

Summary
Background
microRNAs negatively regulate gene expression at the post-transcriptional level. Mounting evidence shows that miR expression is deregulated in human cancers including head and neck squamous cell carcinoma (HNSCC). Epigenetically silenced tumor suppressor miRs may be re-expressed upon treatment with histone deacetylases inhibitors. Suberoylanilide Hydroxamic Acid (SAHA) is a histone deacetylase inhibitor that is currently being investigated in clinical trials for HNSCC. We hypothesized that SAHA will re-express a set of tumor suppressor miRs and enhance the efficacy of cisplatin and radiation in HNSCC.
Results
In this study, miR expression profile was utilized to identify the tumor suppressor miRs that are re-expressed following SAHA treatment in HNSCC. Our data demonstrated that two tumor suppressor miRs, miR-107 and miR-138, were significantly up-regulated in CAL27 and SCC25 cell lines, following SAHA treatment. In addition to this, treatment with SAHA in a dose dependent manner significantly inhibited the cell proliferation, cell migration, and anchorage dependent clonogenic survival in CAL27 and SCC25 cell lines, respectively. Further, the expression of several oncogenes, PKCε, HIF1β, CDK6, and RhoC were down regulated in response to SAHA treatment. Additionally, we demonstrated that the combination treatment with SAHA and a chemotherapeutic drug cisplatin caused a significant reduction of cell growth compared to the single agent treatment.
Conclusion
Our data indicate that SAHA treatment results in reactivation of the silenced tumor suppressor miRs. Furthermore, this study emphasizes the usefulness of this drug as a novel combination therapy for HNSCC patients.
Keywords: Suberoylanilide hydroxamic acid, HDAC inhibitor, Tumor suppressor miR, Head and neck cancer, PKCε, RhoC, miR-107, miR-138
Introduction
Head and neck squamous cell carcinoma (HNSCC) is one of the most prevalent cancers worldwide [24,32]. HNSCC has traditionally been treated with a multi-disciplinary approach involving surgery, radiation- and/or chemotherapy. However, despite intensive treatment, the overall 5-year survival rate has remained stagnant at about 50%. The mortality is not only due to loco-regional failure but also because of disease recurrence and distant metastasis to the lungs and bones [24,39,48]. Better understanding of the molecular mechanisms that drive HNSCC is needed to identify alternative therapeutic options for the HNSCC population.
microRNAs (miRs) are endogenous, small non-coding RNA molecules (18–25 nucleotides long; single-stranded) that, in general, function in RNA silencing and regulate gene expression at the post-transcriptional level. miRs usually bind imperfectly to the 3′-untranslated region of the cognate mRNA and results in reduced translation and/or degradation of mRNA [6]. A single miR is able to target numerous genes to regulate biological processes globally. Therefore, several biological processes such as cell proliferation, cell migration, apoptosis, differentiation, development, immunity, and metabolism are regulated by miRs [17,49]. Importantly, miRs are dysregulated in numerous cancers and can function as oncogenes or tumor suppressors [7,38].
Deacetylation of histones promotes tumorigenesis by repressing genes that inhibit cell cycle progression, differentiation, apoptosis, cell adhesion and inducing expression of genes involved in angiogenesis, cell migration, and invasion. miRs, like protein coding genes, can also be silenced epigenetically and re-expressed by chromatin modifiers [5,25]. Histone deacetylase (HDAC) inhibitors reverse cellular transformation by altering expression of the genes involved in these pathways. HDAC inhibitors have recently shown promise as therapeutic agents for cancer. Although, favorable responses to these compounds as single agents have been demonstrated in several hematological malignancies [10,31,34,44], their efficacy has also been evaluated in multiple clinical trials with a wide variety of solid tumors [36]. Suberoylanilide hydroxamic acid (SAHA) is a FDA approved histone deacetylase inhibitor that is currently under study in HNSCC [10,31]. To increase the efficacy of SAHA-mediated cell cytotoxicity, we explored the miRs and associated genes that are altered upon SAHA treatment. To identify candidate tumor suppressor miRs that are silenced by an epigenetic mechanism in HNSCC, we performed microRNA microarray analysis in two cell lines treated with SAHA. Here, we show that miR-107 and miR-138 are such genes that are reactivated in human HNSCC cells, and their activation by SAHA suppresses tumor cell growth by downregulating their oncogenic targets PKCε, HIF1β, CDK6, and RhoC, which are involved in crucial signaling pathways promoting tumorigenesis.
Materials and methods
Cell culture and treatment with drugs
Human head and neck cancer cell lines namely CAL27, and SCC25, were procured from ATCC (Rockville, MD, USA). CAL27 and SCC25 cells were cultured in Dulbecco Modified Eagle Medium and DMEM/F12 (1:1) medium, respectively containing 10% fetal bovine serum (High clone, UT, USA). Primary keratinocytes (ATCC) were cultured in primary keratinocyte growing media (Life Technologies, NY, USA) supplemented with 5 μg/ml hydrocortisone, 5 μg/ml insulin, 10 ng/ml EGF, 2 μg/ml bovine pituitary extract, and 10−9 M triiodothyronine.
HNSCC or primary keratinocyte cells were treated with DMSO or SAHA (Cayman Chemical) for 72 h. For combination treatment with SAHA and cis-pt, cells were treated with DMSO or SAHA for 24 h followed by 48 h with cis-pt (Sigma Chemical Co, MO, USA) at different doses as indicated. Media was changed every day.
