NaOH (1 mL/g of LiAlH4) and EtOAc (3 mL/g of LiAlH4). The resulting suspension was filtered through a pad of Celite and the Celite washed with warm EtOAc (30 mL) and the eluent concentrated. The residue was purified via flash chromatography (SiO2, 1:49, MeOH:DCM to 1:20, MeOH:DCM) to afford the desired product as oils.
(2-Ethylphenyl)methanamine (6b)
Yield 653 mg (79%) as a colorless oil. 1H NMR (400 MHz, MeOD) δ 6.77−6.69 (m, 1H), 6.63−6.56 (m, 2H), 6.58−6.48 (m, 1H), 4.51 (s, 2H), 2.07 (q, J = 7.6 Hz, 2H), 0.60 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 143.24, 129.80, 129.39, 129.10, 127.47, 126.97, 42.31, 26.37, 15.96. HRSM (ESI) m/z [M + NH4]+ for C9H17N2 153.1392, found 153.1395.
(2-Ethoxyphenyl)methanamine (6c)
Yield 215 mg (59%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.25−7.16 (m, 2H), 6.90 (t, J = 7.4 Hz, 1H), 6.85 (d, J = 8.5 Hz, 1H), 4.07 (q, J = 7.6, 7.1 Hz, 2H), 3.82 (s, 2H), 1.73 (br s, 2H), 1.44 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 156.82, 131.81, 128.52, 128.05, 120.37, 111.07, 63.33, 42.88, 14.98. HRMS (ESI) m/z [M + H]+ for C9H14NO 152.1075, found 152.1077.
(2-Isopropoxyphenyl)methanamine (6d)
Yield 243 mg (59%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.19 (t, J = 6.6 Hz, 2H), 6.92−6.83 (m, 2H), 4.59 (hept, J = 6.0 Hz, 1H), 3.79 (s, 2H), 1.74 (br s, 2H), 1.36 (d, J = 6.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 155.74, 132.68, 128.74, 127.95, 120.24, 112.53, 69.73, 43.07, 22.23 (2C). MS (EI) m/z [M]+ for C10H15NO 165.1, found 165.2.
(2-Propoxyphenyl)methanamine (6e)
Yield 265 mg (67%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.24−7.16 (m, 2H), 6.90 (t, J = 7.4 Hz, 1H), 6.85 (d, J = 8.5 Hz, 1H), 3.96 (t, J = 6.4 Hz, 2H), 3.83 (s, 2H), 1.90−1.78 (m, 4H), 1.06 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 156.94, 131.72, 128.50, 128.08, 120.33, 111.02, 69.26, 42.85, 22.70, 10.78. MS (EI) m/z [M]+ for C10H15NO 165.1, found: 165.2.
General Procedure for Staudinger Reduction
PPh3 (1.1 equiv) was added to a stirred solution of the benzyl azide (1 equiv) in THF:H2O (0.1 M, 10:1) and stirred at rt for 12 h. The reaction mixture was concentrated, and the residue was purified via flash chromatography (SiO2, 1:49, MeOH:DCM to 1:20, MeOH:DCM) to afford the desired product as oils.
(2-Methoxy-4-methylphenyl)methanamine (6f)
Yield 99 mg (73%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.08 (d, J = 7.4 Hz, 1H), 6.72 (d, J = 7.4 Hz, 1H), 6.69 (s, 1H), 3.83 (s, 3H), 3.77 (s, 2H), 2.34 (s, 3H), 1.87 (br s, 2H). 13C NMR (126 MHz, CDCl3) δ 157.32, 138.16, 128.47, 120.96, 111.24, 74.80, 55.12, 42.39, 21.55. HRMS (ESI) m/z [M + H]+ for C9H14NO 152.1075, found 152.1076.
General Procedure for Oxime Reduction
Aryl oximes (e.g., 66a–66e, 0.7 M in THF, 1 equiv) were added dropwise to a stirred solution of LiAlH4 (0.5 M in dry THF, 5 equiv) at 0 °C. The reaction mixture warmed to rt and stirred for 12 h. H2O (1 mL/g of LiAlH4) was added dropwise to quench the excess hydride, followed by 4 M NaOH (1 mL/g of LiAlH4) and EtOAc (3 mL/g of LiAlH4). The resulting suspension was then filtered through a pad of Celite and the Celite washed with warm EtOAc (30 mL), and the eluent was concentrated. The residue was purified via flash chromatography (SiO2, 1:49, MeOH:DCM to 1:20, MeOH:DCM) to afford the desired products as oils.
(5-Chlorofuran-2-yl)methanamine (6g)
Yield 93 mg (47%), yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.12 (d, J = 3.2 Hz, 1H), 6.06 (d, J = 3.2 Hz, 1H), 3.76 (d, J = 0.8 Hz, 2H), 1.58 (br s, 2H). 13C NMR (126 MHz, CDCl3) δ 156.24, 156.23, 135.05, 107.49, 106.62, 39.35. HRMS (ESI) m/z [M + H]+ for C5H7ClNO 132.0216, found 132.0221.
Thiazol-2-ylmethanamine (6h)
Yield 42 mg (9%) as a red oil. 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 3.1 Hz, 1H), 7.26 (d, J = 3.2 Hz, 1H), 4.20 (s, 2H), 1.80 (br, s, 2H). 13C NMR (126 MHz, CDCl3) δ 174.02, 142.60, 118.56, 44.04. HRMS (ESI) m/z [M + H]+ for C4H7N2S 115.0330, found 115.0331.
(5-Methylthiophen-2-yl)methanamine (6i)
Yield 72 mg (16%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 6.70 (d, J = 3.4 Hz, 1H), 6.57 (dd, J = 3.3, 1.3 Hz, 1H), 3.96 (s, 2H), 2.72 (br s, 2H), 2.44 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 143.88, 138.89, 124.79, 124.03, 41.20, 15.40. HRMS (ESI) m/z [M + H]+ for C6H10NS 128.0534, found 128.0532.
(3-Vinylthiophen-2-yl)methanamine (6j)
Yield 152 mg (42%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.20−7.07 (m, 1H), 6.73 (dd, J = 17.5, 11.0 Hz, 1H), 5.54 (dd, J = 17.4, 1.3 Hz, 1H), 5.24 (dd, J = 11.0, 1.3 Hz, 1H), 4.07 (s, 1H), 2.03 (s, 1H). 13C NMR (126 MHz, CDCl3) δ 142.46, 135.23, 128.76, 125.43, 123.24, 114.19, 38.92. HRMS (ESI) m/z [M + H]+ for C7H10NS 140.0534, found 140.0531.
(3-Vinylfuran-2-yl)methanamine (6k)
Yield 57 mg (24%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.29 (dd, J = 2.0, 0.7 Hz, 1H), 6.57 (dd, J = 17.3, 10.7 Hz, 1H), 6.50 (d, J = 2.0 Hz, 1H), 5.42 (dd, J = 17.4, 1.4 Hz, 1H), 5.14 (dd, J = 10.8, 1.4 Hz, 1H), 3.85 (s, 2H), 1.67 (br s, 2H). 13C NMR (126 MHz, CDCl3) δ 152.77, 141.68, 126.24, 119.34, 113.23, 107.72, 37.05. HRMS (ESI) [M + H]+ for C7H10NO 124.0762, found 124.0768.
(3-Ethylthiophen-2-yl)methanamine (6l)
Pd/C 10% (5 mol %) was added to a stirred solution of (3-vinylthiophen-2-yl)methanamine (6k, 50 mg, 0.36 mmol) in MeOH (5 mL) and stirred at rt under a hydrogen atmosphere for 12 h. The reaction mixture was filtered through a pad of Celite, and the eluent was concentrated to produce the title compound which was used without further purification. Yield 45 mg (89%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 5.1 Hz, 1H), 6.87 (d, J = 5.2 Hz, 1H), 4.56 (s, 2H), 4.05 (s, 2H), 2.61 (q, J = 7.6 Hz, 2H), 1.20 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 141.48, 134.90, 128.62, 123.84, 37.77, 21.46, 15.29. HRMS (ESI) m/z [M + H]+ for C7H12NS 147.0690, found 147.0692.
(5-Chlorothiophen-2-yl)methanamine (6m)
A solution of thiophene-2-ylmethanamine (1 mL, 9.7 mmol, 1 equiv) in AcOH:Et2O (9:1, 0.5 M) was cooled to 5 °C followed by the dropwise addition of SO2Cl2 (1.18 mL, 14.6 mmol, 1.5 equiv), maintaining the reaction temperature under 20 °C. The reaction was stirred at rt for 1 h, at which time Et2O (20 mL) was added and stirred for an additional 30 min. The precipitate (hydrochloride salt of desired product) was filtered and the solid washed with Et2O. The isolated solid was dissolved in DCM (25 mL) and washed with sat’d NaHCO3 (2 × 30 mL). The organic layer was separated, dried (Na2SO4), and concentrated. The residue was purified via flash chromatography (SiO2, 1:49 MeOH:DCM to 1:20 MeOH:DCM) to provide the title compound. Yield 658 mg (37%) as a yellow oil. 1H NMR (400 MHz, MeOD) δ 5.51 (d, J = 3.0 Hz, 1H), 5.40 (d, J = 3.8 Hz, 1H), 2.69 (s, 2H), 1.73 (br s, 2H). 13C NMR (126 MHz, MeOD) δ 134.70, 132.71, 130.62, 127.93, 38.70. HRMS (ESI) m/z [M + H]+ for C5H6ClNS 148.9990, found 148.9988.
Isoxazol-5-ylmethanamine (6n)
Yield 159 mg (77%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 6.13 (s, 1H), 4.00 (s, 2H), 1.53 (br, s, 2H). 13C NMR (126 MHz, CDCl3) δ 172.47, 150.28, 100.04, 37.62. MS (EI) m/z [M]+ for C4H6N2O 98.0, found 98.1.
(3,5-Dimethylfuran-2-yl)methanamine (6o)
Yield 98 mg (65%) as a yellow oil. 1H NMR (400 MHz, MeOD) δ 5.78 (s, 1H), 3.70 (s, 2H), 2.17 (s, 3H), 1.92 (s, 3H). 13C NMR (126 MHz, MeOD) δ 152.01, 147.87, 118.48, 110.06, 36.39, 13.41, 9.78. HRMS (ESI) m/z [M + H]+ for C7H12NO 126.0919, found 126.0924.
(3-Chlorothiophen-2-yl)methanamine (6p)
Yield 113 mg (59%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.15 (dd, J = 5.2, 1.2 Hz, 1H), 6.86 (dd, J = 5.3, 1.1 Hz, 1H), 3.98 (s, 2H), 1.79 (br s, 2H). 13C NMR (126 MHz, CDCl3) δ 148.24, 124.45, 123.90, 118.33, 41.51. HRMS (ESI) m/z [M + H]+ for C5H7ClNS 147.9988, found 147.9990.
(4-Chlorothiophen-2-yl)methanamine (6q)
Yield 79 mg (67%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 6.96 (s, 1H), 6.78 (s, 1H), 3.99 (s, 2H), 1.71 (br s, 2H). 13C NMR (126 MHz, CDCl3) δ 148.24, 124.45, 123.90, 118.33, 41.51. HRMS (ESI) m/z [M + H]+ for C5H7ClNS 147.9988, found 147.9995.
General Procedure for Alkylation of 2-Hydroxybenzonitrile
Alkyl iodides (3.7 mmol, 1.1 equiv) and K2CO3 (930 mg, 6.7 mmol, 2 equiv) were added to a stirred solution of 2-hydroxybenzonitrile (400 mg, 3.4 mmol) in DMF (20 mL) at rt and stirred for 4 h. The reaction was quenched with the addition of H2O (20 mL) and EtOAc (40 mL). The organic layer was washed with water (5 × 40 mL), dried (Na2SO4), filtered, and concentrated. The residue was purified by column chromatography (SiO2, 4:1 Hex:EtOAc) to afford the desired alkyl ethers as colorless oils.
2-Ethoxybenzonitrile (7a)
Yield 360 mg (71%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.63−7.45 (m, 2H), 7.04−6.89 (m, 2H), 4.15 (q, J = 7.1 Hz, 2H), 1.48 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 161.02, 134.65, 134.17, 120.91, 116.99, 112.49, 102.36, 64.99, 14.91. MS (EI) m/z [M]+ for C9H9NO 147.1, found 147.1; [M + H]+ for C9H10NO 148.1, found 148.1.
