

Abstract
Background
Development of biologically relevant models from gene expression data notably, microarray data has become a topic of great interest in the field of bioinformatics and clinical genetics and oncology. Only a small number of gene expression data compared to the total number of genes explored possess a significant correlation with a certain phenotype. Gene selection enables researchers to obtain substantial insight into the genetic nature of the disease and the mechanisms responsible for it. Besides improvement of the performance of cancer classification, it can also cut down the time and cost of medical diagnoses.
Methods
This study presents a modified Artificial Bee Colony Algorithm (ABC) to select minimum number of genes that are deemed to be significant for cancer along with improvement of predictive accuracy. The search equation of ABC is believed to be good at exploration but poor at exploitation. To overcome this limitation we have modified the ABC algorithm by incorporating the concept of pheromones which is one of the major components of Ant Colony Optimization (ACO) algorithm and a new operation in which successive bees communicate to share their findings.
Results
The proposed algorithm is evaluated using a suite of ten publicly available datasets after the parameters are tuned scientifically with one of the datasets. Obtained results are compared to other works that used the same datasets. The performance of the proposed method is proved to be superior.
Conclusion
The method presented in this paper can provide subset of genes leading to more accurate classification results while the number of selected genes is smaller. Additionally, the proposed modified Artificial Bee Colony Algorithm could conceivably be applied to problems in other areas as well.
Electronic supplementary material
The online version of this article (doi:10.1186/s12920-016-0204-7) contains supplementary material, which is available to authorized users.
Keywords: Gene selection, Microarray, Cancer classification, Artificial bee colony algorithm, Evolutionary algorithm
Background
Gene expression studies have paved the way for a more comprehensive understanding of the transcriptional dynamics afflicted on a cell under different biological stresses [1–4]. The application of microarrays as a robust and amenable system to record transcriptional profiles across a range of differing species has been growing exponentially. In particular, the evaluation of human expression profiles in both health and in disease has implications for the development of clinical bio-markers for diagnosis as well as prognosis. Hence, diagnostic models from gene expression data provide more accurate, resource efficient, and repeatable diagnosis than the traditional histopathology [5]. Indeed, microarray data is now being used in clinical applications as it is possible to predict the treatment of human diseases by analyzing gene expression data [2, 6–9]. However, one of the inherent issues with gene expression profiles are their characteristically high-dimensional noise, contributing to possible high false positive rates. This is further compounded during analysis of such data whereby the use of all genes may potentially hinder the classifier performance by masking the contribution of the relevant genes [10–15]. This has led to a critical need for the development of analytical tools and methodologies which are able to select a small subset of genes both from a practical and qualitative perspective. As a result the selection of discriminatory genes is essential to improve the accuracy and also to decrease the computational time and cost [16].
The classification of gene expression samples involves feature selection and classifier design. However, noisy, irrelevant, and misleading attributes make the classification task complicated, given they can contain erroneous correlations. A reliable selection method of relevant genes for sample classification is needed in order to increase classification accuracy and to avoid incomprehensibility. The task of gene selection is known as feature selection in artificial intelligence domain. Feature selection has class-labeled data and attempts to determine which features best distinguish among the classes. The genes are considered to be the features that describe the cell. The goal is to select a minimum subset of features that achieves maximum classification performance and to discard the features having little or no effect. These selected features can then be used to classify unknown data. Feature selection can thus be considered as a principle pre-processing tool when solving classification problems [17, 18]. Theoretically, feature selection problems are NP-hard. Performing an extensive search is impossible as the computational time and cost would be excessive [19].
Gene selection methods can be divided into two categories [20]: filter methods, and wrapper or hybrid methods. Detail review on gene selection methods can be found in [20–25]. A gene selection method is categorized as a filter method if it is carried out independently from a classification procedure. In filter approach instead of searching the feature space, selection is done based on statistical properties. Due to lower computational time and cost most previous gene selection techniques in the early era of microarrays analysis have used the filter method. Many filters provide a feature ranking rather than an explicit best feature subset. The top ranking features are chosen manually or via cross-validation [26–28] while the remaining low ranking features are eliminated. Bayesian Network [29], t-test [30], Information Gain (IG) and Signal-to-Noise-Ratio (SNR) [5, 31], Euclidean Distance [32, 33], etc. are the examples of filter methods that are usually considered as individual gene-ranking methods. Filter methods generally rely on a relevance measure to assess the importance of genes from the data, ignoring the effects of the selected feature subset on the performance of the classifier. This may however result in the inclusion of irrelevant and noisy genes in a gene subset. Research shows that, rather than acting independently, genes in a cell interact with one another to complete certain biological processes or to implement certain molecular functions [34].
While the filter methods handle the identification of genes independently, a wrapper or hybrid method on the other hand, implements a gene selection method merging with a classification algorithm. In the wrapper methods [35] a search is conducted in the space of genes, evaluating the fitness of each found gene subset. Fitness is determined by training the specific classifier to be used only with the found gene subset and then approximating the accuracy percentage of the classifier. The hybrid methods usually obtain better predictive accuracy estimation than the filter methods [36–39], since the genes are selected by considering and optimizing the correlations among genes. Therefore, several hybrid methods have been implemented to select informative genes for binary and multi-class cancer classification in recent times [37, 40–50]. However, its computational cost must be taken into account [39]. Notably, filter methods have also been used as a preprocessing step for wrapper methods, allowing a wrapper to be used on larger problem instances.
Recently many diverse population based methods have been developed for investigating gene expression data to select a small subset of informative genes from the data for cancer classification. Over the time a number of variants and hybrids of Particle Swarm Optimization (PSO) have been proposed to solve the gene selection problem. The Combat Genetic Algorithm (CGA) [51, 52] has been embedded within the Binary Particle Swarm Optimization (BPSO) in [44] which serves as a local optimizer at each iteration to improve the solutions of the BPSO. The algorithm has succeeded to achieve high classification accuracy albeit at the cost of unacceptably large size of the selected gene set. Although both PSO and CGA perform well as global optimizer, the proposed algorithm has failed to obtain satisfactory results because of not considering minimization of selected gene size as an objective. Also Li et al. [41] presented a hybridization of BPSO and Genetic Algorithm (GA). However, its performance is not satisfactory enough. Shen et al. [40] discussed incorporation of Tabu Search (TS) in PSO as a local improvement procedure to maintain the population diversity and prevent steering to misleading local optima. Obtained accuracy by their hybrid algorithm is sufficient but they did not provide any discussion at all about the number of genes selected. Again BPSO has been embedded in TS by Chuang et al. [42] to prevent TS form getting trapped in local optima which helps in achieving satisfactory accuracy for some of the datasets. However, to attain that accuracy their algorithm needs to select prohibitively high number of genes. An improved binary particle swarm optimization (IBPSO) is proposed by Chuang et al. [43] which achieves good accuracy for some of the datasets but, again, selects high number of genes. Recently, Mohamad et al. [37] have claimed to enhance the original BPSO algorithm by minimizing the probability of gene to be selected, resulting in the selection of only the most informative genes. They have obtained good classification accuracy with low number of selected genes for some of the datasets. But the number of iterations to achieve the target accuracy is higher than ours, which will be reported in the Results and discussion Section of this paper. A simple modified ant colony optimization (ACO) algorithm is proposed by Yu et al. in [53], which associates two pheromone components with each gene rather than a single one as follows. One component determines the effect of selecting the gene whether the other determines the effect of not selecting it. The algorithm is evaluated using five datasets. It is able to select small number of genes and accuracy is also reasonable. Random forest algorithm for classifying microarray data [54] has obtained good accuracy for some datasets but not for all. Notably, the number of selected genes by the random forest classification algorithm in [54] has been found to be high for some of the datasets. A new variable importance measure based on the difference of proximity matrix has been proposed for gene selection using random forest classification by Zhou et al. [55]. Although it fails to achieve the highest accuracy for any dataset, their algorithm is able to select small number of genes and achieve satisfactory accuracy for all the datasets. Recently, Debnath and Kurita [56] have proposed an evolutionary SVM classifier that adds features in each generation according to the error-bound values for the SVM classifier and frequency of occurrence of the gene features to produce a subset of potentially informative genes.
In this paper, we propose a modified artificial bee colony algorithm to select genes for cancer classification. The Artificial Bee Colony (ABC) algorithm [57], proposed by Karaboga in 2005, is one of the most recent swarm intelligence based optimization technique, which simulates the foraging behavior of a honey bee swarm. The search equation of ABC is reported to be good at exploration but poor at exploitation [58, 59]. To overcome this limitation we have modified the ABC algorithm by incorporating the concept of pheromone which is one of the major components of the Ant Colony Optimization (ACO) algorithm [60, 61] and a new operation in which successive bees communicate to share their results. Even though researchers are unable to establish whether such a communication indeed involve information transfer or not, it is known that foraging decisions of outgoing workers, and the probability to find a recently-discovered food source, are influenced by the interactions [62–67]. Indeed, there is a notable proof that for harvester ants, management of foraging activity is guided by ant encounters [68–71]. Even the mere instance of an encounter may provide some information, such as, the magnitude of the colony’s foraging activity, and may therefore influence the probability of food collection by ants [72–74].
We believe that the selection of genes by our system provide us some interesting clue towards the importance and contribution of that set of particular genes for the respective cancer disease. To elaborate, our system has identified that for diffuse large B-cell lymphoma (DLBCL) only three (3) genes are informative enough to decide about the cancer. Now, this could turn out to be a string statement with regards to the set of genes identified for a particular cancer and we believe further biological validation is required before making such a string claim. We do plan to work towards validation of these inferences.
During the last decade, several algorithms have been developed depending on different intelligent behaviors of honey bee swarms [57, 75–85]. Among those, ABC is the one which has been most widely studied on and applied to solve the real world problems, so far. Comprehensive study on ABC and other bee swarm algorithms can be found in [86–89]. The algorithm has the advantage of sheer simplicity, high flexibility, fast convergence, and strong robustness which can be can be used for solving multidimensional and multimodal optimization problems [90–92]. Since the ABC algorithm was proposed in 2005, it has been applied in many research fields, such as flow shop scheduling problem [93, 94], parallel machines scheduling problem [95], knapsack problem [96], traveling salesman problem [97], quadratic minimum spanning tree problem [98], multiobjective optimization [99, 100], generalized assignment problem [101], neural network training [102], and synthesis [103], data clustering [104], image processing [105], MR brain image classification [106], coupled ladder network [107], wireless sensor network [108], vehicle routing [109], nurse rostering [110], computer intrusion detection [111], live virtual machine migration [112], etc. Studies [86, 113] have indicated that ABC algorithms have high search capability to find good solutions efficiently. Besides, excellent performances has been reported by ABC for a considerable number of problems [98, 100, 114]. Karaboga and Basturk [113] tested for five multidimensional numerical benchmark functions and compared the ABC performance with that of Differential Evolution (DE), Particle Swarm Optimization (PSO) and Evolutionary Algorithm (EA). The study concluded that ABC gets out of a local minimum more efficiently for multivariable and multimodal function optimization and outperformed DE, PSO and EA.
However, it has been observed that the ABC may occasionally stop proceeding toward the global optimum even though the population has not converged to a local optimum [86]. Research [58, 59, 115] shows that the solution search equation of ABC algorithm is good at exploration but unsatisfactory at exploitation. For the population based algorithms the exploration and the exploitation abilities are both necessary features. The exploration ability refers to the ability to investigate the various unknown regions to discover the global optimum in solution space, while the exploitation ability refers to the ability to apply the knowledge of the previous good solutions to find better solutions. The exploration ability and the exploitation ability contradict to each other, so that the two abilities should be well balanced to achieve good performance on optimization problems. As a result, several improvements of ABC have been proposed over the time. Baykasoglu et al. [101] incorporated the ABC algorithm with shift neighborhood searches and greedy randomized adaptive search heuristic and applied it to the generalized assignment problem. Pan et al. [93] proposed a Discrete Artificial Bee Colony (DABC) algorithm with a variant of iterated greedy algorithm with total weighted earliness and tardiness penalties criterion. Li et al. [116] used a hybrid Pareto-based ABC algorithm to solve flexible job shop-scheduling problems. In the proposed algorithm, each food sources is represented by two vectors, the machine assignment and the operation scheduling. Wu et al. [117] combined Harmony Search (HS) and the ABC algorithm to construct a hybrid algorithm. Comparison results show that the hybrid algorithm outperforms ABC, HS, and other heuristic algorithms. Kang et al. [118] anticipated a Hooke Jeeves Artificial Bee Colony algorithm (HJABC) for numerical optimization. HJABC integrates a new local search named ‘modus operandi’ which is based on Hooke Jeeves method (HJ) [119] with the basic ABC. Opposition Based Lévy Flight ABC is developed by Sharma et al. [120]. Lévy flight based random walk local search is proposed and incorporated with ABC to find the global optima. Szeto et al. [109] proposed an enhanced ABC algorithm. The performance of the new approach is tested on two sets of standard benchmark instances. Simulation results show that the newly proposed algorithm outperforms the original ABC and several other existing algorithms. Chaotic Search ABC (CABC) is introduced by Yan et al. [121] to solve the premature convergence issue of ABC by increasing the number of scout and rational using of the global optimal value and chaotic Search. Again a Scaled Chaotic ABC (SCABC) method is proposed in [106] based on fitness scaling strategy and chaotic theory. Based on the Rossler attractor of chaotic theory a novel Chaotic Artificial Bee Colony (CABC) is developed in [122] to improve the performance of ABC. An Improved Artificial Bee Colony (IABC) algorithm is proposed in [123] to improve the optimization ability of ABC. The paper introduces an improved solution search equation in employee and scout bee phase using the best and the worst individual of the population. In addition, the initial population is generated by the piecewise logistic equation which employs chaotic systems to enhance the global convergence. Inspired by Differential Evolution (DE), Gao et al. [124] proposed an improved solution search equation. In order to balance the exploration of the solution search equation of ABC and the exploitation of the proposed solution search equation, a selective probability is introduced. In addition, to enhance the global convergence, when producing the initial population, both chaotic systems and opposition based learning methods are employed. Kang et al. [91] proposed a Rosenbrock ABC (RABC) algorithm which combines Rosenbrock’s rotational direction method with the original ABC. There are two alternative phases of RABC: the exploration phase realized by ABC and the exploitation phased completed by the Rosenbrock method. Tsai et al. [125] introduced the Newtonian law of universal gravitation in the onlooker phase of the basic ABC algorithm in which onlookers are selected based on a roulette wheel to maximize the exploitation capacity of the solutions in this phase and the strategy is named as Interactive ABC (IABC). The IABC introduced the concept of universal gravitation into the consideration of the affection between employed bees and the onlooker bees. The onlooker bee phase is altered by biasing the direction towards random bee according to its fitness. Zhu and Kwong [115] utilized the search information of the global best solution to guide the search of ABC to improve the exploitation capacity. The main idea is to apply the knowledge of the previous good solutions to find better solutions. Reported results show that the new approach achieves better results than the original ABC algorithm. Banharnsakun et al. [126] modified the search pattern of the onlooker bees such that the solution direction is biased toward the best-so-far position. Therefore, the new candidate solutions are similar to the current best solution. Li et al. [58] proposed an improved ABC algorithm called I-ABC, in which the best-so-far solution, inertia weight, and acceleration coefficients are introduced to modify the search process. The proposed method is claimed to have an extremely fast convergence speed. Gbest guided position update equations are introduced in Expedited Artificial Bee Colony (EABC) [127]. Jadon et al. [128] proposed an improved ABC named as ABC with Global and Local Neighborhoods (ABCGLN) which concentrates to set a trade off between the exploration and exploitation and therefore increases the convergence rate of ABC. In the proposed strategy, a new position update equation for employed bees is introduced where each employed bee gets updated using best solutions in its local and global neighborhoods as well as random members from these neighborhoods. With a motivation to balance exploration and exploitation capabilities of ABC, Bansal et al. [129] presents an self adaptive version of ABC named as SAABC. In this adaptive version, to give more time to potential solutions to improve themselves, the parameter ‘limit’, of ABC is modified self adaptively based on current fitness values of the solutions. This setting of ‘limit’ makes low fit solutions less stable, which helps in exploration. Also to enhance the exploration, number of scout bees are increased. To achieve an improved ABC based approach with better global exploration and local exploitation ability, a novel heuristic approach named PS-ABC is introduced by Xu et al. [112]. The method utilizes the binary search idea and the Boltzmann selection policy to achieve the uniform random initialization and thus to make the whole PSABC approach have a better global search potential and capacity at the very beginning. To obtain more efficient food positions Sharma et al. [130] introduced two new mechanisms for the movements of scout bees. In the first method, the scout bee follows a non-linear (quadratic) interpolated path while in the second one, scout bee follows Gaussian movement. The first variant is named as QABC, while the second variant is named as GABC. Numerical results and statistical analysis of benchmark unconstrained, constrained and real life engineering design problems indicate that the proposed modifications enhance the performance of ABC. In order to improve exploitation capability of ABC a new search pattern is proposed by Xu et al. [131] for both employed and onlooker bees. In the new approach, some best solutions are utilized to accelerate the convergence speed. A solution pool is constructed by storing some best solutions of the current swarm. New candidate solutions are generated by searching the neighborhood of solutions randomly chosen from the solution pool. Kumar et al. [97] added crossover operators to the ABC as the operators have a better exploration property. Ji et al. [96] developed a new ABC algorithm combining chemical communication way and behavior communication way based on researches of entomologists. The new ABC algorithm introduces a novel communication mechanism among bees. In order to have a better coverage and a faster convergence speed, a modified ABC algorithm introducing forgetting and neighbor factor (FNF) in the onlooker bee phase and backward learning in the scout bee phase is proposed by Yu et al. [108]. Bansal et al. [132] introduced Memetic ABC (MeABC) in order to balance between diversity and convergence capability of the ABC. A new local search phase is integrated with the basic ABC to exploit the search space identified by the best individual in the swarm. In the proposed phase, ABC works as a local search algorithm in which, the step size that is required to update the best solution, is controlled by Golden Section Search (GSS) [133] approach. In the memetic search phase new solutions are generated in the neighborhood of the best solution depending upon a newly introduced parameter, perturbation rate. Kumar et al. [134] also proposed memetic search strategy to be used in place of employed bee and onlooker bee phase. Crossover operator is applied to two randomly selected parents from current swarm. After crossover operation two new offspring are generated. The worst parent is replaced by the best offspring, other parent remains same. The experimental result shows that the proposed algorithm performs better than the basic ABC without crossover in terms of efficiency and accuracy. Improved onlooker bee phase with help of a local search strategy inspired by memetic algorithm to balance the diversity and convergence capability of the ABC is proposed by Kumar et al. [135]. The proposed algorithm is named as Improved Onlooker Bee Phase in ABC (IOABC). The onlooker bee phase is improved by introducing modified GSS [133] process. Proposed algorithm modifies search range of GSS process and solution update equation in order to balance intensification and diversification of local search space. Rodriguez et al. [95] combined two significant elements with the basic scheme. Firstly, after producing neighboring food sources (in both the employed and onlooker bees phases), a local search is applied with a predefined probability to further improve the quality of the solutions. Secondly, a new neighborhood operator based on the iterated greedy constructive-destructive procedure [136, 137] is proposed. For further discussion please refer to the available reviews on ABC [138]. Several algorithms have been introduced that incorporates idea of ACO or PSO in bee swarm based algorithms. But our approach is unique and different from others. Hybrid Ant Bee Colony Algorithm (HABC) [139] considers pheromone for each candidate solution. On the other hand we incorporated pheromone for each gene (solution components). Our approach to find neighboring solution is different from basic ABC. But HABC follows the same neighbor production mechanism as basic ABC. In our algorithm pheromone deposition is done after each bee stage. While selecting a potential candidate solution we depend on its fitness, but HABC selects a candidate depending upon its pheromone value. Most importantly in our algorithm scout bees make use of the pheromone while exploring to find new food source. Ji et al. [96] proposed an artificial bee colony algorithm merged with pheromone. In this paper scouts are guided by pheromone along with some heuristic information while we only make use of pheromone. The paper updates pheromone only in the employed bee stage while we update pheromone in all the bee stages. Pheromone laying is done by depositing a predefined constant amount. But amount of pheromone we have deposited is a function of fitness measures. Kefayat et al. [140] proposed a hybrid of ABC and ACO. Inside loop contains the ABC and outside loop is ACO without any modification. ABC is applied in the inner loop to optimize a certain constraint (source size) for each ant. Zhu et al. [115] uses ABC in a problem with continuous space. We indirectly guide scout through the best found solutions whether this paper guides the employed and onlooker bees.