Real time RT-PCR
Total RNA was isolated from SAHA treated HNSCC cell lines (CAL 27 and SCC25) as well as from primary keratinocytes using TRIzol reagent (Life Technologies, NY, USA). Expression of mature miRs, miR-107, miR-138, and RNU44 were determined using the Applied Biosystems 7900HT Fast Real-Time PCR System with validated TaqMan gene expression assays (Applied Biosystems, Foster City, CA). The levels of mature microRNAs were normalized to RNU44 using 2−(−ΔΔ)Ct method [27].
Cell proliferation assay
Cell proliferation assay was performed in CAL27 and SCC25 in the absence (control) or presence of SAHA (1 μM) for up to 72 h. The assay was conducted on xCELLigence system according to the manufacturer's protocol (Roche Applied Sciences, Indianapolis, IN).
Cell motility assay
Cell motility assays were carried on in 6 well cell culture plates. At ∼80% confluence, cells were washed with PBS and a fine scratch in the form of a groove was made and immediately photographed. We designated this time as the 0 h. Next, cells were supplemented with complete medium and allowed to grow in the absence or presence of various concentrations of SAHA. A migration of cells from the edge of the groove towards the center was monitored microscopically at 40× magnifications after 24 h to assess the extent of scratched area covered. The width of the scratch was measured at 0 h and after 24 h to calculate the percentage of the gap covered by the cells in a 24 h time period.
Clonogenic survival assay
The clonogenic survival assay was performed according to the method of Franken et al. [12]. Briefly, equal number of SAHA treated or untreated cells were allowed to grow in culture plates for two weeks. At the termination of the assay, cells were rinsed with PBS followed by fixing with 4% paraformaldehyde. Colonies formed were stained with 0.5% (w/v) crystal violet prepared in 25% (v/v) methanol and finally rinsed with water and air-dried.
Western blot analysis
Equal amount of control and SAHA-treated cell lysates were mixed with Laemmli's buffer [20], boiled, and separated on SDS–polyacrylamide gels. Proteins were transferred onto PVDF membranes and probed with polyclonal PKCε, HIF1β, CDK6, and RhoC antibodies (Cell Signaling, MA, USA) followed by anti-rabbit/mouse HRP-conjugated secondary antibodies. Tubulin was used as a loading control. Membranes were visualized using ECL detection system (Amersham Life Science, NJ, USA).
Cell viability assay
Cells were pretreated either with DMSO or SAHA alone for 24 h followed by combined treatment with SAHA+ cis-pt for 48 h. Media was changed every day. At the completion of the treatment cells were exposed to gamma radiation (2 Gy), allowed overnight to recover from chemo-radiation shock. Next, equal number of cells were plated in 96 well plates and allowed to grow for 96 h. Cell viability assay was conducted on these cells using CellTiterGlo kit (Promega, WI, USA).
β-Galactosidase staining of primary keratinocyte
Primary keratinocyte cells were plated in six well plates, 24 h later treated with DMSO or SAHA (at the doses mentioned) for 72 h. Cells were then washed with PBS and stained with β-Galactosidase staining solution using Senescence β-Galactosidase staining kit (Cell Signaling) following the manufacturer's protocol.
Statistical analysis
Statistical analyses were performed using Student's t-test. The mean was reported with Standard deviation (±SD). Differences were considered to be statistically significant when p values were less than 0.05.
Results
Tumor suppressor miRs are significantly upregulated in SAHA-treated head and neck cancer cells
Several studies including ours have demonstrated that growth of cancer cells could be inhibited by activating miRs and tumor suppressor genes through treatment with inhibitors of DNMTs or HDACs alone or in combination [1,3,7,8,13,22]. Numerous tumor suppressor miRs including miR-107 and miR-138 are known to be downregulated in head and neck cancer [9,16,26]. To explore whether those tumor suppressor miRs are epigenetically silenced and could be re-expressed in head and neck tumors, we performed quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) in two cell lines (CAL27 and SCC25) upon SAHA treatment. Previous studies in our laboratory [9,16] and others [26,41] demonstrated that both miR-107 and miR-138 are downregulated in primary HNSCC tumors and cell lines as well. We have shown that miR-107 directly targets PKCε, an overexpressed pro-tumorigenic protein and emerging biomarker [14], and inhibits the activity of this protein by binding at the 3′UTR of its mRNA [9]. Furthermore, studies by Lee et al. [21] demonstrated that miR-107 promoter is methylated and suppressed in pancreatic cancer cells, which could be reversed with chromatin-modifying agents such as 5-aza-2′-deoxycytidine (5-Aza-dC) or the HDAC inhibitor, trichostatin A, alone or in combination. miR-138 targets RhoC [16,18], a pro-metastatic oncogene, that is constitutively expressed in primary HNSCC tissues and most of the head and neck cancer cell lines [16]. As these two miRs are crucial for HNSCC development, we examined whether expression of these two miRs was altered upon SAHA treatment. Indeed, both of these tumor suppressor miRs were significantly upregulated in the SAHA-treated cell lines (Fig. 1A–D). Real-time PCR analysis demonstrated a significant increase in miR-107 (Fig. 1A and B) and miR-138 (Fig. 1C and D) in both SAHA-treated cell lines compared to the respective untreated controls. Taken together, these findings suggest the SAHA plays a significant role in upregulation of validated tumor suppressor miRs in HNSCC cell lines.