2-Isopropoxybenzonitrile (7b)
Yield 405 mg (71%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.57 (dt, J = 7.7, 1.5 Hz, 1H), 7.52 (td, J = 7.9, 7.3, 1.5 Hz, 1H), 6.99 (dd, J = 8.2, 5.2 Hz, 2H), 4.68 (hept, J = 6.1 Hz, 1H), 1.43 (d, J = 6.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 159.92, 134.11, 133.94, 120.45, 116.77, 113.64, 102.99, 71.78, 21.86 (2C). MS (EI) m/z [M]+ for C10H11NO 161.1, found 161.1; [M + H]+ for C10H12NO 162.1, found 162.1.
2-Propoxybenzonitrile (7c)
Yield 385 mg (71%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 7.8 Hz, 1H), 7.50 (td, J = 8.1, 7.4, 1.5 Hz, 1H), 7.00−6.93 (m, 2H), 4.03 (t, J = 6.5 Hz, 2H), 1.88 (h, J = 7.0 Hz, 2H), 1.08 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 160.83, 134.27, 133.76, 120.52, 116.56, 112.19, 102.00, 70.45, 22.33, 10.46. MS (EI) m/z [M]+ for C10H11NO 161.1, found 161.1; [M + H]+ for C10H12NO 162.1, found 162.1.
General Procedure for the Reduction of Carboxylic Acids
The aromatic carboxylic acids (e.g., 68, 0.7 M in THF, 1 equiv) were added dropwise to a stirred solution of LiAlH4 (0.5 M in THF, 5 equiv) at 0 °C. The reaction warmed to rt and stirred for 12 h. H2O (1 mL/g of LiAlH4) was added, to quench the excess hydride, then 4 M NaOH (1 mL/g of LiAlH4) and EtOAc (3 mL/g of LiAlH4). The resulting suspension was filtered through a pad of Celite then the Celite was washed with warm EtOAc and the eluent concentrated. The residue was purified via flash chromatography (SiO2, 3:10 EtOAc:hexanes to 3:5 EtOAc:hexanes) to provide the desired alcohol as colorless oils:
(2-Methoxy-4-methylphenyl)methanol (8)
Yield 187 mg (50%), colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 7.5 Hz, 1H), 6.78 (dd, J = 7.5, 0.8 Hz, 1H), 6.75 (s, 1H), 4.32 (s, 2H), 3.86 (s, 3H), 2.38 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 157.58, 140.13, 130.11, 121.13, 120.80, 111.52, 55.33, 50.01, 21.71. HRSM (ESI) m/z [M]+ for C9H12O2 152.0837, found 152.0846.
Methyl 4,6-Bis((tert-butyldimethylsilyl)oxy)-3-chloro-2-(3-oxopropyl)benzoate (9)
9 was synthesized following procedures detailed in refs 28 and 35. Yield 1.8 g (68%), white amorphous solid. 1H NMR (500 MHz, chloroform-d) δ 9.80 (s, 1H), 6.31 (s, 1H), 3.84 (s, 3H), 2.96−2.92 (m, 2H), 2.80−2.74 (m, 2H), 1.02 (s, 9H), 0.95 (s, 9H), 0.23 (s, 6H), 0.21 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 201.47, 168.36, 153.48, 151.94, 138.10, 121.44, 118.81, 110.15, 109.97, 52.69, 43.83, 26.00 (3C), 25.83 (3C), 18.74, 18.43, −3.96 (2C), −4.00 (2C). HRMS (ESI) m/z [M + H]+ for C23H40ClO5Si2 487.2103, found 487.2120.
Methyl 3-Chloro-2-(2-(1-(4-fluorobenzyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (10)
Yield 22 mg (21%), white amorphous solid. 1H NMR (500 MHz, MeOD) δ 7.07−6.94 (m, 5H), 6.86 (d, J = 1.4 Hz, 1H), 6.29 (s, 1H), 5.00 (s, 2H), 3.69 (s, 3H), 3.22−3.19 (m, 2H), 2.84 (m, 2H). 13C NMR (126 MHz, MeOD) δ 171.22, 164.76, 162.81, 160.69, 158.81, 148.82, 141.37, 134.18, 130.01, 129.94, 127.28, 121.71, 116.75, 116.58, 112.63 (d, J = 558.4 Hz), 103.34, 52.85, 31.45, 27.26. HRMS (ESI) m/z [M + H] for C20H19ClFN2O4 405.1017, found 405.1009.
Methyl 3-Chloro-2-(2-(1-(4-chlorobenzyl)-1H-imidazol-2-yl)-ethyl)-4,6-dihydroxybenzoate (11)
Yield 28 mg, (25%), white amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.24 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 1.4 Hz, 1H), 6.90 (d, J = 8.4 Hz, 2H), 6.78 (d, J = 1.4 Hz, 1H), 6.40 (s, 1H), 4.97 (s, 2H), 3.81 (s, 3H), 3.46−3.40 (m, 2H), 2.89−2.80 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.63, 162.31, 157.66, 147.56, 141.56, 134.60, 134.02, 129.19 (2C), 127.92 (2C), 127.05, 120.00, 114.57, 106.08, 102.68, 52.59, 30.87, 29.69, 26.04 HRMS (ESI) m/z [M + H] for C20H19Cl2N2O4 421.0722, found 421.0714.
Methyl 2-(2-(1-(4-Bromobenzyl)-1H-imidazol-2-yl)ethyl)-3-chloro-4,6-dihydroxybenzoate (12)
Yield 45 mg (38%), off-white amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.36 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 1.4 Hz, 1H), 6.82 (d, J = 8.5 Hz, 2H), 6.77 (d, J = 1.5 Hz, 1H), 6.35 (s, 1H), 4.93 (s, 2H), 3.78 (s, 3H), 3.42−3.34 (m, 2H), 2.84−2.77 (m, 2H). 13C NMR (101 MHz, CDCl3, MeOD) δ 170.23, 161.42, 157.64, 147.25, 141.09, 134.83, 131.69 (2C), 127.85 (2C), 126.55, 121.59, 119.68, 114.27, 102.14, 99.99, 52.03, 30.38, 25.69. HRMS (ESI) m/z [M + H]+ for C20H19BrClN2O4 465.0217, found 465.0237. tR = 4.17 min, 95.3%.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(4-iodobenzyl)-1H-imidazol-2-yl)ethyl)benzoate (13)
Yield 22 mg (35%), white amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD): δ 7.59−7.57 (dd, J = 11.6 Hz, 2H), 7.14−7.11 (dd, J = 10.0 Hz, 2H), 7.05 (s, 1H), 6.86 (s, 1H), 6.44 (s, 1H), 5.14 (s, 2H), 3.87 (s, 3H), 3.48−3.44 (t, J = 16.8, 2H), 2.91−2.87 (t, J = 18.4 Hz, 2H.). 13C NMR (125 MHz, CDCl3, MeOD): δ 170.6, 162.4, 157.6, 147.6, 141.5, 138.1, 128.4, 127.2, 120.0, 114.6, 106.1, 102.7, 93.6, 52.6, 52.2, 49.0, 30.9, 26.1, 25.2, 20.1. HRMS (ESI) m/z [M + H]+ for C20H18ClIN2O4 513.0020, found 513.0070.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(4-methylbenzyl)-1H-imidazol-2-yl)ethyl)benzoate (14)
Yield 44 mg (43%), off-white amorphous solid. 1H NMR (500 MHz, MeOD) δ 7.15 (d, J = 7.9 Hz, 2H), 7.03 (d, J = 1.4 Hz, 1H), 7.00−6.96 (m, 2H), 6.94 (d, J = 1.5 Hz, 1H), 6.39 (s, 1H), 5.05 (s, 2H), 3.77 (s, 3H), 3.37−3.30 (m, 2H), 2.97−2.89 (m, 2H), 2.31 (s, 3H). 13C NMR(125 MHz, MeOD) δ 171.3, 160.9, 159.0, 148.8, 141.5, 138.9, 135.1, 130.5 (2C), 127.9 (2C), 127.1, 121.8, 115.0, 110.1, 103.3, 52.8, 50.1, 31.5, 27.3, 21.1. HRMS (ESI) m/z [M + H]+ for C21H22ClN2O4 401.1268, found 401.1266.
Methyl 3-Chloro-2-(2-(1-(4-ethylbenzyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (15)
Yield 37 mg (39%), off white amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.14 (d, J = 8.1 Hz, 2H), 7.11−7.08 (m, 1H), 6.98−6.91 (m, 2H), 6.85 (d, J = 1.5 Hz, 1H), 6.44 (s, 1H), 4.96 (s, 3H), 3.54−3.45 (m, 2H), 3.04 (t, J = 7.9 Hz, 2H), 2.59 (q, J = 7.6 Hz, 2H), 1.17 (td, J = 7.6, 1.0 Hz, 3H). 13C NMR (125 MHz, CDCl3, MeOD) δ 170.6, 162.1, 158.2, 147.2, 145.0, 140.9, 132.1, 128.8 (2C), 127.2 (2C), 124.2, 120.6, 114.9, 106.2, 103.0, 50.0, 49.9, 30.7, 28.6, 25.6, 15.6. HRMS (ESI) m/z [M + H]+ for C22H24ClN2O4 415.1425, found 415.1432.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(4-isopropylbenzyl)-1H-imidazol-2-yl)ethyl)benzoate (16)
Yield 25 mg (22%), white amorphous solid. 1H NMR (500 MHz, CDCl3) δ 7.13 (d, J = 8.1 Hz, 2H), 7.03 (s, 1H), 6.90 (d, J = 8.2 Hz, 2H), 6.80 (d, J = 1.5 Hz, 1H), 6.49 (s, 1H), 4.95 (s, 2H), 3.79 (s, 3H), 3.50 (t, J = 7.9 Hz, 2H), 3.00−2.92 (m, 2H), 2.82 (hept, J = 6.9 Hz, 1H), 1.16 (d, J = 7.0 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 170.63, 162.91, 157.32, 149.25, 147.18, 127.20 (2C), 126.81 (2C), 120.19, 114.50, 106.24, 103.00, 99.98, 52.76, 49.56, 33.81, 30.77, 29.72, 23.93 (2C). HRMS (ESI) m/z [M + H]+ for C23H26ClN2O4 429.1581, found 429.1588.
Methyl 3-Chloro-2-(2-(1-(4-cyanobenzyl)-1H-imidazol-2-yl)-ethyl)-4,6-dihydroxybenzoate (17)
Yield 37 mg (39%), off white amorphous solid. 1H NMR (500 MHz, CDCl3) δ 7.79 (q, J = 7.8, 6.0 Hz, 2H), 7.61−7.36 (m, 2H), 7.20−7.12 (m, 1H), 7.05 (s, 1H), 6.57 (s, 1H), 5.44−5.25 (m, 2H), 4.00 (s, 3H), 3.57 (q, J = 7.7 Hz, 2H), 3.02 (t, J = 7.9 Hz, 2H). 13C NMR (125 MHz, CDCl3): 170.4, 161.5, 158.0, 147.8, 142.0, 141.2, 132.8, (2C), 127.7, 127.4, 127.0 (2C), 120.2, 118.2, 114.5, 111.6, 102.4, 52.4, 52.3, 30.8, 26.0. HRMS (ESI) m/z [M + H]+ for C21H19ClN3O4 412.1064, found 412.1049.