Methods
Gene expression profiles provide a dynamic means to molecularly characterise the state of a cell and so has great potential as clinical diagnostic and prognostic tool. However, in comparison to the number of genes involved which often exceeds several thousands, available training datasets generally have a fairly small sample size for classification. Hence, inclusion of redundant genes decreases the quality of classification thus increasing false positive rates. To overcome this problem one of the approaches in practice is to search for the informative genes along with applying a filter beforehand. Use of confidently pre-filtering makes it possible to get rid of the majority of redundant noisy genes. Consequently, the underlying method to search the informative genes becomes easier and efficient with respect to time and cost. Finally, to evaluate the fitness of the selected gene subset a classifier is utilized. The selected genes are used as features to classify the testing samples. The inputs and outputs of the method are:
Input: G={G1,G2,…,Gn}, a vector of vectors, where n is the number of genes and Gi={gi,1,gi,2,…,gi,N} is a vector of gene expressions for the ith gene where N is the sample size. So, gi,j is the expression level of the ith gene in the jth sample.
Output: R={R1,R2,…,Rm}, the indices of the genes selected in the optimal subset. Where m is the selected gene size.
The gene selection method starts with a preprocessing step followed by a gene selection algorithm. Finally the classification is done. In what follows, we will describe these steps in detail.
Preprocessing
To make the experimental data suitable for our algorithm and to help the algorithm run faster the preprocessing step is incorporated. The preprocessing step contains the following two stages:
Normalization
Prefilter
Normalization Normalizing the data ensures the allocation of equal weight to each variable by the fitness measure. Without normalization, the variable with the largest scale will dominate the fitness measure [141]. Therefore, normalization reduces the training error, thereby proving the accuracy for the classification problem [142]. The expression levels for each gene are normalized at this step to [0, 1] using the standard procedure which is shown in Eq. 1 below.
| 1 |
Here, among all the expression levels of the gene in consideration, value_max is the maximum original value, value_min is the minimum original value, upper (lower) is 1 (0) and x is the normalized expression level. So for all gene after normalization, value_max will be 1 and value_min will be 0.
Prefilter Gene expression data are characteristically multi faceted given the inherent biological complexity such networks reside in. The huge number of genes causes great computational complexity in wrapper methods when searching for significant genes. Before applying other search methods it is thus prudent to reduce gene subset space by pre-selecting a smaller number of informative genes based on some filtering criteria. Several filter methods have been proposed in the literature which can be used to preprocess data. These include Signal-to-Noise Ratio (SNR) and Information Gain (IG) [5, 31], t-test [30], Bayesian Network [29], Kruskal-Wallis non-parametric analysis of variance (ANOVA) algorithm [143–145], F-test (ratio of in between group variance to within group variance) [146, 147], BW ratio [148], Euclidean Distance [32, 33], etc. After the prefilter stage, we get a ranking of the genes based on the applied statistical methods.
Because of the nature of gene expression data the selected statistical method should be able to deal with high dimensional small sample sized data. According to the assumption of the data characteristics two types of filtering methods exist, namely, parametric and non parametric. Both types of filtering techniques have been employed individually in our proposed algorithm for the sake of comparison. Among many alternatives, in our work, Kruskal Wallis [143–145] and F-test [146, 147] are employed individually to rank the genes. Notably, the former is a non parametric method and the latter is a parametric one.
Kruskal Wallis (KW) The Kruskal-Wallis rank sum test (named after William Kruskal and W. Allen Wallis) is an extension of the Mann-Whitney U or Wilcoxon Sum Rank test [149, 150] for comparing two or more independent samples that may have different sample sizes [143–145]. The Kruskal-Wallis rank sum test (KWRST) is the non-parametric equivalent to the one-way Analysis of Variance (ANOVA). It compares several populations on the basis of independent random samples from each population by determining whether the samples belong to the same distribution. Assumptions for the Kruskal-Wallis test are that within each sample, the observations are independent and identically distributed and the samples are independent of each other. It makes no assumptions about the distribution of the data (e.g., normality or equality of variance) [151, 152]. According to the results found by Deng et al. [153], the assumption about the data distribution often does not hold in gene expression data. The Kruskal-Wallis test is in fact very convenient for microarray data because it does not require strong distributional assumptions [154], it works well on small samples [155], it is suited for multiclass problems, and its p-values can be calculated analytically. The Kruskal-Wallis test is utilized to determine p-values of each gene. The genes are then sorted in increasing order of the p-values. The lower the p-value of a gene, the higher the rank of the gene. The steps of the Kruskal-Wallis test are given below:
-
Step 1For each gene expression vector Gi,
- We rank all gene expression levels across all classes. We Assign any tied values the average of the ranks they would have received if they had not been tied.
-
We calculate the test statistics Ki for gene expression vector Gi of the ith gene, which is given by Eq. 2 below:
2 Here, for the ith gene,N is the sample size,Ci is the number of different classes,is the number of expression levels that are from class k, andis the mean of the ranks of all expression level measurements for class k. - If ties are found while ranking data in the ith gene, correction of ties must be done. For this correction, Ki is divided by: , where Ti is the number of groups of different tied ranks for the ith gene and tj is the number of ties within group j.
- Finally the p-value for the ith gene, pi is approximated by , where refers to the critical chi-square value. To compute the p-values, necessary functions of the already implemented package from https://svn.win.tue.nl/trac/prom/browser/Packages/Timestamps/Trunk/src/edu/northwestern/at/utils/math/are incorporated in our method.
-
Step 2
After the p-values for all the genes are calculated, we rank each gene Gi according to pi. The lower the p-value of a gene, the higher is its ranking.
Kruskal-Wallis is used as a preprocessing step in many gene selection algorithms [156–158]. Kruskal-Wallis test is utilized to rank and pre-select genes in the two-stage gene selection algorithm proposed by Duncan et al. [158]. In the proposed method the number of genes selected from the ranked genes is optimized by cross-validation on the training set. Wang et al. [157] applied Kruskal-Wallis rank sum test to rank all the genes for gene reduction. Obtained results from their study indicate that gene ranking with Kruskal-Wallis rank sum test is very effective. To select an initial informative subset of tumor related genes Kruskal-Wallis rank sum test is utilized by Wang et al. [156].
Besides applying Kruskal-Wallis in prefiltering stage the use of the algorithm for gene selection is also well studied [159, 160]. Chen et al. [160] studied application of different test statistics including Kruskal-Wallis for gene selection. Lan et al. [159] applied Kruskal-Wallis to rank the genes. Finally the top ranked genes are selected as features for the target task classifier. The proposed filter is claimed to be suitable as a preprocessing step for an arbitrary classification algorithm.
Like many other non-parametric tests Kruskal-Wallis uses data rank rather than raw values to calculate the statistic. However, by ranking the data some information about the magnitude of differences between scores is lost. For this reason a parametric method called F-test has been applied separately from Kruskal-Wallis to prefilter the genes. Notably, replacing original scores with ranks does not naturally lead to performance reduction; it rather can result in a better performance at best and a slight degradation at worst.
F-test Another approach to identify the genes that are correlated to the target classes from gene expression data is by using the F-test [146, 147]. F-test is one of the most widely used supervised feature selection methods. The key idea is to find a subset of features, such that the distances between the data points in different classes are as large as possible, while the distances between the data points in the same class are as small as possible in the data space spanned by the selected features. It uses variations among means to estimate variations among individual measurements. F-score for a gene is the ratio of in between group variance to within group variance, where each class label forms a group. The steps to compute the F-score are given below:
-
For each gene expression vector Gi, we compute the Fisher score (i.e., F-Score). The fisher score for the ith gene is given by Eq. 3 below.
3 Here for the ith gene,
μi is the mean for all the gene expression levels corresponding to the ith gene,
and are mean and standard deviation of the kth class respectively,
Ci is the number of classes, and
is the number of samples associated with the kth class.
After computing the Fisher score for each genes, genes are sorted according to the F- score. The higher the F-score of a gene, the higher is its rank.
F-test has been proved to be effective for determining the discriminative power of genes [161]. Use of F-test either as a sidekick in gene selection [158, 162] or as a stand-alone gene selection tool [163] both are practiced in the literature. Duncan et al. [158] used F-test as one of the ranking schemes to preselect the genes. Guo et al. [163] proposed a privacy preserving algorithm for gene selection using F-criterion. The proposed method can be used in other feature selection problems. Au et al. [162] implemented F-test as a criterion function in their proposed algorithm to solve the problem of gene selection. Cai et al. [164] pre-selected top 1,000 genes from each dataset according to Fisher’s ratio. To guide the search their method evaluated discriminative power of features independently according to Fisher criterion. Salem et al. [165] reduced the total number of genes in the input dataset to a smaller subset using F-score.
The F-score is computed independently for each gene, which may lead to a suboptimal subset of features. Generally, the F-test is sensitive to non-normality [166, 167]. Thus the preferred test to use with microarray data is the Kruskal-Wallis test rather than the F-test since the parametricity assumption of data distribution often does not hold for gene expression data [153].
Pre-selection of genes The top ranked genes will enter the next phase. After the genes are ranked according to the statistical method in use, we need to calculate the number of genes to nominate for the next stage. There could be two ways to determine the number of genes to be selected in this stage. Select according to p In this approach we predetermine a threshold and select all the genes that have statistics calculated by Kruskal-Wallis (F-test) below (above) the threshold. This approach generally tends to select comparatively large number of genes [157]. To determine a suitable threshold value we have conducted scientific parameter tuning in the range of [ 0,1]. The analysis is presented in the Additional file 1. Select according to n Another approach is to select a predetermined number of top ranked genes. The number of genes selected from the ranked genes can be either fixed or optimized by cross-validation on the training set. EPSO [37] empirically determined a fixed number (500) and used it for all the datasets. Also several other works in the literature used this approach to preselect genes [41, 53, 156, 158, 168]. Li et al. [41] selected 40 top genes with the highest scores as the crude gene subset using Wilcoxon sum rank test. Yu et al. [53] presented detail information about top 10 marker genes. Initially Wang et al. [156] selected 300 top-ranked genes by KWRST. Duncan et al. [158] considered a set of values for number of top-ranked genes. Based on Fisher’s ratio top 1000 genes are selected by Zhou et al. [168]. But the problem in this approach is that different datasets have different sizes. So a fixed value might not be optimal for all the datasets. Determining a value that is good for all the datasets is not possible. So in this article we have selected a percentage of top ranked genes. As a result number of genes selected will depend on the original size of the dataset. Therefore, when the percentage is set to 0.1, only the top 10 % from the ranked genes are supplied to the next stage. We have scientifically tuned the parameter in the range of [ 0,1]. The analysis is presented in the Additional file 1.
Gene selection
After the preprocessing step only the most informative genes are left. Now they are fed to the search method to further select a smaller subset of informative genes. In this paper as the search method we have used the modified artificial bee colony (mABC) algorithm as described below.
Artificial Bee Colony The Artificial Bee Colony (ABC) algorithm is one of the most recent nature inspired optimization algorithms based on the intelligent foraging behavior of honey bee swarm. ABC algorithm has been proposed by Karaboga in [57] and further developed in [113]. Excellent performances have been exhibited by the ABC algorithm for a considerable number of problems [90–92, 98, 100, 114].
In the ABC algorithm, foraging honey bees are categorized into three groups, namely, employed bees, onlooker bees and scout bees. Each category of honey bees symbolizes one particular operation for generating new candidate solution. Employed bees exploit the food sources. They bring nectar from different food sources to their hive. Onlooker bees wait in the hive for the information on food sources to be shared by the employed bees and search for a food source based on that information. The employed bees whose food sources have been exhausted become scouts and their solutions are abandoned [57]. Then the scout bees search randomly for new food sources near the hive without using any experience. After the scout finds a new food source, it becomes an employed bee again. Every scout is an explorer who does not have any guidance while looking for a new food, i.e., a scout may find any kind of food sources. Therefore, sometimes a scout might accidentally discover a more rich and entirely unknown food source.
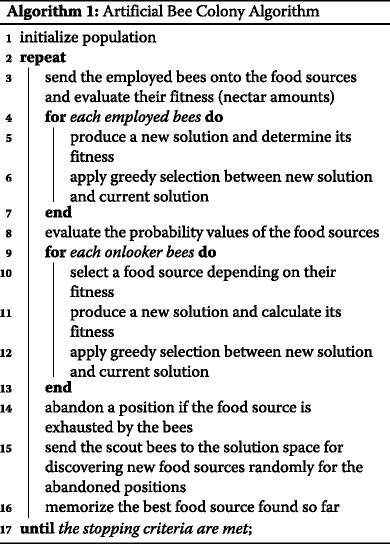
The position of a food source is a possible solution to the optimization problem and the nectar amount of the food source represents the quality of the solution. The bees act as operators over the food sources trying to find the best one among them. Artificial bees attempt to discover the food sources with high nectar amount and finally the one with the highest nectar amount. The onlookers and employed bees carry out the exploitation process in the search space and the scouts control the exploration process. The colony consists of equal number of employed bees and onlooker bees. In the basic form, the number of employed bees is equal to the number of food sources (solutions) thus each employed bee is associated with one and only one food source. For further discussion please refer to the available reviews on ABC [138]. The pseudo-code of the ABC algorithm is presented in Algorithm 1.
Modified ABC algorithm The search equation of ABC is reported to be good at exploration but poor at exploitation [59]. As a result, several improvements of ABC have been proposed over the time [96, 97, 106, 108, 111, 112, 120–122, 124, 125, 127–132, 134]. In employed bee and onlooker bee phase, new solutions are produced by means of a neighborhood operator. In order to enhance the exploitation capability of ABC, a local search method is applied to the solution obtained by the neighborhood operator with a certain probability in [95]. To overcome the limitations of the ABC algorithm, in addition to the approach followed in [95], we have further modified it by incorporating two new components in it. Firstly, we have incorporated the concept of pheromone which is one of the major components of the Ant Colony Optimization (ACO) algorithm [60, 61]. Secondly we have introduced and plugged in a new operation named Communication Operation in which successive bees communicate with each other to share their results. Briefly speaking, the pheromone helps minimizing the number of selected genes while the Communication Operation improves the accuracy. The algorithm is iterated for MAX_ITER times. Each iteration gives a global best solution, gbest. Finally, the gbest of the last iteration, i.e., the gbest with maximum fitness is the output of a single run. It is worth-mentioning that finding a solution with 100 % accuracy is not set as the stopping criteria as further iterations can find a smaller subset with the same accuracy. Ideally, a gene subset containing only one gene with 100 % accuracy is the best possible solution found by any algorithm. The proposed modified ABC is given in Algorithm 11 and the flowchart can be found in Fig. 1. The modified ABC algorithm is described next.