Fig. 1.
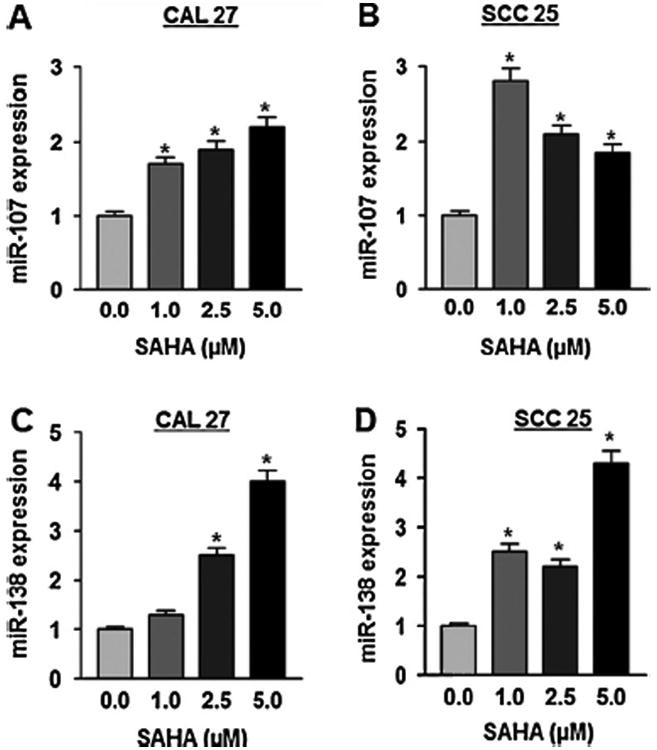
miR-107 and miR-138 are upregulated in SAHA treated HNSCC cell lines. Real time RT-PCR analysis of miR-107, miR-138 and RNU44 was performed with total RNA isolated from CAL27 and SCC25 cell lines treated with DMSO or SAHA (at different doses as indicated in Materials and methods) using validated TaqMan assays. Expression of miR-107/miR-138 was normalized with RNU44. Each assay was performed in triplicate and repeated twice. Data showing multifold increments in miR-107 (A and B), miR-138 (C and D), expressions in SAHA treated CAL 27 and SCC25 cell lines respectively. Data is presented as mean ± SD, *p-value (p ≤ 0.05).
SAHA treatment reduces tumorigenic potential of HNSCC in vitro
Our data demonstrated that SAHA treatment results in upregulation of various miRs in two HNSCC cell lines. To gain insight into the dose response/toxic effect of SAHA we first determined the IC50 (50% growth inhibition) values for these two cell lines. As shown in Fig. 2A, the IC50 values of SAHA for CAL27 and SCC25 were found to be 14.6 and 49.5 μM, respectively. Next, we performed several functional studies to elucidate whether SAHA treatment and consequent miR upregulation could reduce the cancer cell growth. Cell proliferation was evaluated upon treating the cells with SAHA (1 μM) for 72 h in CAL27 and SCC25 lines. The proliferation rate of SAHA treated cells was markedly reduced compared with the untreated counterpart (Fig. 2B). Following SAHA treatment cell proliferation was decreased by 80 ± 3% (p < 0.01) and 90 ± 3% (p < 0.01) in CAL27 and SCC25 cells, respectively. We then performed a cell motility assay to investigate whether SAHA treatment could lead to reduction of migratory properties of HNSCC cell lines. As shown in Fig. 2C, there was a significant decrease in the cell motility in both the cell lines in the presence of different concentrations of SAHA. Moreover, cell migration was reduced in a dose dependent manner. Reduction in cell motility is more pronounced in SCC25 as compared to CAL27 (Fig. 2C). Treatment with SAHA at 5.0 μM inhibited cell migration by 60 ± 4% in CAL27 cells (p < 0.01) (Fig. 2C) and 75 ± 3% in SCC25 cells (p < 0.01) (Fig. 2C). We also determined anchorage dependent colony formation, which is one of the hallmarks of cancer cells. We treated CAL27 and SCC25 cells with two different concentrations of SAHA (1 μM and 2.5 μM). A decrease in colony formation was obtained in a dose dependent manner in both the cell lines tested (Fig. 2D). However, CAL27 was a little more sensitive to SAHA treatment for colony formation ability when compared to SCC25 (Fig. 2D). Colony forming ability was decreased by 81 ± 3% and 88 ± 2%, respectively at 1 μM and 2.5 μM doses of SAHA in CAL27 (p < 0.01) while corresponding inhibitions were 70 ± 3% and 87 ± 1.5% in SCC25 cells (p < 0.01) (Fig. 2D).
Fig. 2.