Methyl 3-Chloro-2-(2-(1-(4-ethynylbenzyl)-1H-imidazol-2-yl)-ethyl)-4,6-dihydroxybenzoate (18)
Yield 37 mg (35%), tan amorphous solid. 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 8.3 Hz, 1H), 7.06 (d, J = 1.4 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 6.86 (d, J = 1.4 Hz, 1H), 6.48 (s, 1H), 5.12 (s, 1H), 3.84 (s, 2H), 3.50 (dd, J = 9.3, 6.7 Hz, 1H), 3.09 (s, 1H), 2.94 (dd, J = 9.0, 7.0 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 170.71, 162.73, 158.41, 147.66, 141.27, 136.69, 132.78, 129.10, 126.79, 126.61, 126.52, 122.09, 120.19, 120.15, 114.98, 105.65, 102.81, 82.85, 52.61, 49.28, 30.86, 26.01. HRMS (ESI) m/z [M + H]+ for C22H20ClN2O4 411.1112, found 411.1121.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(4-(trifluoromethyl)-benzyl)-1H-imidazol-2-yl)ethyl)benzoate (19)
Yield 24 mg (36%), white amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD): δ 7.56−7.55 (dd, J = 9.7 Hz, 2H), 7.15 (s, 1H), 7.10−7.08 (dd, J = 11.3 Hz, 2H), 6.86 (s, 1H), 6.44 (s, 1H), 5.06 (s, 2H), 3.86 (s, 3H), 3.47−3.43 (t, J = 16.3 Hz, 2H), 3.04−3.00 (t, J = 16.3 Hz, 2H). 13C NMR (125 MHz, CDCl3, MeOD) δ 170.2, 162.1, 157.8, 147.3, 140.3, 138.7, 131.1, 130.8, 127.1, 126.3, 126.2, 124.7, 122.6, 120.5, 114.6, 111.4, 106.1, 103.1, 52.9, 30.6, 25.3. HRMS (ESI) m/z [M + H+] for C21H18ClF3N2O4 455.0937, found 455.0972.
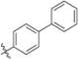
Methyl 2-(2-(1-([1,1′-Biphenyl]-4-ylmethyl)-1H-imidazol-2-yl)-ethyl)-3-chloro-4,6-dihydroxybenzoate (20)
Yield 35 mg (29%), tan amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.54−7.48 (m, 4H), 7.39 (ddd, J = 8.0, 7.0, 1.7 Hz, 2H), 7.31 (td, J = 7.2, 1.4 Hz, 1H), 7.07 (dd, J = 8.3, 1.6 Hz, 2H), 6.98 (s, 1H), 6.86 (s, 1H), 6.41 (s, 1H), 5.07 (s, 2H), 3.83 (s, 3H), 3.52−3.45 (m, 2H), 2.92−2.87 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.80, 162.08, 157.84, 147.65, 141.83, 140.99, 140.24, 135.21, 128.81 (2C), 127.62 (2C), 127.52, 127.16, 127.00 (2C), 126.98 (2C), 120.08, 114.67, 106.06, 102.49, 52.50, 49.64, 30.89, 26.17. HRMS (ESI) m/z [M + H]+ for C26H23ClN2O4 463.1425, found 463.1447.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(4-methoxybenzyl)-1H-imidazol-2-yl)ethyl)benzoate (21)
Yield 25 mg (23%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 6.86−6.84 (m, 1H), 6.82 (d, J = 1.4 Hz, 1H), 6.74−6.69 (m, 4H), 6.31 (s, 1H), 4.85 (s, 2H), 3.72 (s, 3H), 3.65 (s, 3H), 3.38−3.32 (m, 2H), 2.82−2.76 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 174.64, 165.64, 163.22, 162.05, 151.40, 145.46, 135.37, 132.06 (2C), 132.02, 130.37, 123.92, 118.20 (2C), 110.16, 106.36, 99.99, 59.12, 56.29, 34.61, 30.03. HRMS (ESI) m/z [M–H]− for C21H21ClN2O5 415.1061, found 415.1061.
Methyl 3-Chloro-2-(2-(1-(3,4-dichlorobenzyl)-1H-imidazol-2-yl)-ethyl)-4,6-dihydroxybenzoate (22)
Yield 27 mg (23%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.30 (d, J = 8.3 Hz, 1H), 7.05 (d, J = 1.9 Hz, 2H), 6.92 (d, J = 1.7 Hz, 1H), 6.81 (dd, J = 8.3, 2.2 Hz, 1H), 6.32 (s, 1H), 4.94 (s, 2H), 3.76 (s, 3H), 3.36−3.29 (m, 2H), 2.95 (t, J = 7.8 Hz, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 173.55, 164.94, 161.93, 141.26, 137.43, 135.20, 133.07, 130.90, 130.47, 125.31 (2C), 118.43, 116.40, 110.96, 107.00 (2C), 56.54, 53.05, 33.98, 28.86. HRMS (ESI) m/z [M + H]+ for C20H18Cl3N2O4 455.0332, found 455.0354.
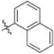
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(naphthalen-1-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (23)
Yield 32 mg (28%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.79 (dd, J = 6.8, 2.5 Hz, 1H), 7.71 (dd, J = 8.0, 3.9 Hz, 2H), 7.48−7.40 (m, 2H), 7.28 (d, J = 1.2 Hz, 1H), 6.93 (d, J = 1.4 Hz, 1H), 6.72 (d, J = 1.5 Hz, 1H), 6.71−6.67 (m, 1H), 6.32 (s, 1H), 5.43 (s, 2H), 3.73 (s, 3H), 3.45−3.35 (m, 2H), 2.94−2.84 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.58, 161.56, 158.02, 147.88, 141.47, 133.57, 131.62, 130.34, 128.92, 128.68, 126.80 (2C), 126.15, 125.35, 123.93, 121.99, 120.36, 114.62, 106.32, 102.38, 52.29, 47.31, 30.74, 26.17. HRMS (ESI) m/z [M + H]+ for C24H21ClN2O4 437.1268, found 437.1250.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(naphthalen-2-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (24)
Yield 28 mg (25%), tan amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ 7.88 (dd, J = 9.1, 4.0 Hz, 2H), 7.83−7.78 (m, 1H), 7.51−7.46 (m, 1H), 7.25 (dd, J = 8.4, 1.9 Hz, 1H), 7.18 (d, J = 1.3 Hz, 1H), 6.85 (d, J = 1.3 Hz, 1H), 6.42 (s, 1H), 5.29 (s, 2H), 3.57 (s, 3H), 2.92−2.86 (m, 2H), 2.77−2.71 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 167.86, 155.00, 154.47, 146.42, 137.73, 135.17, 132.79, 132.21, 128.35, 127.60 (2C), 127.54, 126.80, 126.43 (2C), 126.04, 124.94, 124.84, 120.56, 113.74, 110.78, 101.68, 51.80, 48.38, 29.92, 26.18. HRMS (ESI) m/z [M + H]+ for C24H21ClN2O4 437.1268, found 437.1280.
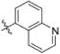
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(quinolin-5-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (25)
Yield 29 mg (25%), white amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 8.87 (dd, J = 4.3, 1.6 Hz, 1H), 8.20−8.16 (m, 1H), 8.00 (d, J = 8.5 Hz, 1H), 7.60 (dd, J = 8.5, 7.2 Hz, 1H), 7.49−7.44 (m, 1H), 6.99 (s, 1H), 6.86−6.83 (m, 1H), 6.76 (s, 1H), 6.36 (s, 1H), 5.48 (s, 2H), 3.83 (s, 3H), 3.48−3.44 (m, 2H), 2.95−2.89 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.55, 161.86, 157.87, 150.21, 147.86, 141.56, 132.32, 131.06, 129.45, 129.37, 127.44, 125.58, 124.79, 121.64, 120.13, 114.59, 106.08, 102.48, 99.99, 52.49, 46.63, 30.93, 26.20. HRMS (ESI) m/z [M + H]+ for C23H21ClN3O4 438.1221, found 438.1207.
Methyl 3-Chloro-2-(2-(1-(3-fluorobenzyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (26)
Yield 26 mg (25%), pale-yellow amorphous solid. 1H NMR (500 MHz, CDCl3) δ 7.29−7.20 (m, 1H), 7.01 (d, J = 1.4 Hz, 1H), 6.92 (m, 1H), 6.82 (d, J = 1.4 Hz, 1H), 6.75 (m, 1H), 6.70−6.58 (m, 1H), 6.46 (s, 1H), 5.01 (s, 2H), 3.80 (s, 3H), 3.52−3.40 (m, 2H), 2.88−2.74 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 157.05, 147.61, 141.78, 130.85, 127.79, 122.12, 120.22 (2C), 115.28, 115.12, 113.69, 113.51, 106.66, 102.98 (2C), 100.13, 53.58, 48.96, 31.01, 26.26. HRMS (ESI) m/z [M + H] for C20H19ClFN2O4 405.1017, found 405.1009.
Methyl 3-Chloro-2-(2-(1-(3-chlorobenzyl)-1H-imidazol-2-yl)-ethyl)-4,6-dihydroxybenzoate (27)
Yield 47 mg (44%), pale-yellow amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.25−7.20 (m, 2H), 7.03 (d, J = 1.6 Hz, 1H), 6.99−6.96 (m, 1H), 6.91−6.84 (m, 2H), 6.40 (s, 1H), 5.00 (s, 2H), 3.82 (s, 3H), 3.49−3.33 (m, 2H), 3.01−2.83 (m, 2H). 13C NMR (125 MHz, CDCl3, MeOD) δ 170.6, 161.9, 158.2, 147.5, 141.1, 137.7, 135.1, 130.5, 128.5, 126.8, 125.9, 124.9, 120.5, 114.8, 106.2, 102.7, 52.6 (2C), 30.6, 25.8. HRMS (ESI) m/z [M + H]+ for C20H19Cl2N2O4 421.0722, found 421.0728.
Methyl 2-(2-(1-(3-Bromobenzyl)-1H-imidazol-2-yl)ethyl)-3-chloro-4,6-dihydroxybenzoate (28)
Yield 23 mg (19%), white amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.38 (m, 1H), 7.19−7.09 (m, 2H), 6.99 (d, J = 1.5 Hz, 1H), 6.91 (m, 1H), 6.84 (d, J = 1.5 Hz, 1H), 6.40 (s, 1H), 5.00 (s, 2H), 3.83 (s, 3H), 3.46−3.38 (m, 2H), 2.92−2.79 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 168.02, 159.25, 155.49, 145.00, 138.77, 135.70, 128.71, 128.01, 126.95, 124.05, 122.60, 120.52, 117.64, 112.15, 103.58, 100.01, 49.95, 46.22, 28.09, 23.35. HRMS (ESI) m/z [M + H] for C20H19BrClN2O4 465.0217, found 465.0225.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(3-iodobenzyl)-1H-imidazol-2-yl)ethyl)benzoate (29)
Yield 4 mg (11%), white amorphous solid. 1H NMR (400 MHz, DMSO-d6): δ 7.67−7.64 (dd, J = 7.05 Hz, 1H), 7.43 (s, 1H), 7.18−7.14 (m, 2H), 7.08−7.06 (dd, J = 9.24 Hz, 1H), 6.86 (s, 1H), 6.47 (s, 1H), 5.14 (s, 2H), 3.68 (s, 3H), 2.92−2.88 (t, J = 16.3 Hz, 2H), 2.73−2.67 (t, J = 18.2 Hz, 2H). 13C NMR (125 MHz, DMSO-d6, MeOD): δ 167.9, 155.0, 154.5, 146.4, 140.2, 137.7, 136.2, 135.1, 130.9, 126.0, 120.6, 113.8, 110.8, 101.8, 99.5, 95.2, 52.0, 47.4, 29.8, 26.1. HRMS (ESI) m/z [M + H+] for C20H18ClIN2O4 513.0023, found 513.0065.
Methyl 2-(2-(1-(3-Methoxybenzyl)-1H-imidazol-2-yl)ethyl)-3-chloro-4,6-dihydroxybenzoate (30)
Yield 32 mg (31%), white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 7.06 (s, 1H), 6.88 (s, 1H), 6.82 (d, J = 9.0 Hz, 1H), 6.62 (d, J = 7.7 Hz, 1H), 6.55 (s, 1H), 6.53 (s, 1H), 5.04 (s, 2H), 3.85 (s, 3H), 3.76 (s, 3H), 3.59−3.51 (m, 2H), 2.95−2.89 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.68, 162.88, 160.13, 157.00, 147.45, 141.62, 137.81, 130.13, 127.12, 120.17, 118.69, 114.33, 113.03, 112.49, 106.43, 102.82, 55.26, 52.64, 49.34, 30.90, 26.09. HRMS (ESI) m/z [M + H]+ for C21H22ClN2O5 417.1217, found 417.1221.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(3-methylbenzyl)-1H-imidazol-2-yl)ethyl)benzoate (31)
Yield 18 mg (17%), tan amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.06 (t, J = 7.6 Hz, 1H), 6.95 (d, J = 7.7 Hz, 1H), 6.85 (d, J = 1.5 Hz, 1H), 6.75 (d, J = 1.5 Hz, 1H), 6.72−6.66 (m, 2H), 6.30 (s, 1H), 4.87 (s, 2H), 3.69 (s, 3H), 3.36−3.30 (m, 2H), 2.81 (dd, J = 9.2, 6.9 Hz, 2H), 2.16 (s, 3H). 13C NMR (126 MHz, CDCl3, MeOD) δ 174.49, 165.52, 161.99, 151.39, 145.16, 142.73, 139.62, 132.79 (2C), 131.28, 129.76, 127.67, 124.32, 118.57, 110.31, 106.43, 56.27, 53.48, 34.48, 29.85, 25.07. HRMS (ESI) m/z [M + H]+ for C21H22ClN2O4 401.1268, found 401.1255.