Fig. 1.
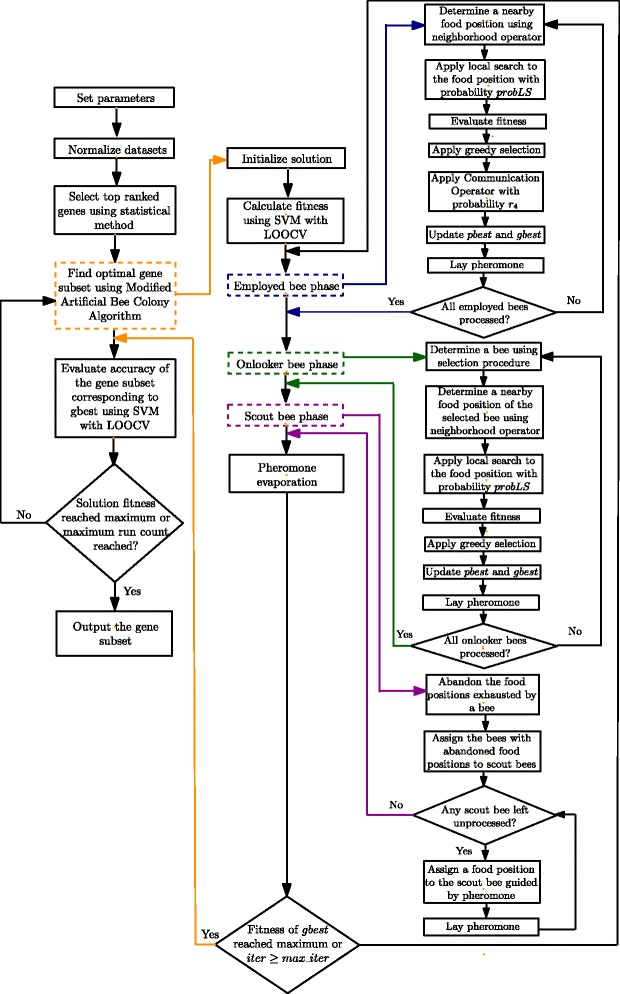
The flowchart of the modified Artificial Bee Colony (mABC) algorithm
Food source positions The position of the food source for the ith bee Si, is represented by vector , where n is the gene size or dimension of the data, , i=1,2,…,m (m is the population size), and d=1,2,…,n. Here, represents that the corresponding gene is selected, while means that the corresponding gene is not selected in the gene subset.
Pheromone We have incorporated the concept of pheromone (borrowed form ACO) to the ABC algorithm as a guide for exploitation. ACO algorithms are stochastic search procedures. The ants’ solution construction is guided by heuristic information about the problem instance being solved and (artificial) pheromone trails, which real ants use as communication media [169] to exchange information on the quality of a solution component. Pheromone helps selecting the most crucial genes. The quantity of pheromone deposited, which may depend on the quantity and quality of the food, guides other ants to the food source. Accordingly, the indirect communication via pheromone trails enables the ants to find shortest paths between their nest and food sources [169]. The gene subset carrying significant information will occur more frequently. Thus the genes in that subset will get reinforced simultaneously which ensures formation of a potential gene subset. The idea of using pheromone is to keep track of the components that are supposed to be good because they were part of a good solution in previous iterations. Because of keeping this information we need less iterations to achieve a target accuracy. Thus, computational time is also reduced.
Pheromone update The (artificial) pheromone trails are a kind of distributed numeric information [170] which is modified by the ants to reflect their experience accumulated while solving a particular problem. The pheromone values are updated using previously generated solutions. The update is focused to concentrate the search in regions of the search space containing high quality solutions. Solution components which are part of better solutions or are used by many ants will receive a higher amount of pheromone, and hence, will be more likely to be used by the ants in future iterations of the algorithm. It indirectly assumes that good solution components construct good solutions. However, to avoid the search getting stuck all pheromone trails are decreased by a factor before getting reinforced again. This mimics the natural phenomenon that, because of evaporation, the pheromone disappears over time unless they are revitalized by more ants. The idea of incorporating pheromone is to keep track of fitness of previous iterations.
The pheromone trails for all the components are represented by the vector P={p1,p2,⋯,pn}, where pi is the pheromone corresponding to the ith gene and n is the total number of genes. To update the pheromone pi corresponding to the ith gene, two steps are followed: pheromone deposition, and pheromone evaporation.
After each step of update, if the pheromone value becomes greater (less) than tmax (tmin), then the value of pheromone is set to tmax (tmin). Use of tmax, tmin is introduced in the Max-Min Ant System (MMAS) presented in [61] to avoid stagnation. The value of tmin is set to 0 and will be kept same throughout. But the value of tmax is updated whenever new global best, gbest solution is found. Pheromone deposition After each iteration the bees acquire new information and update their knowledge of local and global best locations. The best position found so far by the ith bee is known as the pbesti and the best position found so far by all the bees, i.e., the population, is known as the gbest. After each bee completes its tasks in each iteration, pheromone laying is done. The bee deposits pheromone using its knowledge of food locations gained so far. To lay pheromone, the ith bee uses its current location (Xi), the best location found by the bee so far (pbesti), and the best location found so far by all the bees (gbest). This idea is adopted from Particle Swarm Optimization (PSO) metaheuristic [171], where the local and global best locations are used to update the velocity of the current particle. We have also used the current position in pheromone laying to ensure enough exploration though in MMAS [61] only the current best solution is used to update the pheromone. Only the components which are selected in the corresponding solutions get reinforced. Hence, the pheromone deposition by the ith bee utilizes Eq. 4 below:
| 4 |
Here, d=1,2,⋯,n (n is the number of genes),w is the inertia weight, fi is the fitness of Xi, pfi is the fitness of pbesti,gf is the fitness of gbest, is selection of dth gene in Xi, is selection of dth gene in pbesti, is selection of dth gene in gbest, c0, c1, and c2 determines the contribution of fi, pfi, and gf respectively, and r0, r1, r2 are random values in the range of [ 0,1], which are sampled from a uniform distribution.
Here we have, c0+c1+c2=1 and c1=c2. So the individual best and the global best influence the pheromone deposition equally. The value of c0 is set from experimental results presented in the Additional file 1.
The inertia weight is considered to ensure that the contribution of global best and individual best is weighed more in later iterations when they contain meaningful values. To update the value of inertia weight w, two different approaches have been considered. One approach updates the weight so that an initial large value is decreased nonlinearly to a small value [37].
| 5 |
Here, MAX_ITER is the maximum number of iteration and iter is the current iteration.
Another approach is to update the value randomly [172].
| 6 |
Here, r5 is a random value in the range of [ 0,1], which is sampled from a uniform distribution. Performance evaluation of each of these two approach is presented in the Additional file 1. Pheromone evaporation At the end of each iteration, pheromones are evaporated to some extent. The equation for pheromone evaporation is given by Eq. 7:
| 7 |
Here, (1−ρ) is the pheromone evaporation coefficient and pi is the pheromone corresponding to the ith gene and n is the total number of genes. pi(t) represents pheromone value of the ith gene after (t−1)th iteration is completed.
Finally, note that, the value of tmax is updated whenever a new gbest is found. The rationale for such a change is as follows. Over time, as the fitness of gbest increases it also contributes more in the pheromone deposition, which may lead the pheromone values for some of the frequent genes to reach tmax. At that point, the algorithm will fail to store further knowledge about those particular genes. So we need to update the value of tmax after a new gbest is found. This is done using Eq. 8 below.
| 8 |
Here, tmax(g) represents the value of tmax when the gth global best is found by the algorithm.
Communication operator We have incorporated a new operator simulating the communication between the ants in a trail. Even though researchers are unable to establish whether such a communication indeed involves information transfer or not, it is known that foraging decisions of outgoing workers, and their probability to find a recently discovered food source, are influenced by the interactions [62–67]. In fact, there is a large body of evidence emphasizing the role of ant encounters for the regulation of foraging activity particularly for harvester ants [62, 68–71]. Even the mere instance of an encounter may provide information, such as the magnitude of the colony’s foraging activity, and therefore may influence the probability of food collection in ants [72–74].
At each step bees gain knowledge about different components and store their findings by depositing pheromone. After a bee gains new knowledge about the solution components, it share its findings with the successor. So an employed bee gets insight of which components are currently exhibiting excellent performance. Thus a bee obtains idea about food sources from its predecessor. A gene is selected in the current bee if it is selected in its predecessor and pheromone level is greater than a threshold level.
With probability r4 the following communication operator (Eqs. 9 and 10) is applied to each employed bee. The value of r4 is experimentally tuned and the results are presented in the Additional file 1.
| 9 |
Where, for ith bee i>1, d=1,2,⋯,n (n is the number of genes), and
| 10 |
The procedure Communicate(i) to apply the communication operator on ith bee is presented in Algorithm 2.

Initialization Pheromone for all the genes are initialized to tmax. For all the bees food positions are selected randomly. To initialize the ith bee, the function initRandom(Si), given in Algorithm 3, is used. Here we have used a modified sigmoid function that was introduced in [37] to increase the probability of the bits in a food position to be zero. The function is given in Eq. 11 below. It allows the components with high pheromone values to get selected.
| 11 |
Here, x≥0 and sigmoid(x)∈[0,1]

Employed bee phase
To determine a new food position the neighborhood operator is applied to the current food position. Then local search is applied with the probability probLS to the new food position to obtain a better position by exploitation. As local search procedures, Hill Climbing (HC), Simulated Annealing (SA), and Steepest Ascent Hill Climbing with Replacement (SAHCR) are considered. Then greedy selection is applied between the newly found neighbor and the current food position. The performance and comparison among different local search methods are discussed in the Additional file 1. In each iteration the value of gbest, and pbesti are updated using the Algorithm 4.

Onlooker bee phase
At first a food source is selected according to the goodness of the source using a selection procedure. As the selection procedure, Tournament Selection (TS), Fitness-Proportionate Selection (FPS), and Stochastic Universal Sampling (SUS) have been applied individually and the results are discussed in the Additional file 1. To determine a new food position the neighborhood operator is applied to the food position of the selected bee. Then local search is applied with the probability probLS to exploit the food position. As local search methods Hill Climbing, Simulated Annealing, and Steepest Ascent Hill Climbing with Replacement are compared. Then greedy selection is applied between the newly found neighbor and the current food position. In each iteration the value of gbest, and pbesti are updated using the Algorithm 4.
Selection procedure In the onlooker bee phase, an employed bee is selected using a selection procedure for further exploitation. As has been mentioned above, tournament selection, fitness-proportionate selection, and stochastic universal sampling have been applied individually as the selection procedure. Tournament selection In this method the fittest individual is selected among the t individuals picked from the population randomly with replacement [173], where t≥1. Value of t is set to 7 in our algorithm. This selection procedure is simple to implement and easy to understand. The selection pressure of the method directly varies with the tournament size. With the increase of the number of competitors, the selection pressure increases. So selection pressure can easily be adjusted by changing the tournament size. If the tournament size is larger, weak individuals have a smaller chance to be selected. The pseudocode is given in Algorithm 5.
 Fitness-proportionate selection In this approach, individuals are selected in proportion to their fitness [173]. Thus, if an individual has a higher fitness, its probability of getting selected is higher. In fitness-proportionate selection which is also known as roulette wheel selection, even the fittest individual may never be selected. In basic ABC, roulette wheel or fitness-proportionate selection scheme is incorporated. The analogy to a roulette wheel can be envisaged by imagining a roulette wheel in which each candidate solution represents a pocket on the wheel; the size of the pockets are proportionate to the probability of selection of the solution. Selecting N individuals from the population is equivalent to playing N games on the roulette wheel, as each candidate is drawn independently. The pseudocode is given in Algorithm 6.
Fitness-proportionate selection In this approach, individuals are selected in proportion to their fitness [173]. Thus, if an individual has a higher fitness, its probability of getting selected is higher. In fitness-proportionate selection which is also known as roulette wheel selection, even the fittest individual may never be selected. In basic ABC, roulette wheel or fitness-proportionate selection scheme is incorporated. The analogy to a roulette wheel can be envisaged by imagining a roulette wheel in which each candidate solution represents a pocket on the wheel; the size of the pockets are proportionate to the probability of selection of the solution. Selecting N individuals from the population is equivalent to playing N games on the roulette wheel, as each candidate is drawn independently. The pseudocode is given in Algorithm 6.
 Stochastic universal sampling One variant of fitness-proportionate selection is called stochastic universal sampling, which is proposed by James Baker [174]. Where FPS chooses several solutions from the population by repeated random sampling, SUS uses a single random value to sample all of the solutions by choosing them at evenly spaced intervals. This gives weaker members of the population (according to their fitness) a chance to be chosen and thus reduces the unfair nature of fitness-proportional selection methods. In SUS, selection is done in a fitness-proportionate way but biased so that fit individuals always get picked at least once. This is known as a low variance resampling algorithm. SUS is used in genetic algorithms for selecting potentially useful solutions for recombination. The method has become now popular in other venues along with evolutionary computation [173]. The pseudocode is given in Algorithm 7.
Stochastic universal sampling One variant of fitness-proportionate selection is called stochastic universal sampling, which is proposed by James Baker [174]. Where FPS chooses several solutions from the population by repeated random sampling, SUS uses a single random value to sample all of the solutions by choosing them at evenly spaced intervals. This gives weaker members of the population (according to their fitness) a chance to be chosen and thus reduces the unfair nature of fitness-proportional selection methods. In SUS, selection is done in a fitness-proportionate way but biased so that fit individuals always get picked at least once. This is known as a low variance resampling algorithm. SUS is used in genetic algorithms for selecting potentially useful solutions for recombination. The method has become now popular in other venues along with evolutionary computation [173]. The pseudocode is given in Algorithm 7.

Other methods like roulette wheel can have bad performance when a member of the population has a really large fitness in comparison with other members. SUS starts from a small random number, and chooses the next candidates from the rest of population remaining, not allowing the fittest members to saturate the candidate space.
Scout bee
If the fitness of a bee remains the same for a predefined number (limit) of iterations, then it abandons its food position and becomes a scout bee. In basic ABC, it is assumed that only one source can be exhausted in each cycle, and only one employed bee can become a scout. In our modified approach we have removed this restriction. The scout bees are assigned to new food positions randomly. While determining components to form a new food position the solution component with higher pheromone values have higher probability of being selected. The value of limit is experimentally tuned and discussed in the Additional file 1. The variable triali contains the number of times the fitness remains unchanged consecutively for the ith bee. The procedure initRandom(Si) to assign new food positions for scout bees is given in Algorithm 3. In each iteration the value of gbest, and pbesti are updated using the Algorithm 4.
Local search
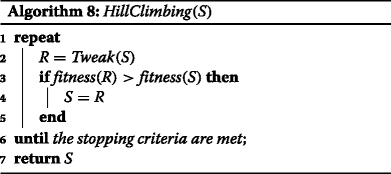
To explore nearby food sources the basic ABC algorithm applies a neighboring operator to the current food source. But in our algorithm we have applied local search to produce a new food position form the current one. In the employed bee and onlooker bee stages, local search is applied with the probability probLS to increase the exploitation ability [95]. The value of probLS is scientifically tuned in the Additional file 1. As has already been mentioned above, as the local search procedures, Hill Climbing (HC), Simulated Annealing (SA), and Steepest Ascent Hill Climbing with Replacement (SAHCR) have been employed as the local search procedure. Depending upon the choice HillClimbing(S) or SimulatedAnnealing(S) or SteepestAscentHillClimbingWithReplacement(S) is called form the method LocalSearch(S). The performance assessment between different local searches and the parameter tuning of the local search methods are discussed in the Additional file 1. Hill climbing Hill climbing is an optimization technique which belongs to the family of local search methods. The algorithm, starting from an arbitrary solution, iteratively tests new candidate solutions in the region of the current solution, and adopt the new ones if they are better. This enables to climb up the hill until local optima is reached. The method does not require to know the strength or direction of the gradient. Hill climbing is good for finding a local optima but it is not necessarily guaranteed to find the global optima. To find a new candidate solution we have applied random tweak to the current solution. The pseudocode is given in Algorithm 8.
 Simulated annealing Annealing is a process in metallurgy where molten metals are slowly cooled to make them reach a state of low energy where they are very strong. Simulated annealing is an analogous optimization method for locating a good approximation to the global optima. It is typically described in terms of thermodynamics. Simulated annealing is a process where the temperature is reduced slowly, starting from mostly exploring by random walk at high temperature eventually the algorithm does only plain hill climbing as it approaches zero temperature. The random movement corresponds to high temperature. Simulated annealing injects randomness to jump out of the local optima. At each iteration the algorithm selects the new candidate solution probabilistically. So the algorithm may sometimes go down hills. The pseudocode is given in Algorithm 9.
Simulated annealing Annealing is a process in metallurgy where molten metals are slowly cooled to make them reach a state of low energy where they are very strong. Simulated annealing is an analogous optimization method for locating a good approximation to the global optima. It is typically described in terms of thermodynamics. Simulated annealing is a process where the temperature is reduced slowly, starting from mostly exploring by random walk at high temperature eventually the algorithm does only plain hill climbing as it approaches zero temperature. The random movement corresponds to high temperature. Simulated annealing injects randomness to jump out of the local optima. At each iteration the algorithm selects the new candidate solution probabilistically. So the algorithm may sometimes go down hills. The pseudocode is given in Algorithm 9.