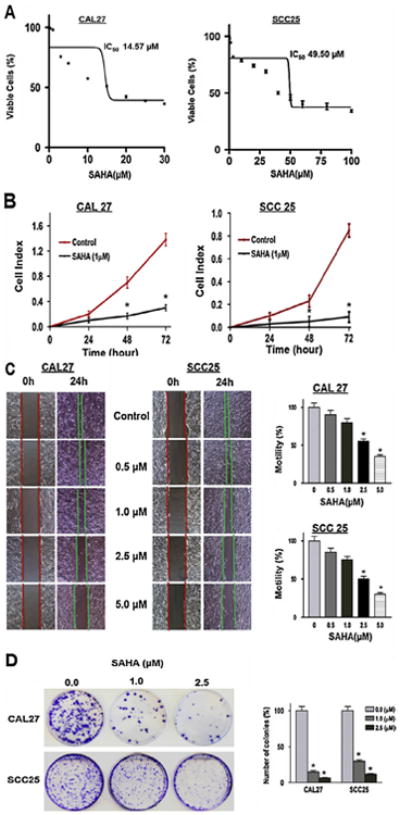
SAHA treatment reduced tumorigenic properties of CAL27 and SCC25 cells in vitro. (A) Determination of IC50. Cells were treated with varying concentrations of SAHA (0–100 μM), and the percentage of viable cells was quantitated after 24 h by CellTiterGlo assay. Representative data from three experimental replicates are shown. Data is presented as mean ± SD. (B) Cell proliferation. Cell proliferation assay was performed using equal number of cells on xCELLigence assay while cells were either treated with DMSO or SAHA (1 μM). Each assay was performed in quadruplicate and repeated twice. Data showing 94% and 97% decrease in cell proliferation upon SAHA treatment in CAL 27 and SCC25, respectively. Data is presented as mean ± SD, (*p-value ≤ 0.05). (C) Cell migration. Cell migration/motility assay was performed using equal number of cells on six well plates while cells were either treated with DMSO or SAHA at the indicated doses (for details see Methods section). Cell motility assay shows the reduction in cell motility from the edges of the wounded cell into the central area in presence of different concentrations of SAHA in CAL27 and SCC25 cell lines. Columns of the bar graphs are representing the area covered by the cells in 24 h time point (magnification 40×). Each assay was performed in triplicate and repeated twice. Data is presented as mean ± SD, (*p-value ≤ 0.05). (D) Colony formation. Clonogenic survival assay was performed using equal number of cells with cells pre-treated with DMSO or SAHA at the indicated doses (for details see Methods section). Colonies were stained with crystal violet and counted. Clonogenic survival assay of different concentration of SAHA treated CAL27 and SCC25 (top). Graphical representation of the colonies formed in control and treated cell lines (bottom). A significant decrease in colony formation is observed in these cell lines (magnification 40×). Each assay was performed in triplicate and repeated twice. Data is presented as mean ± SD, (*p-value ≤ 0.05).
SAHA treatment down-regulates specific miR-targeted oncogenic protein expression in HNSCC cell lines
Because the tumor suppressor role of miR-107 [9,33] and miR-138 [16,33,41] had been well established in HNSCC, we inquired whether the increased expression of these miRs in SAHA-treated cell lines was accompanied by reduced level of their oncogenic target proteins. Therefore, we examined the levels of PKCε and HIF1β, which are targets of miR-107 using the control-and SAHA-treated cell lysates of CAL27 and SCC25. Also, we tested the level of CDK6, which is one of the target proteins of both miR-107 and -138and RhoC, which is the target protein of miR-138. Western blot analysis was performed with the lysates prepared from SAHA treated (24 h) cell lines for the evaluation of target protein expression (Fig. 3A and C). Normalized band intensity corresponding to their respective loading control (tubulin) is shown in Fig. 3B and D. It is clear that there is a significant decrease (70–95%; p < 0.005) in expression of the target proteins upon SAHA treatment (5 μM) in both the HNSCC cell lines (Fig. 3). Moreover, decrease in the level of these proteins was proportional to increased doses of SAHA. While there is increased expression of the indicated miRs (Fig. 1), a concentration dependent decrease in the level of the target proteins (Fig. 3) demonstrates that an inverse association between the expression level of miRs and their respective targets is achieved due to SAHA treatment in HNSCC cell lines. It is, therefore, evident that the anti-tumor effect of SAHA in HNSCC cell lines is achieved at least in part through a resultant decrease in expression of oncogenic proteins due to re-expression of silenced tumor suppressor miRs that target them.
Fig. 3.
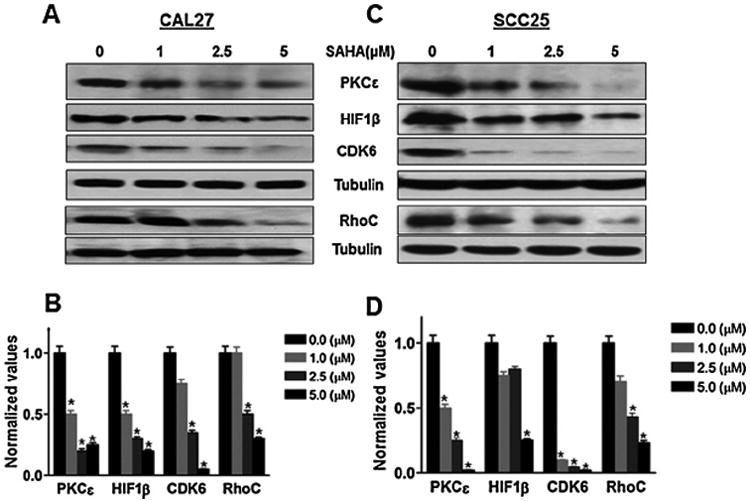
SAHA reduces expression of several oncogenic target proteins of tumor suppressor miRs in HNSCC. Western blots analysis of miRs target proteins which includes PKCε, HIF1β (targets of miR107) and CDK6, RhoC (targets of miR-107 and miR-138) showing decrease in expression after SAHA treatment (24 h) in CAL27 and SCC25 (A and C). Bar graphs (B and D) are the densitometric analysis of the bands using tubulin as the loading control. A remarkable decrease in most of the target proteins can be seen. Western blot was repeated twice.