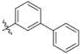
Methyl 2-(2-(1-([1,1′-Biphenyl]-3-ylmethyl)-1H-imidazol-2-yl)-ethyl)-3-chloro-4,6-dihydroxybenzoate (32)
Yield 46 mg (39%), pale-yellow amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.53−7.47 (m, 3H), 7.41 (dd, J = 9.1, 7.3 Hz, 3H), 7.38−7.33 (m, 1H), 7.23 (s, 1H), 7.03 (d, J = 1.3 Hz, 1H), 6.99 (d, J = 7.8 Hz, 1H), 6.89 (d, J = 1.4 Hz, 1H), 6.46 (s, 1H), 5.12 (s, 2H), 3.82 (s, 3H), 3.56−3.46 (m, 2H), 2.97−2.89 (m, 2H). 13C NMR δ (100 MHz, CDCl3): 170.8, 163.0, 157.9, 147.7, 142.4, 141.5, 140.5, 136.6, 129.8 (3C), 129.1(2C), 128.0, 127.3, 127.3, 125.7, 125.6, 120.5, 114.9, 106.3, 103.2, 52.8, 49.9, 31.1, 26.2. HRMS (ESI) m/z [M − H]− for C26H22ClN2O4 461.1268, found 461.1259.
Methyl 3-Chloro-2-(2-(1-(2-fluorobenzyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (33)
Yield 23 mg (22%), white amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.27−7.23 (m, 1H), 7.06−6.98 (m, 2H), 6.94 (d, J = 1.4 Hz, 1H), 6.81 (d, J = 1.4 Hz, 1H), 6.78 (dd, J = 7.5, 1.7 Hz, 1H), 6.40 (s, 1H), 5.05 (s, 2H), 3.82 (s, 3H), 3.64−3.38 (m, 2H), 3.05−2.68 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.80, 162.21, 157.79, 147.67, 141.77, 130.04, 128.42, 127.27, 124.63, 123.62, 123.50, 119.93, 115.60 (d, J = 21.0 Hz), 114.66, 106.02, 102.55, 52.51, 43.46, 30.83, 26.04. HRMS (ESI) m/z [M + H] for C20H18ClFN2O4 405.1029, found 405.1017.
Methyl 2-(2-(1-(2-Chlorobenzyl)-1H-imidazol-2-yl)ethyl)-3-chloro-4,6-dihydroxybenzoate (34)
Yield 23 mg (21%), white amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.27 (s, 1H), 7.24−7.16 (m, 2H), 7.01 (d, J = 1.6 Hz, 1H), 6.94 (dd, J = 1.9, 1.0 Hz, 1H), 6.84 (m, 2H), 6.38 (s, 1H), 4.98 (s, 2H), 3.80 (s, 3H), 3.45−3.35 (m, 2H), 2.94−2.85 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.58, 162.43, 157.43, 147.63, 141.17, 133.48, 132.58, 129.83, 129.62, 127.72, 127.52, 126.18, 120.16, 114.51, 106.20, 102.85, 52.73, 47.48, 30.89, 25.78. HRMS (ESI) m/z [M + H]+ for C20H19Cl2N2O4 421.0722, found 421.0714.
Methyl 2-(2-(1-(2-Bromobenzyl)-1H-imidazol-2-yl)ethyl)-3-chloro-4,6-dihydroxybenzoate (35)
Yield 26 mg (22%), white amorphous solid. 1H NMR (500 MHz, CDCl3) δ 7.54 (dd, J = 8.0, 1.3 Hz, 1H), 7.24−7.20 (m, 1H), 7.15 (td, J = 7.7, 1.7 Hz, 1H), 7.10 (d, J = 1.6 Hz, 1H), 6.78 (d, J = 1.6 Hz, 1H), 6.67−6.57 (m, 1H), 6.48 (s, 1H), 5.06 (s, 2H), 3.85 (s, 3H), 3.48−3.33 (m, 2H), 3.06−2.89 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.47, 162.64, 157.77, 147.55, 140.60, 133.23 (2C), 130.08, 128.23 (2C), 122.57, 120.27 (2C), 114.71, 105.94, 103.08, 52.85, 50.15, 30.78, 25.58. HRMS (ESI) m/z [M + H] for C20H19BrClN2O4 465.0217, found 465.0231.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(2-methylbenzyl)-1H-imidazol-2-yl)ethyl)benzoate (36)
Yield 32 mg (31%), tan amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.25 (s, 1H), 7.08−7.02 (m, 2H), 6.88−6.83 (m, 1H), 6.66−6.58 (m, 1H), 6.47 (t, J = 6.7 Hz, 1H), 6.29 (s, 1H), 4.87 (s, 2H), 3.72 (s, 3H), 3.35−3.25 (m, 2H), 2.81 (dd, J = 10.9, 5.1 Hz, 2H), 2.10 (s, 3H). 13C NMR (126 MHz, CDCl3, MeOD) δ 174.43, 165.45, 161.97, 151.53, 145.12, 139.32, 137.71, 134.49, 132.09, 130.45, 130.31, 129.87, 124.09, 118.52, 110.38, 106.41, 56.28, 51.65, 34.50, 29.86, 22.59. HRMS (ESI) m/z [M + H]+ for C21H22ClN2O4 401.1268, found 401.1261.
Methyl 2-(2-(1-(2-Aminobenzyl)-1H-imidazol-2-yl)ethyl)-3-chloro-4,6-dihydroxybenzoate (37)
Yield 20 mg (19%), white amorphous solid. 1H NMR (500 MHz, MeOD) δ 7.07 (td, J = 7.8, 1.6 Hz, 1H), 6.98 (dd, J = 7.8, 1.4 Hz, 2H), 6.78 (dd, J = 8.0, 1.2 Hz, 1H), 6.65 (td, J = 7.5, 1.2 Hz, 1H), 6.52 (dd, J = 7.6, 1.5 Hz, 1H), 6.39 (s, 1H), 5.03 (s, 2H), 3.79 (s, 3H), 3.37−3.33 (m, 2H), 3.01−2.96 (m, 2H). 13C NMR (126 MHz, MeOD) δ 169.90, 159.44, 157.47, 147.55, 145.00, 140.02, 128.56, 127.00, 125.07, 120.41, 120.32, 117.92, 116.00, 113.55, 108.77, 101.92, 51.46, 45.78, 29.89, 25.71. HRMS (ESI) m/z [M + H] for C20H20ClN3O4 402.1221, found 402.1230.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(2-methoxybenzyl)-1H-imidazol-2-yl)ethyl)benzoate (38)
Yield 46 mg (42%), tan amorphous solid. 1H NMR (500 MHz, CDCl3) δ 7.26−7.23 (m, 1H), 6.97 (d, J = 1.4 Hz, 1H), 6.89−6.84 (m, 2H), 6.83 (d, J = 1.4 Hz, 1H), 6.70 (dd, J = 7.8, 1.7 Hz, 1H), 6.44 (s, 1H), 5.03 (s, 2H), 3.84 (s, 3H), 3.81 (s, 3H), 3.49−3.44 (m, 2H), 2.94−2.90 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 170.96, 162.35, 157.84, 156.56, 147.74, 141.91, 129.33, 127.59, 126.69, 124.66, 120.72, 120.13, 114.71, 110.27, 105.90, 102.51, 55.27, 52.50, 44.85, 30.85, 26.04. HRMS (ESI) m/z [M + H]+ for C21H22ClN2O5 417.1217, found 417.1211.
Methyl 3-Chloro-2-(2-(1-(2-ethylbenzyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (39)
Yield 33 mg (31%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.43 (d, J = 2.1 Hz, 1H), 7.40 (d, J = 3.0 Hz, 1H), 7.32−7.28 (m, 1H), 7.26 (d, J = 7.7 Hz, 1H), 6.83 (d, J = 7.7 Hz, 1H), 6.79 (s, 1H), 6.55 (s, 1H), 4.98 (s, 2H), 4.02 (s, 3H), 3.68 (t, J = 7.3 Hz, 2H), 3.45 (t, J = 7.1 Hz, 3H), 2.52 (q, J = 7.5 Hz, 3H), 1.19 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, CDCl3, MeOD) δ 169.85, 161.95, 158.20, 146.18, 145.33, 142.11, 138.91, 129.92, 129.32, 128.17, 127.04, 120.75, 119.05, 114.84, 106.28, 103.49, 53.17, 30.86, 30.04, 25.20, 24.25, 14.49. HRMS (ESI) m/z [M + H]+ for C22H24ClN2O4 415.1425, found 415.1419. tR = 4.27 min, 95.3%.
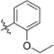
Methyl 3-Chloro-2-(2-(1-(2-ethoxybenzyl)-1H-imidazol-2-yl)-ethyl)-4,6-dihydroxybenzoate (40)
Yield 44 mg (39%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.18 (t, J = 8.0 Hz, 1H), 6.88 (d, J = 1.9 Hz, 1H), 6.83−6.76 (m, 3H), 6.67 (d, J = 7.6 Hz, 1H), 6.38 (s, 1H), 4.99 (s, 2H), 3.97 (q, J = 7.7, 6.8 Hz, 2H), 3.80 (s, 3H), 3.45−3.39 (m, 2H), 2.88 (t, J = 8.1 Hz, 2H), 1.32 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.97, 162.15, 157.97, 155.99, 147.73, 141.95, 129.25, 127.68, 126.65, 124.63, 120.46, 120.13, 114.73, 111.01, 105.90, 102.43, 63.51, 52.45, 44.95, 30.86, 26.12, 14.69. HRMS (ESI) m/z [M + H]+ for C22H24ClN2O5 431.1374, found 431.1378. tR = 2.71 min, 98.3%.
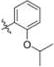
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(2-isopropoxybenzyl)-1H-imidazol-2-yl)ethyl)benzoate (41)
Yield 18 mg (16%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.21 (t, J = 8.0 Hz, 1H), 6.95 (s, 1H), 6.86−6.78 (m, 3H), 6.72 (d, J = 7.5 Hz, 1H), 6.44 (s, 1H), 5.00 (s, 2H), 4.56 (hept, J = 10.8, 5.3 Hz, 1H), 3.85 (s, 3H), 3.52−3.45 (m, 2H), 2.97−2.87 (m, 2H), 1.27 (d, J = 6.0 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 170.97, 162.15, 157.97, 156.00, 147.73, 141.95, 129.25, 127.68, 126.65, 124.63, 120.46, 120.13, 114.73, 111.01, 105.90, 102.43, 99.99, 63.51, 52.45, 44.95, 30.86, 26.12, 14.69. HRMS (ESI) m/z [M + H]+ for C23H26ClN2O5 445.1530, found 445.1521. tR = 2.70 min, 97.7%.