 Steepest ascent hill climbing with replacement This method samples all around the original candidate solution by tweaking n times. Best outcome of the tweaks is considered as the new candidate solution. The current candidate solution is replaced by the new one rather than selecting the best one between the new candidate solution and the current solution. The best found solution is saved in a separate variable. The pseudocode is given in Algorithm 10.
Steepest ascent hill climbing with replacement This method samples all around the original candidate solution by tweaking n times. Best outcome of the tweaks is considered as the new candidate solution. The current candidate solution is replaced by the new one rather than selecting the best one between the new candidate solution and the current solution. The best found solution is saved in a separate variable. The pseudocode is given in Algorithm 10.

Neighborhood operator In the solution we need only the informative genes to be selected. So we discard the uninformative ones from the solution. By this way we will get a small set of informative genes. To find a nearby food position we first find the genes which are selected in the current position. A number of selected genes (at least one) are dropped from the current solution. We get rid of the genes which tend to appear less potential. If the current solution has zero selected genes then we rather select a possibly informative gene. The parameter nd determines the percentage of selected genes to be removed. The value of nd is experimentally tuned in the Additional file 1.
Let Xe={0,1,1,0,1,0,0,1,1,0,1,1,1,0,0,1,0,1,0,0} is a candidate solution with gene size, n=20 and the number of selected gene is 10 (ten). So if nd=0.3 we will randomly pick 3 (three) genes which are currently selected in the current candidate solution (Xe) and change them to 0. Let the indices 2 (two), 8 (eight), and 15 (fifteen) are randomly selected. So , , and will become zero. Nearby food position of the current candidate solution (Xe), found after applying the neighborhood operator will be (changes are shown in boldface font). Please note that we adopt zero-based indexing.
Tweak operator The tweak operation is done by the method Tweak(S). Here, one of the genes is picked randomly and selection of that gene is flipped. So if the gene is selected, after tweak it will be dropped and vice versa. For example let Xe={0,1,1,0,1,0,0,1,1,0,1,1,1,0,0,1,0,1,0,0} is a candidate solution with gene size, n=20 and the number of selected gene is 10 (ten). Let randomly the index 6 (six) is selected. So the tweaked food position of the current candidate solution (Xe), found after applying the tweak operator will be (change is shown in boldface font). Please note that we adopt zero-based indexing.
Fitness Our fitness function has been designed to consider both the classification accuracy and the number of selected genes. The higher the accuracy of an individual the higher is its fitness. On the other hand small number of selected genes yields good solution. So if the percentage of genes that are not selected is higher the fitness will be higher. The value gives the percentage of genes that are not selected in Si. The tradeoff between weight of accuracy and selected gene size is given by w1. Higher value of w1 means accuracy is prioritized more than the selected gene size. So, finally the fitness of the ith bee (Si) is determined according to Eq. 12.
| 12 |
Here, w1 sets the tradeoff between the importance of accuracy and selected gene size, Xi is the food position corresponding to Si, accuracy(Xi) is the LOOCV (Leave One Out Cross Validation) classification accuracy using SVM (to be discussed shortly), and nsi is the number of currently selected genes in Si.
Accuracy To assess the fitness of a food position we need the classification accuracy of the gene subset. The predictive accuracy of a gene subset obtained from the modified ABC is calculated by an SVM with LOOCV (Leave One Out Cross Validation). The higher the LOOCV classification accuracy, the better the gene subset. SVM is very robust with sparse and noisy data. SVM has been found suitable for classifying high dimensional and small-sample sized data [142, 175]. Also SVM is reported to perform well for gene selection for cancer classification [20, 176].
The noteworthy implementations of SVM include SVM light [177], LIBSVM [178], mySVM [179], and BSVM [180, 181]. We have included LIBSVM as the implementation of SVM. For a multi-class SVM, we have utilized the OVO (“one versus one") approach, which is adapted in the LIBSVM [178]. The replacement of dot product by a nonlinear kernel function [182] yields a nonlinear mapping into a higher dimensional feature space [183]. A kernel can be viewed as a similarity function. It takes two inputs and outputs how similar they are. There are four basic kernels for SVM: linear, polynomial, radial basic function (RBF), and sigmoid [184]. The effectiveness of SVM depends on the selection of kernel, the kernel’s parameters, and the soft margin parameter C. Uninformed choices may result in extreme reduction of performance [142]. Tuning SVM is more of an art than an exact science. Selection of a specific kernel and relevant parameters can be achieved empirically. For the SVM, the penalty factor C and Gamma are set to 2000, 0.0001, respectively as adopted in Li et al. [41]. Use of linear and RBF kernel and their parameter tuning is discussed in the Additional file 1.
As classifier for both binary class and multi class gene selection methods, use of SVM is present in [23, 37, 41, 42, 45, 54, 153, 157, 164, 165, 185–200].
Cross-validation is believed to be a good method for selecting a subset of features [201]. LOOCV is in one extremity of k-fold cross validation, where k is chosen as the total number of examples. For a dataset with N examples, N numbers of experiments are performed. For each experiment the classifier learns on N−1 examples and is tested on the remaining one example. In the LOOCV method, a single observation from the original sample is selected as the validation data, and the remaining observations serve as training data. This process is repeated so that each observation in the sample is used once as the validation data. So every example is left out once and a prediction is made for that example. The average error is computed by finding number of misclassification and used to evaluate the model. The beauty of the LOOCV is that despite of the number of generations it will generate the same result each time, thus repetition is not needed.
Pseudocode for the modified ABC algorithm Finally, the pseudocode of our modified ABC algorithm used in this article is given in Algorithm 11 and the flowchart of the proposed gene selection method using Algorithm 11 is given in Fig. 1.

Results and discussion
The algorithm is iterated for MAX_ITER times to obtain an optimal gene subset. Then the gene subset is classified using SVM with LOOCV to find the accuracy of the subset which gives the performance outcome of a single run. Now to find the performance of our approach and to tune the parameters the algorithm is run multiple times (at least 15 times). Finally the average of accuracy along with the number of selected genes from all the runs for a single parameter combination presents the performance of that parameter combination. In this section the performance of our method will be presented using ten publicly available datasets. Different parameters are tuned to enhance the performance of the algorithm using one of the datasets. Parameter tuning and the contribution of different parameters are discussed in the Additional file 1. Comparison with previous methods that used the same datasets is discussed in this section. We have also presented comparison between different known heuristics methods in this section. Four different parameter settings according to different criteria have been proposed in this paper. Performance comparison for all the parameter settings is given in this section. In all cases the optimal results (maximum accuracy and minimum selected gene size) are highlighted using boldface font.
Datasets
Brief attribute summary of the datasets are presented in Table 1. The datasets contains both binary and multi class high dimensional data. The online supplement to the datasets [192] used in this paper is available at http://www.gems-system.org. The datasets are distributed as Matlab data files (.mat). Each file contains a matrix, the columns consist of diagnosis (1st column) and genes, and the rows are the samples.
Table 1.
Attributes of the datasets used for experimental evaluation
| Name of the dataset | Sample size | Number of genes | Number of classes | Reference |
|---|---|---|---|---|
| 9_Tumors | 60 | 5,726 | 9 | [209] |
| 11_Tumors | 174 | 12,533 | 11 | [196] |
| Brain_Tumor1 | 90 | 5,920 | 5 | [210] |
| Brain T umor2 | 50 | 10,367 | 4 | [211] |
| DLBCL | 77 | 5,469 | 2 | [212] |
| Leukemia1 | 72 | 5,327 | 3 | [5] |
| Leukemia2 | 72 | 11,225 | 3 | [213] |
| Lung_Cancer | 203 | 12,600 | 5 | [214] |
| Prostate_Tumor | 102 | 10,509 | 2 | [215] |
| SRBCT | 83 | 2,308 | 4 | [216] |
Optimized parameter values
While selecting the optimized parameter setting (Table 5) we have considered other factors besides the obtained performance.
Table 5.
All the proposed parameter values after tuning
| Parameter | First | Second | Third |
|---|---|---|---|
| probLS | 0.7 | 0.4 | 0.7 |
| ρ | 0.8 | 0.8 | 0.8 |
| w | 1.4 | 1.4 | 1.4 |
| w 1 | 0.85 | 0.85 | 0.85 |
| th n | 0.065 | 0.03 | 0.03 |
| sahc_iter | 12 | 16 | 16 |
| sahc_tweak | 9 | 15 | 15 |
| hc_iter | N/A | 10 | N/A |
| sa_iter | 10 | N/A | N/A |
| t | 5 | N/A | N/A |
| schedule | 0.5 | N/A | N/A |
| tmax | 5 | 5 | 5 |
| tmin | 0 | 0 | 0 |
| c 0 | 0.6 | 0.5 | 0.5 |
| MAX_ITER | 20 | 20 | 20 |
| limit | 35 | 100 | 100 |
| nd | 0.035 | 0.02 | 0.035 |
| PS | 25 | 40 | 40 |
| r 4 | 0.5 | 0.7 | 0.7 |
| ls e | SA | HC | SAHCR |
| ls o | SAHCR | SAHCR | SAHCR |
| Selection method | Tournament selection | Tournament selection | Stochastic universal sampling |
| kernel | Linear | Linear | Linear |
| wt | Equation 5 | Equation 5 | Equation 5 |
| uph | True | True | True |
| prefilter | Kruskal-Wallis | Kruskal-Wallis | Kruskal-Wallis |
After analyzing the results (Table S3 in Additional file 1), we have decided to use 0.5 as the value of r4 in our final experiments. Probability value of 0.7 for local search has been used to ensure that too much exploitation is not done despite that the value of 1.0 gives the highest accuracy (Table S5 in Additional file 1). The value of nd is set to 0.035 as it demonstrates a good enough accuracy with tolerable gene set size among all the values considered for the parameter (Table S6 in Additional file 1). Population size is kept at 25 which shows an acceptable level of accuracy (Table S9 in Additional file 1). We have selected SAHCR as the local search method at the onlooker bee stage and SA at the employed bee stage to ensure both exploration and exploitation. The value 12 is set as iteration count of SAHCR as it shows acceptable accuracy (Table S19 in Additional file 1). The value is kept small because increased iteration count increases the algorithm running time. The value 9 is considered as the final value for number of tweaks in SAHCR (Table S20 in Additional file 1). The value 0.065 is selected as the percentage of genes to be preselected in the preprocessing stage despite that the value 0.03 gives the best accuracy (Table S21 in Additional file 1). This is done because choosing 0.03 might possess the risk of discarding informative genes in the prefiltering step for other datasets. The value 0.6 is set for c0 as it shows good results (Table S23 in Additional file 1). The obtained accuracy is highest for the limit value 100 (Table S25 in Additional file 1). But high value of limit may result in less exploration. Thus, we recommend limit = 35 for this parameter setting after considering the experimental outcomes. Among TS and FPS, TS is considered as the selection method.
The optimized parameter values are listed in Table 2. From the obtained results (Table 7) we can conclude that the algorithm performs consistently for all datasets based on the standard deviation for accuracy (maximum 0.01) and number of selected gene (maximum 5.64) for optimized parameter settings. Our algorithm in fact has achieved satisfactory accuracy even for the default parameter settings albeit with a high standard deviation for the number of selected genes for most of the cases. The main reason for high standard deviation in the selected gene size for the default parameter setting can be attributed to the high default value of c0 and low default value of limit.
Table 2.
Optimized parameter values after tuning
| Parameter | Optimized value |
|---|---|
| probLS | 0.7 |
| ρ | 0.8 |
| w | 1.4 |
| w 1 | 0.85 |
| th n | 0.065 |
| sahc_iter | 12 |
| sahc_tweak | 9 |
| sa_iter | 14 |
| t | 5 |
| schedule | 0.5 |
| tmax | 5 |
| tmin | 0 |
| c 0 | 0.6 |
| MAX_ITER | 20 |
| limit | 35 |
| nd | 0.035 |
| PS | 25 |
| r 4 | 0.5 |
| ls e | SA |
| ls o | SAHCR |
| Selection method | Tournament selection |
| kernel | Linear |
| wt | Equation 5 |
| uph | True |
| prefilter | Kruskal-Wallis |
Table 7.
Experimental results of mABC using default, optimized (first), second and third parameter settings for different datasets
| Dataset name | Evaluation | Default | PS1 | PS2 | PS3 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Criteria | Best | Avg. | S. D. | Worst | Best | Avg. | S. D. | Worst | Best | Avg. | S. D. | Worst | Best | Avg. | S. D. | Worst | ||||
| 9_Tumors | Accuracy | 90.0 | 84.60 | 0.02 | 80.0 | 100 | 98.65 | 0.01 | 96.67 | 100 | 99.11 | 0.01 | 98.33 | 100 | 99.63 | 0.01 | 98.33 | |||
| # Genes | 41 | 50.03 | 52.21 | 37 | 30 | 34.73 | 5.64 | 35 | 21 | 24 | 4 | 26 | 21 | 25.5 | 3.75 | 24 | ||||
| 11_Tumors | Accuracy | 94.25 | 93.08 | 0.01 | 91.38 | 100 | 99.50 | 0.0 | 98.85 | 100 | 99.45 | 0.0 | 98.85 | 100 | 99.7 | 0.0 | 99.43 | |||
| # Genes | 33 | 37.15 | 5.08 | 23 | 42 | 47.27 | 7.79 | 47 | 30 | 30.75 | 4.15 | 30 | 32 | 33.86 | 3.44 | 34 | ||||
| Brain_Tumor1 | Accuracy | 100 | 95.85 | 0.02 | 93.33 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | |||
| # Genes | 13 | 16.53 | 5.78 | 16 | 12 | 16.87 | 2.85 | 20 | 10 | 11.2 | 1.08 | 13 | 8 | 10.58 | 1.17 | 13 | ||||
| Brain_Tumor2 | Accuracy | 100 | 98.09 | 0.01 | 96 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | |||
| # Genes | 11 | 17.6 | 5.82 | 16 | 7 | 10.52 | 1.72 | 15 | 6 | 7.47 | 1.19 | 9 | 6 | 7.35 | 1.0 | 9 | ||||
| DLBCL | Accuracy | 100 | 100 | 0 | 100 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | |||
| # Genes | 4 | 7.67 | 1.77 | 11 | 3 | 4.05 | 0.78 | 5 | 13 | 3.6 | 0.51 | 4 | 3 | 3.33 | 0.49 | 4 | ||||
| Leukemia1 | Accuracy | 100 | 100 | 0 | 100 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | |||
| # Genes | 5 | 8.39 | 1.94 | 12 | 4 | 5.67 | 0.73 | 7 | 4 | 4.4 | 0.51 | 5 | 3 | 3.87 | 0.35 | 4 | ||||
| Leukemia2 | Accuracy | 100 | 100 | 0 | 100 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | |||
| # Genes | 6 | 10.64 | 2.57 | 15 | 4 | 6.29 | 0.98 | 8 | 3 | 3.87 | 0.74 | 5 | 3 | 4.06 | 0.64 | 5 | ||||
| Lung_Cancer | Accuracy | 99.01 | 98.17 | 0.01 | 97.04 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | |||
| # Genes | 27 | 24.59 | 6.94 | 16 | 14 | 23.31 | 5.14 | 32 | 9 | 11.43 | 1.34 | 14 | 9 | 12.44 | 1.5 | 14 | ||||
| Prostate_Tumor | Accuracy | 100 | 96.95 | 0.01 | 95.10 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | |||
| # Genes | 11 | 15.33 | 6.35 | 19 | 5 | 10.73 | 3.15 | 16 | 5 | 6.59 | 0.91 | 8 | 5 | 6.5 | 0.82 | 8 | ||||
| SRBCT | Accuracy | 100 | 100 | 0 | 100 | 100 | 100 | 0.0 | 100 | 100 | ||||||||||
| textbf100 | 0.0 | 100 | 100 | 100 | 0.0 | 100 | ||||||||||||||
| # Genes | 6 | 7.47 | 0.94 | 9 | 5 | 5.59 | 0.51 | 6 | 4 | 4.27 | 0.46 | 5 | 4 | 4 | 0.0 | 4 | ||||
Best results (maximum accuracy and minimum selected gene size) are highlighted using boldface font
The Fig. 2 shows the distribution of obtained accuracy in optimized parameter settings for the dataset 9_Tumors and 11_Tumors. For all other datasets our method obtained 100 % accuracy in all the runs. The horizontal axis represents the accuracy and the vertical axis represents the percentage of time corresponding accuracy is obtained among all the runs. Similarly the Fig. 3 represents the distribution of selected gene size in optimized parameter settings for all the datasets. The horizontal axis represents the selected gene size and the vertical axis represents the percentage of time corresponding gene size is obtained among all the runs.
Fig. 2.
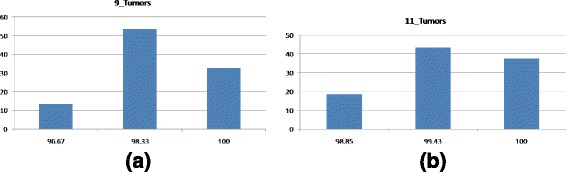
Distribution of classification accuracy using first (optimized) parameter setting for the dataset a 9_Tumors; b 11_Tumors
Fig. 3.
Distribution of number of times selected gene size fall in a specific range using the first (optimized) parameter setting a 9_Tumors; b 11_Tumors; c Brain_Tumor1; d Brain_Tumor2e Leukemia1; f Leukemia2; gDLBCL; h Lung_Cancer; i Prostate_Tumor; jSRBCT
Comparison with different metaheuristics
To compare our method with different metaheuristics we have considered ABC, GA, and ACO. In Additional file 1 performance of GA and ACO respectively for different parameter combination is discussed. Finally, the tuned parameter values are considered to run the experiments for comparing with our proposed method. For ABC, optimized parameter values found for mABC are considered (Table S31 in Additional file 1). Table 3 shows comparison between our work and the evolutionary algorithms in consideration. For this comparison we have considered the 9_Tumors dataset. The results are presented in Table 3. From the results we can see that ABC performs significantly better than GA and ACO. Our experimental results support the study done by Karaboga et al. [86]. Finally, from Table 3 we can see that our proposed mABC performs better than other metaheuristics in consideration according to both the constraints.