The combination of SAHA with chemo-and/or radiation increased the cytotoxicity of HNSCC cells
A common modality of treating various cancers including HNSCC is using a drug combination with DNA damaging chemo-or radio-therapeutic agents [42]. To assess whether SAHA alone or in combination with cis-pt (chemo), and/or radiation could have a more potent effect on HNSCC cytotoxicity, we performed colony formation and cell viability assays after the combination treatments. We treated CAL27 cell line with different doses of SAHA and cis-pt alone or in combination followed by with or without radiation. Colony forming ability was significantly reduced upon treatment with SAHA as a single agent (Fig. 4). The reduction in colony formation in CAL27 was (81 ± 2% and 86 ± 1.5% at 1 μM and 2.5 μM, respectively; Fig. 4A and B). Exposure to radiation following SAHA treatment caused remarkable reduction (92 ± 2% in CAL27) of clonogenic survival compared to SAHA (1 μM) as single agent. Cis-pt alone or in combination with radiation showed similar result to that of SAHA (Fig. 4). On the other hand, combined treatment with SAHA and cis-pt (each at 1 μM) followed by radiation almost completely abrogated the clonogenic survival (Fig. 4A and B). In these experiments, a small increased dose of SAHA (2.5 μM) in combination with a very low concentration of cis-pt (0.5–1.0 μM) caused total inhibition of colony formation without any radiation exposure. These data demonstrated that SAHA potentiates the activity of cis-pt and/or radiation on cell viability. We also investigated whether normal keratinocytes are affected to the same extent as the HNSCC cells when treated similarly. To compare them with the HNSCC cell lines, we performed a cell viability assay. As shown in Fig. 4C, the cancer cell line (CAL27) displayed a significant decrease in cell viability when treated with SAHA alone. Cell death was more prominent when the viability was tested in combination of SAHA/cis-pt followed by exposure to gamma rays (70–95% decrease in viability). This data corroborated very well with the clonogenic survival assay. Interestingly, primary keratinocytes did not show a robust decrease in cell viability when compared to the cancer cells under similar treatment conditions (Fig. 4D). It should be noted that SAHA did not promote senescence in primary keratinocytes (Supplemental Fig. S1). These results suggest that the combination of SAHA and/or cis-pt/radiation could be used for effective HNSCC therapy in the clinic. Furthermore, the specificity of the chemo-radiation therapy towards cancer cells indicates that these drug combinations may be more selective to ablate cancer cells than normal cells.
Fig. 4.
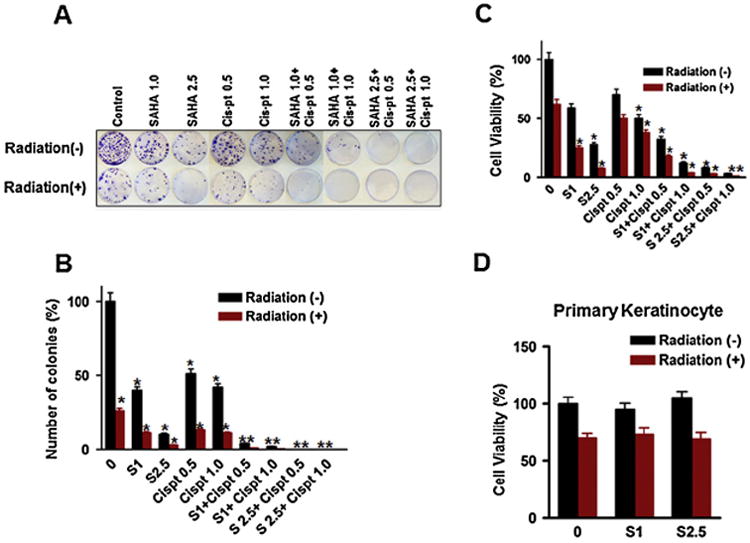
Combined treatment of SAHA and cis-pt and/or radiation abrogates colony forming ability and significantly reduces cell viability of HNSCC cell lines. Colony formation assay was performed in CAL27 cell line treated with SAHA alone or in combination with cis-pt and/or radiation as described in Methods section. Colony formation and quantitative bar graphs of: CAL27 (A and B). Cell viability assay on CAL 27 (C) and primary keratinocytes (D) showing the cell viability potential in the presence of various concentrations and combinations of SAHA and cis-pt and also in the absence or in the presence of radiation (for details see Methods section). Axis is labeled S (SAHA) followed by numeric values (μg/ml concentration). Negative sign (−) represents no radiation, while positive sign (+) indicates presence of radiation. Each assay was performed in triplicate and repeated twice. Data is presented as mean ± SD (*p-value ≤ 0.05).
Discussion
Inhibitors of HDACs have appeared as a very effective class of therapeutics for several cancers. Among the HDAC inhibitors, SAHA is the first FDA approved drug that was initially used successfully in the clinic for treating cutaneous T-cell lymphoma [2,10,31]. In addition, albeit SAHA had been evaluated to target HDACs; it was also found to be very effective on other epigenetic machineries including, acetylation, demethylation of non-HDACs, and miR expression [2,22]. Consistent with these abilities described, in a recent study we have shown that SAHA could reverse cisplatin resistance and potentiate its anti-tumor effect, when used in combination, by dampening down the cancer stem cell factor nanog and thereby blocking tumor metastasis [19]. In this present study, SAHA was shown to reactivate 2 key tumor suppressor miRs and thus, provide a novel mechanism of action for the anti-cancer activity of SAHA in HNSCC.