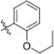
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(2-propoxybenzyl)-1H-imidazol-2-yl)ethyl)benzoate (42)
Yield 34 mg (29%), tan amorphous solid. 1H NMR (500 MHz, CDCl3) δ 7.29−7.26 (m, 1H), 7.02 (d, J = 1.4 Hz, 1H), 6.90−6.85 (m, 3H), 6.76−6.73 (m, 1H), 6.49 (s, 1H), 5.10 (s, 2H), 3.95 (t, J = 6.5 Hz, 2H), 3.84 (s, 3H), 3.56−3.50 (m, 2H), 3.03−2.96 (m, 2H), 1.80 (qt, J = 7.4, 6.5 Hz, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 170.91, 162.91, 157.83, 156.11, 147.58, 141.58, 129.36, 127.76, 126.49, 124.55, 120.56, 120.15, 114.73, 111.04, 105.90, 102.72, 69.53, 52.55, 45.00, 30.85, 26.04, 22.57, 10.61. HRMS (ESI) m/z [M + H]+ for C23H26ClN2O5 445.1530, found 445.1539. tR = 2.73 min, 98.0%.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(2-(trifluoromethyl)-benzyl)-1H-imidazol-2-yl)ethyl)benzoate (43)
Yield 25 mg (21%), white amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.65 (d, J = 7.7 Hz, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.36 (t, J = 7.7 Hz, 1H), 7.02 (s, 1H), 6.79 (s, 1H), 6.57 (d, J = 7.8 Hz, 1H), 6.39 (s, 1H), 5.25 (s, 2H), 3.85 (s, 3H), 3.50−3.41 (m, 2H), 2.93−2.79 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.72, 162.08, 157.95, 148.07, 141.52, 134.95, 132.62, 127.97, 127.61, 126.91, 126.26 (q, J = 5.6 Hz), 125.20, 123.03, 120.21, 114.70, 105.88, 102.50, 52.46, 45.88, 30.87, 25.83. HRMS (ESI) m/z [M + H]+ for C21H19ClF3N2O4 455.0985, found 455.0979.
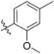
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(2-methoxy-4-methylbenzyl)-1H-imidazol-2-yl)ethyl)benzoate (44)
Yield 45 mg (40%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.28 (d, J = 2.1 Hz, 1H), 6.94 (d, J = 7.6 Hz, 1H), 6.85 (d, J = 2.1 Hz, 1H), 6.76 (d, J = 7.3 Hz, 1H), 6.69 (s, 1H), 6.56 (s, 1H), 4.81 (s, 2H), 3.99 (s, 3H), 3.74 (s, 3H), 3.63 (t, J = 7.4 Hz, 2H), 3.44 (t, J = 7.4 Hz, 2H), 2.34 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 169.87, 161.98, 157.11, 151.39, 145.68, 142.01, 140.11, 129.96, 129.53, 121.73, 121.63, 120.51, 118.55, 112.14, 111.93, 55.44, 54.21, 46.62, 30.48, 29.73, 23.96, 21.75. HRMS (ESI) m/z [M + H]+ for C22H24ClN2O5 431.1374, found 431.1370.
Methyl 3-Chloro-2-(2-(1-(furan-3-ylmethyl)-1H-imidazol-2-yl)-ethyl)-4,6-dihydroxybenzoate (45)
Yield 40 mg (41%), off-white amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.30 (d, J = 1.6 Hz, 1H), 7.20 (s, 1H), 6.84 (dd, J = 2.7, 1.4 Hz, 1H), 6.78 (d, J = 1.4 Hz, 1H), 6.36 (d, J = 2.0 Hz, 1H), 6.15 (d, J = 1.9 Hz, 1H), 4.80 (s, 2H), 3.78 (d, J = 1.9 Hz, 3H), 3.39 (tt, J = 8.9, 2.5 Hz, 2H), 2.95−2.80 (m, 2H). 13C NMR (125 MHz, CDCl3, MeOD): δ 170.8, 161.8, 158.1, 147.3, 144.1, 141.6, 141.6, 140.1, 126.7, 121.0, 119.6, 114.7, 109.4, 106.2, 102.5, 52.4, 41.0, 30.8, 26.1. HRMS (ESI) m/z [M + H]+ for C18H18ClN2O5 377.0904, found 377.0905.
Methyl 3-Chloro-2-(2-(1-(furan-2-ylmethyl)-1H-imidazol-2-yl)-ethyl)-4,6-dihydroxybenzoate (46)
Yield 44 mg (45%), off-white amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.31 (dt, J = 2.4, 1.2 Hz, 1H), 6.87 (dd, J = 2.4, 1.4 Hz, 1H), 6.83 (d, J = 1.5 Hz, 1H), 6.40 (d, J = 1.8 Hz, 1H), 6.26 (dt, J = 3.5, 1.7 Hz, 1H), 6.18 (d, J = 3.3 Hz, 1H), 4.94 (d, J = 1.6 Hz, 2H), 3.82 (d, J = 1.9 Hz, 3H), 3.46 (ddd, J = 10.2, 6.1, 2.0 Hz, 2H), 2.95 (ddd, J = 9.8, 5.9, 1.5 Hz, 2H). 13C NMR (125 MHz, CDCl3, MeOD) δ 170.9, 162.0, 158.2, 149.1, 147.4, 143.2, 141.8, 126.9, 119.7, 114.8, 110.6, 108.8, 106.2, 102.6, 52.6, 42.6, 30.8, 26.1. HRMS (ESI) m/z [M + H]+ for C18H18ClN2O5 377.0904, found 377.0911.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-((5-methylfuran-2-yl)-methyl)-1H-imidazol-2-yl)ethyl)benzoate (47)
Yield 46 mg (46%), off white amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 6.87 (d, J = 1.4 Hz, 1H), 6.84 (d, J = 1.4 Hz, 1H), 6.41 (s, 1H), 6.08 (d, J = 3.1 Hz, 1H), 5.84 (dd, J = 3.0, 1.2 Hz, 1H), 4.88 (s, 2H), 3.84 (s, 3H), 3.49−3.42 (m, 2H), 3.08−2.93 (m, 2H), 2.18 (d, J = 1.0 Hz, 3H). 13C NMR (125 MHz, CDCl3, MeOD) δ 171.0, 162.2, 158.3, 153.1, 147.3, 147.1, 141.9, 126.7, 119.7, 114.9, 109.7, 106.5, 106.1, 102.6, 52.6, 42.7, 30.9, 26.2, 13.5. HRMS (ESI) m/z [M + H]+ for C19H20ClN2O5 391.1061, found 391.1066. tR = 3.05 min, 95.1%.
Methyl 3-Chloro-2-(2-(1-((5-chlorofuran-2-yl)methyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (48)
Yield 50 mg (47%), white amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 6.91 (s, 2H), 6.31 (s, 1H), 6.23 (d, J = 3.4 Hz, 1H), 5.98 (d, J = 3.3 Hz, 1H), 4.87 (s, 2H), 3.73 (s, 3H), 3.37−3.27 (m, 2H), 2.99 (t, J = 7.9 Hz, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 173.88, 165.00, 161.93, 151.08, 150.68, 143.82, 141.62, 124.59, 118.45, 116.26, 111.28 (2C), 106.80 (2C), 56.40, 46.86, 34.09, 29.14. HRMS (ESI) m/z [M + H]+ for C18H17Cl2N2O5 411.0515, found 411.0527. tR = 4.15 min, 95.5%.
Methyl 3-Chloro-2-(2-(1-((3,5-dimethylfuran-2-yl)methyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (49)
Yield 36 mg (25%), off-white amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.28 (d, J = 2.0 Hz, 1H), 6.92 (s, 1H), 6.44 (s, 1H), 5.77 (s, 1H), 4.80 (s, 2H), 3.89 (s, 3H), 3.59−3.48 (m, 2H), 3.37 (t, J = 7.1 Hz, 2H), 2.11 (s, 3H), 1.92 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 169.87, 161.98, 157.11, 151.39, 145.68, 140.11, 129.96, 121.63, 120.51, 118.85, 118.55, 112.14, 111.93, 55.44, 54.21, 46.62, 30.48, 29.73, 23.96, 21.75. HRMS (ESI) m/z [M + H]+ for C20H22ClN2O5 405.1217, found 405.1210. tR = 3.24 min, 95.5%.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(thiophen-2-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (50)
Yield 37 mg (37%), yellow amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.23 (dd, J = 5.1, 1.3 Hz, 1H), 6.95 (d, J = 1.5 Hz, 1H), 6.92 (dd, J = 5.1, 3.5 Hz, 1H), 6.87 (dd, J = 3.5, 1.9 Hz, 2H), 6.43 (s, 1H), 5.17 (d, J = 0.9 Hz, 2H), 3.84 (s, 3H), 3.56−3.44 (m, 2H), 3.01−2.92 (m, 2H). 13C NMR (125 MHz, CDCl3, MeOD) δ 170.9, 162.1, 158.2, 147.3, 141.7, 138.5, 127.2, 126.9, 126.4, 126.2, 119.7, 114.9, 106.2, 102.7, 52.6, 44.7, 30.9, 26.2. HRMS (ESI) m/z [M + H]+ for C18H18ClN2O4S 393.0676, found 393.0674.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(thiophen-3-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (51)
Yield 28 mg (27%), yellow amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.25 (d, J = 4.8 Hz, 1H), 6.98−6.92 (m, 2H), 6.83 (d, J = 1.5 Hz, 1H), 6.81−6.78 (m, 1H), 6.37 (s, 1H), 4.97 (s, 2H), 3.79 (s, 3H), 3.43−3.37 (m, 2H), 2.95−2.88 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 174.48, 165.67, 162.02, 151.06, 145.03, 140.31, 131.26, 130.04, 129.39, 126.69, 124.00, 118.61, 110.15, 106.53, 56.40, 49.27, 34.49, 29.68. HRMS (ESI) m/z [M + H]+ for C18H18ClN2O4S 393.0676, found 393.0693.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-((5-methylthiophen-2-yl)-methyl)-1H-imidazol-2-yl)ethyl)benzoate (52)
Yield 23 mg (22%), off-white amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.34 (d, J = 1.8 Hz, 1H), 7.02−6.96 (m, 1H), 6.79 (d, J = 4.1 Hz, 1H), 6.60 (d, J = 2.7 Hz, 1H), 6.48 (s, 1H), 5.03 (s, 2H), 3.95 (s, 3H), 3.58 (t, J = 7.2 Hz, 2H), 3.39 (t, J = 7.2 Hz, 2H), 2.41 (s, 3H). 13C NMR (126 MHz, CDCl3, MeOD) δ 169.83, 161.87, 158.19, 145.63, 138.77, 131.10, 129.14 (2C), 125.60 (2C), 120.60, 119.05, 106.34, 103.54, 53.15, 46.03, 30.05, 24.21, 15.28. HRMS (ESI) m/z [M + H]+ for C19H20ClN2O4S 407.0832, found 407.0839.
Methyl 3-Chloro-2-(2-(1-((5-chlorothiophen-2-yl)methyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (53)
Yield 20 mg (18%), yellow amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 6.95 (s, 1H), 6.87 (s, 1H), 6.68 (d, J = 3.5 Hz, 1H), 6.63 (d, J = 3.5 Hz, 1H), 6.37 (s, 1H), 5.03 (s, 2H), 3.81 (s, 3H), 3.47−3.37 (m, 2H), 3.02−2.92 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 174.29, 165.57, 162.00, 150.88, 144.73, 140.23, 134.89, 130.21, 130.09, 129.35, 123.74, 118.59, 110.29, 106.65, 56.47, 48.84, 34.48, 29.63. HRMS (ESI) m/z [M + H]+ for C18H17Cl2N2O4S 427.0286, found 427.0292.
Methyl 3-Chloro-2-(2-(1-((4-chlorothiophen-2-yl)methyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (54)
Yield 23 mg (21%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 6.97 (s, 1H), 6.90 (s, 1H), 6.83 (s, 1H), 6.67 (s, 1H), 6.38 (s, 1H), 5.07 (s, 2H), 3.81 (s, 3H), 3.45−3.39 (m, 2H), 2.88 (t, J = 8.2 Hz, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.61, 161.82, 158.12, 147.29, 141.48, 139.55, 127.34, 126.18, 125.05, 120.14, 119.50, 114.71, 106.03, 102.51, 52.44, 44.42, 30.76, 26.05. HRMS (ESI) m/z [M + H]+ for C18H17Cl2N2O4S 427.0286, found 427.0290.