Table 3.
Comparative experimental results of the best subsets produced by mABC and other evolutionary methods for the dataset 9_Tumors
| Evaluation | GA | ACO | ABC | mABC [This work] | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Criteria | Best | Avg. | S.D. | Worst | Best | Avg. | S.D. | Worst | Best | Avg. | S.D. | Worst | Best | Avg. | S.D. | Worst | |||
| Accuracy | 85 | 82.56 | 0.01 | 81.67 | 85 | 83.09 | 0.01 | 81.67 | 91.67 | 87.67 | 0.02 | 85 | 100 | 98.65 | 0.01 | 96.67 | |||
| # Genes | 275 | 276.8 | 12.07 | 301 | 266 | 271.59 | 11.03 | 279 | 57 | 75.67 | 14.18 | 97 | 30 | 34.73 | 5.64 | 35 | |||
Best results (maximum accuracy and minimum selected gene size) are highlighted using boldface font
Comparison with existing methods
Table 4 shows comparison between our work and other works in literature including EPSO for the datasets 9_Tumors, 11_Tumors, Brain_Tumor1, Brain_Tumor2, DLBCL, Leukemia1, Leukemia2, Lung_Cancer, Prostate_Tumor, and SRBCT. The optimized parameter setting listed in Table 2 are used to run the algorithm for at least 15 times. Obtained accuracy (both average and best) by our approach for all the datasets are better or at least equal to the accuracy achieved by EPSO.
Table 4.
Comparative experimental results of the best subsets produced by mABC and other methods for different datasets
| Dataset name | Evaluation | EPSO | IBPSO | TS-BPSO | BPSO-CGA | Random forest | mABC | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Criteria | [37] | [43] | [42] | [44] | [55] | [This work] | |||||||||||
| Best | Avg. | S.D. | Best | Best | Best | Avg. | S.D. | Best | Avg. | S.D. | |||||||
| 9_Tumors | Accuracy | 76.67 | 75.00 | 1.11 | 78.33 | 81.63 | - | 88.0 | 0.037 | 100 | 98.65 | 0.01 | |||||
| # Genes | 251 | 247.10 | 9.65 | 1280 | 2941 | - | 130 | - | 30 | 34.73 | 5.64 | ||||||
| 11_Tumors | Accuracy | 96.55 | 95.40 | 0.61 | 93.10 | 97.35 | - | - | - | 100 | 99.50 | 0.00 | |||||
| # Genes | 243 | 237.70 | 9.66 | 2948 | 3206 | - | - | - | 42 | 47.27 | 7.79 | ||||||
| Brain_Tumor1 | Accuracy | 93.33 | 92.11 | 0.82 | 94.44 | 95.89 | 91.4 | 93.6 | 0.007 | 100 | 100 | 0.0 | |||||
| # Genes | 8 | 7.5 | 2.51 | 754 | 2913 | 456 | 86 | - | 12 | 16.87 | 2.85 | ||||||
| Brain_Tumor2 | Accuracy | 94 | 92.4 | 1.27 | 94.00 | 92.65 | - | - | - | 100 | 100 | 0.0 | |||||
| # Genes | 4 | 6.0 | 1.83 | 1197 | 5086 | - | - | - | 7 | 10.52 | 1.72 | ||||||
| DLBCL | Accuracy | 100 | 100 | 0 | 100 | 100 | - | 94.6 | 0.021 | 100 | 100 | 0.0 | |||||
| # Genes | 3 | 4.7 | 0.82 | 1042 | 2671 | - | 21 | - | 3 | 4.05 | 0.78 | ||||||
| Leukemia1 | Accuracy | 100 | 100 | 0 | 100 | 100 | 100 | 98.1 | 0.006 | 100 | 100 | 0.0 | |||||
| # Genes | 2 | 3.20 | 0.63 | 1034 | 2577 | 300 | 21 | - | 4 | 5.67 | 0.73 | ||||||
| Leukemia2 | Accuracy | 100 | 100 | 0 | 100 | 100 | - | - | - | 100 | 100 | 0.0 | |||||
| # Genes | 4 | 6.8 | 2.2 | 1292 | 5609 | - | - | - | 4 | 6.29 | 0.98 | ||||||
| Lung_Cancer | Accuracy | 96.06 | 95.67 | 0.31 | 96.55 | 99.52 | - | - | - | 100 | 100 | 0.0 | |||||
| # Genes | 7 | 8.3 | 2.11 | 1897 | 6958 | - | - | - | 14 | 23.31 | 5.14 | ||||||
| Prostate_Tumor | Accuracy | 99.02 | 97.84 | 0.62 | 92.16 | 95.45 | 93.7 | - | - | 100 | 100 | 0.0 | |||||
| # Genes | 5 | 6.6 | 2.17 | 1294 | 5320 | 795 | - | - | 5 | 10.73 | 3.15 | ||||||
| SRBCT | Accuracy | 100 | 99.64 | 0.58 | 100 | 100 | 100 | 99.7 | 0.003 | 100 | 100 | 0.0 | |||||
| # Genes | 7 | 14.90 | 13.03 | 431 | 1084 | 880 | 42 | - | 5 | 5.59 | 0.51 | ||||||
Best results (maximum accuracy and minimum selected gene size) are highlighted using boldface font
For 9_Tumors, with the optimized parameter values, our algorithm has achieved 100 % accuracy in 32.7 % runs (Fig. 2a). Average accuracy obtained by this work for this dataset is significantly better than EPSO. Also number of selected genes to achieve the accuracy is remarkably lower than EPSO. Our method selected at least 73.28 % less genes than other methods. It may appear that the reason for exceptionally better performance for 9_Tumors dataset is that the parameter values are optimized specially for this dataset. But even the worst performances for both default and optimized parameter values (Table 7) by this work are better than that of EPSO. For the dataset 11_Tumors, in 23.8 % runs our method obtained the highest (100 %) accuracy (Fig. 2b). The average accuracy obtained by our approach is better than other methods. The average no. of selected genes size is significantly better than previous methods. Our approach obtained at least 40.36 % less gene than previous methods. For Brain_Tumor1, and Brain_Tumor2 the obtained accuracy is better than EPSO and other methods with 100 % accuracy in all the runs. But the number of selected gene on average is little higher than EPSO. For DLBCL both our works and EPSO have achieved 100 % accuracy with 0 standard deviation. But on average number of selected gene is smaller in our algorithm though the best result by both the approaches are the same. For Leukemia1, and Leukemia2 our method has achieved highest (100 %) accuracy like EPSO. But our obtained marginally larger amount of genes than EPSO in the best obtained result for Leukemia1 (2 more). And for Leukemia2 our proposed method selected same number of genes as EPSO in the best obtained result. Also for Leukemia2 dataset, average number of genes selected is smaller in this work. For Lung_Cancer dataset, our algorithm achieved the highest (100 %) accuracy which is better than other methods. But the selected gene size for both the best and the average result is little higher than EPSO. For the dataset Prostate_Tumor, this work has exhibited better performance according to accuracy. Our method has obtained highest (100 %) accuracy with zero standard deviation which is the best accuracy obtained so far by any methods. But average number of selected genes is little higher in our method though best selected gene size is same as EPSO. For SRBCT our method has shown better performance in all cases (according to both accuracy and the number of selected genes for both optimized and default parameter values). For this dataset best result achieved by our method selected only 5 genes (better than EPSO) while obtained 100 % accuracy (same as EPSO). Even the worst results obtained by our algorithm in optimized parameter setting (Table 2) is better than the best result achieved by EPSO. Also our method exhibits more consistent performance according the lower standard deviation than EPSO for both accuracy (maximum 0.01) and selected gene size (maximum 5.64) for most of the datasets. So, in summary, for all the datasets gained accuracy and standard deviation of accuracy by our method is better or equal to the accuracy obtained by EPSO. However, for some cases the number of selected genes is a little higher. To obtain the stated results we have used only 20 iterations while EPSO used 500 iterations [37].
Further tuning of parameters
Tuning with full factorial combination would have allowed us to find the best parameter settings. But it will require enormous computational time. To demonstrate this hypothesis we have done the experiments using another two parameter settings (Table 5) formed from two different viewpoints. While selecting the optimized parameter setting (Table 2) we have considered many other factors besides the performance. So, we have configured two other parameter settings where the performance is considered as the major criterion of value selection along with running time. The last parameter settings (given in Table 5) is created after further tuning is done. Comparison between all the parameter settings including the default one is given in Table 7. From the obtained results (Table 7) it is clear that further tuning can improve results for all the datasets.
Second parameter setting
To propose the second parameter setting we have considered the performance as the major criterion along with the running time for selecting parameter values. High probability of local search increases performance (Table S5 in Additional file 1). But higher probability will also result in increased running time and little exploration. So while preparing this parameter combination we decided to keep the probability low allowing enough exploration, but other values are selected considering the performance outcome mainly. The probability value of 0.4 is selected for local search to prevent too much exploitation and decrease running time. The value 0.03 is selected as the percentage of genes to be preselected in the preprocessing because it gives the best accuracy. The value 0.6 is set for c0 as it shows the best results. The obtained accuracy is highest for the limit value 100 which is selected for this parameter. The value of nd is set to 0.02 as it demonstrates a good enough accuracy. Population size is increased to 40 which gives the best accuracy and small number of selected genes. The value of r4 is increased to 0.7, as increased application of communication operator improves the results. HC is selected as local search method in employed bee stage and SAHCR is selected as local search method in onlooker bee stage because this combination needs comparatively less running time but gives considerably good results. Also the iteration and tweak count for the local search method SAHCR is increased. The main idea behind proposing this parameter setting is to improve performance than the previous parameter setting with decreased running time. So we have selected low probability of local search but for other parameters mainly we have selected values which give best performance.
Third parameter setting
The third parameter settings is proposed after further tuning is done for one of the parameters named “Selection method". To find if further performance upgrade is possible by considering new values we have considered another selection method named Stochastic Universal Sampling (SUS). The results are given in Table 6. From the results we can see that the newly considered method SUS performs better than the others.
Table 6.
Performance outcome for different values of parameter selection method
| Values | Accuracy | No. of selected gene | ||
|---|---|---|---|---|
| Avg. | S.D. | Avg. | S.D. | |
| Fitness proportionate selection | 84.2 | 0.03 | 41.43 | 47.47 |
| Tournament selection | 84.74 | 0.03 | 53.33 | 53.65 |
| Stochastic universal sampling | 85.42 | 0.03 | 35.19 | 5.49 |
Best results (maximum accuracy and minimum selected gene size) are highlighted using boldface font
So, finally, we have proposed another parameter settings which considers the performance as the main criterion to select the parameter values. So, we have kept the probability of the local search high (0.7). This setup takes the highest time to run. Because the probability of local search is kept high and SAHCR is used as local search method for both the stages. SAHCR as local search for both the stages performs the best (Table S14 in Additional file 1). SUS is used as the selection method in onlooker bee stage. Other parameter values are same as optimized (first) parameter setting. The parameter settings is given in Table 5.
Comparison between different parameter settings
Comparison between all the parameter settings including the default one is given in Table 7. For all the parameter settings the best, average, standard deviation (S.D.), and the worst results are reported. First we will present the comparison between results achieved by the default and the first parameter settings. Next we will compare the results obtained by second and third parameter settings with the first parameter settings.
In all cases the first parameter setting exhibits better results according to both accuracy and the number of selected genes than the default parameter setting. For the first parameter setting we can conclude that the algorithm performs consistently for all datasets based on the standard deviation for accuracy (maximum 0.01) and number of selected gene (maximum 5.64). Our algorithm in fact has achieved satisfactory accuracy even for the default parameter setting albeit with a high standard deviation for the number of selected genes for most of the cases. The main reason for high standard deviation in the selected gene size for the default parameter setting can be attributed to the high default value of c0 and low default value of limit.
The best, average, worst, and standard deviation obtained using the first, second, and third parameter settings for all the datasets are given in Table 7. Now we will present comparison between these parameter settings. For the 9_Tumors dataset, best obtained accuracy for all the parameter settings is same (100 % accuracy). But the selected gene size (21) for the best results are same for newly proposed two parameter settings which is better (30 % lower) than the selected gene size (30) obtained by the proposed first parameter settings. For the 11_Tumors dataset obtained average accuracy by third parameter setting is better than other parameter settings. Best obtained accuracy by all three parameter setting is 100 %. But selected gene size for best result is better in the second parameter settings. Also the second parameter setting selected lower (at least 9.18 % lower) number of genes on average than others. Second and third proposed parameter settings selected much lower (at least 33.89 % lower) number of genes on average than the first parameter setting for this dataset. For all other datasets obtained accuracy by all three parameter settings in all the runs is 100 %. For the dataset Brain_Tumor1, the best and the average number of selected genes are better for the third parameter setting. Last two parameter settings obtained at least 33.61 % lower selected genes size than the average number of selected genes by the first parameter setting. The average number of selected genes by the third parameter setting is better (5.54 % lower) than the average number of genes selected by the second parameter setting. And the maximum number of selected genes (13) is same for the last two parameter settings. For the dataset Brain_Tumor2, minimum (6) and maximum (9) number of selected genes are same for the second and third parameter settings. But selected gene size on average is smaller (1.61 % lower) for the third parameter settings than the average selected gene size by the second parameter settings. For both the second and third parameter settings selected gene size on average is lower (at least 28.99 % lower) than that of first parameter setting. For the dataset DLBCL, minimum number of selected gene size (3) is same for all three parameter settings. But average selected gene size obtained by the last two parameter combinations are at least 11.11 % lower than the average selected gene size obtained by the first parameter setting. The maximum number of genes selected (4) by the second and the third parameter settings are same. But on average selected gene size by the third parameter setting is 7.5 % lower than the average number of genes selected by the second parameter setting. For the Leukemia1 dataset, best minimum number of selected genes (3) is obtained by the third parameter setting. On average the last two proposed parameter settings selected at least 22.4 % lower number of genes than the first parameter setting. Also the average number of genes selected by the third parameter setting is 12.05 % lower than the average number of selected genes by the second parameter setting. For the dataset Leukemia2, minimum number of selected genes (3) by both the second and the third parameter setting is same, which is better than the minimum number of selected genes (4) by the first parameter setting. Also the maximum number of selected genes (5) by the second and the third parameter setting are the same, which is lower than the maximum number of genes selected (8) by the first parameter setting. Average number of selected genes by the second and third parameter settings is at least 35.45 % lower than the average selected gene size by the first parameter setting. But for this datasets the average number of selected gene size is minimum for the second parameter setting, which is 4.68 % lower than the average number of selected gene size by the third parameter setting. For the Lung_Cancer dataset, minimum number of selected genes (9) by the second and the third parameter settings is 35.71 % smaller than the minimum number of selected genes (14) obtained by the first parameter setting. Also the maximum number of selected genes (14) by the second and the third parameter settings is 56.25 % lower than the maximum number of selected genes (32) by the first parameter setting. Note that the best obtained gene set size (14) by the first parameter setting for this dataset is same as the worst obtained gene set size (14) by the last two proposed parameter combinations. Also the second and third parameter settings obtained average selected gene size at least 46.63 % smaller than the average number of selected genes by the first parameter setting. But the average selected gene size by the second parameter setting is better (8.12 % lower) than the average selected gene size by the third parameter setting. For the Prostate_Tumor dataset, the minimum number of selected genes (5) is same for all the parameter settings. But the worst obtained gene subset size (8) by the second and the third parameter settings is better (50 % lower) than the worst obtained gene subset size (16) by the first parameter setting. Also the average number of selected genes by the first parameter setting is at least 62.82 % higher than the average number of selected genes by the last two parameter settings. Again the average number of genes selected by the third parameter setting is better (1.37 % lower) than the average number of genes selected by the second parameter setting. For the dataset SRBCT, minimum number of genes selected (4) by the last two parameter setting is better than the minimum number of genes selected (5) by the first parameter setting. The average number of genes selected by the second and third parameter settings are at least 23.61 % better than the average number of genes selected by the first parameter setting. Moreover selected gene size by the third parameter setting is same in all the run, thus standard deviation is zero. So average number of selected genes for the third parameter setting is 6.32 % lower than the average number of genes selected by the second parameter setting. For the datasets 9_Tumors, 11_Tumors, Leukemia2, and Lung_Cancer obtained average number of selected genes is better for the second parameter setting. In all other cases considering both the accuracy and the selected gene size the third parameter setting performed comparatively better.