We concentrated on miRs as currently there is considerable interest focused on the potential therapeutic application of anti-miRs (against oncogenic miRs) and miR mimetics (for tumor suppressor miRs) against cancer because of their small size, stability, ability for targeted delivery and pleotropic functions reducing the possibility of developing resistance [15]. Published works have clearly shown that tumor suppressor miRs are epigenetically silenced in various cancer types including HNSCC [4,8,28,29,40]. Therefore, restoration of these silenced miRs through treatment with epigenetic drugs may perhaps have high potential for cancer therapies. Although several reports are available showing re-expression of tumor suppressor miRs using HDAC inhibitors as a single agent or combination with DNMT inhibitors in a variety of malignancies, none of the studies involved miR activation by HDAC inhibitors in HNSCC. Here, for the first time, we demonstrated that several tumor suppressor miRs were reactivated upon SAHA treatment in HNSCC. Consistent with our work, other groups have reported SAHA-mediated alteration of miR expression profiles in lung, breast, liver, and pancreatic cancer [8,22,30,47]. Our results revealed that SAHA treatment profoundly increased the expression of miR-107. This is consistent with our previous observation, where we showed that miR-107 is dramatically downregulated and functions as a tumor suppressor in HNSCC [9,33]. Furthermore, several other researchers in various cancers have demonstrated reduced expression and tumor suppressor function of miR-107 and its overexpression diminished tumorigenic potential [11,23,46]. Likewise, the expression level of another tumor suppressor miR, miR-138 increased significantly in SAHA-treated cells. Moreover, miR138 is under-expressed and functions as a tumor suppressor in HNSCC [16,18,26]. Further, we have observed an increased expression of miR-138 with a concomitant decrease in its target proteins RhoC and CDK6 [35] upon treatment of HNSCC cell lines with 5-Aza-dc (data not shown). Therefore, we have demonstrated that SAHA reactivated potential tumor suppressor miRs in HNSCC.
In our study, SAHA treatment was very effective in modulating the tumorigenic potential of HNSCC lines. Cell proliferation, cell migration, and colony forming ability of the HNSCC cell lines were significantly blocked even when SAHA was used as a single agent. We have previously shown that miR-107 dampened down the tumorigeneicity of HNSCC by targeting PKCε, HIF1β, and CDK6 that are highly expressed and play important roles in tumorigenesis [9,33]. Moreover, PKCε modulates the activation of well-recognized players in HNSCC such as, Akt, Stat3, and Rho GTPases [14]. Thus, expression of miR-107 reduces the cancer initiating cell population and suppresses the expression profile of core stemness transcription factors. Therefore, our results strongly suggest that the anti-tumor effect of SAHA is due to the resultant reactivation of miR-107 and concomitant decrease in the level of its target proteins. Accordingly, our studies lead to the notion that re-activation of miR107 could be beneficial as an anti-cancer therapeutic for HNSCC. We also demonstrated that in SAHA treated cells the miR-138 level is increased in association with significant decrease in the expression of its target proteins, RhoC and CDK6. However, miR-138 had been documented to exert its anti-tumor effect upon directly targeting several key oncogenic proteins (including RhoC and CDK6) and thereby blocking multiple cellular pathways in HNSCC and other cancers [18,37,45]. In line with these reports, restoring miR-138 expression led to suppression of cell invasion, migration, EMT, caused cell cycle arrest and induced apoptosis. Enforced expression of miR-138 suppressed expression of 194 proteins among which 51 are direct targets that play crucial roles in these cellular pathways. Consequently, SAHA-mediated inhibition of tumorigenic potential, at least in part, is due to the resultant expression of miR-138. Taken together, our study established that SAHA exerts its anti-tumor effect by restoring the epigenetically silenced tumor suppressor miRs.
Previous studies have shown that several HNSCC patients specifically those who are human papillomavirus (HPV) negative, usually have more aggressive disease, and develop resistance to chemotherapy. We recently demonstrated the anti-tumor effect of SAHA in HPV negative cis-pt resistant HNSCC [19]. Consistent with this observation, our results showed that SAHA alone reduced the colony forming ability and cell viability of HPV negative CAL27 cell line significantly. In addition, we demonstrated that combination of SAHA along with cis-pt and/or radiation completely abrogated the cell growth and viabilities. Accumulating evidence suggests a critical role of miR-138 in DNA damage and chemoresistance [43]. miR-138 directly targets histone H2AX, one of the major players of DNA damage response pathways. Our study further shows that SAHA reactivated miR-138, which in turn sensitized the HNSCC cells to DNA damaging agent cis-pt and/or radiation. Our results, therefore, suggest another new direction for treatment with SAHA along with cis-pt and/or radiation to obtain a better therapeutic index.
In summary, we have identified that miR suppression plays a significant role in HNSCC progression. SAHA significantly re-expressed two tumor suppressor miRs, miR-107 and miR-138, with accompanied reduction of the levels of recognized oncogenes, PKCε, HIF1ε, CDK6, and RhoC. In addition, the combination of SAHA and cis-pt coupled with radiation therapy decreased the potential of cancer cell survival with minimal effect on normal keratinocytes. Our work provide key evidence that reactivation of tumor-suppressor miRs through SAHA treatment may be partly responsible to enhance the efficacy of standard chemo-/radiation therapy in HNSCC.