Methyl 3-Chloro-2-(2-(1-((3-chlorothiophen-2-yl)methyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (55)
Yield 17 mg (15%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.21 (dd, J = 5.9, 2.3 Hz, 1H), 6.90 (d, J = 2.1 Hz, 1H), 6.86 (dt, J = 5.2, 1.8 Hz, 2H), 6.41 (s, 1H), 5.09 (s, 2H), 3.84 (s, 3H), 3.54−3.45 (m, 2H), 2.96 (dd, J = 11.9, 5.4 Hz, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.79, 162.08, 158.04, 147.22, 141.70, 131.26, 127.92, 127.32, 125.17, 124.20, 119.32, 114.80, 105.96, 102.55, 52.52, 42.02, 30.79, 26.15. HRMS (ESI) m/z [M + H]+ for C18H16Cl2N2O4S 427.0286, found 427.0278.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(isoxazol-5-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (56)
Yield 15 mg (15%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 8.17 (s, 1H), 6.96 (s, 1H), 6.89 (s, 1H), 6.42 (s, 1H), 6.04 (s, 1H), 5.17 (s, 2H), 3.88 (s, 3H), 3.50−3.41 (m, 2H), 2.97−2.89 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.55, 166.32, 157.95, 150.41, 147.60, 141.46, 127.82, 119.70, 114.64, 106.11, 102.64, 102.47, 102.14, 52.57, 41.22, 30.85, 25.99. HRMS (ESI) [M + H]+ for C17H17ClN3O5 378.0857, found 378.0866.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(thiazol-2-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (57)
Yield 41 mg (40%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.72 (d, J = 3.2 Hz, 1H), 7.30 (d, J = 3.3 Hz, 1H), 6.99 (s, 1H), 6.95 (s, 1H), 6.43 (s, 1H), 5.35 (s, 2H), 3.87 (s, 3H), 3.51−3.46 (m, 2H), 2.98−2.91 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.72, 165.84, 162.13, 157.91, 147.63, 143.06, 141.59, 127.84, 120.27, 119.78, 114.71, 106.01, 102.63, 52.57, 46.84, 30.83, 26.06. HRMS (ESI) m/z [M + H]+ for C17H17ClN3O4S 394.0628, found 394.0630.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-((3-vinylthiophen-2-yl)-methyl)-1H-imidazol-2-yl)ethyl)benzoate (58)
Yield 35 mg (32%), tan amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.19 (d, J = 5.3 Hz, 1H), 7.15 (d, J = 5.3 Hz, 1H), 7.01 (s, 1H), 6.77 (s, 1H), 6.58 (dd, J = 17.3, 10.9 Hz, 1H), 6.45 (s, 1H), 5.56 (d, J = 17.3 Hz, 1H), 5.31 (d, J = 11.0 Hz, 1H), 5.11 (s, 2H), 3.87 (s, 3H), 3.58−3.49 (m, 2H), 3.07 (t, J = 7.8 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 170.94, 163.17, 156.14, 142.75, 140.72, 139.77, 138.46, 130.54, 128.69, 128.46, 126.69, 126.54, 126.04, 113.85, 106.68, 102.61, 52.59, 38.44, 33.98, 32.45. HRMS (ESI) m/z [M + H]+ for C20H20ClN2O4S 419.0832, found 419.0844.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-((3-vinylfuran-2-yl)-methyl)-1H-imidazol-2-yl)ethyl)benzoate (59)
Yield 35 mg (33%), off-white amorphous solid. 1H NMR (400 MHz, CDCl3, MeOD) δ 7.30 (d, J = 2.0 Hz, 1H), 6.84 (d, J = 1.8 Hz, 1H), 6.51 (d, J = 2.0 Hz, 1H), 6.49 (s, 1H), 6.48−6.41 (m, 1H), 5.50 (d, J = 17.3 Hz, 1H), 5.26 (d, J = 11.0 Hz, 1H), 4.94 (s, 2H), 3.91 (s, 3H), 3.59 (t, J = 7.6 Hz, 2H), 3.22 (t, J = 7.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 170.35, 162.14, 157.96, 146.62, 143.64 (2C), 124.53 (2C), 123.65, 119.92 (2C), 116.57, 114.78, 108.33 (2C), 108.24, 106.19, 103.08, 52.84, 41.07, 30.43, 25.17. HRMS (ESI) m/z [M + H]+ for C20H20ClN2O5 403.1061, found 403.1069.
Methyl 3-Chloro-2-(2-(1-((3-ethylthiophen-2-yl)methyl)-1H-imidazol-2-yl)ethyl)-4,6-dihydroxybenzoate (60)
Yield 43 mg (39%), tan amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 7.16 (d, J = 5.2 Hz, 1H), 6.91 (d, J = 1.4 Hz, 1H), 6.86 (d, J = 5.2 Hz, 1H), 6.73 (d, J = 1.5 Hz, 1H), 6.45 (s, 1H), 5.04 (s, 2H), 3.86 (s, 3H), 3.56−3.50 (m, 2H), 3.00−2.95 (m, 2H), 2.53 (q, J = 7.6 Hz, 2H), 1.12 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 170.80, 162.20, 157.88, 147.06, 142.21, 141.84, 130.82, 128.69, 126.96, 124.60, 119.11, 114.70, 106.03, 102.57, 52.57, 42.34, 30.78, 26.25, 21.49, 14.92. HRMS (ESI) m/z [M + H]+ for C20H22ClN2O4S 421.0989, found 421.0995.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(pyridin-2-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (61)
Yield 15 mg (15%), tan amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.57 (d, J = 5.0 Hz, 1H), 7.68 (td, J = 7.7, 1.9 Hz, 1H), 7.13 (d, J = 1.5 Hz, 1H), 6.99 (d, J = 1.6 Hz, 1H), 6.89 (d, J = 7.9 Hz, 1H), 6.51 (s, 1H), 5.23 (s, 2H), 3.89 (s, 3H), 3.52 (t, J = 7.9 Hz, 2H), 3.05 (t, J = 8.0 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 170.50, 162.80, 157.65, 149.98 (2C), 147.44, 137.50 (3C), 123.42, 121.11, 120.70, 114.63, 106.01, 103.24, 52.94, 51.53, 30.62, 25.40. HRMS (ESI) m/z [M–H]− for C19H18ClN3O4 386.0908, found 386.0899.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(pyridin-3-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (62)
Yield 25 mg (25%), yellow amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 8.44 (d, J = 4.3 Hz, 1H), 8.30 (s, 1H), 7.32 (d, J = 6.0 Hz, 1H), 7.31−7.24 (m, 1H), 6.98 (s, 1H), 6.83 (s, 1H), 6.38 (s, 1H), 5.06 (s, 2H), 3.83 (s, 3H), 3.48−3.37 (m, 2H), 2.90−2.84 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 174.42, 165.67, 162.04, 152.92, 152.83, 151.54, 145.18, 138.88, 136.30, 131.13, 128.14, 123.84, 118.57, 110.07, 106.52, 56.40, 50.83, 34.70, 29.95. HRMS (ESI) m/z [M – H]− C19H18ClN3O4 386.0908, found 386.0903.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(pyridin-4-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (63)
Yield 32 mg (30%), yellow amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 8.44−8.29 (m, 2H), 6.95 (d, J = 1.4 Hz, 1H), 6.88−6.82 (m, 2H), 6.81 (d, J = 1.5 Hz, 1H), 6.30 (s, 1H), 5.03 (s, 2H), 3.78 (s, 3H), 3.40−3.28 (m, 2H), 2.78−2.71 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 170.39, 161.48, 157.93, 149.56 (2C), 147.76, 146.40, 141.07, 127.39, 121.31 (2C), 120.18, 114.46, 106.24, 102.43, 52.34, 30.70, 25.91, 25.11, 19.88. HRMS (ESI) m/z [M–H]− C19H18ClN3O4 386.0908, found 386.0912.
Methyl 3-Chloro-4,6-dihydroxy-2-(2-(1-(pyrimidin-5-ylmethyl)-1H-imidazol-2-yl)ethyl)benzoate (64)
Yield 45 mg (45%), white amorphous solid. 1H NMR (500 MHz, CDCl3, MeOD) δ 9.09 (s, 1H), 8.46 (s, 2H), 7.05 (s, 1H), 6.92 (s, 1H), 6.40 (s, 1H), 5.12 (s, 2H), 3.86 (s, 3H), 3.52−3.36 (m, 2H), 3.00−2.79 (m, 2H). 13C NMR (126 MHz, CDCl3, MeOD) δ 174.09, 165.38, 161.94, 159.48, 159.42, 159.38, 151.44, 144.71, 134.07, 130.84, 123.84, 118.47, 110.37, 106.66, 56.46, 48.81, 34.66, 29.80. HRMS (ESI) m/z [M – H]− C18H17ClN4O4 387.0860, found 387.0861.
5-Chlorofuran-2-carbaldehyde (65)
5-Nitrofuran-2-carbaldehyde (730 mg, 5.1 mmol) was stirred in conc HCl (40 mL) at 40 °C for 15 h. The reaction was poured into H2O (80 mL) and extracted with EtOAc (3 × 50 mL). The organic layers were combined, dried (Na2SO4), and concentrated. The residue was purified via flash chromatography (SiO2, 1:4 EtOAc:hexanes) to provide the title compound. Yield 266 mg (40%), yellow oil. 1H NMR (400 MHz, CDCl3) δ 9.53 (s, 1H), 7.23 (d, J = 3.6 Hz, 1H), 6.42 (d, J = 3.6 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 176.37, 152.03, 144.15, 122.67, 109.77. HRMS (ESI) m/z [M + H]+ for C5H4ClO2 130.9900, found 130.9900.
General Procedure for Oxime Formation
NH2OH·HCl (3 equiv) and NaOAc (3 equiv) were added to a stirred solution of aromatic aldehydes (1 equiv) in MeOH (0.2 M) and stirred for 8 h at rt. The reaction was quenched with the addition of H2O (20 mL) and EtOAc (20 mL). The organic layer was washed with H2O (2 × 20 mL), separated, dried (Na2SO4), and concentrated. The residue was purified via flash chromatography (1:10 EtOAc:hexanes to 1:3 EtOAc:hexanes) to provide the desired products as amorphous solids:
5-Chlorofuran-2-carbaldehyde Oxime (66a)
Yield 225 mg (75%) as a white amorphous solid, 1:1 mixture of isomers. 1H NMR (400 MHz, CDCl3) δ 7.91 (s, 1H), 7.46 (s, 1H), 7.35 (d, J = 3.5 Hz, 1H), 6.61 (d, J = 3.4 Hz, 1H), 6.36 (d, J = 3.5 Hz, 1H), 6.26 (d, J = 3.4 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 146.42, 144.40, 139.54, 139.11, 135.93, 121.19, 114.68, 109.78, 109.37, 108.45. HRMS (ESI) m/z [M + H]+ for C5H5ClNO2 146.0009, found 146.0003.
Thiazole-2-carbaldehyde Oxime (66b)
Yield 510 mg (91%), white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.37 (s, 1H), 7.86 (d, J = 3.2 Hz, 1H), 7.33 (dd, J = 3.3, 0.9 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 144.69, 143.24, 142.28, 140.70, 123.45, 119.96. HRMS (ESI) m/z [M + H]+ for C4H5N2OS 129.0123, found 129.0124.
5-Methylthiophene-2-carbaldehyde Oxime (66c)
Yield 491 mg (76%), white amorphous solid. 1H NMR (500 MHz, CDCl3) δ 7.62 (s, 1H), 7.21 (d, J = 3.7 Hz, 1H), 6.77 (dd, J = 3.6, 1.1 Hz, 1H), 2.53 (d, J = 1.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 146.84, 141.47, 132.33, 128.93, 124.83, 15.32. HRMS (ESI) m/z [M + H]+ for C6H8NOS 142.0327, found 142.0330.
3-Vinylthiophene-2-carbaldehyde oxime (66d)
Yield 407 mg (95%), white amorphous solid, 2:1 mixture of isomers. 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.36 (s, 2H), 7.92 (s, 2H), 7.87 (s, 1H), 7.30 (d, J = 5.3 Hz, 2H), 7.25 (d, J = 5.4 Hz, 2H), 7.20 (d, J = 5.3 Hz, 1H), 7.01 (d, J = 5.4 Hz, 2H), 6.84 (dd, J = 17.4, 10.9 Hz, 1H), 5.65 (d, J = 17.3 Hz, 1H), 5.38 (d, J = 11.0 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 144.35, 143.69, 140.75, 130.66, 130.27, 130.16, 128.09, 127.53, 127.10, 125.61, 116.88, 113.85. HRMS (ESI) m/z [M + H]+ for C7H8NOS 154.0327, found 154.0331.