Conclusions
Microarray technology allows producing databases of cancerous tissues based on gene expression data [202]. Available training datasets for cancer classification generally have a fairly small sample size compared to the number of genes involved and consists of multiclass categories. The sample size is likely to remain small at least for the near future due to the expense of microarray sample collection [203]. The huge number of genes causes grave computational overhead and poor predictive accuracy in wrapper methods when searching for significant genes. So to select small subsets of relevant genes involved in different types of cancer remains a challenge. So we apply a statistical method in preprocessing step to filter out the noisy genes. Then a search method is utilized for further selection of smaller subset of informative genes. Selection of pertinent genes enable researchers to obtain significant insight into the genetic nature of the disease and the mechanisms responsible for it [183, 204]. Recent research has demonstrated that one of the most important applications of microarrays technology is cancer classification [205, 206]. Biomarker discovery in high-dimensional microarray data helps studying the biology of cancer [207]. When a large number of noisy, redundant genes are filtered the performance of cancer classification is improved [208]. Besides, gene selection can also cut down the cost of medical diagnoses. We believe that the selection of genes by our system provide us some interesting clue towards the importance and contribution of that set of particular genes for the respective cancer disease. To elaborate, our system has identified that for diffuse large B-cell lymphoma (DLBCL) only three (3) genes are informative enough to decide about the cancer. Now, this could turn out to be a string statement with regards to the set of genes identified for a particular cancer and we believe further biological validation is required before making such a string claim. We do plan to work towards validation of these inferences. To this end we believe that our method presented in this paper is a significant contribution and would be useful in medical diagnosis as well as for further research.
Acknowledgements
Part of this research work was carried out when Mohammad Sohel Rahman was on a Sabbatical Leave from BUET.
Declaration
The publication fee for this article is waived according to Open Access Waiver fund of BioMed Central.
This article has been published as part of BMC Medical Genomics Vol 9 Suppl 2 2016: Selected articles from the IEEE International Conference on Bioinformatics and Biomedicine 2015: medical genomics. The full contents of the supplement are available online at http://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-9-supplement-2.
Authors’ contributions
JM and MR conceived the study. JM devised and implemented the algorithms and conducted the experiments. JM, MR, RS and MK analysed and interpreted the results. The research work was supervised by MR and MK JM wrote the manuscript. All authors reviewed and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional file
Gene Selection for Cancer Classification With the Help of Bees. The supplementary file is available in the link: https://goo.gl/APTj0n. (PDF 417 kb)
Contributor Information
Johra Muhammad Moosa, Email: johra.moosa@gmail.com.
Rameen Shakur, Email: rameen.shakur@sanger.ac.uk.
Mohammad Kaykobad, Email: kaykobad@cse.buet.ac.bd.
Mohammad Sohel Rahman, Email: sohel.kcl@gmail.com.
References
- 1.Pavlidis S, Payne AM, Swift S. Multi-membership gene regulation in pathway based microarray analysis. Algorithms Mol Biol. 2011;6(1):1–22. doi: 10.1186/1748-7188-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brazma A, Vilo J. Gene expression data analysis. FEBS Lett. 2000;480(1):17–24. doi: 10.1016/S0014-5793(00)01772-5. [DOI] [PubMed] [Google Scholar]
- 3.Fodor SP. DNA sequencing: Massively parallel genomics. Science. 1997;277(5324):393–5. doi: 10.1126/science.277.5324.393. [DOI] [Google Scholar]
- 4.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270(5235):467–70. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 5.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–7. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 6.Chang JC, Hilsenbeck SG, Fuqua SA. The promise of microarrays in the management and treatment of breast cancer. Breast Cancer Res. 2005;7(3):100. doi: 10.1186/bcr1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeffrey SS, Lønning PE, Hillner BE. Genomics-based prognosis and therapeutic prediction in breast cancer. J Nat Compr Cancer Netw. 2005;3(3):291–300. doi: 10.6004/jnccn.2005.0016. [DOI] [PubMed] [Google Scholar]
- 8.Lønning PE, Sørlie T, Børresen-Dale AL. Genomics in breast cancer–therapeutic implications. Nat Clin Prac Oncol. 2005;2(1):26–33. doi: 10.1038/ncponc0072. [DOI] [PubMed] [Google Scholar]
- 9.Park C, Cho SB. Genetic search for optimal ensemble of feature-classifier pairs in DNA gene expression profiles. In: Neural Networks, 2003. Proceedings of the International Joint Conference On. IEEE: 2003. p. 1702–1707.
- 10.Li W. The-more-the-better and the-less-the-better. Bioinformatics. 2006;22(18):2187–8. doi: 10.1093/bioinformatics/btl189. [DOI] [PubMed] [Google Scholar]
- 11.Li W, Yang Y. Methods of Microarray Data Analysis: Papers from CAMDA ’00. Boston, MA: Springer US; 2002. How many genes are needed for a discriminant microarray data analysis. [Google Scholar]
- 12.Stephanopoulos G, Hwang D, Schmitt WA, Misra J, Stephanopoulos G. Mapping physiological states from microarray expression measurements. Bioinformatics. 2002;18(8):1054–63. doi: 10.1093/bioinformatics/18.8.1054. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen DV, Rocke DM. Multi-class cancer classification via partial least squares with gene expression profiles. Bioinformatics. 2002;18(9):1216–26. doi: 10.1093/bioinformatics/18.9.1216. [DOI] [PubMed] [Google Scholar]
- 14.Bicciato S, Luchini A, Di Bello C. PCA disjoint models for multiclass cancer analysis using gene expression data. Bioinformatics. 2003;19(5):571–8. doi: 10.1093/bioinformatics/btg051. [DOI] [PubMed] [Google Scholar]
- 15.Tan Y, Shi L, Tong W, Gene Hwang G, Wang C. Multi-class tumor classification by discriminant partial least squares using microarray gene expression data and assessment of classification models. Comput Biol Chem. 2004;28(3):235–43. doi: 10.1016/j.compbiolchem.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Tinker AV, Boussioutas A, Bowtell DD. The challenges of gene expression microarrays for the study of human cancer. Cancer Cell. 2006;9(5):333–9. doi: 10.1016/j.ccr.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Choudhary A, Brun M, Hua J, Lowey J, Suh E, Dougherty ER. Genetic test bed for feature selection. Bioinformatics. 2006;22(7):837–42. doi: 10.1093/bioinformatics/btl008. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Yang J, Teng X, Xia W, Jensen R. Feature selection based on rough sets and particle swarm optimization. Pattern Recognit Lett. 2007;28(4):459–71. doi: 10.1016/j.patrec.2006.09.003. [DOI] [Google Scholar]
- 19.Cover TM, Van Campenhout JM. On the possible orderings in the measurement selection problem. Syst Man Cybernet IEEE Trans. 1977;7(9):657–61. doi: 10.1109/TSMC.1977.4309803. [DOI] [Google Scholar]
- 20.George G, Raj VC. Review on feature selection techniques and the impact of SVM for cancer classification using gene expression profile. International J Comp Science & Engineering Survey (IJCSES) 2011;2(3):26–38. [Google Scholar]
- 21.Ahmade F, Norwawi NM, Deris S, Othman NH. Information Technology, 2008. ITSim 2008. International Symposium On. Kuala Lumpur, Malaysia: IEEE; 2008. A review of feature selection techniques via gene expression profiles. [Google Scholar]
- 22.Saeys Y, Inza I, Larrañaga P. A review of feature selection techniques in bioinformatics. Bioinformatics. 2007;23(19):2507–17. doi: 10.1093/bioinformatics/btm344. [DOI] [PubMed] [Google Scholar]
- 23.Lu Y, Han J. Cancer classification using gene expression data. Inform Syst. 2003;28(4):243–68. doi: 10.1016/S0306-4379(02)00072-8. [DOI] [Google Scholar]
- 24.Guyon I, Elisseeff A. Feature Extraction. Berlin, Heidelberg: Springer Berlin Heidelberg; 2006. An introduction to feature extraction. [Google Scholar]
- 25.Lazar C, Taminau J, Meganck S, Steenhoff D, Coletta A, Molter C, de Schaetzen V, Duque R, Bersini H, Nowe A. A survey on filter techniques for feature selection in gene expression microarray analysis. IEEE/ACM Trans Comput Biol Bioinform (TCBB) 2012;9(4):1106–19. doi: 10.1109/TCBB.2012.33. [DOI] [PubMed] [Google Scholar]
- 26.Geisser S, Vol. 55. Predictive Inference: CRC Press; 1993.
- 27.Kohavi R. Proceedings of the 14th International Joint Conference on Artificial Intelligence. IJCAI’95. volume 2. San Francisco, CA, USA: Morgan Kaufmann Publishers Inc.; 1995. A study of cross-validation and bootstrap for accuracy estimation and model selection. [Google Scholar]
- 28.Devijver PA, Kittler J. Pattern Recognition: A Statistical Approach. London: Prentice-Hall; 1982. [Google Scholar]
- 29.Giallourakis C, Henson C, Reich M, Xie X, Mootha VK. Disease gene discovery through integrative genomics. Annu Rev Genomics Hum Genet. 2005;6:381–406. doi: 10.1146/annurev.genom.6.080604.162234. [DOI] [PubMed] [Google Scholar]
- 30.Press WH, Flannery BP, Teukolsky SA, Vetterling WT. Numerical recipes. Cambridge: Cambridge university press; 1990. [Google Scholar]
- 31.Wang Z. Neuro-fuzzy modeling for microarray cancer gene expression data. First year transfer report, University of Oxford. 2005.
- 32.Cho SB, Won HH. Machine learning in DNA microarray analysis for cancer classification. In: Proceedings of the First Asia-Pacific Bioinformatics Conference on Bioinformatics 2003. APBC ’03. Volume 19. Darlinghurst, Australia, Australia: 2003. p. 189–98.
- 33.Hu H, Li J, Wang H, Daggard G. Knowledge-Based Intelligent Information and Engineering Systems: 10th International Conference, KES 2006, Bournemouth, UK, October 9-11, 2006. Proceedings, Part I. Berlin, Heidelberg: Springer Berlin Heidelberg; 2006. Combined gene selection methods for microarray data analysis. [Google Scholar]
- 34.Liu Z, Magder LS, Hyslop T, Mao L. Survival associated pathway identification with group lp penalized global AUC maximization. Algorithms Mol Biol. 2010;5:30. doi: 10.1186/1748-7188-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohavi R, John GH. Wrappers for feature subset selection. Artif Intell. 1997;97(1):273–324. doi: 10.1016/S0004-3702(97)00043-X. [DOI] [Google Scholar]
- 36.Zhang H, Ho T, Kawasaki S. Wrapper feature extraction for time series classification using singular value decomposition. Int J Knowl Syst Sci. 2006;3:53–60. [Google Scholar]
- 37.Mohamad MS, Omatu S, Deris S, Yoshioka M, Abdullah A, Ibrahim Z. An enhancement of binary particle swarm optimization for gene selection in classifying cancer classes. Algorithms Mol Biol. 2013;8(1):1–11. doi: 10.1186/1748-7188-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inza I, Larrañaga P, Blanco R, Cerrolaza AJ. Filter versus wrapper gene selection approaches in DNA microarray domains. Artif Intell Med. 2004;31(2):91–103. doi: 10.1016/j.artmed.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 39.Karegowda AG, Jayaram M, Manjunath A. Feature subset selection problem using wrapper approach in supervised learning. Int J Comput Appl. 2010;1(7):13–7. [Google Scholar]
- 40.Shen Q, Shi WM, Kong W. Hybrid particle swarm optimization and tabu search approach for selecting genes for tumor classification using gene expression data. Comput Biol Chem. 2008;32(1):53–60. doi: 10.1016/j.compbiolchem.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 41.Li S, Wu X, Tan M. Gene selection using hybrid particle swarm optimization and genetic algorithm. Soft Comput. 2008;12(11):1039–48. doi: 10.1007/s00500-007-0272-x. [DOI] [Google Scholar]
- 42.Chuang LY, Yang CH, Yang CH. Tabu search and binary particle swarm optimization for feature selection using microarray data. J Comput Biol. 2009;16(12):1689–703. doi: 10.1089/cmb.2007.0211. [DOI] [PubMed] [Google Scholar]
- 43.Chuang LY, Chang HW, Tu CJ, Yang CH. Improved binary PSO for feature selection using gene expression data. Comput Biol Chem. 2008;32(1):29–38. doi: 10.1016/j.compbiolchem.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 44.Chuang LY, Yang CH, Li JC, Yang CH. A hybrid BPSO-CGA approach for gene selection and classification of microarray data. J Comput Biol. 2012;19(1):68–82. doi: 10.1089/cmb.2010.0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu JJ, Cutler G, Li W, Pan Z, Peng S, Hoey T, Chen L, Ling XB. Multiclass cancer classification and biomarker discovery using GA-based algorithms. Bioinformatics. 2005;21(11):2691–7. doi: 10.1093/bioinformatics/bti419. [DOI] [PubMed] [Google Scholar]
- 46.Li L, Darden TA, Weingberg C, Levine A, Pedersen LG. Gene assessment and sample classification for gene expression data using a genetic algorithm/k-nearest neighbor method. Comb Chem High Throughput Screen. 2001;4(8):727–39. doi: 10.2174/1386207013330733. [DOI] [PubMed] [Google Scholar]
- 47.Mohamad MS, Omatu S, Deris S, Yoshioka M. Trends and Applications in Knowledge Discovery and Data Mining: PAKDD 2013 International Workshops: DMApps, DANTH, QIMIE, BDM, CDA, CloudSD, Gold Coast, QLD, Australia, April 14-17, 2013, Revised Selected Papers. Berlin, Heidelberg: Springer Berlin Heidelberg; 2013. A Constraint and Rule in an Enhancement of Binary Particle Swarm Optimization to Select Informative Genes for Cancer Classification. [Google Scholar]
- 48.Mohamad MS, Omatu S, Deris S, Yoshioka M. Particle swarm optimization with a modified sigmoid function for gene selection from gene expression data. Artif Life Robotics. 2010;15(1):21–4. doi: 10.1007/s10015-010-0757-z. [DOI] [Google Scholar]
- 49.Chuang LY, Yang CH, Wu KC, Yang CH. A hybrid feature selection method for DNA microarray data. Comput Biol Med. 2011;41(4):228–37. doi: 10.1016/j.compbiomed.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 50.Khushaba RN, Al-Ani A, Al-Jumaily A. Feature subset selection using differential evolution and a statistical repair mechanism. Expert Syst Appl. 2011;38(9):11515–26. doi: 10.1016/j.eswa.2011.03.028. [DOI] [Google Scholar]
- 51.Erol OK, Eksin I. A new optimization method: big bang–big crunch. Adv Eng Softw. 2006;37(2):106–11. doi: 10.1016/j.advengsoft.2005.04.005. [DOI] [Google Scholar]
- 52.Eksin I, Erol OK. Evolutionary algorithm with modifications in the reproduction phase. IEE Proc-Softw. 2001;148(2):75–80. doi: 10.1049/ip-sen:20010503. [DOI] [Google Scholar]
- 53.Yu H, Gu G, Liu H, Shen J, Zhao J. A modified ant colony optimization algorithm for tumor marker gene selection. Genomics Proteomics Bioinformatics. 2009;7(4):200–8. doi: 10.1016/S1672-0229(08)60050-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Díaz-Uriarte R, De Andres SA. Gene selection and classification of microarray data using random forest. BMC Bioinformatics. 2006;7(1):3. doi: 10.1186/1471-2105-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Q, Hong W, Luo L, Yang F. Gene selection using random forest and proximity differences criterion on DNA microarray data. J Convergence Inform Technol. 2010;5(6):161–70. doi: 10.4156/jcit.vol5.issue6.17. [DOI] [Google Scholar]
- 56.Debnath R, Kurita T. An evolutionary approach for gene selection and classification of microarray data based on SVM error-bound theories. Biosystems. 2010;100(1):39–46. doi: 10.1016/j.biosystems.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 57.Karaboga D. An idea based on honey bee swarm for numerical optimization. Technical report, Technical report-tr06, Erciyes university, engineering faculty, computer engineering department. 2005. http://mf.erciyes.edu.tr/abc/pub/tr06_2005.pdf.
- 58.Li G, Niu P, Xiao X. Development and investigation of efficient artificial bee colony algorithm for numerical function optimization. Appl Soft Comput. 2012;12(1):320–32. doi: 10.1016/j.asoc.2011.08.040. [DOI] [Google Scholar]
- 59.Murugan R, Mohan M. Artificial bee colony optimization for the combined heat and power economic dispatch problem. ARPN J Eng Appl Sci. 2012;7(5):597–604. [Google Scholar]
- 60.Dorigo M, Maniezzo V, Colorni A. The ant system: An autocatalytic optimizing process. Technical report. 1991.
- 61.Stützle T, Hoos HH. MAX–MIN ant system. Future Generation Comput Syst. 2000;16(8):889–914. doi: 10.1016/S0167-739X(00)00043-1. [DOI] [Google Scholar]
- 62.Bollazzi M, Roces F. Information needs at the beginning of foraging: grass-cutting ants trade off load size for a faster return to the nest. PloS One. 2011;6(3):17667. doi: 10.1371/journal.pone.0017667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roces F. Olfactory conditioning during the recruitment process in a leaf-cutting ant. Oecologia. 1990;83(2):261–2. doi: 10.1007/BF00317762. [DOI] [PubMed] [Google Scholar]
- 64.Howard JJ, Henneman LM, Cronin G, Fox JA, Hormiga G. Conditioning of scouts and recruits during foraging by a leaf-cutting ant, Atta colombica. Anim Behav. 1996;52(2):299–306. doi: 10.1006/anbe.1996.0175. [DOI] [Google Scholar]
- 65.Roces F. Odour learning and decision-making during food collection in the leaf-cutting antacromyrmex lundi. Insectes Sociaux. 1994;41(3):235–9. doi: 10.1007/BF01242294. [DOI] [Google Scholar]
- 66.Dussutour A, Beshers S, Deneubourg JL, Fourcassie V. Crowding increases foraging efficiency in the leaf-cutting ant atta colombica. Insectes Sociaux. 2007;54(2):158–65. doi: 10.1007/s00040-007-0926-9. [DOI] [Google Scholar]
- 67.Farji-Brener A, Amador-Vargas S, Chinchilla F, Escobar S, Cabrera S, Herrera M, Sandoval C. Information transfer in head-on encounters between leaf-cutting ant workers: food, trail condition or orientation cues? Anim Behav. 2010;79(2):343–9. doi: 10.1016/j.anbehav.2009.11.009. [DOI] [Google Scholar]
- 68.Detrain C, Deneubourg JL, Jarau S, Hrncir M. Social cues and adaptive foraging strategies in ants. Food Exploitation by Social Insects: Ecological, Behavioral, and Theoretical Approaches. 2009:29–52. CRC Press: Contemporary Topics in Entomology Series, Boca Raton, USA.