Supplementary Material
Acknowledgments
This work was supported, in part, by The John Young Memorial Grant from The Joan Bisesi Fund for Head and Neck Oncology Research (to JD), The Slomin Foundation, and Arthur G. James Cancer Hospital and Richard J. Solove Research Institute, The Ohio State University Comprehensive Cancer Center. Authors thank Dr. James Lang for critical reading of the manuscript.
Role of funding source: None of the funding agency had any involvement in the study design, analysis, data interpretation, writing, and in the decision to submit the article for publication.
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
Appendix A. Supplementary material: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.oraloncology.2016.02.015.
References
- 1.Baylin SB. Novartis Foundation Symposium. Chichester, New York: John Wiley; 1999. Reversal of gene silencing as a therapeutic target for cancer-roles for DNA methylation and its interdigitation with chromatin. [PubMed] [Google Scholar]
- 2.Boumber Y, Issa J. Epigenetics in cancer: what's the future. Oncology. 2011;25:220–6. [PubMed] [Google Scholar]
- 3.Cameron EE, Bachman KE, Myöhänen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–7. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 4.Choudhry H, Catto JW. MicroRNA and Cancer. Springer; 2011. Epigenetic regulation of microRNA expression in cancer. [DOI] [PubMed] [Google Scholar]
- 5.Cortez CC, Jones PA. Chromatin, cancer and drug therapies. Mutat Res/Fundam Mol Mech Mutagen. 2008;647:44–51. doi: 10.1016/j.mrfmmm.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cowland JB, Hother C, Gronbaek K. MicroRNAs and cancer. APMIS. 2007;115:1090–106. doi: 10.1111/j.1600-0463.2007.apm_775.xml.x. [DOI] [PubMed] [Google Scholar]
- 7.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–14. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datta J, Kutay H, Nasser MW, Nuovo GJ, Wang B, Majumder S, et al. Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Res. 2008;68:5049–58. doi: 10.1158/0008-5472.CAN-07-6655. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Datta J, Smith A, Lang JC, Islam M, Dutt D, Teknos TN, et al. MicroRNA-107 functions as a candidate tumor-suppressor gene in head and neck squamous cell carcinoma by downregulation of protein kinase Cvarepsilon. Oncogene. 2012;31:4045–53. doi: 10.1038/onc.2011.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–9. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng L, Xie Y, Zhang H, Wu Y. MiR-107 targets cyclin-dependent kinase 6 expression, induces cell cycle G1 arrest and inhibits invasion in gastric cancer cells. Med Oncol. 2012;29:856–63. doi: 10.1007/s12032-011-9823-1. [DOI] [PubMed] [Google Scholar]
- 12.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–9. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 13.Ghoshal K, Datta J, Majumder S, Bai S, Dong X, Parthun M, et al. Inhibitors of histone deacetylase and DNA methyltransferase synergistically activate the methylated metallothionein I promoter by activating the transcription factor MTF-1 and forming an open chromatin structure. Mol Cell Biol. 2002;22:8302–19. doi: 10.1128/MCB.22.23.8302-8319.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorin MA, Pan Q. Protein kinase C epsilon: an oncogene and emerging tumor biomarker. Mol Cancer. 2009;8 doi: 10.1186/1476-4598-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haussecker D, Kay MA. MiR-122 continues to blaze the trail for microRNA therapeutics. Mol Ther. 2010;18:240–2. doi: 10.1038/mt.2009.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Islam M, Datta J, Lang JC, Teknos TN. Down regulation of RhoC by microRNA-138 results in de-activation of FAK, Src and Erk1/2 signaling pathway in head and neck squamous cell carcinoma. Oral Oncol. 2014;50:448–56. doi: 10.1016/j.oraloncology.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jansson MD, Lund AH. MicroRNA and cancer. Molecular oncology. 2012;6:590–610. doi: 10.1016/j.molonc.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang L, Liu X, Kolokythas A, Yu J, Wang A, Heidbreder CE, et al. Downregulation of the Rho GTPase signaling pathway is involved in the microRNA-138-mediated inhibition of cell migration and invasion in tongue squamous cell carcinoma. Int J Cancer. 2010;127:505–12. doi: 10.1002/ijc.25320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar B, Yadav A, Lang JC, Teknos TN, Kumar P. Suberoylanilide hydroxamic acid (SAHA) reverses chemoresistance in head and neck cancer cells by targeting cancer stem cells via the downregulation of nanog. Genes Cancer. 2015;6:169. doi: 10.18632/genesandcancer.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Lee EM, Shin S, Cha HJ, Yoon Y, Bae S, Jung JH, et al. Suberoylanilide hydroxamic acid (SAHA) changes microRNA expression profiles in A549 human non-small cell lung cancer cells. Int J Mol Med. 2009;24:45–50. doi: 10.3892/ijmm_00000204. [DOI] [PubMed] [Google Scholar]