3-Vinylfuran-2-carboxaldehyde Oxime (66e)
Yield 105 mg (48%), white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.40 (d, J = 1.6 Hz, 1H), 6.76 (dd, J = 17.4, 10.9 Hz, 1H), 6.62 (d, J = 1.9 Hz, 1H), 5.59 (dd, J = 17.5, 1.2 Hz, 1H), 5.32 (dd, J = 10.8, 1.2 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 144.28, 143.04, 139.55, 126.52, 125.37, 116.55, 108.93. HRMS (ESI) m/z [M + H]+ for C7H8NO2 138.0555, found 138.0549.
5-(Bromomethyl)isoxazole (67)
N-Bromosuccinimide (2.14 g, 12 mmol, 1 equiv) and benzoyl peroxide (290 mg, 1.2 mmol, 0.1 equiv) were added to a stirred solution of 5-methylisoxazole (1 g, 12 mmol) in CCl4 (50 mL). The reaction was heated to 80 °C for 5 h then concentrated. The resulting residue was redissolved in EtOAc (30 mL) and washed with H2O (3 × 20 mL). The organic layer was separated, dried (Na2SO4), and concentrated. The residue was purified via flash chromatography (SiO2, 1:10 EtOAc:hexanes to 3:10 EtOAc:hexanes) to provide the title compound. Yield 725 mg (38%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.22 (d, J = 1.6 Hz, 1H), 6.33 (d, J = 1.7 Hz, 1H), 4.49 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 166.90, 150.56, 103.07, 18.34. MS (EI) m/z [M]+ for C4H4BrNO 160.9, found 161.0; [M – Br]+ for C4H4NO 82.0, found 82.0.
3,5-Dimethylfuran-2-carboxylic Acid (68)
A solution of 3-methylfuran-2-carboxylic acid (1 g, 8 mmol, 1 equiv) in THF (3 mL) was added to a stirred solution of LDA (1 M in THF, 16 mmol, 2.1 equiv) at −78 °C and stirred at this temperature for 15 min followed by the addition of iodomethane (1 mL, 16 mmol, 2 equiv). The reaction was stirred for an additional 15 min at −78 °C before removal of the ice bath and slowly warming to room temperature. The reaction was quenched with the addition of saturated NH4Cl (50 mL). The organic layer was separated, and the aqueous layer was extracted with EtOAc (2 × 40 mL). The organic layers were combined, dried (Na2SO4), and concentrated. The residue was purified via flash chromatography (SiO2, 3:10 EtOAc:hexanes to 3:5 EtOAc:hexanes) to provide the title compound. Yield 774 mg (69%) as a white amorphous solid. 1H NMR (400 MHz, CDCl3) δ 12.35 (br s, 1H), 6.00 (s, 1H), 2.31 (s, 3H), 2.30 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 164.38, 157.04, 138.03, 135.34, 112.39, 13.97, 11.83. HRMS (ESI) m/z [M + H]+ for C7H9O3 141.0552, found 141.0551.
(3,5-Dimethylfuran-2-yl)methanol (69a)
Yield 575 mg (82%), colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.80 (s, 1H), 4.52 (s, 2H), 2.24 (s, 3H), 1.99 (s, 3H), 1.62 (br s, 1H). 13C NMR (126 MHz, CDCl3) δ 145.18, 124.22, 115.31, 108.96, 61.40, 13.55, 9.68. MS (EI) m/z [M + H]+ for C7H11O2 127.1, found 127.1.
(3-Chlorothiophen-2-yl)methanol (69b)
Yield 240 mg (36%), yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.25 (dd, J = 5.4, 1.1 Hz, 1H), 6.90 (dd, J = 5.4, 1.1 Hz, 1H), 4.81 (dd, J = 6.1, 1.3 Hz, 2H), 2.04 (br s, 1H). 13C NMR (126 MHz, CDCl3) δ 135.98, 127.80, 124.60, 123.00, 57.60. MS (EI) m/z [M]+ for C5H5ClOS 148.0, found 148.0.
(4-Chlorothiophen-2-yl)methanol (69c)
Yield 165 mg (25%), yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.05 (s, 1H), 6.88 (s, 1H), 4.77 (d, J = 5.8 Hz, 2H), 1.92 (br s, 1H). 13C NMR (126 MHz, CDCl3) δ 144.73, 125.54, 124.79, 119.88, 60.09. MS (EI) m/z [M]+ for C5H5ClOS 148.0, found 148.0; [M + H]+ for C5H6ClOS 149.0, found 149.0.
Fluorescence Polarization
Assay was performed in 96-well format in black, flat bottom plates (Santa Cruz Biotechnology) with a final volume of 100 μL. 25 μL of assay buffer (20 mM HEPES, pH 7.3, 50 mM KCl, 5 mM MgCl2, 20 mM Na2MoO4, 2 mM DTT, 0.1 mg/mL BGG, and 0.01% NP-40) containing 6 nM FITC-GDA (fluorescent tracer, stock in DMSO and diluted in assay buffer) and 50 μL of assay buffer containing 10 nM of either Grp94 or Hsp90α were added to each well. Compounds were tested in triplicate wells (1% DMSO final concentration). For each plate, wells containing buffer only (background), tracer in buffer only (low polarization control), and protein and tracer in buffer with 1% DMSO (high polarization control) were included. Plates were incubated at 4 °C with rocking for 24 h. Polarization values (in mP units) was measured at 37 °C with an excitation filter at 485 nm and an emission filter at 528 nm. Polarization values were correlated to % tracer bound and compound concentrations. The concentration at which the tracer was 50% displaced by the inhibitor was determined using Graphpad Prism.
X-ray Crystallography
The gene for the truncated N-terminal domain of cGrp94, NΔ41 (amino acid residues 69–337, with residues 287–327 substituted with GGGG, as in Dollins et al.47) was synthesized and codon-optimized by DNA2.0 (Menlo Park, CA). The construct was designed with an N-terminal maltose binding protein (MBP) fusion, which is cleavable by tobacco etch virus (TEV) protease. Fidelity of the finalized MBR-TEV-NΔ41 plasmid was confirmed by Eurofins MWG Operon. The plasmid for MBP-TEV-NΔ41 was transformed in Escherichia coli BL21(DE3). Cells were grown in Luria–Bertani broth supplemented with 100 mg/L ampicillin (Amresco) at 37 °C with shaking at 225 rpm. Once cells reached an OD600 = 0.6–0.8, they were induced with 0.5 mM isopropyl-β-D-1-thiogalactopyranoside (Calbiochem) and were harvested by centrifugation after an additional 4 h of growth with shaking. Cells were solubilized in phosphate buffer (PBS, 10 mM Na2HPO4, 10 mM KH2PO4, 0.2 M NaCl, pH 7.2) containing protease inhibitor cocktail (Complete EDTA-free tablets, Roche), lysed with passage through a French Press, and the resulting lysate was clarified by ultra-centrifugation. The MBP-TEV-NΔ41 fusion protein was purified using a 20 mL column packed with amylose resin (New England BioLabs) equilibrated with PBS. Protein was eluted with a buffer containing PBS supplemented with 1 mM EDTA and 10 mM maltose. MBP-TEV-NΔ41 was further fractionated with a prep grade Superdex 75 column (GE Healthcare) equilibrated with PBS. MBP-TEV-NΔ41 was cleaved by TEV protease (prepared in house using pRK793 plasmid48), and the protease was removed by passing the mixture over a 5 mL HisTrap HP column (GE Healthcare) equilibrated with 50 mM Tris buffer with 0.5 M NaCl at pH 8.0. The mixture of MBP and NΔ41 were separated by amylose affinity purification, followed by a Superdex 75 polishing step, as above. Any fractions of NΔ41 containing trace amounts of MBP were subjected to a final round of amylose affinity purification. The NΔ41 protein purity was assessed by SDS-PAGE concentrated to 30 mg/mL in a 100 mM Tris buffer at pH 7.5, as quantified by the Bradford assay (reagents from Amresco) using BSA as a standard. Crystals of NΔ41 were grown by the handing drop method by equilibration against a reservoir solution containing 35% PEG400, 100 mM Tris at pH 7.5, and 120 mM MgCl2. Crystals were soaked for ~30 min in a solution of mother liquor containing 10 mM compound 48 (diluted from a 100 mM stock prepared in DMSO). After soaking, 100% glycerol was added to the drop containing the crystals (to a total concentration of 25%) and the crystals were cryocooled in liquid nitrogen. Diffraction data were collected at the Advanced Photon Source, Argonne National Laboratories Beamline Southeast Region Collaborateve Access Team (SER-CAT) 22-BM. Data were processed using XDS/XSCALE.49 The initial model was obtained by molecular replacement using the polypeptide chain of the Grp94-RDA structure (PDB 2GFD) the search model in Phaser.50 The model was iteratively built and refined suing Coot and Phenix refine.51 PEG 400 and glycerol ligand models were prepared by inputting the corresponding SMILES string, and additional restraints were generated with eLBOW in Phenix refine.50 The model of 48 was prepared using the PRODRG2 server, with additional restraints generated using eLBOW.52 The structure has been deposited in the Protein Data Bank with the PDB accession code 5IN9.
Wound-Healing Scratch Assay
MDA-MB-231 cells (ATCC) were grown to confluence and seeded at 200,000 cells/well/mL in a 12-well plate and grown to confluence. Each well was scratched twice with a 20–200 μL pipet tip. Compounds were then added (0.25% DMSO final concentration) and 0 h pictures were taken with an Olympus IX-71 microscope (10× objective) and the plates were returned to the incubator until 24 h pictures were taken. Images were processed and % wound closure was determined using ImageJ. All experiments were performed in quadruplicate.
Anti-Proliferative Assay
MDA-MB-231 cells were grown to confluence and seeded at 2000 cells/well/0.1 mL in a 96-well plate and placed back in the incubator for 24 h. Compounds or vehicle were administered at the desired concentrations (1% DMSO final concentration) and incubated for 72 h. The % viable cells was determined using the MTS/PMS cell proliferation kit (Promega) per the manufacturer’s instructions. Cells treated with vehicle were normalized to 100% proliferation and values adjusted accordingly.
Western Blot Analysis
MDA-MB-231 cells were grown to confluence and seeded at 200,000 cells/mL in a 10 cm dish and placed back in the incubator for 24 h. Compounds or vehicle were administered at the desired concentrations (0.25% DMSO final concentration) and incubated for 24 h. Cells were then harvested in cold PBS and lysed using MPER (Thermo Scientific) supplemented with protease inhibitors (Roche) according to the manufacturer’s instructions. Cell lysates were obtained by centrifugation at 14000g for 15 min at 4 °C. Protein concentrations for each sample were determined using the Pierce BCA assay kit following the manufacturer’s directions. Equal amounts of proteins (20 μg) were separated using gel electrophoresis under reducing conditions (10% acrylamide gels) then transferred to PVDF membranes and immunoblotted with the corresponding primary antibodies. Membranes were then incubated with the correct HRP-labeled secondary antibody, developed with a chemiluminescent substrate, and visualized.
Supplementary Material
Acknowledgments
This work was supported by grants from The National Institutes of Health to CA109265 (B.S.J.B.), EY024232 (B.S.J.B. and C.A.D.), and EY021205 (R.L.L.). Support for the NMR instrumentation was provided by NIH Shared Instrumentation Grants (S10OD016360, S10RR024664) NSF Major Research Instrumentation Grant (0320648).
ABBREVIATIONS
- Grp94
glucose-regulated protein 94
- Hsp90 and Hsp70
heat shock protein 90 and 70
- ER
endoplasmic reticulum
- TLR
toll-like receptor
- IGF
insulin-like growth factor
- NECA
N-ethylcarboxamideadenosine
- RDA
radamide
- GDA
geldanamycin
- POAG
primary open angle glaucoma
- ERAD
endoplasmic reticulum associated degradation
- TM
trabecular meshwork
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00085.
1H and 13C NMR spectra for all new compounds and HPLC traces for most active compounds (PDF) Molecular formula strings (CSV)
Notes
The authors declare no competing financial interest.