- 69.Gordon DM. Ant Encounters: Interaction Networks and Colony Behavior. New Jersey, USA: Princeton University Press; 2010. [Google Scholar]
- 70.Greene MJ, Gordon DM. Interaction rate informs harvester ant task decisions. Behav Ecol. 2007;18(2):451–5. doi: 10.1093/beheco/arl105. [DOI] [Google Scholar]
- 71.Schafer RJ, Holmes S, Gordon DM. Forager activation and food availability in harvester ants. Anim Behav. 2006;71(4):815–22. doi: 10.1016/j.anbehav.2005.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Torres-Contreras H, Vasquez RA. Spatial heterogeneity and nestmate encounters affect locomotion and foraging success in the ant dorymyrmex goetschi. Ethology. 2007;113(1):76–86. doi: 10.1111/j.1439-0310.2006.01302.x. [DOI] [Google Scholar]
- 73.Gordon DM, Mehdiabadi NJ. Encounter rate and task allocation in harvester ants. Behav Ecol Sociobiol. 1999;45(5):370–7. doi: 10.1007/s002650050573. [DOI] [Google Scholar]
- 74.Gordon DM, Paul RE, Thorpe K. What is the function of encounter patterns in ant colonies? Anim Behav. 1993;45(6):1083–100. doi: 10.1006/anbe.1993.1134. [DOI] [Google Scholar]
- 75.Teodorovic D. Transport modeling by multi-agent systems: a swarm intelligence approach. Transport Planning Technol. 2003;26(4):289–312. doi: 10.1080/0308106032000154593. [DOI] [Google Scholar]
- 76.Teodorović D, Dell’Orco M. Bee colony optimization–a cooperative learning approach to complex transportation problems. In: Advanced OR and AI Methods in Transportation: Proceedings of 16th Mini–EURO Conference and 10th Meeting of EWGT (13–16 September 2005).–Poznan: Publishing House of the Polish Operational and System Research. Poland: 2005. p. 51–60.
- 77.Tereshko V. Parallel Problem Solving from Nature PPSN VI: 6th International Conference Paris, France, September 18–20, 2000 Proceedings. Berlin, Heidelberg: Springer Berlin Heidelberg; 2000. Reaction-diffusion model of a honeybee colony’s foraging behaviour. [Google Scholar]
- 78.Tereshko V, Lee T. How information-mapping patterns determine foraging behaviour of a honey bee colony. Open Syst Inform Dyn. 2002;9(02):181–93. doi: 10.1023/A:1015652810815. [DOI] [Google Scholar]
- 79.Tereshko V, Loengarov A. Collective decision making in honey-bee foraging dynamics. Comput Inform Syst. 2005;9(3):1. [Google Scholar]
- 80.Lucic P, Teodorovic D. Transportation modeling: an artificial life approach. In: Tools with Artificial Intelligence, 2002.(ICTAI 2002). Proceedings. 14th IEEE International Conference On. IEEE: 2002. p. 216–23.
- 81.Drias H, Sadeg S, Yahi S. Computational Intelligence and Bioinspired Systems. Berlin, Heidelberg: Springer Berlin Heidelberg; 2005. Cooperative bees swarm for solving the maximum weighted satisfiability problem. [Google Scholar]
- 82.Benatchba K, Admane L, Koudil M. Using bees to solve a data-mining problem expressed as a max-sat one. In: Artificial Intelligence and Knowledge Engineering Applications: A Bioinspired ApproachApproach: First International Work-Conference on the Interplay Between Natural and Artificial Computation, IWINAC 2005, Las Palmas, Canary Islands, Spain, June 15-18, 2005, Proceedings, Part II. Berlin, Heidelberg: Springer Berlin Heidelberg: 2005. p. 212–20, doi:10.1007/11499305_22.
- 83.Wedde HF, Farooq M, Zhang Y. Ant Colony Optimization and Swarm Intelligence: 4th International Workshop, ANTS 2004, Brussels, Belgium, September 5-8, 2004. Proceedings. Berlin, Heidelberg: Springer Berlin Heidelberg; 2004. BeeHive: An efficient fault-tolerant routing algorithm inspired by honey bee behavior. [Google Scholar]
- 84.Yang XS. Engineering optimizations via nature-inspired virtual bee algorithms. In: Artificial Intelligence and Knowledge Engineering Applications: A Bioinspired Approach: First International Work-Conference on the Interplay Between Natural and Artificial Computation, IWINAC 2005, Las Palmas, Canary Islands, Spain, June 15-18, 2005, Proceedings, Part II. Berlin, Heidelberg: Springer Berlin Heidelberg: 2005. p. 317–23, doi:10.1007/11499305_33.
- 85.Pham D, Ghanbarzadeh A, Koc E, Otri S, Rahim S, Zaidi M. The bees algorithm-a novel tool for complex optimisation problems. In: Proceedings of the 2nd Virtual International Conference on Intelligent Production Machines and Systems (IPROMS 2006): 2006. p. 454–9.
- 86.Karaboga D, Akay B. A comparative study of artificial bee colony algorithm. Appl Math Comput. 2009;214(1):108–32. [Google Scholar]
- 87.Karaboga D, Akay B. A survey: algorithms simulating bee swarm intelligence. Artif Int Rev. 2009;31(1–4):61–85. doi: 10.1007/s10462-009-9127-4. [DOI] [Google Scholar]
- 88.Bitam S, Batouche M, Talbi E. -g. Parallel & Distributed Processing, Workshops and Phd Forum (IPDPSW), 2010 IEEE International Symposium On. Atlanta, GA: IEEE; 2010. A survey on bee colony algorithms. [Google Scholar]
- 89.Karaboga D, Gorkemli B, Ozturk C, Karaboga N. A comprehensive survey: artificial bee colony (ABC) algorithm and applications. Artif Int Rev. 2014;42(1):21–57. doi: 10.1007/s10462-012-9328-0. [DOI] [Google Scholar]
- 90.Karaboga D, Basturk B. A powerful and efficient algorithm for numerical function optimization: artificial bee colony (ABC) algorithm. J Global Optimization. 2007;39(3):459–71. doi: 10.1007/s10898-007-9149-x. [DOI] [Google Scholar]
- 91.Kang F, Li J, Ma Z. Rosenbrock artificial bee colony algorithm for accurate global optimization of numerical functions. Inform Sci. 2011;181(16):3508–31. doi: 10.1016/j.ins.2011.04.024. [DOI] [Google Scholar]
- 92.Davidović T, Ramljak D, Šelmić M, Teodorović D. Bee colony optimization for the p-center problem. Comput Oper Res. 2011;38(10):1367–76. doi: 10.1016/j.cor.2010.12.002. [DOI] [Google Scholar]
- 93.Pan QK, Tasgetiren MF, Suganthan PN, Chua TJ. A discrete artificial bee colony algorithm for the lot-streaming flow shop scheduling problem. Inform Sci. 2011;181(12):2455–68. doi: 10.1016/j.ins.2009.12.025. [DOI] [Google Scholar]
- 94.Pan QK, Wang L, Mao K, Zhao JH, Zhang M. An effective artificial bee colony algorithm for a real-world hybrid flowshop problem in steelmaking process. Automation Sci Eng IEEE Trans. 2013;10(2):307–22. doi: 10.1109/TASE.2012.2204874. [DOI] [Google Scholar]
- 95.Rodriguez FJ, García-Martínez C, Blum C, Lozano M. Parallel Problem Solving from Nature - PPSN XII: 12th International Conference, Taormina, Italy, September 1-5, 2012, Proceedings, Part II. Berlin, Heidelberg: Springer Berlin Heidelberg; 2012. An artificial bee colony algorithm for the unrelated parallel machines scheduling problem. [Google Scholar]
- 96.Ji J, Wei H, Liu C, Yin B. Artificial bee colony algorithm merged with pheromone communication mechanism for the 0–1 multidimensional knapsack problem. Math Problems Eng. 2013;2013:13. [Google Scholar]
- 97.Pandey S, Kumar S. Enhanced Artificial Bee Colony Algorithm and It’s Application to Travelling Salesman Problem. HCTL Open International Journal of Technology Innovations and Research. 2013; 2.
- 98.Sundar S, Singh A. A swarm intelligence approach to the quadratic minimum spanning tree problem. Inform Sci. 2010;180(17):3182–91. doi: 10.1016/j.ins.2010.05.001. [DOI] [Google Scholar]
- 99.Omkar S, Senthilnath J, Khandelwal R, Naik GN, Gopalakrishnan S. Artificial Bee Colony (ABC) for multi-objective design optimization of composite structures. Appl Soft Comput. 2011;11(1):489–99. doi: 10.1016/j.asoc.2009.12.008. [DOI] [Google Scholar]
- 100.Akbari R, Hedayatzadeh R, Ziarati K, Hassanizadeh B. A multi-objective artificial bee colony algorithm. Swarm Evol Comput. 2012;2:39–52. doi: 10.1016/j.swevo.2011.08.001. [DOI] [Google Scholar]
- 101.Baykasoglu A, Ozbakir L, Tapkan P. Artificial bee colony algorithm and its application to generalized assignment problem. Swarm Intelligence: Focus on Ant and particle swarm optimization. 2007:113–144. Itech Education and Publishing, Vienna, Austria.
- 102.Karaboga D, Basturk B. Foundations of Fuzzy Logic and Soft Computing: 12th International Fuzzy Systems Association World Congress, IFSA 2007, Cancun, Mexico, June 18-21, 2007. Proceedings. Berlin, Heidelberg: Springer Berlin Heidelberg; 2007. Artificial bee colony (ABC) optimization algorithm for solving constrained optimization problems. [Google Scholar]
- 103.Kumbhar PY, Krishnan S. Use of Artificial Bee Colony (ABC) algorithm in artificial neural network synthesis. Int J Adv Eng Sci Technol. 2011;11(1):162–71. [Google Scholar]
- 104.Yan X, Zhu Y, Zou W, Wang L. A new approach for data clustering using hybrid artificial bee colony algorithm. Neurocomputing. 2012;97:241–50. doi: 10.1016/j.neucom.2012.04.025. [DOI] [Google Scholar]
- 105.Xu C, Duan H. Artificial bee colony (ABC) optimized edge potential function (EPF) approach to target recognition for low-altitude aircraft. Pattern Recognit Lett. 2010;31(13):1759–72. doi: 10.1016/j.patrec.2009.11.018. [DOI] [Google Scholar]
- 106.Zhang Y, Wu L, Wang S. Magnetic resonance brain image classification by an improved artificial bee colony algorithm. Progress Electromagnet Res. 2011;116:65–79. doi: 10.2528/PIER11031709. [DOI] [Google Scholar]
- 107.Mukherjee P, Satish L. Construction of equivalent circuit of a single and isolated transformer winding from FRA data using the ABC algorithm. Power Deliv IEEE Trans. 2012;27(2):963–70. doi: 10.1109/TPWRD.2011.2176966. [DOI] [Google Scholar]
- 108.Yu X, Zhang J, Fan J, Zhang T. A Faster Convergence Artificial Bee Colony Algorithm in Sensor Deployment for Wireless Sensor Networks. Int J Distrib Sensor Netw. 2013;2013:9. [Google Scholar]
- 109.Szeto W, Wu Y, Ho SC. An artificial bee colony algorithm for the capacitated vehicle routing problem. Eur J Oper Res. 2011;215(1):126–35. doi: 10.1016/j.ejor.2011.06.006. [DOI] [Google Scholar]
- 110.Todorovic N, Petrovic S. Bee colony optimization algorithm for nurse rostering. Syst Man Cybernet Syst IEEE Trans. 2013;43(2):467–73. doi: 10.1109/TSMCA.2012.2210404. [DOI] [Google Scholar]
- 111.Zhang Z. Efficient Computer Intrusion Detection Method based on Artificial Bee Colony Optimized Kernel Extreme Learning Machine. TELKOMNIKA Indonesian J Electrical Eng. 2014;12(3):1954–9. [Google Scholar]
- 112.Xu G, Ding Y, Zhao J, Hu L, Fu X. A Novel Artificial Bee Colony Approach of Live Virtual Machine Migration Policy Using Bayes Theorem. Sci World J. 2013;2013:13. doi: 10.1155/2013/369209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Karaboga D, Basturk B. On the performance of artificial bee colony (ABC) algorithm. Appl Soft Comput. 2008;8(1):687–97. doi: 10.1016/j.asoc.2007.05.007. [DOI] [Google Scholar]
- 114.Kashan MH, Nahavandi N, Kashan AH. DisABC: A new artificial bee colony algorithm for binary optimization. Appl Soft Comput. 2012;12(1):342–52. doi: 10.1016/j.asoc.2011.08.038. [DOI] [Google Scholar]
- 115.Zhu G, Kwong S. Gbest-guided artificial bee colony algorithm for numerical function optimization. Appl Math Comput. 2010;217(7):3166–73. [Google Scholar]
- 116.Li J-Q, Pan Q-K, Xie S-X, Wang S. A hybrid artificial bee colony algorithm for flexible job shop scheduling problems. Int J Comput Commun Control. 2011;6(2):286–96. doi: 10.15837/ijccc.2011.2.2177. [DOI] [Google Scholar]
- 117.Wu B, Qian C, Ni W, Fan S. Hybrid harmony search and artificial bee colony algorithm for global optimization problems. Comput Math Appl. 2012;64(8):2621–34. doi: 10.1016/j.camwa.2012.06.026. [DOI] [Google Scholar]
- 118.Kang F, Li J, Ma Z, Li H. Artificial bee colony algorithm with local search for numerical optimization. J Softw. 2011;6(3):490–7. doi: 10.4304/jsw.6.3.490-497. [DOI] [Google Scholar]
- 119.Hooke R, Jeeves TA. “Direct Search” Solution of Numerical and Statistical Problems. J ACM (JACM) 1961;8(2):212–29. doi: 10.1145/321062.321069. [DOI] [Google Scholar]
- 120.Sharma H, Bansal JC, Arya K. Opposition based lévy flight artificial bee colony. Memetic Comput. 2013;5(3):213–27. doi: 10.1007/s12293-012-0104-0. [DOI] [Google Scholar]
- 121.Yan G, Li C. An effective refinement artificial bee colony optimization algorithm based on chaotic search and application for pid control tuning. J Comput Inf Syst. 2011;7(9):3309–16. [Google Scholar]
- 122.Zhang Y, Wu L. Face pose estimation by chaotic artificial bee colony. Int J Digital Content Technol Appl. 2011;5(2):55–63. doi: 10.4156/jdcta.vol5.issue2.7. [DOI] [Google Scholar]
- 123.Liu H, Gao L, Kong X, Zheng S. Control and Decision Conference (CCDC), 2013 25th Chinese. Guiyang, China: IEEE; 2013. An improved artificial bee colony algorithm. [Google Scholar]
- 124.Gao W-F, Liu S-Y. A modified artificial bee colony algorithm. Comput Oper Res. 2012;39(3):687–97. doi: 10.1016/j.cor.2011.06.007. [DOI] [Google Scholar]
- 125.TSai PW, Pan JS, Liao BY, Chu SC. Enhanced artificial bee colony optimization. Int J Innov Comput Inform Control. 2009;5(12):5081–092. [Google Scholar]
- 126.Banharnsakun A, Achalakul T, Sirinaovakul B. The best-so-far selection in artificial bee colony algorithm. Appl Soft Comput. 2011;11(2):2888–901. doi: 10.1016/j.asoc.2010.11.025. [DOI] [Google Scholar]
- 127.Jadon SS, Bansal JC, Tiwari R, Sharma H. Proceedings of the Third International Conference on Soft Computing for Problem Solving: SocProS 2013, Volume 2. New Delhi: Springer India; 2014. Expedited Artificial Bee Colony Algorithm. [Google Scholar]
- 128.Jadon SS, Bansal JC, Tiwari R, Sharma H. Artificial bee colony algorithm with global and local neighborhoods. Int J Syst Assur Eng Manag. 2014:1–13.