- 22.Lee J, Huang S. Cancer epigenetics: mechanisms and crosstalk of a HDAC inhibitor, vorinostat. Chemotherapy. 2013;2 doi: 10.4172/2167-7700.1000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee KH, Lotterman C, Karikari C, Omura N, Feldmann G, Habbe N, et al. Epigenetic silencing of MicroRNA miR-107 regulates cyclin-dependent kinase 6 expression in pancreatic cancer. Pancreatology. 2009;9:293–301. doi: 10.1159/000186051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 25.Liep J, Rabien A, Jung K. Feedback networks between microRNAs and epigenetic modifications in urological tumors. Epigenetics. 2012;7:315–25. doi: 10.4161/epi.19464. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Chen Z, Yu J, Xia J, Zhou X. MicroRNA profiling and head and neck cancer. Comp Funct Genom. 2009;837514 doi: 10.1155/2009/837514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setién F, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–9. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 29.Meng F, Glaser SS, Francis H, Yang F, Han Y, Stokes A, et al. Epigenetic regulation of miR-34a expression in alcoholic liver injury. Am J Pathol. 2012;181:804–17. doi: 10.1016/j.ajpath.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nalls D, Tang SN, Rodova M, Srivastava RK, Shankar S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS One. 2011;6:e24099. doi: 10.1371/journal.pone.0024099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–15. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 32.Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol. 2001;2:533–43. doi: 10.1016/S1470-2045(01)00486-7. [DOI] [PubMed] [Google Scholar]
- 33.Piao L, Zhang M, Datta J, Xie X, Su T, Li H, et al. Lipid-based nanoparticle delivery of Pre-miR-107 inhibits the tumorigenicity of head and neck squamous cell carcinoma. Mol Ther. 2012;20:1261–9. doi: 10.1038/mt.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410–7. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu S, Huang D, Yin D, Li F, Li X, Kung Hf, et al. Suppression of tumorigenicity by microRNA-138 through inhibition of EZH2-CDK4/6-pRb-E2F1 signal loop in glioblastoma multiforme. Biochim Biophys Acta (BBA)-Mol Basis Disease. 2013;1832:1697–707. doi: 10.1016/j.bbadis.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 36.Qiu T, Zhou L, Zhu W, Wang T, Wang J, Shu Y, et al. Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials. Future Oncol. 2013;9:255–69. doi: 10.2217/fon.12.173. [DOI] [PubMed] [Google Scholar]
- 37.Song T, Zhang X, Wang C, Wu Y, Cai W, Gao J, et al. MiR-138 suppresses expression of hypoxia-inducible factor 1alpha (HIF-1alpha) in clear cell renal cell carcinoma 786-O cells. Asian Pac J Cancer Prev. 2011;12:1307–11. [PubMed] [Google Scholar]
- 38.Suzuki H, Maruyama R, Yamamoto E, Kai M. DNA methylation and microRNA dysregulation in cancer. Mol Oncol. 2012;6:567–78. doi: 10.1016/j.molonc.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takes RP, Rinaldo A, Silver CE, Haigentz M, Jr, Woolgar JA, Triantafyllou A, et al. Distant metastases from head and neck squamous cell carcinoma Part I. Basic aspects Oral Oncol. 2012;48:775–9. doi: 10.1016/j.oraloncology.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 40.Tsai KW, Hu LY, Wu CW, Li SC, Lai CH, Kao HW, et al. Epigenetic regulation of miR-196b expression in gastric cancer. Genes Chromosom Cancer. 2010;49:969–80. doi: 10.1002/gcc.20804. [DOI] [PubMed] [Google Scholar]
- 41.Tu HF, Lin SC, Chang KW. MicroRNA aberrances in head and neck cancer: pathogenetic and clinical significance. Curr Opin Otolaryngol Head Neck Surg. 2013;21:104–11. doi: 10.1097/MOO.0b013e32835e1d6e. [DOI] [PubMed] [Google Scholar]
- 42.Tuttle S, Hertan L, Daurio N, Porter S, Kaushick C, Li D, et al. The chemopreventive and clinically used agent curcumin sensitizes HPV-but not HPV+ HNSCC to ionizing radiation, in vitro and in a mouse orthotopic model. Cancer Biol Ther. 2012;13:575–84. doi: 10.4161/cbt.19772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Taniguchi T. MicroRNAs and DNA damage response: implications for cancer therapy. Cell Cycle. 2013;12:32–42. doi: 10.4161/cc.23051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485–91. doi: 10.1200/JCO.2010.28.9066. [DOI] [PubMed] [Google Scholar]
- 45.Xiqiang L, Cheng W, Zujian C, Yi J, Yun W, Antonia K, et al. MicroRNA-138 suppresses epithelial-mesenchymal transition in squamous cell carcinoma cell lines. Biochem J. 2011;440:23–31. doi: 10.1042/BJ20111006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamakuchi M, Lotterman CD, Bao C, Hruban RH, Karim B, Mendell JT, et al. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci. 2010;107:6334–9. doi: 10.1073/pnas.0911082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang H, Lan P, Hou Z, Guan Y, Zhang J, Xu W, et al. Histone deacetylase inhibitor SAHA epigenetically regulates miR-17-92 cluster and MCM7 to upregulate MICA expression in hepatoma. Br J Cancer. 2015;112:112–21. doi: 10.1038/bjc.2014.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yi JS, Kim JS, Lee JH, Choi SH, Nam SY, Kim SY, et al. 18F-FDG PET/CT for detecting distant metastases in patients with recurrent head and neck squamous cell carcinoma. J Surg Oncol. 2012;106:708–12. doi: 10.1002/jso.23185. [DOI] [PubMed] [Google Scholar]
- 49.Zhang C. Novel functions for small RNA molecules. Curr Opin Mol Ther. 2009;11:641. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.