References
- 1.Li J, Buchner J. Structure, function and regulation of the Hsp90 machinery. Biomed J. 2013;36:106–117. doi: 10.4103/2319-4170.113230. [DOI] [PubMed] [Google Scholar]
- 2.Centenera MM, Fitzpatrick AK, Tilley WD, Butler LM. Hsp90: still a viable target in prostate cancer. Biochim Biophys Acta, Rev Cancer. 2013;1835:211–218. doi: 10.1016/j.bbcan.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Liu B, Staron M, Hong F, Wu BX, Sun S, Morales C, Crosson CE, Tomlinson S, Kim I, Wu D, Li Z. Essential roles of Grp94 in gut homeostasis via chaperoning canonical Wnt pathway. Proc Natl Acad Sci U S A. 2013;110:6877–6882. doi: 10.1073/pnas.1302933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suntharalingam A, Abisambra JF, O’Leary JC, Koren J, Zhang B, Joe MK, Blair LJ, Hill SE, Jinwal UK, Cockman M, Duerfeldt AS, Tomarev S, Blagg BS, Lieberman RL, Dickey CA. Glucose-regulated protein 94 triage of mutant myocilin through endoplasmic reticulum-associated degradation subverts a more efficient autophagic clearance mechanism. J Biol Chem. 2012;287:40661–40669. doi: 10.1074/jbc.M112.384800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hua Y, White-Gilbertson S, Kellner J, Rachidi S, Usmani SZ, Chiosis G, Depinho R, Li Z, Liu B. Molecular chaperone gp96 is a novel therapeutic target of multiple myeloma. Clin Cancer Res. 2013;19:6242–6251. doi: 10.1158/1078-0432.CCR-13-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kusuma BR, Zhang L, Sundstrom T, Peterson LB, Dobrowsky RT, Blagg BS. Synthesis and evaluation of novologues as C-terminal Hsp90 inhibitors with cytoprotective activity against sensory neuron glucotoxicity. J Med Chem. 2012;55:5797–5812. doi: 10.1021/jm300544c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hall JA, Forsberg LK, Blagg BS. Alternative approaches to Hsp90 modulation for the treatment of cancer. Future Med Chem. 2014;6:1587–1605. doi: 10.4155/fmc.14.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des. 2013;19:347–365. doi: 10.2174/138161213804143725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Gorska M, Popowska U, Sielicka-Dudzin A, Kuban-Jankowska A, Sawczuk W, Knap N, Cicero G, Bucchieri F, Wozniak M. Geldanamycin and its derivatives as Hsp90 inhibitors. Front Biosci Landmark Ed. 2012;17:2269–2277. doi: 10.2741/4050. [DOI] [PubMed] [Google Scholar]
- 11.Khandelwal A, Crowley VM, Blagg BS. Natural product inspired N-terminal Hsp90 inhibitors: from bench to bedside? Med Res Rev. 2016;36:92–118. doi: 10.1002/med.21351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhat R, Tummalapalli SR, Rotella DP. Progress in the discovery and development of heat shock protein 90 (Hsp90) inhibitors. J Med Chem. 2014;57:8718–8728. doi: 10.1021/jm500823a. [DOI] [PubMed] [Google Scholar]
- 13.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta, Mol Cell Res. 2012;1823:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimamura T, Perera SA, Foley KP, Sang J, Rodig SJ, Inoue T, Chen L, Li D, Carretero J, Li YC, Sinha P, Carey CD, Borgman CL, Jimenez JP, Meyerson M, Ying W, Barsoum J, Wong KK, Shapiro GI. Ganetespib (STA-9090), a nongeldanamycin HSP90 inhibitor, has potent antitumor activity in in vitro and in vivo models of non-small cell lung cancer. Clin Cancer Res. 2012;18:4973–4985. doi: 10.1158/1078-0432.CCR-11-2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garon EB, Finn RS, Hamidi H, Dering J, Pitts S, Kamranpour N, Desai AJ, Hosmer W, Ide S, Avsar E, Jensen MR, Quadt C, Liu M, Dubinett SM, Slamon DJ. The HSP90 inhibitor NVP-AUY922 potently inhibits non-small cell lung cancer growth. Mol Cancer Ther. 2013;12:890–900. doi: 10.1158/1535-7163.MCT-12-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson LB, Eskew JD, Vielhauer GA, Blagg BS. The hERG channel is dependent upon the Hsp90α isoform for maturation and trafficking. Mol Pharmaceutics. 2012;9:1841–1846. doi: 10.1021/mp300138n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen B, Piel WH, Gui L, Bruford E, Monteiro A. The HSP90 family of genes in the human genome: insights into their divergence and evolution. Genomics. 2005;86:627–637. doi: 10.1016/j.ygeno.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 18.Taldone T, Patel PD, Patel M, Patel HJ, Evans CE, Rodina A, Ochiana S, Shah SK, Uddin M, Gewirth D, Chiosis G. Experimental and structural testing module to analyze paralogue-specificity and affinity in the Hsp90 inhibitors series. J Med Chem. 2013;56:6803–6818. doi: 10.1021/jm400619b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song HY, Dunbar JD, Zhang YX, Guo D, Donner DB. Identification of a protein with homology to Hsp90 that binds the type 1 tumor necrosis factor receptor. J Biol Chem. 1995;270:3574–3581. [PubMed] [Google Scholar]
- 20.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–276. doi: 10.1038/nrc3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stothert AR, Suntharalingam A, Huard DJ, Fontaine SN, Crowley VM, Mishra S, Blagg BS, Lieberman RL, Dickey CA. Exploiting the interaction between Grp94 and aggregated myocilin to treat glaucoma. Hum Mol Genet. 2014;23:6470–6480. doi: 10.1093/hmg/ddu367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. J Biol Chem. 2003;278:48330–48338. doi: 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- 23.Marzec M, Eletto D, Argon Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta, Mol Cell Res. 2012;1823:774–787. doi: 10.1016/j.bbamcr.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stothert AR, Fontaine SN, Sabbagh JJ, Dickey CA. Targeting the ER-autophagy system in the trabecular meshwork to treat glaucoma. Exp Eye Res. 2016;144:38–45. doi: 10.1016/j.exer.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weekes MP, Antrobus R, Talbot S, Hor S, Simecek N, Smith DL, Bloor S, Randow F, Lehner PJ. Proteomic plasma membrane profiling reveals an essential role for gp96 in the cell surface expression of LDLR family members, including the LDL receptor and LRP6. J Proteome Res. 2012;11:1475–1484. doi: 10.1021/pr201135e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, Que NS, Taldone T, Finotti P, Stephani RA, Gewirth DT, Chiosis G. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013;9:677–684. doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel HJ, Patel PD, Ochiana SO, Yan P, Sun W, Patel MR, Shah SK, Tramentozzi E, Brooks J, Bolaender A, Shrestha L, Stephani R, Finotti P, Leifer C, Li Z, Gewirth DT, Taldone T, Chiosis G. Structure-activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum Hsp90 paralog Grp94. J Med Chem. 2015;58:3922–3943. doi: 10.1021/acs.jmedchem.5b00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duerfeldt AS, Peterson LB, Maynard JC, Ng CL, Eletto D, Ostrovsky O, Shinogle HE, Moore DS, Argon Y, Nicchitta CV, Blagg BS. Development of a Grp94 inhibitor. J Am Chem Soc. 2012;134:9796–9804. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muth A, Crowley V, Khandelwal A, Mishra S, Zhao J, Hall J, Blagg BS. Development of radamide analogs as Grp94 inhibitors. Bioorg Med Chem. 2014;22:4083–4098. doi: 10.1016/j.bmc.2014.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosser MF, Nicchitta CV. Ligand interactions in the adenosine nucleotide-binding domain of the Hsp90 chaperone, GRP94. I. Evidence for allosteric regulation of ligand binding. J Biol Chem. 2000;275:22798–22805. doi: 10.1074/jbc.M001477200. [DOI] [PubMed] [Google Scholar]
- 31.Immormino RM, Dollins DE, Shaffer PL, Soldano KL, Walker MA, Gewirth DT. Ligand-induced conformational shift in the N-terminal domain of GRP94, an Hsp90 chaperone. J Biol Chem. 2004;279:46162–46171. doi: 10.1074/jbc.M405253200. [DOI] [PubMed] [Google Scholar]
- 32.Wassenberg JJ, Reed RC, Nicchitta CV. Ligand interactions in the adenosine nucleotide-binding domain of the Hsp90 chaperone, GRP94. II. Ligand-mediated activation of GRP94 molecular chaperone and peptide binding activity. J Biol Chem. 2000;275:22806–22814. doi: 10.1074/jbc.M001476200. [DOI] [PubMed] [Google Scholar]
- 33.Immormino RM, Metzger LE, IV, Reardon PN, Dollins DE, Blagg BS, Gewirth DT. Different poses for ligand and chaperone in inhibitor-bound Hsp90 and GRP94: implications for paralog-specific drug design. J Mol Biol. 2009;388:1033–1042. doi: 10.1016/j.jmb.2009.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park KD, Morieux P, Salome C, Cotten SW, Reamtong O, Eyers C, Gaskell SJ, Stables JP, Liu R, Kohn H. Lacosamide isothiocyanate-based agents: novel agents to target and identify lacosamide receptors. J Med Chem. 2009;52:6897–6911. doi: 10.1021/jm9012054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clevenger RC, Blagg BS. Design, synthesis, and evaluation of a radicicol and geldanamycin chimera, radamide. Org Lett. 2004;6:4459–4462. doi: 10.1021/ol048266o. [DOI] [PubMed] [Google Scholar]
- 36.Hadden MK, Blagg BS. Synthesis and evaluation of radamide analogues, a chimera of radicicol and geldanamycin. J Org Chem. 2009;74:4697–4704. doi: 10.1021/jo900278g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naci D, Vuori K, Aoudjit F. α2β1 integrin in cancer development and chemoresistance. Semin Cancer Biol. 2015;35:145–153. doi: 10.1016/j.semcancer.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 39.Wu S, Hong F, Gewirth D, Guo B, Liu B, Li Z. The molecular chaperone gp96/GRP94 interacts with Toll-like receptors and integrins via its C-terminal hydrophobic domain. J Biol Chem. 2012;287:6735–6742. doi: 10.1074/jbc.M111.309526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haidari M, Zhang W, Caivano A, Chen Z, Ganjehei L, Mortazavi A, Stroud C, Woodside DG, Willerson JT, Dixon RA. Integrin α2β1 mediates tyrosine phosphorylation of vascular endothelial cadherin induced by invasive breast cancer cells. J Biol Chem. 2012;287:32981–32992. doi: 10.1074/jbc.M112.395905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dejeans N, Glorieux C, Guenin S, Beck R, Sid B, Rousseau R, Bisig B, Delvenne P, Buc Calderon P, Verrax J. Overexpression of GRP94 in breast cancer cells resistant to oxidative stress promotes high levels of cancer cell proliferation and migration: implications for tumor recurrence. Free Radical Biol Med. 2012;52:993–1002. doi: 10.1016/j.freeradbiomed.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 42.Ghosh S, Shinogle HE, Galeva NA, Dobrowsky RT, Blagg BS. Endoplasmic reticulum resident heat shock protein 90 (Hsp90) isoform glucose-regulated protein 94 (Grp94) regulates cell polarity and cancer cell migration by affecting intracellular transport. J Biol Chem. 2016 doi: 10.1074/jbc.M115.688374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tamm ER, Braunger BM, Fuchshofer R. Intraocular pressure and the mechanisms involved in resistance of the aqueous humor flow in the trabecular meshwork outflow pathways. Prog Mol Biol Transl Sci. 2015;134:301–314. doi: 10.1016/bs.pmbts.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 44.Donegan RK, Lieberman RL. Discovery of molecular therapeutics for glaucoma: challenges, successes, and promising directions. J Med Chem. 2016;59:788–809. doi: 10.1021/acs.jmedchem.5b00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hill SE, Donegan RK, Lieberman RL. The glaucoma-associated olfactomedin domain of myocilin forms polymorphic fibrils that are constrained by partial unfolding and peptide sequence. J Mol Biol. 2014;426:921–935. doi: 10.1016/j.jmb.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 47.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the Hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tropea JE, Cherry S, Waugh DS. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol Biol. 2009;498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- 49.Kabsch W, editor. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.