- 129.Bansal JC, Sharma H, Arya KV, Deep K, Pant M. Self-adaptive artificial bee colony. Optimization. 2014;63(10):1513–1532. doi: 10.1080/02331934.2014.917302. [DOI] [Google Scholar]
- 130.Sharma TK, Pant M. Improvised Scout Bee Movements in Artificial Bee Colony. International Journal of Modern Education and Computer Science. 2014;6(1):1. doi: 10.5815/ijmecs.2014.01.01. [DOI] [Google Scholar]
- 131.Xu Y, Fan P, Yuan L. A simple and efficient artificial bee colony algorithm. Math Probl Eng. 2013;2013:9. [Google Scholar]
- 132.Bansal JC, Sharma H, Arya K, Nagar A. Memetic search in artificial bee colony algorithm. Soft Comput. 2013;17(10):1911–28. doi: 10.1007/s00500-013-1032-8. [DOI] [Google Scholar]
- 133.Kiefer J. Sequential minimax search for a maximum. Proc Am Math Soc. 1953;4(3):502–6. doi: 10.1090/S0002-9939-1953-0055639-3. [DOI] [Google Scholar]
- 134.Kumar S, Sharma VK, Kumari R. An Improved Memetic Search in Artificial Bee Colony Algorithm. Int J Comput Sci Inform Technol (0975–9646) 2014;5(2):1237–47. [Google Scholar]
- 135.Kumar S, Sharma VK, Kumari R. Improved Onlooker Bee phase in artificial bee colony algorithm. International Journal of Computer Applications. 2014;90(6):31–39. doi: 10.5120/18054-8967. [DOI] [Google Scholar]
- 136.Jacobs LW, Brusco MJ. Note: A local-search heuristic for large set-covering problems. Naval Res Logistics (NRL) 1995;42(7):1129–40. doi: 10.1002/1520-6750(199510)42:7<1129::AID-NAV3220420711>3.0.CO;2-M. [DOI] [Google Scholar]
- 137.Ruiz R, Stützle T. A simple and effective iterated greedy algorithm for the permutation flowshop scheduling problem. Eur J Oper Res. 2007;177(3):2033–49. doi: 10.1016/j.ejor.2005.12.009. [DOI] [Google Scholar]
- 138.Verma BK, Kumar D. A review on Artificial Bee Colony algorithm. Int J Eng Technol. 2013;2(3):175–86. doi: 10.14419/ijet.v2i3.1030. [DOI] [Google Scholar]
- 139.Shah H, Ghazali R, Nawi NM. Emerging Trends and Applications in Information Communication Technologies: Second International Multi Topic Conference, IMTIC 2012, Jamshoro, Pakistan, March 28-30, 2012. Proceedings. Berlin, Heidelberg: Springer Berlin Heidelberg; 2012. Hybrid ant bee colony algorithm for volcano temperature prediction. [Google Scholar]
- 140.Kefayat M, Ara AL, Niaki SN. A hybrid of ant colony optimization and artificial bee colony algorithm for probabilistic optimal placement and sizing of distributed energy resources. Energy Conversion Manag. 2015;92:149–61. doi: 10.1016/j.enconman.2014.12.037. [DOI] [Google Scholar]
- 141.Tahir MA, Bouridane A, Kurugollu F. Simultaneous feature selection and feature weighting using Hybrid Tabu Search K-nearest neighbor classifier. Pattern Recognit Lett. 2007;28(4):438–46. doi: 10.1016/j.patrec.2006.08.016. [DOI] [Google Scholar]
- 142.Hsu CW, Chang CC, Lin CJ, et al.A practical guide to support vector classification. 2003. Department of Computer Science, National Taiwan University. http://www.csie.ntu.edu.tw/%7Ecjlin/papers/guide/guide.pdf.
- 143.Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952;47(260):583–621. doi: 10.1080/01621459.1952.10483441. [DOI] [Google Scholar]
- 144.Corder GW, Foreman DI. Nonparametric Statistics for Non-statisticians: a Step-by-step Approach: John Wiley & Sons; 2009.
- 145.Siegel S. Nonparametric statistics for the behavioral sciences. New York, NY, US: McGraw-hill; 1956. [Google Scholar]
- 146.Norton BJ, Strube MJ. Guide for the Interpretation of One-way Analysis of Variance. Phys Therapy. 1985;65(12):1888–96. doi: 10.1093/ptj/65.12.1888. [DOI] [PubMed] [Google Scholar]
- 147.Bishop CM. Neural networks for pattern recognition. USA: Oxford university press; 1995. [Google Scholar]
- 148.Dudoit S, Fridlyand J, Speed TP. Comparison of discrimination methods for the classification of tumors using gene expression data. J Am Stat Assoc. 2002;97(457):77–87. doi: 10.1198/016214502753479248. [DOI] [Google Scholar]
- 149.Wilcoxon F. Individual comparisons by ranking methods. Biometrics bulletin. 1945;1(6):80–83. doi: 10.2307/3001968. [DOI] [Google Scholar]
- 150.Lehmann EL, D’Abrera HJ. Nonparametrics: Statistical Methods Based on Ranks. San Francisco: Springer; 2006. [Google Scholar]
- 151.Hollander M, Wolfe DA, Chicken E. Nonparametric Statistical Methods. United States: John Wiley & Sons; 2013. [Google Scholar]
- 152.Gibbons JD, Chakraborti S. International Encyclopedia of Statistical Science. Berlin, Heidelberg: Springer Berlin Heidelberg; 2011. Nonparametric Statistical Inference. [Google Scholar]
- 153.Deng L, Pei J, Ma J, Lee DL. A rank sum test method for informative gene discovery. In: Proceedings of the Tenth ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. ACM: 2004. p. 410–9.
- 154.Deng L, Ma J, Pei J. Rank sum method for related gene selection and its application to tumor diagnosis. Chin Sci Bull. 2004;49(15):1652–7. doi: 10.1007/BF03184138. [DOI] [Google Scholar]
- 155.Emmert-Streib F, Dehmer M. Medical Biostatistics for Complex Diseases. United States: John Wiley & Sons; 2010. [Google Scholar]
- 156.Wang SL, Li XL, Fang J. Finding minimum gene subsets with heuristic breadth-first search algorithm for robust tumor classification. BMC Bioinformatics. 2012;13(1):178. doi: 10.1186/1471-2105-13-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang S, Li X, Zhang S. Advanced Intelligent Computing Theories and Applications. With Aspects of Theoretical and Methodological Issues: 4th International Conference on Intelligent Computing, ICIC 2008 Shanghai, China, September 15-18, 2008 Proceedings. Berlin, Heidelberg: Springer Berlin Heidelberg; 2008. Neighborhood rough set model based gene selection for multi-subtype tumor classification. [Google Scholar]
- 158.Duncan WE. Gene Set Based Ensemble Methods for Cancer Classification. PhD thesis. The University of Tennessee Knoxville; 2013.
- 159.Lan L, Vucetic S. Improving accuracy of microarray classification by a simple multi-task feature selection filter. Int J Data Mining Bioinformatics. 2011;5(2):189–208. doi: 10.1504/IJDMB.2011.039177. [DOI] [PubMed] [Google Scholar]
- 160.Chen D, Liu Z, Ma X, Hua D. Selecting genes by test statistics. BioMed Res Int. 2005;2005(2):132–8. doi: 10.1155/JBB.2005.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Leung Y, Hung Y. A multiple-filter-multiple-wrapper approach to gene selection and microarray data classification. IEEE/ACM Trans Comput Biol Bioinform (TCBB) 2010;7(1):108–17. doi: 10.1109/TCBB.2008.46. [DOI] [PubMed] [Google Scholar]
- 162.Au WH, Chan KC, Wong AK, Wang Y. Attribute clustering for grouping, selection, and classification of gene expression data. IEEE/ACM Trans Comput Biol Bioinform (TCBB) 2005;2(2):83–101. doi: 10.1109/TCBB.2005.17. [DOI] [PubMed] [Google Scholar]
- 163.Guo S, Zhong S, Zhang A. Bioinformatics and Bioengineering (BIBE), 2014 IEEE International Conference on. Boca Raton, FL: IEEE; 2014. Privacy Preserving Calculation of Fisher Criterion Score for Informative Gene Selection. [Google Scholar]
- 164.Cai R, Hao Z, Yang X, Huang H. A new hybrid method for gene selection. Pattern Anal Appl. 2011;14(1):1–8. doi: 10.1007/s10044-010-0180-z. [DOI] [Google Scholar]
- 165.Salem DA, Seoud R, Ali HA. DMCA: A Combined Data Mining Technique for Improving the Microarray Data Classification Accuracy. In: 2011 International Conference on Environment and Bioscience: 2011. p. 36–41.
- 166.Box GEP. Non-normality and tests on variances. Biometrika. 1953;40(3/4):318–335. doi: 10.2307/2333350. [DOI] [Google Scholar]
- 167.Markowski CA, Markowski EP. Conditions for the effectiveness of a preliminary test of variance. Am Stat. 1990;44(4):322–6. [Google Scholar]
- 168.Zhou X, Mao K. LS bound based gene selection for DNA microarray data. Bioinformatics. 2005;21(8):1559–64. doi: 10.1093/bioinformatics/bti216. [DOI] [PubMed] [Google Scholar]
- 169.Deneubourg JL, Aron S, Goss S, Pasteels JM. The self-organizing exploratory pattern of the argentine ant. J Insect Behav. 1990;3(2):159–68. doi: 10.1007/BF01417909. [DOI] [Google Scholar]
- 170.Dorigo M, Bonabeau E, Theraulaz G. Ant algorithms and stigmergy. Future Generation Comput Syst. 2000;16(8):851–71. doi: 10.1016/S0167-739X(00)00042-X. [DOI] [Google Scholar]
- 171.Kennedy J. Encyclopedia of Machine Learning. Boston, MA: Springer US; 2010. Particle swarm optimization. [Google Scholar]
- 172.Hamidi J. Control System Design Using Particle Swarm Optimization (PSO) Int J Soft Comput Eng. 2012;1(6):116–9. [Google Scholar]
- 173.Luke S, Vol. 113. Essentials of Metaheuristics: Lulu Com; 2013.
- 174.Baker JE. Reducing bias and inefficiency in the selection algorithm. In: Proceedings of the Second International Conference on Genetic Algorithms: 1987. p. 14–21.
- 175.Wang J, Du H, Liu H, Yao X, Hu Z, Fan B. Prediction of surface tension for common compounds based on novel methods using heuristic method and support vector machine. Talanta. 2007;73(1):147–56. doi: 10.1016/j.talanta.2007.03.037. [DOI] [PubMed] [Google Scholar]
- 176.Lee JW, Lee JB, Park M, Song SH. An extensive comparison of recent classification tools applied to microarray data. Comput Stat Data Anal. 2005;48(4):869–85. doi: 10.1016/j.csda.2004.03.017. [DOI] [Google Scholar]
- 177.Joachims T. Svmlight: Support vector machine. SVM-Light Support Vector Machine, University of Dortmund. 1999; 19(4). http://svmlight.joachims.org/, 2015-02-01.
- 178.Chang CC, Lin CJ. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol (TIST) 2011;2(3):27. [Google Scholar]
- 179.Rüping S. mysvm–a support vector machine: University of Dortmund, Computer Science; 2004. http://www-ai.cs.uni-dortmund.de/SOFTWARE/MYSVM/index.html. 2015-02-01.
- 180.Hsu CW, Lin CJ. Bsvm. 2006. https://www.csie.ntu.edu.tw/%7Ecjlin/bsvm/.
- 181.Hsu CW, Lin CJ. BSVM-2.06. 2009. https://www.csie.ntu.edu.tw/%7Ecjlin/bsvm/.
- 182.Vapnik VN, Vapnik V. Statistical Learning Theory. New York: Wiley; 1998. [Google Scholar]
- 183.Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Mach Learn. 2002;46(1–3):389–422. doi: 10.1023/A:1012487302797. [DOI] [Google Scholar]
- 184.Pirooznia M, Deng Y. Svm classifier–a comprehensive java interface for support vector machine classification of microarray data. BMC Bioinformatics. 2006;7(Suppl 4):25. doi: 10.1186/1471-2105-7-S4-S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang JG, Deng HW. Gene selection for classification of microarray data based on the Bayes error. BMC Bioinformatics. 2007;8(1):370. doi: 10.1186/1471-2105-8-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jäger J, Sengupta R, Ruzzo WL. Improved gene selection for classification of microarrays. In: Proceedings of the Eighth Pacific Symposium on Biocomputing: 3–7 January 2003; Lihue, Hawaii: 2002. p. 53–64. [DOI] [PubMed]
- 187.Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang CH, Angelo M, Ladd C, Reich M, Latulippe E, Mesirov JP, et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Nat Acad Sci. 2001;98(26):15149–54. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Saravanan V, Rangasamy M. An Efficient statistical model based classification algorithm for classifying cancer gene expression data with minimal gene subsets. Int J Cyber Soc Educ. 2009;2(2):51–66. [Google Scholar]
- 189.Wang L, Chu F, Xie W. Accurate cancer classification using expressions of very few genes. IEEE/ACM Trans Comput Biol Bioinform (TCBB) 2007;4(1):40–53. doi: 10.1109/TCBB.2007.1006. [DOI] [PubMed] [Google Scholar]
- 190.Furey TS, Cristianini N, Duffy N, Bednarski DW, Schummer M, Haussler D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16(10):906–14. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- 191.Garcia-Nieto J, Alba E, Apolloni J. Hybrid DE-SVM approach for feature selection: application to gene expression datasets. In: Logistics and Industrial Informatics, 2009. LINDI 2009. 2nd International. IEEE: 2009. p. 1–6.
- 192.Statnikov A, Aliferis CF, Tsamardinos I, Hardin D, Levy S. A comprehensive evaluation of multicategory classification methods for microarray gene expression cancer diagnosis. Bioinformatics. 2005;21(5):631–43. doi: 10.1093/bioinformatics/bti033. [DOI] [PubMed] [Google Scholar]
- 193.Tu CJ, Chuang LY, Chang JY, Yang CH, et al. Feature selection using PSO-SVM. IAENG Int J Comput Sci. 2007;33(1):111–6. [Google Scholar]
- 194.Omar N, bin Othman MS, et al.Particle Swarm Optimization Feature Selection for Classification of Survival Analysis in Cancer. Int J Innov Comput. 2013; 2(1).
- 195.Mallika R, Saravanan V. An svm based classification method for cancer data using minimum microarray gene expressions. World Acad Sci Eng Technol. 2010;4:485–9. [Google Scholar]
- 196.Su AI, Welsh JB, Sapinoso LM, Kern SG, Dimitrov P, Lapp H, Schultz PG, Powell SM, Moskaluk CA, Frierson HF, et al. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 2001;61(20):7388–93. [PubMed] [Google Scholar]
- 197.Xu J, Sun L, Gao Y, Xu T. An ensemble feature selection technique for cancer recognition. Bio-med Mater Eng. 2014;24(1):1001–8. doi: 10.3233/BME-130897. [DOI] [PubMed] [Google Scholar]
- 198.Yang S, Naiman DQ. Multiclass cancer classification based on gene expression comparison. Stat Appl Genet Mol Biol. 2014;13(4):477–96. doi: 10.1515/sagmb-2013-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Statnikov A, Aliferis CF, Tsamardinos I. Methods for multi-category cancer diagnosis from gene expression data: a comprehensive evaluation to inform decision support system development. Medinfo. 2004;11(Pt 2):813–7. [PubMed] [Google Scholar]
- 200.Wang Y, Tetko IV, Hall MA, Frank E, Facius A, Mayer KF, Mewes HW. Gene selection from microarray data for cancer classification-a machine learning approach. Comput Biol Chem. 2005;29(1):37–46. doi: 10.1016/j.compbiolchem.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 201.Breiman L, Spector P. Submodel selection and evaluation in regression. the X-random case. International statistical review/revue internationale de Statistique. 1992;60(3):291–319. [Google Scholar]
- 202.Knudsen S. A Biologist’s Guide to Analysis of DNA Microarray Data. United States: John Wiley & Sons; 2011. [Google Scholar]
- 203.Dougherty ER. Small sample issues for microarray-based classification. Comp Funct Genomics. 2001;2(1):28–34. doi: 10.1002/cfg.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wang Y, Makedon FS, Ford JC, Pearlman J. Hykgene: a hybrid approach for selecting marker genes for phenotype classification using microarray gene expression data. Bioinformatics. 2005;21(8):1530–7. doi: 10.1093/bioinformatics/bti192. [DOI] [PubMed] [Google Scholar]
- 205.Cho JH, Lee D, Park JH, Lee IB. New gene selection method for classification of cancer subtypes considering within-class variation. FEBS Lett. 2003;551(1):3–7. doi: 10.1016/S0014-5793(03)00819-6. [DOI] [PubMed] [Google Scholar]
- 206.Li T, Zhang C, Ogihara M. A comparative study of feature selection and multiclass classification methods for tissue classification based on gene expression. Bioinformatics. 2004;20(15):2429–37. doi: 10.1093/bioinformatics/bth267. [DOI] [PubMed] [Google Scholar]
- 207.Balmain A, Gray J, Ponder B. The genetics and genomics of cancer. Nat Genet. 2003;33:238–44. doi: 10.1038/ng1107. [DOI] [PubMed] [Google Scholar]
- 208.Mohamad M, Omatu S, Yoshioka M, Deris S, et al. A cyclic hybrid method to select a smaller subset of informative genes for cancer classification. Int J Innov Comput Inform Control. 2009;5(8):2189–202. [Google Scholar]
- 209.Staunton JE, Slonim DK, Coller HA, Tamayo P, Angelo MJ, Park J, Scherf U, Lee JK, Reinhold WO, Weinstein JN, et al. Chemosensitivity prediction by transcriptional profiling. Proc Nat Acad Sci. 2001;98(19):10787–92. doi: 10.1073/pnas.191368598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415(6870):436–42. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 211.Nutt CL, Mani D, Betensky RA, Tamayo P, Cairncross JG, Ladd C, Pohl U, Hartmann C, McLaughlin ME, Batchelor TT, et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003;63(7):1602–7. [PubMed] [Google Scholar]
- 212.Shipp MA, Ross KN, Tamayo P, Weng AP, Kutok JL, Aguiar RC, Gaasenbeek M, Angelo M, Reich M, Pinkus GS, et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8(1):68–74. doi: 10.1038/nm0102-68. [DOI] [PubMed] [Google Scholar]
- 213.Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30(1):41–7. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 214.Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Nat Acad Sci. 2001;98(24):13790–5. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D’Amico AV, Richie JP, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1(2):203–9. doi: 10.1016/S1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 216.Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu CR, Peterson C, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7(6):673–9. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]